

Summary
Mycobacterium tuberculosis (Mtb) evades host clearance by inhibiting autophagy. MicroRNA-25 (miR-25) expression was significantly up-regulated in the lung tissues of mice infected with Bacillus Calmette-Guerin (BCG) and macrophages infected with Mtb or BCG, especially in the early stages of infection. MiR-25 can significantly increase the survival of Mtb and BCG in macrophages. We validated that miR-25 targets the NPC1 protein located on the lysosomal membrane, resulting in damage to lysosomal function, thereby inhibiting autophagolysosome formation and promoting the survival of Mtb and BCG. Consistently, mice lacking miR-25 exhibited more resistant to BCG infection. In addition, we found that Rv1759c induces the expression of miR-25 through NFKB inhibitor zeta (NFKBIZ). This study demonstrates that the role of miR-25 during Mtb infection contributes to a better understanding of the pathogenesis of tuberculosis (TB).
Subject areas: Biological sciences, Molecular biology, Molecular interaction
Graphical abstract
Highlights
-
•
Mtb up-modulates miR-25 expression especially in the early stage of infection
-
•
miR-25 targeting NPC1 impairs autophagic flux in macrophages
-
•
Mice lacking miR-25 exhibits more resistant to BCG infection
-
•
Rv1759c regulates miR-25 expression and Mtb survival via NFKBIZ
Biological sciences; Molecular biology; Molecular interaction
Introduction
Tuberculosis (TB) is an infectious disease that seriously threatens human health. In 2019, there were an estimated 10 million new TB cases worldwide, and approximately 1.4 million people died from TB, including 208,000 people with HIV (Geneva: World Health Organization, 2020). Approximately one-quarter of the population in the world is infected with M. tuberculosis (Mtb), and 10% develop active tuberculosis. Because of the survival of Mtb in macrophages, there is still a risk of turning latent TB into active TB and causing TB to spread when immunity is reduced (Ahmad, 2011). Intracellular survival contributes to the pathogenicity and latency of Mtb (Cambier et al., 2014). However, the mechanism of Mtb resistance to host clearance is still not entirely clear, and it is critical to deeply explore the survival mechanism of Mtb in macrophages.
Autophagy is an intracellular degradation system that can effectively remove damaged organelles, misfolded proteins, and invading pathogenic microorganisms (Mizushima and Komatsu, 2011), and it plays an important role in the fight against bacteria, including Mtb (Gutierrez et al., 2004; Huang and Brumell, 2014). When macroautophagy, referred to as autophagy, was activated, double-layer membrane autophagosomes including bacteria were formed, transported to and fused with the lysosome to degrade intracellular bacteria (Levine et al., 2011). Mtb can inhibit the process of autophagy by preventing the fusion of autophagosomes and lysosomes to enhance the survival ability of macrophages (BoseDasgupta and Pieters, 2018; Bussi and Gutierrez, 2019).
MicroRNAs (miRNAs) are a type of noncoding RNA of 18–22 nucleotides that participate in gene posttranscriptional regulation (Bartel, 2004). MiRNAs play important roles in regulating autophagy in the progression of Mtb infection. MiR-33 and miR-33∗ have been demonstrated to be up-regulated during Mtb infection which can increase the survival of Mtb by down-regulating autophagy-related gene 5 (ATG5), autophagy-related gene 12 (ATG12), lysosome-associated membrane protein 1 (LAMP1), transcription factors forkhead box protein O3 (FOXO3) and transcription factor EB (TFEB) (Ouimet et al., 2016). Similarly, miR-155 was up-regulated during Mtb infection by targeting autophagy effector autophagy-related gene 3 (ATG3) to inhibit autophagy and promote the intracellular survival of Mtb (Etna et al., 2018). Another study reported that miR-144∗ targets the autophagy protein DNA damage regulated autophagy modulator 2 (DRAM2), which plays an important role in the maturation of phagosomes and weakens the antibacterial effect of Mtb in human monocytes and macrophages (Kim et al., 2017). MiR-27a has been reported in the literature to promote the survival of Mtb in murine primary peritoneal macrophages by targeting the Ca2+ transporter calcium channel voltage-dependent alpha 2/delta subunit 3 (Cacna2d3) located in the endoplasmic reticulum (ER) to inhibit the formation of autophagosomes (Liu et al., 2018). More recently, miR-889 was found to be able to target tumor necrosis factor (TNF)-like weak apoptosis inducer (TWEAK) to inhibit autophagy, thereby maintaining the survival of Mtb (Chen et al., 2020). However, the mechanism by which miRNAs regulate the autophagy process and affect the survival of Mtb has not been completely elucidated.
In this study, we found that the expression of miR-25 was increased in Mtb/Bacillus Calmette-Guerin (BCG)-infected macrophages and BCG-infected mouse lungs. MiR-25 directly targets the lysosomal cholesterol transporter NPC1, impairs autophagosome-lysosome fusion and promotes intracellular survival of Mtb.
Results
miR-25 expression is up-regulated by BCG and Mtb infection
To evaluate the levels of miRNAs in response to BCG infection in vivo, we detected miRNAs’ expression in lung tissues of BCG-infected C57/BL6 mice by using quantitative real-time PCR (qRT-PCR). The results showed that miR-25, miR-93 and miR-106b of the miR-106b-25 cluster and miR-146 were up-regulated at 3 and 7 days post infection (3 dpi and 7 dpi) whereas other miRNAs were only up-regulated at 7 dpi. At 3 dpi, the expression of miR-25 increased to approximately 20 times, which was the highest multiple of expression change, and the expression change of miR-146a was the slightest. At 7 dpi, the expression of miR-146a increased approximately 12-fold, which was the highest multiple of expression change, and the expression change of miR-106b was the slightest (Figures 1A and 1B). In addition, we examined miRNAs’ expression in THP-1 cells infected with BCG and H37Rv. The results showed that miR-25, miR-93 and miR-106b were highly expressed during BCG and H37Rv infections. MiR-20a was only down-regulated during BCG infection. MiR-429 and miR-146a were only up-regulated during H37Rv infection. MiR-119a, miR-199b and miR-320 were down-regulated regardless of BCG or H37Rv infection (Figure 1C). MiR-146a was up-regulated significantly during Mtb infection, which was consistent with previous studies (Etna et al., 2018; Malardo et al., 2016; Vegh et al., 2015; Kathirvel et al., 2020; Zhou et al., 2016). The above findings suggest that the miR-106b-25 cluster were significantly up-regulated in BCG-infected mouse lungs and BCG/H37Rv-infected macrophages. Because the up-regulation of miR-25 in mouse lung tissues was higher than that of miR-93 and miR-106bat 3 dpi and the function of miR-25 in regulating Mtb infection was not revealed, we chose miR-25 for further study. We examined miR-25 expression in BCG-infected THP-1 cells with different multiplicities of infections (MOIs) and found that with increasing MOI, the miR-25 expression levels gradually increased (Figure 1D). Similarly, we detected the expression of miR-25 in THP-1 cells infected with H37Rv at different MOIs. Because of the difference in virulence between H37Rv and BCG, we used lower MOIs. The results showed that when THP-1 cells infected with H37Rv at an MOI of 1, the expression of miR-25 increased significantly. As the MOI increased, the expression level of miR-25 gradually increased (Figure 1F). We detected miR-25 expression in THP-1 cells infected with BCG or H37Rv at different time points. The results showed that miR-25 increased by approximately 13, 5 and 4 times at 6, 12 and 24 h post BCG infection (6 hpi, 12 hpi and 24 hpi), respectively (Figure 1E). Also, the results showed that miR-25 increased by approximately 8, 13 and 5 times at 6, 12 and 24 h post H37Rv infection, respectively (Figure 1G). This demonstrated that the expression of miR-25 was significantly up-regulated in the early stages of infection, and the high expression of miR-25 gradually weakened with the extension of the infection time, which is similar to the result of BCG infection in vivo. These results prove that miR-25 is up-regulated during BCG and Mtb infection and may play a role in the early stages of infection.
Figure 1.

The miR-25 level is induced by Bacillus Calmette–Guérin and Mtb infection
(A and B) Quantitative real-time PCR (qRT-PCR) detection of the expression of miRNAs in the lungs of the control group or mice (n = 3) infected with 4.4 × 105 CFU BCG via tail vein for 3 days (A) and 7 days (B).
(C) qRT-PCR detection of the expression of miRNAs in THP-1 cells of the control group or cells infected with BCG or H37Rv.
(D) qRT-PCR detection of the expression of miR-25 in THP-1 cells infected with BCG at the indicated MOIs for 24 h.
(E) qRT-PCR detection of the expression of miR-25 in THP-1 cells infected with BCG for 6, 12 and 24 h.
(F) qRT-PCR detection of the expression of miR-25 in THP-1 cells infected with H37Rv at the indicated MOIs for 24 h.
(G) qRT-PCR detection of the expression of miR-25 in THP-1 cells infected with H37Rv for 6, 12 and 24 h. Data are presented as means ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.005, ns, not significant.
miR-25 promotes intracellular survival of BCG and Mtb
To explore the function of miR-25 during BCG infection, we examined the effect of miR-25 on the intracellular survival of BCG in macrophages. THP-1 cells pre-transfected with miR-25 mimic or miR-25 inhibitor were infected with BCG at an MOI of 20, cells were lysed at 6 hpi, 12 hpi and 24 hpi, and the survival of the bacteria in the cells was detected by colony-forming unit (CFU) assay. The effect of miR-25 mimic or inhibitor on cell viability was detected, and it was found that there were no significant differences between miR-25 mimic or inhibitor and their respective control groups (Figure S1A). The expression level of miR-25 after transient transfection was detected by qRT-PCR, and the results confirmed that the miR-25 mimic significantly increased miR-25 expression in THP-1 cells whereas the miR-25 inhibitor significantly reduced the miR-25 expression level (Figure S1B). CFU results showed that the miR-25 mimic significantly increased BCG survival in THP-1 cells. At 6 hpi, 12 hpi and 24 hpi, the number of surviving bacteria in the miR-25 mimic treatment group was approximately 1.5 times that of the control group (Figure 2A). The miR-25 inhibitor reduced the survival of BCG at three time points. At 6 hpi, the number of intracellular bacteria in the miR-25 inhibitor group had a downward trend compared with the control group whereas at 12 hpi and 24 hpi, the number of intracellular bacteria significantly decreased by 0.5 times (Figure 2B). In addition, we constructed a miR-25-overexpressing THP-1 cell line (miR-25 THP-1) and a miR-25-knockdown THP-1 cell line (sh-miR-25 THP-1). MiR-25 expression levels in miR-25 THP-1 and sh-miR-25 THP-1 cells were identified by using qRT-PCR, and the results showed that miR-25 was significantly increased in miR-25 THP-1 cells but significantly decreased in sh-miR-25 THP-1 cells (Figures S1C and S1D). The same amounts of miR-25 THP-1, WT THP-1, sh-NC THP-1 or sh-miR-25 THP-1 cells were seeded into 6-well plates, and cells were subsequently infected with BCG for CFU analysis at different time points. The results were consistent with previously revealed CFU analysis. The up-regulated expression of miR-25 promotes the survival of BCG in cells (Figure S1E). In contrast, the down-regulated expression of miR-25 reduced the intracellular survival of BCG (Figure S1F). Compared with the WT THP-1 group, the intracellular survival ratio of miR-25 THP-1 cells increased by 3 times at 6 hpi and approximately 1.5 times at 12 hpi and 24 hpi. Compared with the sh-NC THP-1 group, the intracellular survival rate of sh-miR-25 THP-1 cells was reduced by approximately 0.5 times after infection. To explore whether miR-25 plays the same role in the H37Rv infection process, we detected the survival of the bacteria in H37Rv-infected THP-1 cells pre-transfected with miR-25 mimic or miR-25 inhibitor at an MOI of 5 at 6 hpi, 12 hpi and 24 hpi, respectively. The results showed that miR-25 mimic promote intracellular survival of H37Rv, while miR-25 inhibitor reduce the intracellular survival of H37Rv (Figures 2C and 2D), which was consistent with the results demonstrated during BCG infection. We also demonstrated that H37Rv has a higher survival rate in miR-25 THP-1, and a lower survival rate in sh-miR-25 THP-1 cells (Figures S1G and S1H). These results prove that miR-25 can increase the intracellular survival of BCG and Mtb.
