

Abstract
Neutralizing antibodies (nAbs) that target the SARS-CoV-2 spike protein have received emergency use approval for treatment of COVID-19. However, with the emergence of variants of concern, there is a need for new treatment options. We report a format that enables modular assembly of bi-paratopic tetravalent nAbs with antigen-binding sites from two distinct nAbs. The tetravalent nAb purifies in high yield and exhibits biophysical characteristics that are comparable to those of clinically used therapeutic antibodies. The tetravalent nAb binds to the spike protein trimer at least 100-fold more tightly than bivalent IgGs (apparent KD < 1 pM) and neutralizes a broad array of SARS-CoV-2 pseudoviruses, chimeric viruses, and authentic viral variants with high potency. Together, these results establish the tetravalent diabody-Fc-Fab as a robust, modular platform for rapid production of drug-grade nAbs with potencies and breadth of coverage that greatly exceed those of conventional bivalent IgGs.
Keywords: antibody, SARS-CoV-2, neutralization, passive immunotherapy, bispecific, bi-paratopic, virus variant, S protein, RBD, mutational escape
Graphical abstract
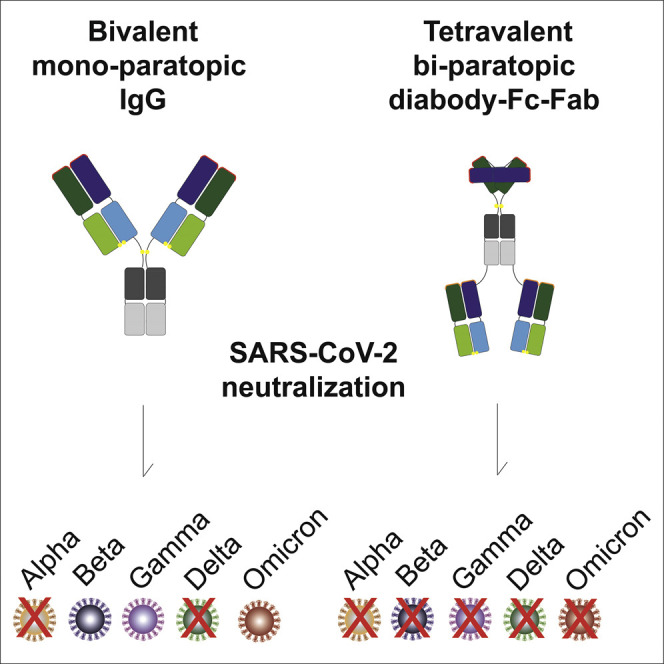
Miersch et al. use modular design principles to develop a tetravalent antibody format that can combine two antibodies into a single molecule. They show that a bi-paratopic tetravalent antibody targeting SARS-CoV-2 behaves like conventional IgGs (e.g., yield, purity, stability, solubility) but potently and broadly inhibits variants that resist IgG drugs.
Introduction
SARS-CoV-2, the causative agent of the ongoing COVID-19 pandemic, relies on its surface spike glycoprotein (S protein) to mediate interaction with and entry into host cells (Hoffmann et al., 2020). The surface of the virus particle is studded with 25–100 copies of S protein homotrimers, and each S protein consists of two subunits (Klein et al., 2020; Walls et al., 2020). The N-terminal subunit (S1) mediates host cell recognition, whereas the C-terminal subunit (S2) mediates membrane fusion and host cell entry. The S1 domain itself contains a distinct N-terminal domain (NTD) followed by a receptor-binding domain (RBD) that is responsible for host cell recognition through interactions with the human cell-surface protein angiotensin-converting enzyme 2 (ACE2).
Most neutralizing antibodies (nAbs) that arise in response to SARS-CoV-2 infection target the S1 subunit, and while the most potent of these bind to the RBD and compete with ACE2 (Brouwer et al., 2020; Garrett Rappazzo et al., 2021; Tortorici et al., 2020), other RBD-binding antibodies are also neutralizing (Pinto et al., 2020; VanBlargan et al., 2022). Cloning and recombinant production of these nAbs have led to the development of several drugs that have proven successful for inhibiting virus replication in patients and have gained emergency approval for therapy (Gottlieb et al., 2021; Razonable et al., 2021). Structural studies have shown that the most potent neutralizing immunoglobulins G (IgGs) bind to epitopes that overlap with the ACE2 epitope and take advantage of their bivalent nature to achieve high affinity by binding simultaneously to two of the three RBDs within the S protein trimer (Miersch et al., 2021; Yan et al., 2021).
Despite the significant clinical success of bivalent nAbs for treatment of COVID-19, they suffer from two major limitations. First, even nAbs that are highly potent in vitro must be administered at high doses, presumably at least in part due to low penetrance of IgGs to sites within the lungs where SARS-CoV-2 infects and replicates (Hart et al., 2001). Second, IgG drugs that were derived from nAbs that arose in response to the original strain of the virus have proven to be ineffective against many variants of concern (VoC) that have arisen since. The emergence of B.1.1.529 (Omicron) and its subvariants highlights this, as most existing nAbs—including several authorized for clinical use—have proven to be ineffective or substantially less potent against these VoC (Cao et al., 2022; VanBlargan et al., 2022). Moreover, there is some evidence that single-agent nAb therapy may promote the evolution of resistant variants (Jensen et al., 2021; Weisblum et al., 2020). Notably, most VoC that resist current therapeutic nAbs contain mutations within the RBD, which disrupt binding to nAbs but not to ACE2 (Greaney et al., 2021; McCallum et al., 2021; Planas et al., 2021; Zhou et al., 2021).
Although limitations on potency and breadth of coverage are inherent with single-agent nAb drugs, several options for alleviating these problems have been explored. Cocktails of two nAbs, ideally binding to non-overlapping epitopes, have been shown to be more potent and resistant to virus escape; nonetheless, the complexity and expense of combining two nAbs make this approach challenging (Baum et al., 2020; Hansen et al., 2020; Weinreich et al., 2021), and even cocktails lose efficacy against the highly divergent B.1.1.529 variant (VanBlargan et al., 2022). Alternatively, small modular antibody (Ab) variable domains or non-Ab scaffolds have been assembled as multimers that can simultaneously engage all three RBDs on an S protein trimer and thus enhance potency beyond bivalent binding (Cao et al., 2020a; Hunt et al., 2021; Kayabolen et al., 2021; Walser et al., 2021; Xu et al., 2021). In another approach, a bispecific, bivalent IgG—with each arm targeting a different epitope on the RBD—has been shown to neutralize VoC that escape from conventional IgGs (De Gasparo et al., 2021).
We previously explored approaches that expand the valency of conventional bivalent IgGs without introducing non-Ab scaffolds that may compromise drug development due to issues of immunogenicity, developability, and/or manufacturing. We recently isolated a human nAb (15033) against SARS-CoV-2 from a synthetic phage-displayed library, and engineered a variant nAb (15033-7, or 33-7 for brevity) with changes in the light chain that enhanced potency (Miersch et al., 2021). We then constructed tetravalent nAbs by fusing additional copies of the Fab 33-7 to the N- or C-terminus of the IgG 33-7 and showed that they exhibited enhanced potency against an original SARS-CoV-2 isolate and an evolved VoC. Importantly, the tetravalent nAbs could be produced at high yields with the same methods used for bivalent IgGs, without compromising the biophysical properties required for effective drug development.
We now report a tetravalent Ab format that expands on our earlier work and provides the advantage of enabling facile, modular assembly of tetravalent, bi-paratopic nAbs containing two distinct paratopes targeting the SARS-CoV-2 RBD. To achieve this, we exploited the modularity of Ab variable regions and converted an IgG (Figure 1A) to a functionally equivalent diabody-Fc format (D-Fc; Figure 1B), consisting of heavy and light chain (VH and VL, respectively) domains linked together in a single polypeptide, to which we could add additional paratopes to form tetravalent Abs. The facile fusion of Fab arms to the C terminus of the homodimeric D-Fc format enables the generation of tetravalent diabody-Fc-Fab (D-Fc-F) molecules (Figure 1C). Because the diabody paratope does not require a separate light chain, this format is modular, as Fabs can be added without any issues of the Fab light chain interfering with the diabody or vice versa. By adding a Fab paratope to its corresponding D-Fc or a Fab paratope from a different nAb, either mono-paratopic or bi-paratopic tetravalent D-Fc-F molecules can be assembled.
Figure 1.