Figure 2.

miR-25 promotes the intracellular survival of BCG and Mtb
(A–D) THP-1 cells pre-transfected with control mimic, miR-25 mimic, control inhibitor or miR-25 inhibitor were infected with BCG or H37Rv for a specified period of time, and then CFU detectionwas performed.
(E) WT or miR-25−/− mice were infected with BCG for 7 days, and the lungs, livers and spleens were subjected to CFU assay, (F) lungs were treated with H&E staining, scale bar, 200 μm; the WT or miR-25−/− mice were infected with PBS for 7 days, (F) H&E staining in lungs of WT or miR-25−/−mice, scale bar, 200 μm. Data are presented as means ± SD . ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.005, ns, not significant.
miR-25 increases susceptibility to BCG infection
To evaluate the role of miR-25 in the process of BCG infection in vivo, we generated miR-25−/− mice by using the CRISPR-Cas9 genome editing method (Wang et al., 2013). We used qRT-PCR to measure the expression of members of the miR-25-93-106b cluster, and the results showed that miR-25 expression was significantly decreased (Figure S1I) but that miR-93 and miR-106b expression did not change significantly (Figure S1J). We infected WT mice or miR-25−/− mice with BCG via tail vein injection. At 7 dpi, CFU analysis found that miR-25−/− mice had lower bacterial loads in lung, spleen and liver than WT mice (Figure 2E). Pathological analysis showed that the lung tissues of WT mice and miR-25−/− mice treated with PBS at 7 dpi were normal (Figure 2F). BCG-infected WT mice had thickened alveolar walls and exuded inflammatory cells. The miR-25−/− mice showed slight signs of inflammation in the lungs (Figure 2F). These results suggest that miR-25 increases the sensitivity of mice to BCG infection.
miR-25 regulates autophagy pathway during BCG and Mtb infection
Previous studies have shown that the intracellular survival of Mtb is closely related to autophagy. It was demonstrated that enhancing the level of cell autophagy is beneficial to the elimination of intracellular Mtb (Alvarez-Jiménez et al., 2018; Pahari et al., 2020). To explore whether miR-25 affects the intracellular survival of BCG through the autophagy pathway, THP-1 cells pre-transfected with miR-25 mimic or inhibitor were infected with BCG and then treated with the autophagy inhibitor Bafilomycin A1 (BAF-A1) (Yoshimori et al., 1991). The CFU analysis results at 24 hpi showed that there was no significant difference in BCG intracellular survival between the different treatment groups (Figure S2A). These results suggest that miR-25 affects the intracellular survival of BCG through the autophagy pathway. To further explore the relationship between the function of miR-25 and autophagy, we detected the autophagy marker protein microtubule-associated protein 1 light chain 3 (MAP1 LC3/LC3) (Tanida et al., 2008). The conversion of cytoplasmic LC3-I to LC3-II located in the inner and outer autophagosome membranes indicates the formation of autophagosomes (Slobodkin and Elazar, 2013). The western blot results showed that the amount of LC3-II in the cell increased when miR-25 was overexpressed (Figures 3A and S2B) whereas the accumulation of LC3-II in the cell decreased when miR-25 was silenced (Figure 3B and S2C). These results showed that when miR-25 increases, the number of intracellular autophagosomes increases, and the intracellular survival of BCG increases. P62 is an autophagy substrate that can be used to detect autophagic flux (Pankiv et al., 2007; Mizushima et al., 2010), and the western blot results showed that the miR-25 mimic lead to increased accumulation of P62 (Figure 3A). The miR-25 inhibitor reduced the amount of P62 compared with the inhibitor control group only at 12 hpi and 24 hpi, which may be because the effect of the miR-25 inhibitor was not as strong as that of the miR-25 mimic (Figure 3B). The same function was verified during H37Rv infection: the miR-25 mimic increased the accumulation of LC3-II whereas the miR-25 inhibitor had the opposite effect (Figures 3C and 3D). The above results indicate that miR-25 affects the survival of BCG and Mtb through the autophagy pathway and may prevent the fusion stage of autophagosomes and lysosomes. Furthermore, we used immunofluorescence (IF) to detect the co-localization of the autophagosome marked with LC3 protein and the lysosome marked with LAMP1 protein and measured the Pearson correlation coefficient (PCC). PCC is a standard statistical analysis that can measure the strength of the linear relationship between two variables. In 1992, Mendes used PCC to improve the analysis of fluorescence co-localization. The basic principle is that the physical relationship between molecules is represented by a statistical relationship(Barlow et al., 2010). The value of the PCC ranges from −1 to 1, which is closer to 1, indicating a stronger co-localization relationship (Dunn et al., 2011). We found that co-localization decreased after miR-25 mimic treatment, and co-localization increased after miR-25 inhibitor treatment (Figures 2E and 2G). The results of the co-localization in the cell line were similar to the above results. The co-localization of autophagosomes and lysosomes was reduced in miR-25 THP-1 cells compared with WT THP-1 cells, whereas increased in sh-miR-25 THP-1 cells compared with sh-NC THP-1 cells (Figures S2D and S2F). The same function was verified during H37Rv infection: the miR-25 mimic decreased the co-localization of LC3-labeled autophagosomes and LAMP1-labeled lysosomes whereas the miR-25 inhibitor had the opposite effect (Figures 3F and 3H). During H37Rv infection, the co-localization of autophagosomes and lysosomes in miR-25 THP-1 cells was less than that of WT THP-1 cells whereas the co-localization in sh-miR-25 THP-1 cells was more than that of sh-NC THP-1 cell (Figures S2E and S2G). The mCherry red fluorescence protein-green fluorescence protein-LC3B (mRFP-GFP-LC3B) reporter system, a red and green double fluorescence reporter system, is often used to detect autophagic flux. Because the GFP label is sensitive to pH and RFP is not, red fluorescence indicates the fusion of autophagosomes and lysosomes, yellow fluorescence indicates the co-localization of red fluorescent protein and green fluorescent protein, indicating that autophagic flux was interrupted (Kimura et al., 2007). MRFP-GFP-LC3B THP-1 cells were pre-transfected with miR-25 mimic or inhibitor and infected with BCG to observe the co-localization of red and green protein. The results reveal that the miR-25 mimic prevents the conversion of autophagosomes to autophagolysosomes with more yellow fluorescent signal (Figure S3A). PCC measurements also showed that the co-localization of red and green fluorescent proteins increased (Figure S3C). While the miR-25 inhibitor increases the fusion of autophagosomes and lysosomes, resulting in almost complete quenching of the green fluorescent signal (Figure S3A), PCC measurement also showed that the co-localization of red and green fluorescent proteins was significantly reduced (Figure S3C). The similar results were verified during H37Rv infection: the miR-25 mimic prevents the conversion of autophagosomes to autophagolysosomes, which present more yellow fluorescent and higher value of PCC whereas the miR-25 inhibitor had the opposite effect (Figures S3B and S3D). It was suggested that the miR-25 mimic blocked the fusion of autophagosomes and lysosomes whereas the miR-25 inhibitor promoted autophagic flux. All the results suggested that miR-25 affects the intracellular survival of BCG and Mtb through the autophagy pathway.
Figure 3.

miR-25 reguletes autophagy pathway during BCG and Mtb infection
(A–F) THP-1 cells pre-transfected with control mimic, miR-25 mimic, control inhibitor, miR-25 inhibitor NPC1 siRNA, or NFKBIZ siRNA were infected with BCG or H37Rv for a specified period of time, western blotting of the lysate was performed to detect the amount of LC3 and P62 (A, B, C and D), indirect immunofluorescence (IF) analysis of the co-localization of autophagosomes (LC3, green) and lysosomes (LAMP-1, red) bar, 10 μM (E and F).
(G and H) Pearson correlation coefficients (PCCs) of images of internalized Alexa Fluor 488-LC3 and Alexa Fluor 594-LAMP1 in THP-1 cells. Data are presented as means ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.005, ns, not significant.
miR-25 directly targets NPC1
We predicted the target genes of miR-25 by online softwares (TargetScane, miRDB, miRanda) and crossed target genes with autophagy pathway-related genes. The results indicated that NPC1, one of the cholesterol transport proteins in lysosomes, is a potential target gene of miR-25. We analyzed the binding positions of the NPC1-3′UTR (Figure 4A) and detected whether miR-25 directly binds to the 3′UTR of NPC1. We constructed plasmid pmirGLO-NPC1-WT-UTR containing NPC1 3′UTR, transfected pmirGLO-NPC1-WT-UTR and miR-25 mimic or control mimic into HEK293T cells and detected luciferase activity. The results showed that the relative luciferase was significantly decreased under the influence of the miR-25 mimic (Figure 4B). PmirGLO-NPC1-MUT-UTR contains four nucleotide mutations in the NPC1 3′UTR (from CAAU to ACCG), which are thought to be the binding sites for miR-25. PmirGLO-NPC1-MUT-UTR was constructed to confirm the binding sites of miR-25 and NPC1. PmirGLO-NPC1-MUT-UTR was co-transfected with miR-25 mimic or control mimic into HEK293T cells to measure the relative luciferase activity. The results showed that miR-25 did not influence the relative luciferase activity and indicated that miR-25 directly binds to the 3′UTR of NPC1 (Figure 4B).
Figure 4.

miR-25 directly targets NPC1
(A) Predicted binding sites of miR-25 and NPC1 3′UTR.
(B) Luciferase activity of HEK293T cells transfected with either PmirGLO-NPC1-WT-UTR or PmirGLO-NPC1-MUT-UTR plus control mimic and miR-25 mimic.
(C) THP-1 cells pretreated with control mimic, miR-25 mimic, control inhibitor or miR-25 inhibitor were infected with Bacillus Calmette–Guérin for a specified period of time, and qRT-PCR detection of NPC1 levels was performed,
(D and E) western blotting of the lysate was performed to detect the amount of NPC1. (F and G) THP-1 cells pre-transfected with control siRNA or NPC1 siRNA were infected with BCG or H37Rv for a specified period of time, and then CFU detection was performed, indirect immunofluorescence (IF) analysis of the co-localization of autophagosomes (LC3, green) and lysosomes (LAMP-1, red) bar, 10 μM (H).
(I) Pearson correlation coefficients (PCCs) of images of internalized Alexa Fluor 488-LC3 and Alexa Fluor 594-LAMP1 in THP-1 cells. Data are presented as means ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.005, ns, not significant.