Antibody formats
(A–C) Schematics are shown for (A) bivalent IgG, (B) bivalent diabody-Fc (D-Fc), and (C) bi-paratopic tetravalent diabody-Fc-Fab (D-Fc-F) formats. The paratopes in the IgG and D-Fc molecules are shown in red. In the D-Fc-F molecule, the diabody paratopes are shown in red and the Fab paratopes are shown in orange. Linkers are shown in black, and intermolecular disulfide bonds are shown as yellow spheres. The domains are labeled and colored as follows: Fc, gray (CH2, dark; CH3, light); Fab heavy chain, blue (VH, dark; CH1, light); light chain, green (VL, dark; CL, light).
We demonstrate the feasibility of this strategy, showing that a bi-paratopic D-Fc-F could be produced with yields and purities similar to those of conventional IgGs and exhibited excellent biophysical properties comparable to those of Ab drugs. Moreover, D-Fc-F neutralized a panel of pseudoviruses representing diverse SARS-CoV-2 VoC with potencies that greatly exceeded the component IgGs and proved resilient even against variants (including B.1.1.529) that resisted the IgGs and the mono-paratopic D-Fc-F. Most importantly, neutralization of both chimeric and authentic infectious VoC in independent laboratories validated these findings, confirming both high potency and breadth of coverage.
Results
Characterization of Ab 15036
To complement our previously characterized nAb 33-7, we used well-established phage display methods with a highly validated synthetic human Fab-phage library (Persson et al., 2013) to identify a second Ab that bound to the RBD (see STAR Methods). Ab 15036 (or 36 for brevity) had a different paratope in comparison with 33-7 (Figure 2A). Nonetheless, the two Abs competed with each other for binding to immobilized RBD, as assessed by ELISAs in which binding of Fab-phage was measured in the presence or absence of saturating concentration of IgG protein (Figure 2B). Whereas IgG 33-7 completely blocked phage-displayed Fab 36, IgG 36 only partially blocked phage-displayed Fab 33-7, suggesting that the epitopes for the two Fabs likely overlap but are not identical. Bilayer interferometry (BLI) assays showed that IgG 36 bound to the S protein trimer with a high affinity (apparent KD = 220 pM) that was comparable to the previously determined affinity of IgG 33-7 (apparent KD = 70 pM) (Figures 2C and Table 1 ) (Miersch et al., 2021). Taken together, these results showed that Ab 36 binds with high affinity to an epitope on the RBD that at least partially overlaps the epitope of Ab 33-7, a potent nAb that competes with ACE2.
Figure 2.

Characterization of antibody 36
(A) CDR sequences of Abs 33-7 and 36. Positions are numbered according to the IMGT nomenclature (Lefranc et al., 2003). Only positions that were diversified in the Fab-phage library are shown.
(B) Phage ELISAs for phage-displayed Fab 33-7 (black bars) or Fab 36 (white bars) binding to immobilized RBD in the presence of 250 nM blocking IgG 33-7 or 36 or an isotype control. The binding signal was normalized to the signal in the absence of blocking IgG. Error bars represent the standard deviation of signals obtained from duplicate wells and results are representative of n = 2 independent experiments.
(C) BLI sensor traces (black) for IgG 36 binding to immobilized S protein ectodomain trimer. Indicated concentrations of IgG 36 were allowed to bind for 600 s and dissociation was monitored for an additional 600 s. The curves were globally fit (red) to a 1:1 binding model, and derived binding constants are shown in Table 1.
Table 1.
Affinities and biophysical characteristics of nAbs
| nAb | Binding constantsa |
Pseudovirus inhibitionb |
SEC analysisc |
Yieldd (mg/L) | ||||
|---|---|---|---|---|---|---|---|---|
| kon (105 M−1 s−1) | koff (10−5 s−1) | Apparent KD (pM) | IC50 (ng/mL) | IC50 (pM) | SEC elution volume (mL) | Monodispersity (%) | ||
| Trastuzumab | – | – | – | – | – | 8.7 | 92 | 303 |
| IgG 33-7 | 9.1 | 6.4 | 70 | 16 | 110 | 8.7 | 95 | 165 |
| IgG 36 | 6.2 | 14 | 220 | 80 | 530 | 8.3 | 91 | 167 |
| D-Fc 33-7 | 5.2 | 8.5 | 160 | 30 | 273 | 9.3 | 71 | 88 |
| D-Fc-F 33-7:33-7 | 7.8 | <0.1 | <1 | 9.2 | 44 | 8 | 80 | 190 |
| D-Fc-F 33-7:36 | 8.0 | <0.1 | <1 | 3.6 | 17 | 7.9 | 81 | 189 |
Characterization of D-Fc 33-7
We produced IgG (Figure 1A) and D-Fc (Figure 1B) versions of Ab 33-7 by transient transfection of Expi293F cells and purified the proteins to homogeneity (Figure 3A) by protein-A-affinity chromatography in high yield (165 or 88 mg/L for the IgG or D-Fc, respectively) (Table 1). The gels showed single bands of the appropriate size for intact molecules under non-reducing conditions with intact intermolecular disulfide bonds, whereas under reducing conditions without disulfide bonds, they showed a heavy-chain band and a light-chain band for the IgG, or a single band for a D-Fc monomer. Size-exclusion chromatography (SEC) revealed that the IgG eluted as a predominantly (95%) monodispersed peak with an elution volume nearly identical to that of trastuzumab, whereas the D-Fc eluted as a predominantly (72%) monodispersed peak that was slightly delayed compared with trastuzumab, consistent with its smaller molecular weight (Figure 3B). Affinity measurements by BLI showed that the IgG and D-Fc proteins bound with similar high affinities to the trimeric S protein ectodomain (ECD) (apparent KD = 70 or 160 pM, respectively) (Figure 3C and Table 1). Importantly, in cell-based assays with a pseudovirus displaying the SARS-CoV-2 S protein, the IgG and D-Fc proteins neutralized infection with equivalent potencies (IC50 = 29 or 30 ng/mL, respectively) (Figure 3D).
Figure 3.

Characterization of D-Fc 33-7
(A) SDS-PAGE analysis of IgG and D-Fc versions of Ab 33-7 under non-reducing (middle) or reducing conditions (right).
(B) Analytical gel filtration SEC of IgG 33-7 (dashed blue), D-Fc 33-7 (solid red), and trastuzumab IgG (dotted black).
(C) BLI sensor traces for 5 nM IgG 33-7 (blue) or D-Fc 33-7 (red) binding to immobilized S-protein ECD trimer. Abs were allowed to bind for 600 s, and dissociation was monitored for an additional 600 s. Traces and curve fits (black) for the complete analysis at various concentrations are shown in Figure S1, and the derived binding constants are shown in Table 1.
(D) Neutralization of pseudovirus by IgG 33-7 (blue triangles, dashed line) or D-Fc 33-7 (red circles, solid line). Error bars represent the standard deviation of triplicate measurements and results shown are representative of two independent experiments. Pseudovirus was generated with S protein from the original strain isolated in Wuhan.
Characterization of D-Fc-F proteins
We constructed D-Fc-F proteins (Figure 1C) in which the Fab of either the 33-7 or 36 nAb was fused to the C-terminus of D-Fc 33-7. The resulting tetravalent molecules contained either four copies of the 33-7 paratope (mono-paratopic, named D-Fc-F 33-7:33-7) or two copies of the 33-7 paratope in the diabody head and two copies of the 36 paratope in the Fab arms (bi-paratopic, named D-Fc-F 33-7:36). D-Fc-F 33-7:33-7 and 33-7:36 were purified in similarly high yields (190 or 189 mg/L, respectively) (Figure 4A and Table 1) with the same methods used for purification of IgG and D-Fc proteins. In gel filtration SEC, both proteins exhibited a predominant monodisperse peak (80% and 81% for D-Fc-F 33-7:33-7 or 33-7:36, respectively) that eluted earlier than the IgG trastuzumab, consistent with the larger size of the D-Fc-F protein (Figures 4B and Table 1). BLI analysis with trimeric S protein ECD showed that D-Fc-F proteins bound tightly and exhibited slow off-rates that were beyond the dynamic range of the instrument. Consequently, apparent dissociation constants could not be measured accurately, but were in the subpicomolar range (apparent KD < 1 pM) (Figures 4C and Table 1).
Figure 4.

Characterization of D-Fc-F proteins
(A) SDS-PAGE analysis of D-Fc-F 33-7:33-7 and D-Fc-F 33-7:36 under non-reducing (middle) or reducing conditions (right).
(B) Analytical gel filtration SEC of D-Fc-F 33-7:33-7 (dashed blue), D-Fc-F 33-7:36 (solid red), and trastuzumab IgG (dotted black).