To detect the effect of miR-25 on NPC1 expression levels during BCG infection, THP-1 cells pre-transfected with miR-25 mimic or miR-25 inhibitor were infected with BCG. The qRT-PCR results and western blot results showed that when miR-25 was overexpressed, NPC1 was significantly down-regulated at the transcriptional and protein levels (Figures 4C and 4D), and when THP-1 cells were transfected with miR-25 inhibitor, NPC1 was up-regulated (Figures 4C and 4E). NPC1 expression levels were also detected in miR-25 THP-1 and sh-miR-25 THP-1 cells infected with BCG, indicating the same trend. The expression level of NPC1 in miR-25 THP-1 cells was down-regulated at both mRNA and protein levels compared with WT THP-1 cells (Figures S4A and S4C). The opposite result was verified in sh-miR-25 THP-1 cells compared with sh-NC THP-1 cells (Figures S4B and S4D). The expression of NPC1 in the lung tissue of miR-25−/− mice was significantly increased compared to the expression of NPC1 in WT mice during BCG infection (Figure S4E). This evidence suggests that miR-25 regulates NPC1 expression by directly binding to the 3′UTR of NPC1 during BCG infection. Subsequently, we used western blotting to detect the expression level of NPC1 during THP-1 cells, and the results showed that NPC1 was down-regulated in THP-1 cells infected with BCG, which gradually decreased with increasing MOI (Figure S4F). NPC1 expression was also found in the lungs of mice infected with BCG through the tail vein and was reduced compared to NPC1 expression in mice infected with PBS (Figure S4G). These results demonstrate that miR-25 regulates NPC1 expression during BCG infection.
NPC1 inhibits intracellular survival of BCG and Mtb
To explore the effect of NPC1 on intracellular survival in BCG-infected macrophages, we designed a specific siRNA for NPC1 and demonstrated its effect by detecting NPC1 expression in THP-1 cells transfected with NPC1 siRNA by using qRT-PCR and western blot. The results showed that NPC1 siRNA can significantly reduce the expression of NPC1 (Figures S5A and S5C). There was no significant difference between the effects of NPC1 siRNA and NC siRNA on THP-1 cell viability (Figure S1A). THP-1 cells were transfected with NPC1 siRNA or NC siRNA and infected with BCG, and intracellular survival at different time points post BCG infection was detected. The results showed that the intracellular survival of the NPC1 siRNA transfection group was significantly increased by nearly 2 times compared with that of the control group at 6 hpi. At 12 hpi and 24 hpi, the intracellular survival increased by approximately 1.5 times (Figure 4F). In addition, we constructed the NPC1 knockdown cell line sh-NPC1 THP-1 and examined the expression of NPC1 at the transcriptional and protein levels. The results showed that NPC1 was significantly down-regulated in sh-NPC1 THP-1 cells (Figures S5B and S5D). After BCG infection, the intracellular survival in sh-NPC1 THP-1 or sh-NC THP-1 cells was detected at different time points. The results showed that the survival of BCG in sh-NPC1 THP-1 cells was about 2.5-fold, 1.5-fold and 1.5-fold higher than that of the control group at 6 hpi and 12 hpi, 24 hpi, respectively (Figure S5E). These results suggested that silencing NPC1 increased the viability of BCG in macrophages. Our previous data proved that miR-25 affects the intracellular survival of BCG by blocking the fusion of autophagosomes with lysosomes. To investigate whether NPC1 affects BCG intracellular survival through the autophagy pathway, we detected autophagosome and lysosome co-localization through confocal and PCC assay, and the results showed that when NPC1 was knocked down, autophagosome and lysosome co-localization was decreased (Figures 4H, 4I, S5G, and S5H). MRFP-GFP-LC3B THP-1 cells were transfected with NPC1 siRNA and infected with BCG which can increase the co-localization of red and green fluorescent (Figures S5H and S5I). The same result was verified in the H37Rv infection process. Silencing NPC1 reduced the fusion of autophagosomes and lysosomes and increased H37Rv intracellular survival (Figure 4G and S5F). In addition, we used IF to detect the co-localization of autophagosomes and lysosomes in THP-1 cells transfected with NPC1 siRNA and sh-NPC1 THP-1 cells infected with H37Rv. The results showed that NPC1 silencing reduced the co-localization of autophagosomes and lysosomes compared with the control cells (Figures 3F, 3H, S2E, and S2G). The co-localization of red and green fluorescent in MRFP-GFP-LC3B THP-1 cells were transfected with NPC1 siRNA and infected with H37Rv was similarly with that of BCG infection (Figures S3B and S3D). These results show that NPC1 can inhibit the intracellular survival of BCG and Mtb through the autophagy pathway by affecting the fusion of autophagosomes and lysosomes.
miR-25 regulates BCG and Mtb infection via NPC1
To determine whether miR-25 regulates the intracellular survival of BCG by targeting NPC1, THP-1 cells pre-transfected with NPC1 siRNA plus miR-25 mimic or miR-25 inhibitor were infected with BCG at an MOI of 20, and cells were lysed at 6 hpi, 12 hpi and 24 hpi. The expression level of NPC1 after transient transfection was detected by qRT-PCR and western blot. The results showed that under the effect of NPC1 siRNA, miR-25 mimic or miR-25 inhibitor did not change the expression of NPC1 (Figures S6A, S6B, and S6C). CFU results showed that miR-25 mimic or miR-25 inhibitor had no significant influence on BCG survival in THP-1 cells at different time points when transfected with NPC1 siRNA (Figures 5A and 5B). The western blot results showed that the amount of LC3Ⅱ showed the same trend as the CFU results. In the presence of NPC1 siRNA, the effect of miR-25 mimic increasing LC3Ⅱ or miR-25 inhibitor reducing LC3Ⅱ was weakened (Figures 5E and 5F). We detected autophagic flux in co-transfected THP-1 cells infected with BCG. The confocal microscopy results showed that NPC1 siRNA blocked the effect of the miR-25 mimic or miR-25 inhibitor on the fusion of autophagosomes and lysosomes (Figure 5I). PCC analysis showed that miR-25 did not reduce the co-localization of autophagosomes and lysosomes, and the miR-25 inhibitor did not increase the fusion of autophagosomes and lysosomes (Figure 5K). Almost the same amount of yellow fluorescent signal was present in MRFP-GFP-LC3B THP-1 cells transfected with NPC1 siRNA plus miR-25 mimic or control mimic (Figure S6D). PCC measurements also showed that the co-localization of red and green fluorescent proteins was not significantly increased (Figure S6F). Confocal and PCC analysis showed that miR-25 inhibitor did not promote autophagy flux compared with the control group. The same results were also proved during H37Rv infection. When THP-1 cells were co-transfected with NPC1 siRNA plus miR-25 mimic or inhibitor, the intracellular survival, accumulation of LC3Ⅱ and the fusion of autophagosomes and lysosomes did not change (Figures 5C, 5D, 5G, 5H, 5J, and 5L). MRFP-GFP-LC3B THP-1 cells were co-transfected with NPC1 siRNA plus miR-25 mimic or inhibitor had almost the same amount of yellow fluorescent signal (Figures S6E and S6G). These results indicate that miR-25 inhibits autophagy by targeting NPC1 to promote the intracellular survival of BCG and Mtb.
Figure 5.

NPC1 inhibits intracellular survival of BCG and Mtb
(A–J) THP-1 cells pretreated with control siRNA or NPC1 siRNA were infected with BCG or H37Rv for a specified period of time, and then CFU detection (A–D) was performed, western blotting of the lysate was performed to detect the amount of LC3 (E–H), IF analysis of the co-localization of autophagosomes (LC3, green) and lysosomes (LAMP-1, red) bar, 10 μM (I and J).
(K and L) PCCs of images of internalized Alexa Fluor 488-LC3 and Alexa Fluor 594-LAMP1 in THP-1 cells. Data are presented as means ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.005, ns, not significant.
Rv1759c induces the expression of miR-25 through NFKBIZ
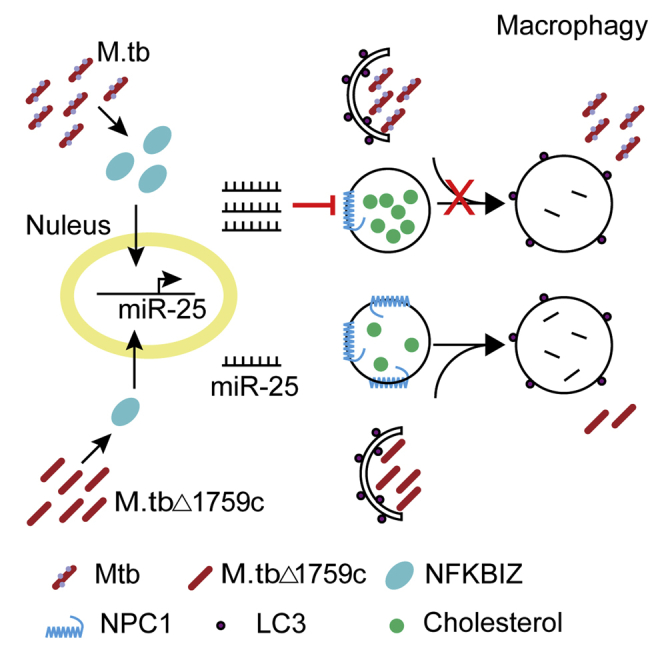
To explore the mechanism by which Mtb up-regulates the expression of miR-25, we used mutant strains of different genes to infect THP-1 macrophages and detected the expression of miR-25. The results showed that the expression of miR-25 was weakened when Rv1759c was deleted (Figure 6A). THP-1 cells were infected with H37Rv or H37RvΔ1759c and lysed at 4 hpi. The results of CFU assay showed that the number of intracellular bacteria of the two strains were almost the same (Figure 6B). We infected THP-1 with H37RvΔ1759c and H37Rv and performed CFU analysis at 24 hpi. The results showed that H37RvΔ1759c showed less survival than H37Rv (Figure 6C). Rv1759c is a member of the polymorphic GC-rich sequence (PE_PGRS) subfamily of proline–glutamic acid/proline–proline–glutamic acid (PE/PPE) family proteins. PE_PGRS proteins play important roles in the pathogenesis and immune escape of Mtb (Xie et al., 2021). It has been proved that the survival rate of Mycobacterium marine in granulomas was reduced after deletion of the PE_PGRS homologous gene (Ramakrishnan et al., 2000). Previous literature proved that PE_PGRS47 could inhibit the autophagy of macrophages and increase Mtb intracellular survival (Saini et al., 2016). Another study proved that PE_PGRS62 can reduce the maturation of phagosomes and the expression of IL-1β (Huang et al., 2012). To reveal the mechanism by which Rv1759c affects miR-25, we extracted RNA from H37Rv-infected and H37RvΔ1759c-infected THP-1 cells at an MOI of 1 at 24 hpi for sequencing. Samples were defined as the H37Rv infection group, H37RvΔ1759c infection group and CELL group. We found NFKB inhibitor zeta (NFKBIZ) regulatory factor of NFκB family, a gene down-regulated in H37Rv vs H37RvΔ1759c and up-regulated in CELL vs H37Rv. We used qRT-PCR to detect the expression of NFKBIZ in 9 samples to confirm the authenticity of the sequencing data. The results demonstrate that the change trends of mRNA expression in the H37Rv infection group and H37RvΔ1759c infection group compared to the CELL group are consistent with the sequencing results (Figures 6D and 6E). Previous studies have shown that NFKBIZ can complete the regulation of downstream factors through the NFκB signaling pathway (Slowikowski et al., 2020; Hörber et al., 2016; Tartey et al., 2014). The expression of NFKBIZ was up-regulated in the H37Rv infection group relative to the CELL group, but there was no difference between the H37RvΔ1759c infection group and the CELL group. The regulation of miR-25 expression by the NFκB signaling pathway has also been reported in the relevant literature (Zeng et al., 2021). These results suggest that the unchanged expression of miR-25 during H37RvΔ1759c infection may be caused by the unchanged expression of NFKBIZ, which affects the normal function of NFκB. We detected the expression level of miR-25 after transfected with NFKBIZ siRNA, and the results showed that silencing NFKBIZ significantly reduced the expression of miR-25 during H37Rv infection (Figure 6F). In addition, we proved that silencing NFKBIZ can reduce the viability of H37Rv in THP-1 cells (Figure 6G), which is the same as the effect of miR-25 inhibitor on the survival of H37Rv in macrophages. The confocal microscopy results showed that NFKBIZ siRNA increased the co-localization of LC3-labeled autophagosomes and LAMP1-labeled lysosomes during H37Rv infection (Figures 3F and 3H). The fusion of autophagosomes and lysosomes was increased in MRFP-GFP-LC3B THP-1 cells transfected with the NFKBIZ siRNA during H37Rv infection, resulting in almost complete quenching of the green fluorescent signal (Figure S3B), PCC measurement also showed that the co-localization of red and green fluorescent proteins was significantly reduced (Figure S3D). These results show that Rv1759c regulate NFκB through NFKBIZ to affect the expression of miR-25.