(C) BLI sensor traces for 5 nM D-Fc-F 33-7:33-7 (blue) and D-Fc-F 33-7:36 (red) binding to immobilized S-protein ECD trimer. Abs were allowed to bind for 600 s, and dissociation was monitored for an additional 600 s. Traces and curve fits (black) for the complete analysis at various concentrations are shown in Figure S1, and the derived binding constants are shown in Table 1.
Inhibition of virus infection in cell-based assays
To compare the efficacy of the D-Fc-F proteins and their component IgG proteins for neutralization of SARS-CoV-2 VoC, we first used mammalian cell infection assays with pseudoviruses consisting of HIV-gag-based, lentivirus-like particles pseudotyped with various SARS-CoV-2 S proteins (Connor et al., 1995). We used a panel of seven pseudoviruses based on the S protein of an early “wild-type” (WT) SARS-CoV-2 isolated in Washington State (strain 2019 n-CoV/USA_WA1/2020) modified to incorporate RBD mutations corresponding to six VoC, including strains B.1.1.7, MB61 (Fiorentini et al., 2021), B.1.351, B.1.1.28 (P.1), B.1.617.1, and B.1.427/429) (see Table S1 for sequences). The two IgGs (33-7 and 36) and two D-Fc-Fs (33-7:33-7 and 33-7:33-6) were assayed against the pseudovirus panel (Figure 5 ), and IC50 values were calculated (Table 2 ).
Figure 5.

Neutralization of SARS-CoV-2 pseudoviruses
Neutralization assays were conducted with HIV-gag-based, lentivirus-like particles pseudotyped with the following SARS-CoV-2 S-proteins: (A) original strain from Wuhan, (B) B.1.1.7, (C) B.1.617.1, (D) B.1.427/429, (E) MB61, (F) B.1.351, and (G) B.1.1.28. The following nAbs were tested: IgG 33-7 (cyan triangles, dashed lines), IgG 36 (black diamonds, dashed lines), D-Fc-F 33-7:33-7 (orange circles, solid lines), and D-Fc-F 33-7:36 (red squares, solid lines). The pseudovirus was pretreated with serial dilutions of nAb, and infection of HEK293T cells stably expressing human ACE2 was measured relative to untreated control. Samples were run in triplicate, and results are representative of two independent experiments. Error bars indicate standard error of the mean. IC50 values were calculated from fit curves and are shown in Table 2.
Table 2.
IC50 values for pseudovirus neutralization by nAbs
| Pseudovirusa |
nAb IC50 (ng/mL)b |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Origin | Pango ID | Greek ID | IgG 33-7 |
IgG 36 |
D-Fc-F 33-7:33-7 |
D-Fc-F 33-7:36 |
||||
| ng/mL | pM | ng/mL | pM | ng/mL | pM | ng/mL | pM | |||
| China | – | – | 16 | 110 | 80 | 530 | 9.2 | 44 | 3.6 | 17 |
| Italy | MB61 | – | 160 | 1,100 | 28 | 190 | 46 | 220 | 5.3 | 25 |
| UK | B.1.1.7 | Alpha | 20 | 130 | 150 | 1,000 | 9.2 | 44 | 1.6 | 8 |
| S. Africa | B.1.351 | Beta | 680 | 4,500 | 1,400 | 9,300 | 2,100 | 10,000 | 5.6 | 27 |
| Brazil | B.1.1.28 | Gamma | 1,600 | 11,000 | 680 | 4,500 | 550 | 2,600 | 3.5 | 17 |
| USA | B.1.427/429 | Epsilon | 20 | 130 | 57 | 380 | 9.1 | 43 | 3.6 | 17 |
| India | B.1.617.1 | Kappa | 32 | 210 | 34 | 230 | 10 | 48 | 0.6 | 3 |
| Botswana | B.1.1.529 | Omicron | NNc | NN | NTd | NT | NT | NT | 26 | 124 |
Pseudovirus variants were generated by introducing RBD mutations corresponding to known variants of interest and variants of concern (Figure S2) into the plasmid encoding S protein (original Wuhan strain) and used for pseudotyping.
Values obtained by luminescent pseudovirus infection assay following exposure of ACE2-HEK293T cells to pseudovirus preincubated with serial dilutions of antibody.
NN, no neutralization observed at concentrations up to 250 nM.
NT, neutralization was not tested.
IgG 33-7 was highly effective against WT (Figure 5A, IC50 = 16 ng/mL), B.1.1.7 (Figure 5B, IC50 = 20 ng/mL), B.1.617.1 (Figure 5C, IC50 = 32 ng/mL), and B.1.427/429 (Figure 5D, IC50 = 20 ng/mL); moderately effective against MB61 (Figure 5E, IC50 = 160 ng/mL); and less effective against B.1.351 (Figure 5F, IC50 = 680 ng/mL) and B.1.1.28 (Figure 5G, IC50 = 1,600 ng/mL). IgG 36 exhibited a similar pattern of activity, but, in general, was less potent than IgG 33-7. The mono-paratopic, tetravalent D-Fc-F 33-7:33-7 was more potent against the five variants that were neutralized by the IgGs—WT, B.1.1.7, B.1.617.1, B.1.427/429, and MB61 (Figures 5A–5E, IC50 = 9–46 ng/mL)—but it also was ineffective against the two variants that resisted the IgGs—B.1.351 and B.1.1.28 (Figures 5F and 5G, IC50 = 2,100 or 550 ng/mL, respectively). In contrast, the bi-paratopic, tetravalent D-Fc-F 33-7:36 potently neutralized the entire pseudovirus panel (IC50 < 10 ng/mL) with only a modest decrease against B.1.1.529 and was the most potent agent in every case (Table 2). Indeed, D-Fc-F 33-7:36 inhibited infection of the variants that resisted the IgGs and D-Fc-F 33-7:33-7, showing >100-fold enhancement in some cases. Indeed, the IC50 values for D-Fc-F 33-7:36 against five of the seven pseudoviruses were virtually identical, suggesting that we may have reached the limits of the dynamic range for this assay, and some potencies may be underestimated.
Finally, we assessed the neutralization potency of D-Fc-F 33-7:36 in cell-based assays with authentic infectious virus in two independent laboratories. At Washington University, we used a panel of seven viruses with S protein variants representing diverse VoC, including authentic isolates of USA_WA1/2020, B.1.1.7, B.1.351, B.1.617.2, AY.1, and B.1.1.529 and a chimeric B.1.1.528 variant generated on the USA_WA1/2020 background. We observed potent neutralization of the USA_WA1/2020 strain and five variants (IC50 = 1.5–9.1 ng/mL, Figure 6A), with a small reduction in activity against the B.1.1.529 BA.1 variant (IC50 = 44 ng/mL). We validated these results at the University of Brescia, where we assessed activity against the B.1617.2 and B.1.1.28 variants with authentic virus and again observed extremely high potencies (IC50 = 8.5 or 12 ng/mL, respectively, Figure 6B). Collectively, these results confirmed that the bi-paratopic tetravalent D-Fc-F 33-7:36 potently inhibits infection of SARS-CoV-2 with broad coverage against VoC, including the B.1.1.529 BA.1 variant.
Figure 6.

Neutralization of SARS-CoV-2 variants by D-Fc-F 33-7:36
(A) Neutralization of authentic virus variants. A focus reduction test was used to evaluate infection of Vero-hACE2-TMPRSS2 cells following exposure to virus variants preincubated with serial dilutions of D-Fc-F 33-7:36. Samples were run in duplicate, and results shown are representative of two independent experiments.
(B) Neutralization of authentic isolates of virus variants. A microneutralization assay was used to evaluate infection of Vero E6 cells following exposure to virus variants preincubated with serial dilutions of D-Fc-F 33-7:36. In both cases, samples were run in duplicate and normalized to infection in the absence of antibody. Error bars show the standard deviation of the mean percent relative infection values. Plots of nAb concentration versus relative infection for each were fit to estimate the IC50 values shown in the keys.
Discussion
The emergence of VoC that resist clinically approved nAbs (Hoffmann et al., 2021; Wang et al., 2021), as well as convalescent plasma and vaccine sera (Wall et al., 2021; Zhou et al., 2021), highlights the need for alternative therapeutics. In particular, the E484K mutation found in the RBDs of the B.1.351 and B.1.528 VoC impairs neutralization by bamlanivimab (Wang et al., 2021) and casirivimab (Kim et al., 2021; Wang et al., 2021). Even without this mutation, the previously dominant B.1.617.2 variant also resists these nAbs to varying degrees (Planas et al., 2021). The extensive set of mutations found in the now dominant B.1.1.529 variant has rendered these Abs and their cocktails ineffective (Cameroni et al., 2022), resulting in the revocation of their emergency use approval for treatment of illness caused by this variant. The nAb sotrovimab was granted emergency use authorization because it targets a conserved epitope outside the receptor-binding motif (Cameroni et al., 2022) and neutralizes the B.1.1.529 BA.1 variant with reasonable potency. However, sotrovimab has since been de-authorized due to lack of potency against the BA.2 variant. Only a cocktail of two nAbs (AZD8895 and AZD1061)—with non-overlapping epitopes that bind the ACE2 binding site (Dong et al., 2021)—remains authorized for emergency clinical use as a pre-exposure prophylactic against COVID-19. Going forward, there is an urgent need for more potent and broadly neutralizing therapeutics that are capable of neutralizing existing VoC and those that will emerge in the future.
The trimeric structure of the SARS-CoV-2 S protein represents a vulnerability that can be exploited to enhance the potency of nAbs with advanced Ab engineering technologies. Structural studies have shown that highly potent neutralizing IgGs take advantage of their two arms to bind simultaneously to two identical epitopes on the RBD, and bivalent binding enhances affinity and consequent potency of neutralization (Einav et al., 2019; Yan et al., 2021). We recently showed that fusing additional Fabs to a neutralizing IgG enhanced potency further and also observed a concomitant improvement in the neutralization of a VoC that resisted the IgG (Miersch et al., 2021). Moreover, structural studies of a tetravalent nAbs in complex with the S protein ectodomain confirmed that the two arms of the IgG bind simultaneously to two epitopes on the RBD and suggested that a third arm can engage additional epitopes, within the same trimer or perhaps within a different trimer. These findings provide a molecular mechanism to explain the enhanced potency and breadth of coverage.
Here, we report a major advance in Ab engineering with a format that enables rapid and facile combination of two distinct paratopes in a tetravalent nAb that retains the favorable biophysical properties of conventional IgG drugs. We started the engineering process by converting our potent neutralizing IgG 33-7 to a D-Fc format, which enabled modular attachment of additional Fabs at the C-terminus to produce tetravalent D-Fc-F proteins. The modular design strategy allowed for facile production of either the mono-paratopic D-Fc-F 33-7:33-7 or the bi-paratopic D-Fc-F 33-7:36, both of which could be produced at yields comparable to or better than those reported for scFv or IgG formats expressed from Expi293F cells (Bertoglio et al., 2021) and exhibited biophysical properties comparable to those of IgGs (Figure 4). SEC analysis revealed that only ∼80% of the D-Fc-F protein was of the expected size, but this fraction could be readily purified by preparative SEC. Moreover, this purified material remained homogeneous (>95% monodispersed) even after 60 days of storage at 37°C (Figure S3), demonstrating the high stability of the purified D-Fc-F protein. The tetravalent nAbs proved to be more potent than either IgG in neutralization assays with a panel of pseudoviruses representing SARS-CoV-2 VoC. Moreover, the bi-paratopic D-Fc-F 33-7:36 exhibited potent and broad neutralization of authentic SARS-CoV-2 viruses including multiple VoC. These studies establish that this tetravalent format represents a promising means for the development of next-generation nAbs for the treatment of COVID-19.
Our results revealed the value of bi-paratopic design, as D-Fc-F 33-7:36 was potently inhibitory against every VoC tested, including those that resisted both IgGs and even the mono-paratopic D-Fc-F 33-7:33-7. These results were not necessarily expected, given that the epitopes of nAbs 33-7 and 36 presumably overlap and exhibit similar activity patterns in the pseudovirus neutralization assays. Nonetheless, the two paratopes function differently in the tetravalent D-Fc-F context, as evidenced by the potency and breadth of coverage exhibited by D-Fc-F 33-7:36 compared with D-Fc-F 33-7:33-7. While the structural basis for the functional enhancement obtained by combiantion of the two paratopes in D-Fc-F 33-7:36 remains to be determined, these results establish the bi-paratopic, tetravalent D-Fc-F format as a platform for drug design to tackle SARS-CoV-2 and perhaps other viruses.
Our approach is agnostic and generally applicable to SARS-CoV-2 and other viruses with similar envelope proteins. Importantly, a priori knowledge of structural information is not required to enable rapid testing of paratope pairs in a high-throughput manner. We and other groups have already identified hundreds of nAbs targeting diverse neutralizing epitopes on the SARS-CoV-2 RBD and other regions of the S protein (Cao et al., 2020b; Hansen et al., 2020; Liu et al., 2020; Miersch et al., 2021; Rogers et al., 2020; Shi et al., 2020; Wan et al., 2020; Wec et al., 2020), including broadly neutralizing nAbs (Rappazzo et al., 2021). Our D-Fc-F format allows modular combination of virtually any Fab with D-Fc 33-7, thus enabling production of hundreds of tetravalent nAbs that can be tested systematically to develop the best possible frontline treatment for disease caused by current VoC and those that may emerge in the future.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-M13-HRP | GE Healthcare | Cat# GE27-9421-01 |
| Anti-human kappa-HRP | Southern Biotech | Cat# 2060-05: RRID:AB_2795720 |
| Phage displayed antibody library | Persson et al., 2013 | NA |
| goat anti-human IgG (HRP-conjugated) | Abcam | Cat# ab6858: RRID:AB_955433 |
| Oligoclonal mixture of anti-SARS-CoV-2 antibodies | Liu et al. (2021) | SARS2-2, SARS2-11, SARS2-16, SARS2-31, SARS2-38, SARS2-57, SARS2-71 |
| Goat anti-mouse IgG (HRP-conjugated) | Sigma | Cat# A5278: RRID:AB_258232 |
| Goat anti-human IgG (HRP-conjugated) | Sigma | Cat# A6029: RRID:AB_258272 |
| Bacterial and virus strains | ||
| DH5 alpha | Thermofisher | Cat# 18265017 |
| Omnimax | Invitrogen | Cat# C854003 |
| CJ236 | Takara | Cat# 3418 |
| M13K07 | NEB | Cat# N0315S |
| 2019 n-CoV/USA_WA1/2020 | CDC | NA |
| B.1.1.7 (Alpha) isolate | (Chen et al., 2021) | NA |
| B.1.351 (Beta) isolate | Washington University | NA |
| B.1.617.2 (Delta) isolate | Washington University | NA |
| AY.1 (Delta+) isolate | Washington University | NA |
| B.1.1.529 (BA.1) (Omicron) isolate | Washington University | NA |
| 2019-nCoV/Italy-Gamma | University of Brescia Medical School | NA |
| 2019-nCoV/Italy-Delta | University of Brescia Medical School | NA |
| Chemicals, peptides, and recombinant proteins | ||
| SARS-CoV2 RBD | This manuscript | NA |
| SARS-CoV2 spike | This manuscript | NA |
| SARS-CoV2 spike | Abclonal | RP01283LQ |
| Neutravidin | ThermoFisher | Cat# 31000 |
| Streptavidin HRP | ThermoFisher | Cat# SNN1004 |
| Biotin | Sigma | Cat# B4501-5G |
| hIgG1 33-7 | This manuscript | NA |
| hIgG1 36 | This manuscript | NA |
| D-Fc 33-7 | This manuscript | NA |
| D-Fc-F 33-7:33-7 | This manuscript | NA |
| D-Fc-F 33-7:36 | This manuscript | NA |
| rProtein A sepharose | GE Healthcare | Cat# 17127903 |
| Ni-NTA affinity resin | Qiagen | Cat# 30230 |
| Sypro orange | Sigma | Cat# S5692 |
| Bovine serum albumin | Bioshop | Cat# ALB001.250 |
| Histidine | Bioshop | Cat# HIS100.100 |
| Sucrose | Bioshop | Cat# SUC700.500 |
| Sodium chloride | Bioshop | Cat# SOD001.10 |