Figure 6.

Rv1759c induces the expression of miR-25 through NFKBIZ
(A–C) (A) qRT-PCR detection of the expression of miR-25 in THP-1 cells infected with H37Rv, H37RvΔphoP , H37RvΔ0818 , H37RvΔ3220c, H37RvΔ3143 or H37RvΔ1759c an MOI of 5. The H37Rv and H37RvΔ1759c were used to infect THP-1 cells at an MOI of 5 according to the abovementioned infection method, and then CFU detection was performed at 0 hpi, 4 hpi and 24 hpi (B and C).
(D) Relative expression levels of NFKBIZ from RNA-seq data.
(E) qRT-PCR detection of the fold changes of NFKBIZ relative to the CELL group. (F) qRT-PCR detection of the expression of miR-25 in THP-1 cells pre-transfected with NC siRNA or NFKBIZ siRNA and infected with H37Rv.
(G) THP-1 cells pre-transfected with NC siRNA or NFKBIZ siRNA were infected with H37Rv for a specified period of time, and then CFU detection was performed. Data are presented as means ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.005, ns, not significant.
Discussion
Mtb is a successful intracellular pathogen that can evade the elimination of host autophagy and establish infection. Accumulating evidence suggests that during Mtb infection, miRNAs can regulate intracellular survival through the autophagy pathway, but the mechanism by which miRNAs affect survival has not been completely clarified. To explore whether there are more miRNAs that affect Mtb intracellular survival, we detected the expression of miRNAs in lung tissues of 3-d and 7-d mice infected with BCG, and THP-1 cells infected with BCG or H37Rv. Some previous studies on miRNAs are consistent with our results. MiR-146a in the lung tissues of mice infected with Mtb (Malardo et al., 2016), and Mtb-infected primary dendritic cells (DCs) was up-regulated (Etna et al., 2018), which is consistent with our conclusions. Previous research conclusions clarified that miR-146a can reduce the production of nitric oxide (NO) in macrophages by targeting tumor necrosis factor (TNF) receptor-associated factor 6 (TRAF6) (Li et al., 2016b), reducing host defense against intracellular BCG. A previous study showed that miR-106b was up-regulated in Mtb-infected human peripheral blood mononuclear macrophages, consistent with our results. MiR-106b targets cathepsin S (CtsS) and increases the survival of Mtb in macrophages, probably by preventing antigen presentation or phagolysosome acidification (Pires et al., 2017). The mechanism of up-regulation of miR-20a in mouse lung tissues has also been demonstrated. MiR-20a in BCG-infected murine macrophages was up-regulated and inhibited autophagy by targeting autophagy-related proteins autophagy-related gene 7 (ATG7) and autophagy-related protein 16-1 (ATG16L1) to increase intracellular survival (Guo et al., 2016). Some miRNAs were only differentially expressed in clinical tuberculosis patients, but the mechanism in the course of Mtb infection is still unclear. The results of previous research are not completely consistent with ours. MiR-199a was down-regulated in the peripheral blood of patients with tuberculous meningitis (TBM) compared with healthy people (Pan et al., 2019). Our results showed that miR-199a was only up-regulated in the lung tissues of mice infected with BCG, with an expression trend opposite to previous studies. Our results showed that miR-199b and miR-320b are up-regulated in mouse lung tissues but down-regulated in THP-1 cells infected with H37Rv. Previous studies have shown that miR-199b is up-regulated in the peripheral blood macrophages of patients with active TB (Zhang et al., 2019) and the serum of patients with multidrug-resistant tuberculosis (MDR-TB) (Wang et al., 2016) compared with healthy individuals, and miR-320b is up-regulated in the serum of patients with TB (Zhang et al., 2013). Studies have shown that miR-429 is down-regulated in exosomes in the pleural effusion of TB patients (Wang et al., 2017). Our results showed that miR-429 was up-regulated in BCG-infected mouse lung tissue and H37Rv-infected THP-1 cells. These results indicate that the immune responses of different hosts to different strains are not exactly the same, and the expression of miRNAs in different types of samples are not exactly the same. We found that miR-186 was up-regulated in mouse lung tissue, while miR-25 was significantly up-regulated in different samples. We chose miR-25 for the next functional study to reveal its regulatory mechanism.
MiR-25 and miR-146a showed significant up-regulation in the lung tissues of infected BCG mice at 3 dpi or 7 dpi, but there were significant differences in the fold change between miR-146a and miR-25. MiR-25 had the highest fold up-regulation at 3 dpi, and miR-146a had the highest fold up-regulation at 7 dpi. MiR-146a is up-regulated in the early and late stages of BCG infection and may play a greater role in the late stages of infection. The expression of miR-25 was the highest at 6 hpi in the BCG in vitro infection experiment and 12 hpi in the H37Rv in vitro infection experiment. CFU analysis results showed that miR-25 mimic had the strongest effect at 6 hpi, and the miR-25 inhibitor had the weakest effect at 6 hpi. The same trend was also shown in the analysis of CFU of cell lines. These results suggest that miR-25 plays a role in the early stages of BCG infection and that there may be more miRNAs that play roles in the early stages of Mtb infection, which may require more attention.
To investigate whether miR-25 affects BCG intracellular survival through the autophagy pathway, we blocked autophagy using the V-ATPase-specific inhibitor BAF-A1 and found that the effect of miR-25 on BCG intracellular survival was abolished (Mauvezin and Neufeld, 2015). The initiation of autophagy, the formation of autophagosomes and the fusion of autophagosomes and lysosomes are the three steps by which autophagy completes the elimination of intracellular pathogens. Previous studies have shown that Mtb can inhibit the clearance of the host by interfering with different stages of autophagy (Garg et al., 2020; Romagnoli et al., 2012; Etna et al., 2018; Guo et al., 2016). We found that when miR-25 was overexpressed, the amount of LC3-II increased, indicating that the number of autophagosomes in macrophages increased, but the elimination of intracellular bacteria was reduced. Confocal experiments showed that overexpression of miR-25 reduced the co-localization of autophagosomes and lysosomes and blocked the fusion of autophagosomes and lysosomes in dual fluorescence detection. When miR-25 was inhibited, the opposite results were presented. These results suggest that miR-25 blocks autophagic flux by reducing the formation of autophagolysosomes. We used bioinformatics prediction and experimental verification to prove that miR-25 regulates the target gene NPC1 related to autophagy. Niemann-Picktype C1 (NPC1) is one of the cholesterol transport proteins in lysosomes, and NPC1 and Niemann-Picktype C2 (NPC2) cooperate to complete the transport of cholesterol in lysosomes. Soluble NPC2 accepts cholesterol from low-density lipoprotein (LDL) and carries it to the N-terminal domain of the membrane protein NPC1, and cholesterol is inserted into the lysosomal membrane by NPC1 to achieve cholesterol transport within the lysosome(Kwon et al., 2009). The loss of function caused by either NPC1 or NPC2 mutations can result in Niemann-Pick disease type C (NPDtype C). Studies have found that autophagy in fibroblasts of patients with NPD-type C is impaired, and in NPC1 mutant or deleted cell lines, autophagolysosome formation is reduced and autophagic flux is blocked (Sarkar et al., 2013). Stimulating autophagy can alleviate the accumulation of cholesterol in cells and can be used as a potential NPD-type C treatment (Sarkar et al., 2014). Previous studies have shown that mutations or deletions of NPC1 impairs lysosomal function, and functional lysosomes are critical to ensure complete autophagy. We examined NPC1 function during Mtb infection and found that under NPC1 silencing in vitro, autophagic flux was blocked during BCG or H37Rv infection and that the intracellular survival rate was increased. In both siRNA-transfected THP-1 cells and sh-NPC1 THP-1 cells, survival rate was higher at 6 hpi than at 24 hpi, supporting the conclusion that miR-25 is an early miRNA during Mtb infection. This result is not only consistent with the function of miR-25 but also consistent with previous studies showing that the mutation or absence of NPC1 disrupts the function of lysosomes and impairs autophagy (Sarkar et al., 2013). NPD-type C disease is a lysosomal storage disease (LSD) caused by lysosomal cholesterol accumulation because of NPC1 mutation. Abnormalities in lysosomes in LSD damage the autophagy pathway by affecting the degradation of autophagy substrates in the cell and leading to the accumulation of autophagosomes and substrates (Myerowitz et al., 2021; Ballabio and Bonifacino, 2020; Settembre et al., 2008; Seranova et al., 2017; Lieberman et al., 2012). The normal function of lysosomes is the key for the host to complete the elimination of intracellular Mtb through the autophagy pathway. A previous study found that NPC1 is down-regulated in BCG infection, which is consistent with our finding (Lee et al., 2010). NPD-type C disease caused by NPC1 mutation is a neurodegenerative disease. The mouse model with NPC1 mutation will show mild cerebellar ataxia and other neurological symptoms from 6 weeks of age and continue to worsen (Colombo et al., 2021; Meneses-Salas et al., 2021; Kurokawa et al., 2021). Therefore, we performed NPC1-related functional verification in vitro.
Rv1759c is a member of the PE/PPE protein family. The literature proves that the family proteins PPE10, PPE60, PPE27, PPE44 and PE13 can regulate the NFκB signaling pathway and change the expression of its downstream target genes, including host cytokines or change the strain intracellular survival (Asaad et al., 2021; Gong et al., 2019; Su et al., 2018; Yang et al., 2017; Yu et al., 2017; Li et al., 2016a). We demonstrated that the deletion of Rv1759c can reduce the survival of Mtb in macrophages. By analyzing RNA-seq data, we found that the expression of the atypical inhibitor of κB (IκB) regulatory factor (NFKBIZ) was unchanged in H37RvΔ1759c-infected cells, whereas it was up-regulated in H37Rv-infected cells. It was reported that NFKBIZ can interact with NFκB protein to promote or inhibit the transcription of NFκB target genes (Grondona et al., 2018; Niida et al., 2012; Kohda et al., 2016). During H37RvΔ1759c infection, the expression of NFκB target genes including cytokines and chemokines, such as IL-6 and IL10, is lower than that of H37Rv infection. These results suggest that NFKBIZ positively regulates the NFκB signaling pathway during Mtb infection. Previous studies showed that miR-25 is regulated by the NFκB signaling pathway. We suppose that miR-25 was regulated by NFKBIZ and affects the intracellular survival of Mtb. We demonstrated that the silencing of NFKBIZ leads to the down-regulation of miR-25 expression and the increase of intracellular survival of Mtb. These results indicate that the effect of Rv1759c on Mtb intracellular survival regulate the expression of miR-25 through NFKBIZ.
Overall, our study demonstrate that Rv1759c regulates miR-25 expression to down-regulate NPC1 in the early stages of infection through NFKBIZ, thereby blocking the fusion of autophagosomes and lysosomes, revealing the strategy by which Mtb inhibits host autophagy. These findings provide candidate molecules for the development of TB therapeutics.