| Penicillin-streptomycin | Gibco | Cat# 15070063 |
| DMEM | Thermofisher | Cat# 11995073 |
| Fetal bovine serum | Gibco | Cat# 12483-020 |
| Lipofectamine 2000 | Thermofisher | Cat# 11668027 |
| Expi293F expression media | Thermofisher | Cat# A1435102 |
| 293Fectin reagent | Thermofisher | Cat# 12347019 |
| FectoPro transfection reagent | Polyplus Transfection | Cat# 116-010 |
| Opti-Mem media | Polyplus Transfection | Cat. # 116-010 |
| DPBS | Thermofisher | Cat# 14190250 |
| Protease inhibitor cocktail | Sigma | Cat# P-8849 |
| Biotin ligase | This paper | NA |
| In-Fusion EcoDry Master Mix | TakaraBio | Cat# 638953 |
| Critical commercial assays | ||
| ONE-Glo EX Luciferase Assay System | Promega | Cat. #8130 |
| Experimental models: Cell lines | ||
| Vero E6 | ATCC | Cat# CRL-1586: RRID:CVCL_0574 |
| HEK293T | ATCC | Cat# CRL-3216: RRID:CVCL_0063 |
| HEK293T-ACE2 | This manuscript | NA |
| HEK293T-ACE2-TMPRS22 | This manuscript | NA |
| Expi293F | ThermoFisher | Cat# A14527 |
| Freestyle 293-F | ThermoFisher | Cat# R79007 |
| Experimental models: Organisms/strains | ||
| Chimeric B.1.1.28 (Gamma) S-protein 2019 n-CoV/USA_WA1/2020 | (Chen et al., 2021) | NA |
| Oligonucleotides | ||
| GGTGGAGGTGGCTCCG | IDT DNA | Db Inv Fwd |
| GGCGACTGCGGCCGC | IDT DNA | Db Inv Rev |
| TCGGGTGGAGGTGGCAGTGATATCCAGAT GACCCAGTCCCCG |
IDT DNA | Db LC Fwd w lnkr |
| CCGGAGCCACCTCCACCTTTGATCTCCACCT TGGTACCCTGT |
IDT DNA | Db LC Rev w lnkr |
| TCGCGGCCGCAGTCGCCGAGGTTCAGCTGG TGGAG |
IDT DNA | HC Fwd Inf |
| ATCACTGCCACCTCCACCCGAGGAGACGGT GACC |
IDT DNA | HC Rev w lnkr Inf |
| GGGGGCTCTGGAGGCGGAACCGGTGAGGTT CAGCTGGTGGAG |
IDT DNA | Fab Fwd w lnkr Inf |
| CCATGGCGGCCAAGCTGGGGATCCTTAT CATGTGTGAGTTTTGTCACAAGATTTGGG |
IDT DNA | Fab Rev w lnkr Inf |
| ACCGGTTCCGCCTCCA | IDT DNA | Fab Inv Fwd |
| TGATAAGGATCCCCAGCTTGGC | IDT DNA | Fab Inv Rev |
| Recombinant DNA | ||
| hIgG1 33 | This manuscript | NA |
| hk 33-7 | This manuscript | NA |
| D-Fc 33-7 | This manuscript | NA |
| D-Fc-F 33-7:33-7 | This manuscript | NA |
| D-Fc-F 33-7:36 | This manuscript | NA |
| HIV Gag vectors | Icosagen AS | N/A |
| pNL4-3.luc.R-E | NIH AIDS Reagent Program | Cat# 3418 |
| PB-cmv-SARS-CoV-2 spike wt 1-1254 (C-Δ19) | This manuscript | NA |
| Software and algorithms | ||
| Curvefitting: Prism 8.4.3 | Graphpad | NA |
| BLI data analysis: Octet Systems software 9.0 | Forte Bio | NA |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Shane Miersch (shane.miersch@utoronto.ca).
Materials availability
Materials generated in this study are available upon request from the lead contact, subject to an MTA.
Experimental model and subject details
Cells
Mammalian cells were maintained in humidified environments at 37°C in 5% CO2 in the indicated media. Vero E6 (ATCC, CRL-1586), Vero-hACE2-TMPRSS2 cells, HEK293T (ATCC, ACS-4500) and HEK293T cells stably overexpressing ACE2 were maintained at 37°C in 5% CO2 in DMEM containing 10% (vol/vol) FBS. Expi293F cells (ThermoFisher, A14528) were maintained at 37°C in 8% CO2 in Expi293F expression media (ThermoFisher, A1435101). Vero-hACE2-TMPRSS2 cell cultures were supplemented with 10 μg mL−1 of puromycin.
Method details
Protein production and purification
The SARS-CoV-2 S-protein ECD was obtained as a kind gift from Dr. James Rini, produced and purified as described (Miersch et al., 2021) or was obtained commercially (Abclonal, RPO1283LQ). Purified proteins were site-specifically biotinylated in a reaction with 200 μM biotin, 500 μM ATP, 500 μM MgCl2, 30 μg/mL BirA, 0.1% (v/v) protease inhibitor cocktail and not more than 100 μM of the protein-AviTag substrate. The reactions were incubated at 30°C for 2 h and biotinylated proteins were purified by size-exclusion chromatography.
Ab production and purification
IgG, D-Fc and D-Fc-F proteins were produced in Expi293F cells (ThermoFisher) by transient transfection, by diluting heavy and light chain expression plasmid DNA in OptiMem serum-free media (Gibco, 31985088) before the addition of and incubation with FectoPro (Polyplus Transfection, 10100007) for 10 min. For IgG and D-Fc-F production, equivalent amounts of plasmids encoding heavy chain or light chain were transfected, whereas for D-Fc production, a single construct encoding the D-Fc protein was transfected. Following addition of the DNA complex to Expi293F cells and a 5-day expression period, Abs were purified using rProtein-A Sepharose (GE Healthcare, 17127904), then buffer exchanged and concentrated using Amicon Ultra-15 Centrifugal Filter devices (Millipore, UFC805096). IgG and D-Fc proteins were stored in PBS (Gibco), and D-Fc-F proteins were stored in 10 mM L-histidine, 0.9% sucrose, 140 mM NaCl, pH 6.0.
Phage display selections
A synthetic, phage-displayed Fab library (Persson et al., 2013) was selected for binding to biotinylated SARS-CoV-2 RBD, as described (Miersch et al., 2021). Individual clones from the same selected phage pool that previously yielded Ab 15033 were screened by phage ELISA to identify additional RBD-binding Fab-phage clones, including the progenitor of Ab 36. The heavy chain of the 36 progenitor was combined with a library of light chains with diversity in CDR-L3, and Fab 36 was isolated following additional rounds of selection, as described (Miersch et al., 2021).
Phage ELISAs
Phage ELISAs were performed, as described (Miersch et al., 2021). Briefly, 96-well Nunc Maxisorp (Sigma-Aldrich, M9410) plates were coated with neutravidin and blocked with PBS, 0.2% BSA. Biotinylated target protein was captured from solution by incubation in neutravidin-coated and BSA-blocked wells for 15 min with shaking at room temperature. Wells were incubated for 1 h with a saturating concentration of IgG in PBS, or with PBS alone, and subsequently, Fab-phage were added and incubated for 30 min. Plates were washed, incubated with an anti-M13 antibody-HRP conjugate (GE Healthcare, 27-9421-01), and developed with TMB substrate (KPL, KP-50-76-03), as described (Miersch et al., 2017).
Construction of genes encoding D-Fc and D-Fc-F proteins
DNA fragments encoding heavy (VH) and light chain (VL) variable domains, (terminating at Ser128 or Lys127, respectively, IMGT numbering throughout (Lefranc et al., 2003)) were amplified by PCR from the IgG expression vectors using oligonucleotide primers reported in the key resources table. D-Fc constructs were constructed by In-Fusion cloning (Takara Bio, USA) to fuse these fragments with an intervening linker (sequence: GGGGS) with VH-linker-VL orientation, and then cloning the resultant insert into the pSCSTa mammalian expression vector, which resulted in the diabody being fused to the N-terminus of the hinge residue Asp6 with an intervening linker (sequence: GGGGSGGGGS). D-Fc-F constructs were assembled by fusing a Fab heavy chain to Fc residue Gly129 at the C-terminus of the D-Fc with an intervening linker (sequence: GGGGSGGGGSGGGTG).
Size exclusion chromatography
Protein samples (50 μg) were injected onto a TSKgel BioAssist G3SWxl column (Tosoh) fitted with a guard column using an NGC chromatography system and a C96 autosampler (Biorad). The column was preequilibrated in a PBS mobile phase and protein retention was monitored by absorbance at 215 nm during a 1.5 CV isocratic elution in PBS.
Biolayer interferometry
The binding kinetics (kon and koff) and apparent affinity (KD) of Abs binding to the S-protein ECD were determined by BLI with an Octet HTX instrument (ForteBio) at 1000 rpm and 25°C. Biotinylated S-protein ECD was captured on streptavidin biosensors from a 2 μg/mL solution to achieve a binding response of 0.4–0.6 nm and unoccupied sites were quenched with 100 μg/mL biotin. Abs were diluted with assay buffer (PBS, 1% BSA, 0.05% Tween 20), and 67 nM of an irrelevant biotinylated protein of similar size was used as negative control. Following equilibration with assay buffer, loaded biosensors were dipped for 600 s into wells containing 3-fold serial dilutions of each Ab starting at 67 nM, and subsequently, were transferred back into assay buffer for 600 s. Binding response data were corrected by subtraction of response from a reference and were fitted with a 1:1 binding model using ForteBio Octet Systems software 9.0.