Limitations of the study
In the present study, we demonstrate that miR-25 targets NPC1 and enhances BCG and H37Rv survival by impairing autophagic flux. A limitation of this study is that, although we indicated the effect of miR-25 on NPC1 protein expression, we did not simultaneously examine the expression of another cholesterol transporter protein NPC2, and the lysosomal membrane protein LAMP1. This study focused on the fusion of autophagosomes and lysosomes, and did not explore the effect of miR-25 on autophagy initiation and autophagic membrane elongation. To better characterize the link to autophagy, more autophagy markers such as Beclin-1, Atg5, p62 in western blot and immunofluorescence assay should be tested.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| mouse anti-β-actin | Cell Signaling Technology | Cat#3700; RRID: AB_2242334 |
| rabbit anti-LC3 | Cell Signaling Technology | Cat#3868; RRID: AB_2137707 |
| rabbit anti-NPC1 | Abcam | Cat#ab134113; RRID:AB_2734695 |
| rat anti-LAMP1 | Abcam | Cat#ab25245; RRID:AB_449893 |
| HRP goat anti-rabbit IgG | Abclone | AS014 |
| HRP goat anti-mouse IgG | Abclone | AS003 |
| Alexa Fluor 594-labeled goat anti-rat IgG | Antgene | ANT035 |
| Alexa Fluor 488-labeled goat anti-rabbit IgG | Antgene | ANT024 |
| Bacterial and virus strains | ||
| M. tuberculosis H37Rv | ATCC | ATCC 27294 |
| BCG Tokyo strain | ATCC | ATCC 35737 |
| M. tuberculosis H37RvΔphoP | This paper | N/A |
| M. tuberculosis H37RvΔ0818 | This paper | N/A |
| M. tuberculosis H37RvΔ3220c | This paper | N/A |
| M. tuberculosis H37RvΔ3143 | This paper | N/A |
| M. tuberculosis H37RvΔ1759c | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| 7H11 agar media | Difco | 212203 |
| 7H9 agar media | Difco | 271310 |
| Baf-A1 | Selleck | S1413 |
| DAPI | Beyotime | C1005 |
| OADC | Difco | 212352 |
| Glycerol | Sigma Aldrich | G5516 |
| Tween 80 | Sigma Aldrich | P8074 |
| RPMI-1640 | Gibco | 31800022 |
| DMEM | Gibco | 12800017 |
| HiPerFect Transfection Reagent | Qiagen | 301705 |
| PMA | Sigma Aldrich | 79346 |
| TRIzol | Life Technologies | 15596026 |
| Lipofectamine™ 2000 Transfection Reagent | Invitrogen | 11668019 |
| RIPA lysis buffer | Beyotime | P0013B |
| Critical commercial assays | ||
| RNeasy minikit | Qiagen | 217004 |
| miRcute Plus miRNA First-Strand cDNA Synthesis Kit | Tiangen | KR21 |
| miRNA using miRcute Plus miRNA qPCR Detection Kit (SYBR Green) | Tiangen | FP411 |
| HiScript II Q Select RT SuperMix for qPCR(+gDNA wiper) | Vazyme | R233 |
| AceQ qPCR SYBR Green Master Mix | Vazyme | Q111 |
| Dual-Luciferase® Reporter (DLR™) Assay System | PROMOGA | E1910 |
| Deposited data | ||
| Raw and analyzed data | This paper | GSE184660 |
| Experimental models: Cell lines | ||
| THP-1 | ATCC | TIB-202 |
| 293T | ATCC | CRL-3216 |
| Experimental models: Organisms/strains | ||
| MiR-25−/− mice | BIOCYTOGEN | N/A |
| C57BL/6 mice | Charles River Labs | N/A |
| Oligonucleotides | ||
| MiR-25 mimic CAUUGCACUUGUCUCGGUCUCUGA |
GenePharma | B02003 |
| control mimic UUCUCCGAACGUGUCACGUTT |
GenePharma | B04002 |
| miR-25 inhibitor UCAGACCGAGACAAGUGCAAUG |
GenePharma | B03001 |
| inhibitor control CAGUACUUUUGUGUAGUACAA |
GenePharma | B04001 |
| NPC1 siRNA GAGGUACAAUUGCGAAUAUTT |
GenePharma | A01001 |
| NFKBIZ siRNA GCCCGAUUCGUUGUCUGAUTT |
GenePharma | A01001 |
| control siRNA UUCUCCGAACGUGUCACGUTT |
GenePharma | B04002 |
| Primers for qPCR see Table S1 | This paper | N/A |
| Recombinant DNA | ||
| pmirGLO | PROMOGA | E1330 |
| psPAX2 | Addgene | 12260 |
| pMD2.G | Addgene | 12259 |
| pLVX-shRNA1 | HedgehogBio | HH-shRNA-044 |
| PLVX-puro | HedgehogBio | HH-LV-048 |
| Software and algorithms | ||
| TargetScan | http://www.targetscan.org/ | N/A |
| miRDB | http://www.mirdb.org/miRDB/ | N/A |
| miRanda | http://www.microrna.org/microrna/home.do | N/A |
| GraphPad Prism 8 | https://www.scienceplus.com/nl/prism-commercieel.html | N/A |
| Image Lab Software | https://www.bio-rad.com/fr-fr/product/image-lab-software?ID=KRE6P5E8Z | N/A |
| ZEN | https://www.zeiss.com/microscopy/int/products/microscope-software/zen-lite.html | N/A |
| UCSC | http://genome.ucsc.edu/ | N/A |
Resource availability
Lead contact
Further information and requests for resources should contact to Prof. Chen Tan (tanchen@mail.hzau.edu.cn).
Materials availability
This study did not generate new unique reagents.
Method details
Antibodies and reagents
Mouse anti-β-actin (Cell Signaling Technology, 3700), rabbit anti-LC3 (Cell Signaling Technology, 3868), rabbit anti-NPC1 (Abcam, ab134113), rat anti-LAMP1 (Abcam, ab25245) antibody, HRP goat anti-rabbit IgG (H+L) (Abclone, AS014), HRP goat anti-mouse IgG (H+L) (Abclone, AS003), Alexa Fluor 594-labeled goat anti-rat IgG (H + L) (Antgene, ANT035) and Alexa Fluor 488-labeled goat anti-rabbit IgG (H + L) (Antgene, ANT024) antibodies were used. Bafilomycin A1 (Baf-A1; Selleck, S1413), DAPI (Beyotime, C1005).
Cell culture and bacterial strains
The Mtb reference strain H37Rv (ATCC 27294) and BCG Tokyo strain (ATCC 35737) were cultured in Middlebrook 7H9 medium (Becton Dickinson, Difco, 271310) supplemented with 10% oleic acid, albumin, dextrose (OADC) (Becton Dickinson, Difco, 212352), 0.5% (v/v) glycerol (Sigma Aldrich, G5516), and 0.05% (v/v) Tween 80 (Sigma Aldrich, P8074). We recombined the homologous allelic substitute (AES) containing Sac B (sucrose counterselection gene) and Hyg (hygromycin resistance) tags on the temperature-sensitive vector (ts). At 30°C, the synthesis of phage was induced. The collected high-titer phage was used to infect H37Rv, and the gene-deleted strain was obtained by homologous recombination (Bardarov et al., 2002; Jain et al., 2014). The phage expressing the dissociation enzyme Tnp R was used for the subsequent elimination of the sac B-hyg tag. H37RvΔphoP, H37RvΔ0818, H37RvΔ3220c, H37RvΔ3143 or H37RvΔ1759c were obtained according to the above method. Bacteria that grew to the logarithmic period were collected, re-suspended in RPMI-1640 medium supplemented with 10% fetal bovine serum and stored at −80°C. After a series of dilutions, bacteria were plated on solid Middle Brook 7H11 medium (Becton Dickinson, Difco, 212203) supplemented with 10% OADC and 0.5% (v/v) glycerol. The colony forming assay was used to count the number of viable bacteria (Li et al., 2017). THP-1 human monocyte macrophages were grown in RPMI-1640 medium supplemented with 10% fetal bovine serum, and HEK293T cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS, which were both cultured at 37°C with 5% CO2.
To knock down miR-25, the shRNA sequence targeting miR-25 was cloned into the pLVX-shRNA1 vector to generate the plasmid pLVX-sh-miR-25. To knock down NPC1, the shRNA sequence targeting NPC1 was cloned into the pLVX-shRNA1 vector to generate the plasmid pLVX-sh-NPC1. The negative control shRNA sequence was cloned into the PLVX-shRNA1 vector to generate the plasmid pLVX-sh-NC. A fragment containing miR-25 from the THP-1 genome was amplified by using primers (miR-25-F: CGGAATTCCACATCTTCTTTCTCCACCAC, miR-25-R: CGGGATCCGGACACGTTCTCTCTGC) and cloned into the pLVX-Puro vector using EcoR1 and Bamh1 to generate PLVX-puro-miR-25. Then, HEK293T cells were transfected with psPAX2, pMD2.G and pLVX-sh-miR-25/pLVX-sh-NPC1/pLVX-sh-NC/pLVX-Puro-miR-25 to produce lentivirus. THP-1 cells were infected with lentivirus and cultured in the presence of 1 μg/mL puromycin (InvivoGen, ant-pr-1) for 7 days. Sh-miR-25 THP-1, sh-NPC1 THP-1 and miR-25 THP-1 cells were identified by RT-PCR.
HEK293T cells were transfected with psPAX2, pMD2.G and pLVX-Puro-RFP-GFP-hLC3B to product lentivirus. THP-1 cells were infected with lentivirus and cultured in the presence of 1 μg/mL puromycin for 7 days. MRFP-GFP-LC3B THP-1 cells were detected by fluorescence microscopy.
The target sequence for sh-miR-25 is TCAGACCGAGACAAGTGCAATG.
The target sequence for sh-NPC1 is CTCCCATCGATAGCAATAT.
The target sequence for sh-NC is GCTTCGCGCCGTAGTCTTA.
Macrophage transfection
THP-1 cells (1.5×106) were seeded in each well of 6-well plates and differentiated with PMA. Then, 50 nM miR-25 mimic, control mimic, miR-25 inhibitor, inhibitor control, NPC1 siRNA or control siRNA was transfected into cells by using HiPerFect Transfection Reagent (Qiagen, 301705) according to the reagent instructions. Cells were transfected 48 h before collection or further experiments.
MiR-25 mimic (5′-CAUUGCACUUGUCUCGGUCUCUGA-3′), control mimic (5′-UUCUCCGAACGUGUCACGUTT-3′), miR-25 inhibitor (5′-UCAGACCGAGACAAGUGCAAUG-3′), inhibitor control (5′-CAGUACUUUUGUGUAGUACAA-3′), NPC1 siRNA (5′-GAGGUACAAUUGCGAAUAUTT-3′), NFKBIZ siRNA (5′-GCCCGAUUCGUUGUCUGAUTT-3′) and control siRNA (5′-UUCUCCGAACGUGUCACGUTT-3′) were purchased from GenePharma.
Macrophage infection
THP-1 cells were differentiated with 100 ng/mL phorbol myristate acetate (PMA) for 48 h and incubated with 1640 medium for another 8 h. Cells were infected with a series of multiplicities of infection (MOIs). At 4 h post infection (4 hpi), cells were washed three times with phosphate-buffered saline (PBS) and incubated at 37°C with fresh medium. Cells were collected with TRIzol reagent (Life Technologies, 15596026) at 0, 6, 12, and 24 h post infection (0 hpi, 6 hpi, 12 hpi and 24 hpi).
Intracellular survival of bacteria
THP-1 cells were activated, transfected and infected with bacteria. At 6 hpi, 12 hpi and 24 hpi, THP-1 cells were lysed with 0.025% Triton X-100 after washing with PBS. The intracellular bacteria were continuously diluted in PBS, and the appropriate dilution was selected for plating and counting. Alternatively, after 4 h of infection, the cells were incubated with fresh medium supplemented with 20 nM bafilomycin A1, and the intracellular survival of bacteria was detected by CFU assay. The experiment had three biological replicates and three technical replicates.
RNA-seq library preparation
The H37Rv strain and H37RvΔ1759c strain were used to infect THP-1 cells at an MOI of 1 according to the above mentioned infection method, and samples were collected at 24 hpi, defined as the H37Rv infection group, H37RvΔ1759c infection group and CELL group. Each group of samples had three multiple replicates. Total RNA of the three groups was isolated by using an RNeasy minikit (Qiagen, 217004) and interrupted to fragments approximately 300 bp in length by using ion interruption. After synthesizing the first strand cDNA and the second strand cDNA, PCR amplification was used to enrich the library fragments, and then the library was subjected to quality inspection by the Agilent 2100 Bioanalyzer. Next-generation sequencing technology (next-generation sequencing, NGS), based on the Illumina HiSeq sequencing platform, was used to perform paired-end (PE) sequencing on these libraries. We defined the genes whose differential expression met |log2FoldChange| > 1 and whose P-value < 0.05 as differentially expressed genes (DEGs). Library construction and sequencing were completed by Shanghai Personalbio Technology Company.
RNA Isolation and Quantitative RT-PCR (qRT-PCR)
Total RNA was isolated from cells and tissues using an RNeasy minikit (Qiagen, 217004). The tailing method for rapid batch detection of miRNA expression, reverse transcription of miRNA using miRcute Plus miRNA First-Strand cDNA Synthesis Kit (Tiangen, KR21), quantitative analysis of miRNA using miRcute Plus miRNA qPCR Detection Kit (SYBR Green) (Tiangen, FP411). (miR-146a-F:5′-TTGAGAACTGAATTCCATGGGTT-3′,miR-20a-F:5′-TAAAGTGCTTATAGTGCAGGTAG-3′,miR-25-F:5′-CATTGCACTTGTCTCGGTCTGA-3′,miR-186-F:5′-CAAAGAATTCTCCTTTTGGGCT-3′,miR-199a-F:5′-CCCAGTATTCAGACTACCTGTTC-3′,miR-199b-F:5′-CCCAGTGTTTAGACTACCTGTTC-3′,miR-320b-F:5′-AAAAGCTGGGTTGAGAGGGCGA-3′,miR-429-F:5′-GTAATACTGTCTGGTAAAACCGT-3′). For more accurate determinate miR-25miR-93 and miR-106b expression, reverse transcription by using HiScript II Q Select RT SuperMix for qPCR(+gDNA wiper) (Vazyme, R233) with reverse transcription primer(GenePharma, F02001), total RNA was reverse-transcribed to cDNA by using HiScript II Q Select RT SuperMix for qPCR(+gDNA wiper) (Vazyme, R233) with Oligo dT, the quantitative of target genes by using AceQ qPCR SYBR Green Master Mix (Vazyme, Q111). The real-time PCR was conducted on a Bio-Rad CFX RT-PCR machine, the β-actin is the reference mRNA for NPC1, U6 small nuclear RNA (U6) is the reference miRNA of miRNA in cells and tissues. The gene expression changes were calculated by the 2-ΔΔCt method. (β-actin-F:5′-TTGGCTTGACTCAGGATT-3′,β-actin-R:5′-CTGTAACAACGCATCTCATAT-3′,NPC1-F:5′-TACCAGACATCAGGCTACTTAACA-3′,NPC1-R:5′-CGATTCCGTAATGCTTCCGATT-3′,NFKBIZ-F:5′-CAAGGTGTGAACATAGAAC-3′,NFKBIZ-R:5′-TCCATCAGACAACGAATC-3′, U6-F:5′-CTCGCTTCGGCAGCACA-3′,U6-R:5′-AACGCTTCACGAATTTGCGT-3′) We summarized the primer sequences used to conduct gene expression analysis using qPCR into an excel table named as Table S1 and uploaded as supplementary data.
Dual luciferase report assay
The reporter plasmid pmirGLO-NPC1-WT-UTR was obtained by amplifying the NPC1-3′UTR using PCR with cDNA and inserting it into the pmirGLO vector. The reporter plasmid pmirGLO-NPC1-UTR-MUT was obtained by inserting a point-mutated NPC1-3UTR amplicon by using fixed-point mutation PCR into the pmirGLO vector. When the fusion degree of HEK293T cells in a 24-well plate was approximately 80%, 200 ng of reporter plasmid pmirGLO-NPC1-UTR/pmirGLO-NPC1-UTR-MUT and miR-25 mimic/control mimic at a final concentration of 100 nm were transfected into cells by using Lip2000. At 24 h posttransfection, the Dual-Luciferase® Reporter (DLR™) Assay System (PROMOGA, E1910) was used to detect the activity of firefly luciferase and Renilla luciferase and calculate the ratio of the firefly luciferase value to the Renilla luciferase value.
(NPC1-UTR-F: 5′-CGAGCTCCCCTCTCGCAGGGCATG-3′,
NPC1-UTR-R: 5′-CCGCTCGAGTAGAATCTCTTCCATTTAGCTT-3′,
MUT-NPC1-F: 5′-AAAGAGCTTTATTAATGACCGAAATTAACTTTGTACACATTTTTATAT-3′, MUT-NPC1-R: 5′-ATATAAAAATGTGTACAAAGTTAATTTCGGTCATTAATAAAGCTCTTT-3′).
Western blot
The cells were lysed with RIPA lysis buffer (Beyotime, P0013B) and incubated for 10 min on ice. The samples were centrifuged at 12,000 rpm for 10minat 4°C. The sample supernatant was added to 5× SDS Loding Buffer to a final concentration of 1× and boiled at 100°C for 10 min. Proteins were separated by SDS-polyacrylamide gel electrophoresis (12%, w/v), and the target proteins were transferred to nitrocellulose membranes (Millipore, ISEQ00010). The membranes were blocked with 5% BSA diluted in TBST for 2hat room temperature and incubated with primary antibodies at 4°C overnight. Membranes were washed with TBST and then incubated with secondary antibodies at room temperature for 1 h. Membranes were washed with TBST, and protein levels were quantified by the Gel imaging system (Bio-Rad, USA). ß-actin was used as loading control. Mouse antiβ-actin and rabbit antiLC3 were diluted 1:1000 in blocking solution, and rabbit antiNPC1, HRP goat anti-rabbit IgG and HRP goat anti-mouse IgG were diluted 1:5000 in blocking solution.
Immunofluorescence
THP-1 cells (2×105) were seeded into a glass-bottomed dish, differenced, transfected and infected according to the above experimental method. Macrophages were fixed with 4% paraformaldehyde at room temperature for 30 min, treated with 1% Triton X-100 diluted in PBS at room temperature for 20 min, and blocked with 5% BSA diluted in PBS at room temperature for 2 h. The cells were incubated with primary antibodies overnight at 4°C, incubated with secondary antibodies at room temperature in the dark for 1 h and incubated with DAPI for 10minat room temperature. A confocal microscope (Carl Zeiss LSM 880 Confocol Microscope) was used to measure the co-localization of LC3 with LAMP1. Rabbit antiLC3, rat antiLAMP1, Alexa Fluor 594-labeled goat anti-rat IgG (H + L) and Alexa Fluor 488-labeled goat anti-rabbit IgG (H + L) (Antgene, ANT024).
A total of 2×105 mRFP-GFP-LC3B THP-1 cells were seeded into a glass-bottomed dish, differenced, transfected and infected according to the above experimental method. Confocal microscopy was used to measure red fluorescence and green fluorescence.
Bioinformatics analysis
Target gene prediction was conducted using the software TargetScan (v7.2, http://www.targetscan.org/), miRDB (http://www.mirdb.org/miRDB/) and miRanda (http://www.microrna.org/microrna/home.do). NPC1 3UTR sequence was from UCSC (http://genome.ucsc.edu/).
Experimental model and subject details
We commissioned BIOCYTOGEN to customize miR-25-deficient mice and obtained F1-positive heterozygous mice(C57BL/6 background). F1-generation mice were bred in the Experimental Animal Center of Huazhong Agricultural University to obtain miR-25−/− homozygous mice. Mice that were genotyped and tested for miR-25 expression were used for subsequent experiments. C57BL/6 mice were purchased from Charles River Labs (Beijing, China). The experimental infection research was carried out according to the Guidelines for the Supervision and Use of Experimental Animals in Hubei Province, and the experimental protocol was approved by the Scientific Ethics Committee of Huazhong Agricultural University (Permit Number: HZAUMO-2019-056).
Six-to eight-week-old female C57BL/6 WT mice were purchased from Charles River Labs. WT mice with similar body weights were reared on the same diet for 3 to 5 days to adapt to the environment. WT mice were divided into a control group (n = 6) and an infection group (n = 6). Mice were infected with 4.4×105 BCG or the same volume of PBS via tail vein injection. At 3- and 7-days post infection (3 dpi and 7 dpi), the mice were euthanized, and lung tissue was collected to detect miRNA expression.
Six- to eight-week-old female C57BL/6 WT mice were purchased from Charles River Labs. Six- to eight-week-old female C57BL/6 miR-25−/−mice were generated in the Experimental Animal Center of Huazhong Agricultural University. WT mice and miR-25−/− mice with similar body weights were reared on the same diet for 3 to 5 days to adapt to the environment. WT mice (n = 6) and miR-25−/− mice (n = 6) were infected with 4.4×105 BCG via tail vein injection. At 7 dpi, the mice were euthanized, and lung tissue was collected to detect bacterial counts. The lung homogenateson was serially diluted in PBS, and the appropriate dilution was selected for plating and counting. The lungs were fixed with 4% paraformaldehyde solution and then in paraffin. The paraffin-embedded lung tissue was sliced at a thickness of 2–3 mm and then stained with hematoxylin and eosin.
Quantification and statistical analysis
Experiments were performed 3 times, data are presented as the means with SD. Statistical analyses were conducted by unpaired Student’s t-test (comparisons of miRNA expression levels between uninfected and infected with BCG mice; comparisons of BCG or H37Rv survival between miR-25 mimic, miR-25 inhibitor, NPC1 siRNA, NFKBIZ siRNA treatment group and their control group; comparisons of BCG survival between WT mice and miR-25−/− mice; comparisons of PCC values between miR-25 mimic, miR-25 inhibitor, NPC1 siRNA during BCG infection, miR-25 inhibitor during H37Rv infection and their control group; comparisons of NPC1 expression levels between miR-25 mimic, miR-25 inhibitor and their control group; comparisons of relative luciferase activity between miR-25 mimic, miR-25 inhibitor and their control group; comparisons of strains survival between H37Rv and H37RvΔ1759c), one-way ANOVA with the Tukey correction for multiple comparisons (comparisons of miR-25 expression levels among different time point infection of BCG and H37Rv; comparisons of NFKBIZ expression levels among CELL, H37Rv and H37RvΔ1759c infection) or one-way ANOVA with the Dunnet correction for multiple comparisons (comparisons of miRNA expression levels among CELL, BCG and H37Rv infection; comparisons of miR-25 expression levels among different MOI infection of BCG and H37Rv; comparisons of PCC values among control mimic, miR-25 mimic, NPC1 siRNA and NFKBIZ siRNA during H37Rv infection; comparisons of miR-25 expression levels among CELL and H37Rv strains with different genes deleted; comparisons of miR-25 expression levels among CELL, H37Rv infection transfected with NC siRNA and H37Rv infection transfected with NFKBIZ siRNA) using GraphPad Prism software 8.0.∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.005, ns, not significant.