Production of pseudoviruses
HEK-293 cells (ATCC) were seeded in a 6-well plate at 3 × 105 cells/well in DMEM (ThermoFisher, 11995-065) supplemented with 10% FBS and 1% penicillin-streptomycin (Gibco, 15140122) and grown overnight at 37°C with 5% CO2. HEK293 cells were co-transfected with 1 μg pNL4-3.luc.R-E- plasmid (luciferase expressing HIV-1 with defective envelop protein) (NIH AIDS Reagent Program, ARP2128) and 0.06 μg CMV-promoter driven plasmid encoding the S-protein using Lipofectamine™ 2000 transfection reagent (ThermoFisher, 11668027). Pseudovirus particles were harvested by collecting supernatant 48 h after transfection and were filter sterilized (0.44 μm, Millipore-Sigma, SLHA033SS).
Pseudovirus infection assays
HEK293T cells stably over-expressing full-length human ACE2 protein were seeded in 96-well white polystyrene microplates (Corning, CLS3610) at 3 × 104 cells/well in DMEM (10% FBS and 1% penicillin-streptomycin) and were grown overnight at 37°C with 5% CO2. Pseudovirus particles were mixed with Ab, incubated at room temperature for 10 min, and added to the cells. The cells were incubated at 37°C with 5% CO2, the medium was replaced with fresh DMEM (10% FBS and 1% penicillin-streptomycin) after 6 h, and again every 24 h up to 72 h. To measure the luciferase signal (pseudovirus entry), DMEM was removed and DPBS (ThermoFisher) was added to cells before mixing with an equal volume of ONE-Glo™ EX Luciferase Assay System (Promega, E8130). Relative luciferase units were measured using a BioTek Synergy Neo plate reader (BioTek Instruments Inc.). The data were analyzed by GraphPad Prism (GraphPad Software, LLC, Version 9.0.2).
Authentic virus infection assays
To evaluate the neutralization of SARS-Cov-2 VoC, the USA_WA1/2020 strain generated by reverse genetics (Xie et al., 2020), authentic isolates of B.1.1.7, B.1.351, B.1.617.2, B.1.617.2+, B.1.1.529 (BA.1) and the chimeric B.1.1.28 (that was generated on the genetic background of WA1/2020) as described (Chen et al., 2021) were evaluated for neutralization using a focus reduction neutralization test and potency was estimated as described (Case et al., 2020a, 2020b; Miersch et al., 2021). All viruses were verified by next-generation sequencing. Alternatively, authentic SARS-CoV-2 strains were used in a micro-neutralization assay as described (Caccuri et al., 2020). In brief, serial four-fold dilutions of antibody, starting from 10 μg/mL, were pre-incubated with 102 focus-forming units per 100 μL at 37°C for 1 h. The antibody-virus mixture was transferred to 96-well tissue culture plates containing Vero-hACE2-TMPRSS2 cell monolayers in duplicate and incubated at 37°C and 5% CO2 for 1 h, subsequently, the cells were overlaid with 1% (wt/vol) methylcellulose in MEM. Plates were harvested at 24 h by removal of overlays and fixation for 20 min with 4% paraformaldehyde in PBS at room temperature. Plates were washed with permeabilization buffer (PBS supplemented with 0.1% saponin and 0.1% BSA) and sequentially incubated overnight with the oligoclonal pool of anti-SARS-Cov-2 Abs (Liu et al., 2021). The plates with Omicron (B.1.1.529) were additionally incubated with the cross-reactive pool of Abs for SARS-CoV-1 that bind to the RBD and compete with Ab (VanBlargan et al., 2022). The plates were incubated with HRP-conjugated anti-mouse (Sigma; A5278)/goat anti-human Abs (Sigma; A6029) and visualized by KPL TrueBlue substrate and quantitated by an Immunospot microanalyzer (Cellular Technologies), as described (VanBlargan et al., 2022) (Manenti et al., 2020).
Quantification and statistical analysis
Plots of antibody concentration versus % relative infection were generated from the mean value of number of technical replicates with error bars shown as the standard deviation of the replicates. Generated curves were fit by non-linear regression in Prism 9 (GraphPad Software, Version 9.0.2) using the variable slope (four-parameter) inhibitor versus response model. Number of independent experiments and technical repeats are indicated in the Figure legends. For BLI, sensorgram plots were globally fit using a 1:1 binding model to obtain the values for the binding kinetics and associated errors using FortéBio Data Analysis Software (FortéBio, Version 9.0.0.15).
Acknowledgments
We thank Dr. James Rini for providing biotinylated S protein, RBD, and the ACE2-expressing HEK293T cells. This study was partially supported by grants from Canadian Institutes of Health Operating Grant COVID-19 Rapid Research Funding Opportunity OV3-170649, an Emergent Ventures/Thistledown Foundation FAST grant, Emergent Ventures/The Mercatus Center FAST (#2161 and #2189), and Temerty Foundation Knowledge Translation Grant-Novel Antibody Tools for COVID-19. Infrastructure was supported by Canada Foundation for Innovation Infrastructure and Operating Grant #IOF-33363, as well as grants from the Rome Foundation (Italy, Prot 317A/I) 592, Italian Ministry of University and Research (FISR2020IP_03161), Regione Lazio, and by US National Institutes of Health grants (R01AI161374, R01AI143292, P01AI120943, and R01 AI157155).
Author contributions
Conceptualization, S.M. and S.S.S.; methodology, S.M. and R.S.; investigation, S.M., N.S., R.S., C.C., J.B.C., G.K.A., A.C., F.C., A.Z., and G.N.; supervision, S.M., M.S.D., and S.S.S.; writing – original draft, S.M., M.S.D., and S.S.S.; writing – review & editing, S.M. and M.S.D.; visualization, S.M.; validation, S.M.; project administration, S.M.; funding acquisition, M.S.D. and S.S.S.
Declaration of interests
S.M. and S.S.S. are inventors on a patent application describing anti-SARS-CoV-2 antibodies and multivalent antibody formats. M.S.D. is a consultant for Inbios, Vir Biotechnology, Senda Biosciences, and Carnival Corporation and is on the Scientific Advisory Boards of Moderna and Immunome. The Diamond laboratory has received funding support in sponsored research agreements from Moderna, Vir Biotechnology, and Emergent BioSolutions.
Published: May 16, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110905.
Supplemental information
Data and code availability
All data reported in this paper will be shared by the lead contact upon reasonable request.