Acknowledgments
We thank Howard Hughes Medical Institute (HHMI) investigator W.R.Jacobs jr providing pYUB004S, phAE159 the specialized transduction system. We thank Professor Zhou Hongbo, Professor Cao Gang, and Professor Guo Aizhen from Huazhong Agricultural University for providing pmirGLO vector, psPAX2 and pMD2.G vextors, and BCG Tokyo strain, respectively.
We thank Dr. Chuan-You Li from Beijing Tuberculosis & Thoracic Tumor Research Institute for kindly providing the Mtb strain. This work was supported by grants from the National Key R&D Program of China [2021YFD1800402], the Natural Science Foundation of Hubei Province [2019CFA095, 2021CFA016].
Author contributions
CT, YX, HC, and XW designed experiments. WD, GW, and JF performed the animal experiments and the in vitro experiments. WD, JF, PL, RW, HL, WL, and CW analyzed data. C.T, Y.X, and W.D wrote the manuscript. All authors reviewed the manuscript and approved the final version of the manuscript.
Declaration of interests
The authors have no conflicts of interest to declare.
Published: May 20, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.104279.
Supplemental information
Data and code availability
-
•
The transcriptional profiling data are available at the GEO database (GEO: GSE184660). We summarized the differential genes information into an excel table named as Table S2 and uploaded as supplementary data.
-
•
This paper does not report original code.
References
- Ahmad S. Pathogenesis, immunology, and diagnosis of latent Mycobacterium tuberculosis infection. Clin. Dev. Immunol. 2011;2011:814943. doi: 10.1155/2011/814943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Jiménez V.D., Leyva-Paredes K., García-Martínez M., Vázquez-Flores L., García-Paredes V.G., Campillo-Navarro M., Romo-Cruz I., Rosales-García V.H., Castañeda-Casimiro J., González-Pozos S., et al. Extracellular vesicles released from Mycobacterium tuberculosis-infected neutrophils promote macrophage autophagy and decrease intracellular mycobacterial survival. Front. Immunol. 2018;9:272. doi: 10.3389/fimmu.2018.00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asaad M., Kaisar Ali M., Abo-Kadoum M.A., Lambert N., Gong Z., Wang H., Uae M., Nazou S.A.E., Kuang Z., Xie J. Mycobacterium tuberculosis PPE10 (Rv0442c) alters host cell apoptosis and cytokine profile via linear ubiquitin chain assembly complex HOIP-NF-κB signaling axis. Int. Immunopharmacol. 2021;94:107363. doi: 10.1016/j.intimp.2020.107363. [DOI] [PubMed] [Google Scholar]
- Ballabio A., Bonifacino J.S. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 2020;21:101–118. doi: 10.1038/s41580-019-0185-4. [DOI] [PubMed] [Google Scholar]
- Bardarov S., Bardarov S., Pavelka M.S., Sambandamurthy V., Larsen M., Tufariello J., Chan J., Hatfull G., Jacobs W.R. Specialized transduction: an efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis. Microbiology (Reading) 2002;148:3007–3017. doi: 10.1099/00221287-148-10-3007. [DOI] [PubMed] [Google Scholar]
- Barlow A.L., Macleod A., Noppen S., Sanderson J., Guérin C.J. Colocalization analysis in fluorescence micrographs: verification of a more accurate calculation of pearson's correlation coefficient. Microsc. Microanal. 2010;16:710–724. doi: 10.1017/S143192761009389X. [DOI] [PubMed] [Google Scholar]
- Bartel D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- BoseDasgupta S., Pieters J. Macrophage-microbe interaction: lessons learned from the pathogen Mycobacterium tuberculosis. Semin. Immunopathol. 2018;40:577–591. doi: 10.1007/s00281-018-0710-0. [DOI] [PubMed] [Google Scholar]
- Bussi C., Gutierrez M.G. Mycobacterium tuberculosis infection of host cells in space and time. FEMS Microbiol. Rev. 2019;43:341–361. doi: 10.1093/femsre/fuz006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambier C.J., Falkow S., Ramakrishnan L. Host evasion and exploitation schemes of Mycobacterium tuberculosis. Cell. 2014;159:1497–1509. doi: 10.1016/j.cell.2014.11.024. [DOI] [PubMed] [Google Scholar]
- Chen D.Y., Chen Y.M., Lin C.F., Lo C.M., Liu H.J., Liao T.L. MicroRNA-889 inhibits autophagy to maintain mycobacterial survival in patients with latent tuberculosis infection by targeting TWEAK. mBio. 2020;11 doi: 10.1128/mBio.03045-19. e03045–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo A., Dinkel L., Müller S.A., Sebastian Monasor L., Schifferer M., Cantuti-Castelvetri L., König J., Vidatic L., Bremova-Ertl T., Lieberman A.P., et al. Loss of NPC1 enhances phagocytic uptake and impairs lipid trafficking in microglia. Nat. Commun. 2021;12:1158. doi: 10.1038/s41467-021-21428-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn K.W., Kamocka M.M., McDonald J.H. A practical guide to evaluating colocalization in biological microscopy. Am. J. Physiol. Cell Physiol. 2011;300:C723–C742. doi: 10.1152/ajpcell.00462.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etna M.P., Sinigaglia A., Grassi A., Giacomini E., Romagnoli A., Pardini M., Severa M., Cruciani M., Rizzo F., Anastasiadou E., et al. Mycobacterium tuberculosis-induced miR-155 subverts autophagy by targeting ATG3 in human dendritic cells. PLoS Pathog. 2018;14:e1006790. doi: 10.1371/journal.ppat.1006790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg R., Borbora S.M., Bansia H., Rao S., Singh P., Verma R., Balaji K.N., Nagaraja V. Mycobacterium tuberculosis calcium pump CtpF modulates the autophagosome in an mTOR-dependent manner. Front. Cell Infect. Microbiol. 2020;10:461. doi: 10.3389/fcimb.2020.00461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geneva: World Health Organization Global tuberculosis report. 2020. https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-tuberculosis-report-2021
- Gong Z., Kuang Z., Li H., Li C., Ali M.K., Huang F., Li P., Li Q., Huang X., Ren S., et al. Regulation of host cell pyroptosis and cytokines production by Mycobacterium tuberculosis effector PPE60 requires LUBAC mediated NF-κB signaling. Cell Immunol. 2019;335:41–50. doi: 10.1016/j.cellimm.2018.10.009. [DOI] [PubMed] [Google Scholar]
- Grondona P., Bucher P., Schulze-Osthoff K., Hailfinger S., Schmitt A. NF-κB activation in lymphoid malignancies: genetics, signaling, and targeted therapy. Biomedicines. 2018;6:38. doi: 10.3390/biomedicines6020038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L., Zhao J., Qu Y., Yin R., Gao Q., Ding S., Zhang Y., Wei J., Xu G. microRNA-20a inhibits autophagic process by targeting ATG7 and ATG16L1 and favors mycobacterial survival in macrophage cells. Front. Cell Infect. Microbiol. 2016;6:134. doi: 10.3389/fcimb.2016.00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez M.G., Master S.S., Singh S.B., Taylor G.A., Colombo M.I., Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- Hörber S., Hildebrand D.G., Lieb W.S., Lorscheid S., Hailfinger S., Schulze-Osthoff K., Essmann F. The atypical inhibitor of NF-κB, IκBζ, controls macrophage interleukin-10 expression. J. Biol. Chem. 2016;291:12851–12861. doi: 10.1074/jbc.M116.718825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J., Brumell J.H. Bacteria-autophagy interplay: a battle for survival. Nat. Rev. Microbiol. 2014;12:101–114. doi: 10.1038/nrmicro3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y., Zhou X., Bai Y., Yang L., Yin X., Wang Z., Zhao D. Phagolysosome maturation of macrophages was reduced by PE_PGRS 62 protein expressing in Mycobacterium smegmatis and induced in IFN-γ priming. Vet. Microbiol. 2012;160:117–125. doi: 10.1016/j.vetmic.2012.05.011. [DOI] [PubMed] [Google Scholar]
- Jain P., Hsu T., Arai M., Biermann K., Thaler D.S., Nguyen A., González P.A., Tufariello J.M., Kriakov J., Chen B., et al. Specialized transduction designed for precise high-throughput unmarked deletions in Mycobacterium Tuberculosis. mBio. 2014;5 doi: 10.1128/mBio.01245-14. e01245–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.K., Lee H.M., Park K.S., Shin D.M., Kim T.S., Kim Y.S., Suh H.W., Kim S.Y., Kim I.S., Kim J.M., et al. MIR144∗ inhibits antimicrobial responses against Mycobacterium tuberculosis in human monocytes and macrophages by targeting the autophagy protein DRAM2. Autophagy. 2017;13:423–441. doi: 10.1080/15548627.2016.1241922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S., Noda T., Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3:452–460. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- Kohda A., Yamazaki S., Sumimoto H. The nuclear protein IκBζ forms a transcriptionally active complex with nuclear factor-κB (NF-κB) p50 and the Lcn2 promoter via the N- and C-terminal ankyrin repeat motifs. J. Biol. Chem. 2016;291:20739–20752. doi: 10.1074/jbc.M116.719302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurokawa Y., Osaka H., Kouga T., Jimbo E., Muramatsu K., Nakamura S., Takayanagi Y., Onaka T., Muramatsu S.I., Yamagata T. Gene therapy in a mouse model of Niemann-Pick disease type C1. Hum. Gene Ther. 2021;32:589–598. doi: 10.1089/hum.2020.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon H.J., Abi-Mosleh L., Wang M.L., Deisenhofer J., Goldstein J.L., Brown M.S., Infante R.E. Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell. 2009;137:1213–1224. doi: 10.1016/j.cell.2009.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B.Y., Jethwaney D., Schilling B., Clemens D.L., Gibson B.W., Horwitz M.A. The Mycobacterium bovis bacille Calmette-Guerin phagosome proteome. Mol. Cell Proteomics. 2010;9:32–53. doi: 10.1074/mcp.M900396-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B., Mizushima N., Virgin H.W. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Li Q., Yu Z., Zhou M., Xie J. Mycobacterium tuberculosis PE13 (Rv1195) manipulates the host cell fate via p38-ERK-NF-κB axis and apoptosis. Apoptosis. 2016;21:795–808. doi: 10.1007/s10495-016-1249-y. [DOI] [PubMed] [Google Scholar]
- Li M., Wang J., Fang Y., Gong S., Li M., Wu M., Lai X., Zeng G., Wang Y., Yang K., Huang X. microRNA-146a promotes mycobacterial survival in macrophages through suppressing nitric oxide production. Sci. Rep. 2016;6:23351. doi: 10.1038/srep23351. Erratum in: Sci Rep. 2016 Apr 22;6:24555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P., Wang R., Dong W., Hu L., Zong B., Zhang Y., Wang X., Guo A., Zhang A., Xiang Y., et al. Comparative proteomics analysis of human macrophages infected with virulent Mycobacterium bovis. Front. Cell Infect. Microbiol. 2017;7:65. doi: 10.3389/fcimb.2017.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman A.P., Puertollano R., Raben N., Slaugenhaupt S., Walkley S.U., Ballabio A. Autophagy in lysosomal storage disorders. Autophagy. 2012;8:719–730. doi: 10.4161/auto.19469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F., Chen J., Wang P., Li H., Zhou Y., Liu H., Liu Z., Zheng R., Wang L., Yang H., et al. MicroRNA-27a controls the intracellular survival of Mycobacterium tuberculosis by regulating calcium-associated autophagy. Nat. Commun. 2018;9:4295. doi: 10.1038/s41467-018-06836-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathirvel M., S S., M S. Expression levels of candidate circulating microRNAs in pediatric tuberculosis. Pathog. Glob. Health. 2020;114:262–270. doi: 10.1080/20477724.2020.1761140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malardo T., Gardinassi L.G., Moreira B.P., Padilha É., Lorenzi J.C., Soares L.S., Gembre A.F., Fontoura I.C., de Almeida L.P., et al. MicroRNA expression signatures in lungs of mice infected with Mycobacterium tuberculosis. Tuberculosis (Edinb) 2016;101:151–159. doi: 10.1016/j.tube.2016.09.003. [DOI] [PubMed] [Google Scholar]
- Mauvezin C., Neufeld T.P. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy. 2015;11:1437–1438. doi: 10.1080/15548627.2015.1066957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meneses-Salas E., Garcia-Forn M., Castany-Pladevall C., Lu A., Fajardo A., Jose J., Wahba M., Bosch M., Pol A., Tebar F., et al. Lack of annexin A6 exacerbates liver dysfunction and reduces lifespan of Niemann-Pick type C protein-deficient mice. Am. J. Pathol. 2021;191:475–486. doi: 10.1016/j.ajpath.2020.12.009. Erratum in: Am J Pathol. 2021 Jul;191(7):1325. [DOI] [PubMed] [Google Scholar]
- Mizushima N., Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- Mizushima N., Yoshimori T., Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myerowitz R., Puertollano R., Raben N. Impaired autophagy: the collateral damage of lysosomal storage disorders. EBioMedicine. 2021;63:103166. doi: 10.1016/j.ebiom.2020.103166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niida T., Isoda K., Kitagaki M., Ishigami N., Adachi T., Matsubara O., Takeda K., Kishimoto T., Ohsuzu F. IκBNS regulates interleukin-6 production and inhibits neointimal formation after vascular injury in mice. Cardiovasc. Res. 2012;93:371–379. doi: 10.1093/cvr/cvr323. [DOI] [PubMed] [Google Scholar]
- Ouimet M., Koster S., Sakowski E., Ramkhelawon B., van Solingen C., Oldebeken S., Karunakaran D., Portal-Celhay C., Sheedy F.J., Ray T.D., et al. Mycobacterium tuberculosis induces the miR-33 locus to reprogram autophagy and host lipid metabolism. Nat. Immunol. 2016;17:677–686. doi: 10.1038/ni.3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahari S., Negi S., Aqdas M., Arnett E., Schlesinger L.S., Agrewala J.N. Induction of autophagy through CLEC4E in combination with TLR4: an innovative strategy to restrict the survival of Mycobacterium tuberculosis. Autophagy. 2020;16:1021–1043. doi: 10.1080/15548627.2019.1658436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan L., Liu F., Zhang J., Li J., Jia H., Huang M., Liu X., Chen W., Ding Z., Wang Y., et al. Genome-wide miRNA analysis identifies potential biomarkers in distinguishing tuberculous and viral meningitis. Front. Cell Infect. Microbiol. 2019;9:323. doi: 10.3389/fcimb.2019.00323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankiv S., Clausen T.H., Lamark T., Brech A., Bruun J.A., Outzen H., Øvervatn A., Bjørkøy G., Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- Pires D., Bernard E.M., Pombo J.P., Carmo N., Fialho C., Gutierrez M.G., Bettencourt P., Anes E. Mycobacterium tuberculosis modulates miR-106b-5p to control cathepsin S expression resulting in higher pathogen survival and poor T-cell activation. Front. Immunol. 2017;8:1819. doi: 10.3389/fimmu.2017.01819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan L., Federspiel N.A., Falkow S. Granuloma-specific expression of Mycobacterium virulence proteins from the glycine-rich PE-PGRS family. Science. 2000;288:1436–1439. doi: 10.1126/science.288.5470.1436. [DOI] [PubMed] [Google Scholar]
- Romagnoli A., Etna M.P., Giacomini E., Pardini M., Remoli M.E., Corazzari M., Falasca L., Goletti D., Gafa V., Simeone R., et al. ESX-1 dependent impairment of autophagic flux by Mycobacterium tuberculosis in human dendritic cells. Autophagy. 2012;8:1357–1370. doi: 10.4161/auto.20881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini N.K., Baena A., Ng T.W., Venkataswamy M.M., Kennedy S.C., Kunnath-Velayudhan S., Carreño L.J., Xu J., Chan J., Larsen M.H., et al. Suppression of autophagy and antigen presentation by Mycobacterium tuberculosis PE_PGRS47. Nat. Microbiol. 2016;1:16133. doi: 10.1038/nmicrobiol.2016.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S., Carroll B., Buganim Y., Maetzel D., Ng A.H., Cassady J.P., Cohen M.A., Chakraborty S., Wang H., Spooner E., et al. Impaired autophagy in the lipid-storage disorder Niemann-Pick type C1 disease. Cell Rep. 2013;5:1302–1315. doi: 10.1016/j.celrep.2013.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S., Maetzel D., Korolchuk V.I., Jaenisch R. Restarting stalled autophagy a potential therapeutic approach for the lipid storage disorder, Niemann-Pick type C1 disease. Autophagy. 2014;10:1137–1140. doi: 10.4161/auto.28623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seranova E., Connolly K.J., Zatyka M., Rosenstock T.R., Barrett T., Tuxworth R.I., Sarkar S. Dysregulation of autophagy as a common mechanism in lysosomal storage diseases. Essays Biochem. 2017;61:733–749. doi: 10.1042/EBC20170055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C., Fraldi A., Jahreiss L., Spampanato C., Venturi C., Medina D., de Pablo R., Tacchetti C., Rubinsztein D.C., Ballabio A. A block of autophagy in lysosomal storage disorders. Hum. Mol. Genet. 2008;17:119–129. doi: 10.1093/hmg/ddm289. [DOI] [PubMed] [Google Scholar]
- Slobodkin M.R., Elazar Z. The Atg8 family: multifunctional ubiquitin-like key regulators of autophagy. Essays Biochem. 2013;55:51–64. doi: 10.1042/bse0550051. [DOI] [PubMed] [Google Scholar]
- Slowikowski K., Nguyen H.N., Noss E.H., Simmons D.P., Mizoguchi F., Watts G.F.M., Gurish M.F., Brenner M.B., Raychaudhuri S. CUX1 and IκBζ (NFKBIZ) mediate the synergistic inflammatory response to TNF and IL-17A in stromal fibroblasts. Proc. Natl. Acad. Sci. U S A. 2020;117:5532–5541. doi: 10.1073/pnas.1912702117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H., Zhang Z., Liu Z., Peng B., Kong C., Wang H., Zhang Z., Xu Y. Mycobacterium tuberculosis PPE60 antigen drives Th1/Th17 responses via Toll-like receptor 2-dependent maturation of dendritic cells. J. Biol. Chem. 2018;293:10287–10302. doi: 10.1074/jbc.RA118.001696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanida I., Ueno T., Kominami E. LC3 and autophagy. Methods Mol. Biol. 2008;445:77–88. doi: 10.1007/978-1-59745-157-4_4. [DOI] [PubMed] [Google Scholar]
- Tartey S., Matsushita K., Vandenbon A., Ori D., Imamura T., Mino T., Standley D.M., Hoffmann J.A., Reichhart J.M., Akira S., Takeuchi O. Akirin2 is critical for inducing inflammatory genes by bridging IκB-ζ and the SWI/SNF complex. EMBO J. 2014;33:2332–2348. doi: 10.15252/embj.201488447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vegh P., Magee D.A., Nalpas N.C., Bryan K., McCabe M.S., Browne J.A., Conlon K.M., Gordon S.V., Bradley D.G., MacHugh D.E., Lynn D.J. MicroRNA profiling of the bovine alveolar macrophage response to Mycobacterium bovis infection suggests pathogen survival is enhanced by microRNA regulation of endocytosis and lysosome trafficking. Tuberculosis (Edinb). 2015;95:60–67. doi: 10.1016/j.tube.2014.10.011. [DOI] [PubMed] [Google Scholar]
- Wang C., Liu C.M., Wei L.L., Shi L.Y., Pan Z.F., Mao L.G., Wan X.C., Ping Z.P., Jiang T.T., Chen Z.L., et al. A group of novel serum diagnostic biomarkers for multidrug-resistant tuberculosis by iTRAQ-2D LC-MS/MS and solexa sequencing. Int. J. Biol. Sci. 2016;12:246–256. doi: 10.7150/ijbs.13805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Yang H., Shivalila C.S., Dawlaty M.M., Cheng A.W., Zhang F., Jaenisch R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153:910–918. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Xu Y.M., Zou Y.Q., Lin J., Huang B., Liu J., Li J., Zhang J., Yang W.M., Min Q.H., et al. Identification of differential expressed PE exosomal miRNA in lung adenocarcinoma, tuberculosis, and other benign lesions. Medicine (Baltimore) 2017;96:e8361. doi: 10.1097/MD.0000000000008361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y., Zhou Y., Liu S., Zhang X.L. PE_PGRS: vital proteins in promoting mycobacterial survival and modulating host immunity and metabolism. Cell Microbiol. 2021;23:e13290. doi: 10.1111/cmi.13290. [DOI] [PubMed] [Google Scholar]
- Yang G., Luo T., Sun C., Yuan J., Peng X., Zhang C., Zhai X., Bao L. PPE27 in Mycobacterium smegmatis enhances mycobacterial survival and manipulates cytokine secretion in mouse macrophages. J. Interferon Cytokine Res. 2017;37:421–431. doi: 10.1089/jir.2016.0126. [DOI] [PubMed] [Google Scholar]
- Yoshimori T., Yamamoto A., Moriyama Y., Futai M., Tashiro Y. Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J. Biol. Chem. 1991;266:17707–17712. [PubMed] [Google Scholar]
- Yu Z., Zhang C., Zhou M., Li Q., Li H., Duan W., Li X., Feng Y., Xie J. Mycobacterium tuberculosis PPE44 (Rv2770c) is involved in response to multiple stresses and promotes the macrophage expression of IL-12 p40 and IL-6 via the p38, ERK, and NF-κB signaling axis. Int. Immunopharmacol. 2017;50:319–329. doi: 10.1016/j.intimp.2017.06.028. [DOI] [PubMed] [Google Scholar]
- Zeng N., Wen Y.H., Pan R., Yang J., Yan Y.M., Zhao A.Z., Zhu J.N., Fang X.H., Shan Z.X. Dickkopf 3: a novel target gene of miR-25-3p in promoting fibrosis-related gene expression in myocardial fibrosis. J. Cardiovasc. Transl. Res. 2021;14:1051–1062. doi: 10.1007/s12265-021-10116-w. [DOI] [PubMed] [Google Scholar]
- Zhang X., Guo J., Fan S., Li Y., Wei L., Yang X., Jiang T., Chen Z., Wang C., Liu J., et al. Screening and identification of six serum microRNAs as novel potential combination biomarkers for pulmonary tuberculosis diagnosis. PLoS One. 2013;8:e81076. doi: 10.1371/journal.pone.0081076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Zhang X., Zhao Z., Zheng Y., Xiao Z., Li F. Integrated bioinformatics analysis and validation revealed potential immune-regulatory miR-892b, miR-199b-5p and miR-582-5p as diagnostic biomarkers in active tuberculosis. Microb. Pathog. 2019;134:103563. doi: 10.1016/j.micpath.2019.103563. [DOI] [PubMed] [Google Scholar]
- Zhou M., Yu G., Yang X., Zhu C., Zhang Z., Zhan X. Circulating microRNAs as biomarkers for the early diagnosis of childhood tuberculosis infection. Mol. Med. Rep. 2016;13:4620–4626. doi: 10.3892/mmr.2016.5097. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The transcriptional profiling data are available at the GEO database (GEO: GSE184660). We summarized the differential genes information into an excel table named as Table S2 and uploaded as supplementary data.
-
•
This paper does not report original code.