This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Baum A., Fulton B.O., Wloga E., Copin R., Pascal K.E., Russo V., Giordano S., Lanza K., Negron N., Ni M., et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science. 2020;369:1014–1018. doi: 10.1126/SCIENCE.ABD0831/SUPPL_FILE/PAP.PDF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoglio F., Fühner V., Ruschig M., Heine P.A., Abassi L., Klünemann T., Rand U., Meier D., Langreder N., Steinke S., et al. A SARS-CoV-2 neutralizing antibody selected from COVID-19 patients binds to the ACE2-RBD interface and is tolerant to most known RBD mutations. Cell Rep. 2021;36:109433. doi: 10.1016/J.CELREP.2021.109433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwer P.J.M., Caniels T.G., van der Straten K., Snitselaar J.L., Aldon Y., Bangaru S., Torres J.L., Okba N.M.A., Claireaux M., Kerster G., et al. Potent neutralizing antibodies from COVID-19 patients define multiple targets of vulnerability. Science. 2020;369:643–650. doi: 10.1126/SCIENCE.ABC5902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caccuri F., Zani A., Messali S., Giovanetti M., Bugatti A., Campisi G., Filippini F., Scaltriti E., Ciccozzi M., Fiorentini S., Caruso A. A persistently replicating SARS-CoV-2 variant derived from an asymptomatic individual. J. Transl. Med. 2020;18:362. doi: 10.1186/S12967-020-02535-1/FIGURES/4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameroni E., Bowen J.E., Rosen L.E., Saliba C., Zepeda S.K., Culap K., Pinto D., VanBlargan L.A., de Marco A., di Iulio J., et al. Broadly neutralizing antibodies overcome SARS-CoV-2 Omicron antigenic shift. Nature. 2022;602:664–670. doi: 10.1038/s41586-021-04386-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L., Goreshnik I., Coventry B., Case J.B., Miller L., Kozodoy L., Chen R.E., Carter L., Walls A.C., Park Y.J., et al. De novo design of picomolar SARS-CoV-2 miniprotein inhibitors. Science. 2020;370:426–431. doi: 10.1126/SCIENCE.ABD9909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y., Su B., Guo X., Sun W., Deng Y., Bao L., Zhu Q., Zhang X., Zheng Y., Geng C., et al. Potent neutralizing antibodies against SARS-CoV-2 identified by high-throughput single-cell sequencing of convalescent patients’ B cells. Cell. 2020;182:73–84.e16. doi: 10.1016/j.cell.2020.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y., Wang J., Jian F., Xiao T., Song W., Yisimayi A., Huang W., Li Q., Wang P., An R., et al. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature. 2022;602:657–663. doi: 10.1038/S41586-021-04385-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case J.B., Bailey A.L., Kim A.S., Chen R.E., Diamond M.S. Growth, detection, quantification, and inactivation of SARS-CoV-2. Virology. 2020;548:39–48. doi: 10.1016/J.VIROL.2020.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case J.B., Rothlauf P.W., Chen R.E., Liu Z., Zhao H., Kim A.S., Bloyet L.M., Zeng Q., Tahan S., Droit L., et al. Neutralizing antibody and soluble ACE2 inhibition of a replication-competent VSV-SARS-CoV-2 and a clinical isolate of SARS-CoV-2. Cell Host Microbe. 2020;28:475–485.e5. doi: 10.1016/J.CHOM.2020.06.021/ATTACHMENT/1473FB09-A200-4124-8413-5A897225E5E7/MMC1.PDF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R.E., Zhang X., Case J.B., Winkler E.S., Liu Y., VanBlargan L.A., Liu J., Errico J.M., Xie X., Suryadevara N., et al. Resistance of SARS-CoV-2 variants to neutralization by monoclonal and serum-derived polyclonal antibodies. Nat. Med. 2021;27:717–726. doi: 10.1038/s41591-021-01294-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor R.I., Chen B.K., Choe S., Landau N.R. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology. 1995;206:935–944. doi: 10.1006/VIRO.1995.1016. [DOI] [PubMed] [Google Scholar]
- Dong J., Zost S.J., Greaney A.J., Starr T.N., Dingens A.S., Chen E.C., Chen R.E., Case J.B., Sutton R.E., Gilchuk P., et al. Genetic and structural basis for SARS-CoV-2 variant neutralization by a two-antibody cocktail. Nat. Microbiol. 2021;6:1233–1244. doi: 10.1038/S41564-021-00972-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einav T., Yazdi S., Coey A., Bjorkman P.J., Phillips R. Harnessing avidity: quantifying the entropic and energetic effects of linker length and rigidity for multivalent binding of antibodies to HIV-1. Cell Syst. 2019;9:466–474.e7. doi: 10.1016/j.cels.2019.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorentini S., Messali S., Zani A., Caccuri F., Giovanetti M., Ciccozzi M., Caruso A. First detection of SARS-CoV-2 spike protein N501 mutation in Italy in August, 2020. Lancet Infect. Dis. 2021;21:e147. doi: 10.1016/S1473-3099(21)00007-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappazzo C.G., Tse L.v., Kaku C.I., Wrapp D., Sakharkar M., Huang D., Deveau L.M., Yockachonis T.J., Herbert A.S., Battles M.B., et al. Broad and potent activity against SARS-like viruses by an engineered human monoclonal antibody. Science. 2021;371:823–829. doi: 10.1126/SCIENCE.ABF4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Gasparo R., Pedotti M., Simonelli L., Nickl P., Muecksch F., Cassaniti I., Percivalle E., Lorenzi J.C.C., Mazzola F., Magrì D., et al. Bispecific IgG neutralizes SARS-CoV-2 variants and prevents escape in mice. Nature. 2021;593:424–428. doi: 10.1038/s41586-021-03461-y. [DOI] [PubMed] [Google Scholar]
- Gottlieb R.L., Nirula A., Chen P., Boscia J., Heller B., Morris J., Huhn G., Cardona J., Mocherla B., Stosor V., et al. Effect of bamlanivimab as monotherapy or in combination with etesevimab on viral load in patients with mild to moderate COVID-19: a randomized clinical trial. JAMA. 2021;325:632–644. doi: 10.1001/JAMA.2021.0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaney A.J., Starr T.N., Gilchuk P., Zost S.J., Binshtein E., Loes A.N., Hilton S.K., Huddleston J., Eguia R., Crawford K.H.D., et al. Complete mapping of mutations to the SARS-CoV-2 spike receptor-binding domain that escape antibody recognition. Cell Host Microbe. 2021;29:44–57.e9. doi: 10.1016/J.CHOM.2020.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen J., Baum A., Pascal K.E., Russo V., Giordano S., Wloga E., Fulton B.O., Yan Y., Koon K., Patel K., et al. Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science. 2020;369:1010–1014. doi: 10.1126/science.abd0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart T.K., Sellers T.S., Maleeff B.E., Eustis S., Schwartz L.W., Bugelski P.J., Herzyk D.J., Cook R.M., Tsui P., Appelbaum E.R., Martin E.C. Preclinical efficacy and safety of mepolizumab (SB-240563), a humanized monoclonal antibody to IL-5, in cynomolgus monkeys. J. Allergy Clin. Immunol. 2001;108:250–257. doi: 10.1067/MAI.2001.116576. [DOI] [PubMed] [Google Scholar]
- Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.H., Nitsche A., et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280.e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Hofmann-Winkler H., Krüger N., Kempf A., Nehlmeier I., Graichen L., Arora P., Sidarovich A., Moldenhauer A.S., Winkler M.S., et al. SARS-CoV-2 variant B.1.617 is resistant to bamlanivimab and evades antibodies induced by infection and vaccination. Cell Rep. 2021;36:109415. doi: 10.1016/J.CELREP.2021.109415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt A.C., Case J.B., Park Y.-J., Cao L., Wu K., Walls A.C., Liu Z., Bowen J.E., Yeh H.-W., Saini S., et al. Multivalent designed proteins protect against SARS-CoV-2 variants of concern. bioRxiv. 2021 doi: 10.1101/2021.07.07.451375. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen B., Luebke N., Feldt T., Keitel V., Brandenburger T., Kindgen-Milles D., Lutterbeck M., Freise N.F., Schoeler D., Haas R., et al. Emergence of the E484K mutation in SARS-COV-2-infected immunocompromised patients treated with bamlanivimab in Germany. Lancet Reg. Health Eur. 2021;8:100164. doi: 10.1016/J.LANEPE.2021.100164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayabolen A., Akcan U., Ozturan D., Sarayloo E., Nurtop E., Ozer B., Sahin G.N., Dogan O., Lack N., Kaya M., et al. Protein scaffold-based multimerization of soluble ACE2 efficiently blocks SARS-CoV-2 infection in vitro. bioRxiv. 2021 doi: 10.1101/2021.01.04.425128. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.J., Jang U.S., Soh S.M., Lee J.Y., Lee H.R. The impact on infectivity and neutralization efficiency of SARS-CoV-2 lineage B.1.351 pseudovirus. Viruses. 2021;13:633. doi: 10.3390/V13040633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein S., Cortese M., Winter S.L., Wachsmuth-Melm M., Neufeldt C.J., Cerikan B., Stanifer M.L., Boulant S., Bartenschlager R., Chlanda P. SARS-CoV-2 structure and replication characterized by in situ cryo-electron tomography. Preprint at bioRxiv. 2020 doi: 10.1101/2020.06.23.167064. 2020.06.23.167064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefranc M.P., Pommié C., Ruiz M., Giudicelli V., Foulquier E., Truong L., Thouvenin-Contet V., Lefranc G. IMGT unique numbering for immunoglobulin and T cell receptor variable domains and Ig superfamily V-like domains. Dev. Comp. Immunol. 2003;27:55–77. doi: 10.1016/S0145-305X(02)00039-3. [DOI] [PubMed] [Google Scholar]
- Liu L., Wang P., Nair M.S., Yu J., Rapp M., Wang Q., Luo Y., Chan J.F.W., Sahi V., Figueroa A., et al. Potent neutralizing antibodies against multiple epitopes on SARS-CoV-2 spike. Nature. 2020;584:450–456. doi: 10.1038/s41586-020-2571-7. [DOI] [PubMed] [Google Scholar]
- Liu Z., VanBlargan L.A., Bloyet L.M., Rothlauf P.W., Chen R.E., Stumpf S., Zhao H., Errico J.M., Theel E.S., Liebeskind M.J., et al. Identification of SARS-CoV-2 spike mutations that attenuate monoclonal and serum antibody neutralization. Cell Host Microbe. 2021;29:477–488.e4. doi: 10.1016/J.CHOM.2021.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manenti A., Maggetti M., Casa E., Martinuzzi D., Torelli A., Trombetta C.M., Marchi S., Montomoli E. Evaluation of SARS-CoV-2 neutralizing antibodies using a CPE-based colorimetric live virus micro-neutralization assay in human serum samples. J. Med. Virol. 2020;92:2096–2104. doi: 10.1002/JMV.25986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCallum M., Bassi J., De Marco A., Chen A., Walls A.C., Di Iulio J., Tortorici M.A., Navarro M.-J., Silacci-Fregni C., Saliba C., et al. SARS-CoV-2 immune evasion by the B.1.427/B.1.429 variant of concern. Science. 2021;373:648–654. doi: 10.1126/SCIENCE.ABI7994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miersch S., Maruthachalam B.V., Geyer C.R., Sidhu S.S. Structure-directed and tailored diversity synthetic antibody libraries yield Novel anti-EGFR antagonists. ACS Chem. Biol. 2017;12:1381–1389. doi: 10.1021/acschembio.6b00990. [DOI] [PubMed] [Google Scholar]
- Miersch S., Li Z., Saberianfar R., Ustav M., Brett Case J., Blazer L., Chen C., Ye W., Pavlenco A., Gorelik M., et al. Tetravalent SARS-CoV-2 neutralizing antibodies show enhanced potency and resistance to escape mutations. J. Mol. Biol. 2021;433:167177. doi: 10.1016/J.JMB.2021.167177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson H., Ye W., Wernimont A., Adams J.J., Koide A., Koide S., Lam R., Sidhu S.S. CDR-H3 diversity is not required for antigen recognition by synthetic antibodies. J. Mol. Biol. 2013;425:803–811. doi: 10.1016/j.jmb.2012.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D., Park Y.J., Beltramello M., Walls A.C., Tortorici M.A., Bianchi S., Jaconi S., Culap K., Zatta F., de Marco A., et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature. 2020;583:290–295. doi: 10.1038/s41586-020-2349-y. [DOI] [PubMed] [Google Scholar]
- Planas D., Veyer D., Baidaliuk A., Staropoli I., Guivel-Benhassine F., Rajah M.M., Planchais C., Porrot F., Robillard N., Puech J., et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature. 2021;596:276–280. doi: 10.1038/s41586-021-03777-9. [DOI] [PubMed] [Google Scholar]
- Razonable R.R., Pawlowski C., O’Horo J.C., Arndt L.L., Arndt R., Bierle D.M., Borgen M.D., Hanson S.N., Hedin M.C., Lenehan P., et al. Casirivimab–Imdevimab treatment is associated with reduced rates of hospitalization among high-risk patients with mild to moderate coronavirus disease-19. EClinicalMedicine. 2021;40:101102. doi: 10.1016/J.ECLINM.2021.101102/ATTACHMENT/32421CA8-2613-4860-B00A-44BC0843404B/MMC1.DOCX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers T.F., Zhao F., Huang D., Beutler N., Burns A., He W.T., Limbo O., Smith C., Song G., Woehl J., et al. Isolation of potent SARS-CoV-2 neutralizing antibodies and protection from disease in a small animal model. Science. 2020;369:956–963. doi: 10.1126/science.abc7520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi R., Shan C., Duan X., Chen Z., Liu P., Song J., Song T., Bi X., Han C., Wu L., et al. A human neutralizing antibody targets the receptor-binding site of SARS-CoV-2. Nature. 2020;584:120–124. doi: 10.1038/s41586-020-2381-y. [DOI] [PubMed] [Google Scholar]
- Tortorici M.A., Beltramello M., Lempp F.A., Pinto D., Dang H.v., Rosen L.E., McCallum M., Bowen J., Minola A., Jaconi S., et al. Ultrapotent human antibodies protect against SARS-CoV-2 challenge via multiple mechanisms. Science. 2020;370:950–957. doi: 10.1126/SCIENCE.ABE3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanBlargan L.A., Errico J.M., Halfmann P.J., Zost S.J., Crowe J.E., Purcell L.A., Kawaoka Y., Corti D., Fremont D.H., Diamond M.S. An infectious SARS-CoV-2 B.1.1.529 Omicron virus escapes neutralization by therapeutic monoclonal antibodies. Nat. Med. 2022;28:490–495. doi: 10.1038/s41591-021-01678-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall E.C., Wu M., Harvey R., Kelly G., Warchal S., Sawyer C., Daniels R., Hobson P., Hatipoglu E., Ngai Y., et al. Neutralising antibody activity against SARS-CoV-2 VOCs B.1.617.2 and B.1.351 by BNT162b2 vaccination. Lancet. 2021;397:2331–2333. doi: 10.1016/S0140-6736(21)01290-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls A.C., Park Y.J., Tortorici M.A., Wall A., McGuire A.T., Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;181:281–292.e6. doi: 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walser M., Rothenberger S., Hurdiss D.L., Schlegel A., Calabro V., Fontaine S., Villemagne D., Paladino M., Hospodarsch T., Neculcea A., et al. Highly potent anti-SARS-CoV-2 multivalent DARPin therapeutic candidates. bioRxiv. 2021 doi: 10.1101/2020.08.25.256339. Preprint at. [DOI] [Google Scholar]
- Wan J., Xing S., Ding L., Wang Y., Gu C., Wu Y., Rong B., Li C., Wang S., Chen K., et al. Human-IgG-neutralizing monoclonal antibodies block the SARS-CoV-2 infection. Cell Rep. 2020;32:107918. doi: 10.1016/j.celrep.2020.107918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P., Nair M.S., Liu L., Iketani S., Luo Y., Guo Y., Wang M., Yu J., Zhang B., Kwong P.D., et al. Antibody resistance of SARS-CoV-2 variants B.1.351 and B.1.1.7. Nature. 2021;593:130–135. doi: 10.1038/s41586-021-03398-2. [DOI] [PubMed] [Google Scholar]
- Wec A.Z., Wrapp D., Herbert A.S., Maurer D.P., Haslwanter D., Sakharkar M., Jangra R.K., Dieterle M.E., Lilov A., Huang D., et al. Broad neutralization of SARS-related viruses by human monoclonal antibodies. Science. 2020;369:731–736. doi: 10.1126/science.abc7424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreich D.M., Sivapalasingam S., Norton T., Ali S., Gao H., Bhore R., Xiao J., Hooper A.T., Hamilton J.D., Musser B.J., et al. Trial Investigators REGEN-COV antibody combination and outcomes in outpatients with covid-19. N. Engl. J. Med. 2021;385:e81. doi: 10.1056/NEJMOA2108163/SUPPL_FILE/NEJMOA2108163_DATA-SHARING.PDF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisblum Y., Schmidt F., Zhang F., DaSilva J., Poston D., Lorenzi J.C.C., Muecksch F., Rutkowska M., Hoffmann H.H., Michailidis E., et al. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. Elife. 2020;9:e61312. doi: 10.7554/ELIFE.61312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X., Muruato A., Lokugamage K.G., Narayanan K., Zhang X., Zou J., Liu J., Schindewolf C., Bopp N.E., Aguilar P.v., et al. An Infectious cDNA Clone of SARS-CoV-2. Cell Host Microbe. 2020;27:841–848.e3. doi: 10.1016/J.CHOM.2020.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., Xu K., Jung S., Conte A., Lieberman J., Muecksch F., Lorenzi J.C.C., Park S., Schmidt F., Wang Z., et al. Nanobodies from camelid mice and llamas neutralize SARS-CoV-2 variants. Nature. 2021;595:278–282. doi: 10.1038/s41586-021-03676-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan R., Wang R., Ju B., Yu J., Zhang Y., Liu N., Wang J., Zhang Q., Chen P., Zhou B., et al. Structural basis for bivalent binding and inhibition of SARS-CoV-2 infection by human potent neutralizing antibodies. Cell Res. 2021;31:517–525. doi: 10.1038/s41422-021-00487-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D., Dejnirattisai W., Supasa P., Liu C., Mentzer A.J., Ginn H.M., Zhao Y., Duyvesteyn H.M.E., Tuekprakhon A., Nutalai R., et al. Evidence of escape of SARS-CoV-2 variant B.1.351 from natural and vaccine-induced sera. Cell. 2021;184:2348–2361.e6. doi: 10.1016/J.CELL.2021.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon reasonable request.
This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.