

Summary
Early Th17 responses are necessary to provide protection against Mycobacterium tuberculosis (Mtb). Mtb impedes Th17 polarization by restricting CD40 co-stimulatory pathway on dendritic cells (DCs). We previously demonstrated that engaging CD40 on DCs increased Th17 responses. However, the molecular mechanisms that contributed to Th17 polarization were unknown. Here, we identify the Notch ligand DLL4 as necessary for Th17 polarization and demonstrate that Mtb limits DLL4 on DCs to prevent optimal Th17 responses. Although Mtb infection induced only low levels of DLL4, engaging CD40 on DCs increased DLL4 expression. Antibody blockade of DLL4 on DCs reduced Th17 polarization in vitro and in vivo. In addition, we show that the Mtb Hip1 protease attenuates DLL4 expression on lung DCs by impeding CD40 signaling. Overall, our results demonstrate that Mtb impedes CD40-dependent DLL4 expression to restrict Th17 responses and identify the CD40-DLL4 pathways as targets for developing new Th17-inducing vaccines and adjuvants for tuberculosis.
Subject areas: Molecular biology, Parasitology
Graphical abstract
Highlights
-
•
Mtb restricts Th17 responses by impairing CD40 signaling on dendritic cells
-
•
Engaging CD40 on DCs increases Notch ligand Dll4 transcript and surface expression
-
•
DLL4 is necessary for polarizing Th17 and multifunctional T cells in the lungs of mice
-
•
Mtb impairs CD40/DLL4 pathway through the Hip1 serine protease immune evasion protein
Molecular biology; Bacteriology
Introduction
Mycobacterium tuberculosis (Mtb) is the causative agent of tuberculosis (TB), a serious global health problem that led to the death of 1.5 million individuals worldwide in 2020 (World Health Organization, 2021) alone. The currently licensed vaccine against TB, Mycobacterium bovis Bacillus Calmette-Guérin (BCG), has poor efficacy against pulmonary TB in adults and children (Rodrigues et al., 1993; Mangtani et al., 2014). Significant barriers that hinder developing more efficacious TB vaccines include our limited understanding of protective immunity against infection and disease, and the Mtb immune evasion mechanisms that impede protective host immune responses. CD4 effector T cell responses are critical for immune control of Mtb. IFN-γ-producing CD4 T helper 1 (Th1) cells are necessary for inducing antimicrobial functions in macrophages (Flynn et al., 1993; Cooper et al., 1993; Newport et al., 1996; Macmicking et al., 1997; Cooper, 2009), but are insufficient for providing protection against TB disease. Studies in animal models and humans from several groups, including our own, have identified important roles for IL-17 and Th17 responses in protective immunity against Mtb (Perreau et al., 2013; Gopal et al., 2014; Okada et al., 2015; Sia et al., 2017; Dijkman et al., 2019; Shanmugasundaram et al., 2020; Ogongo et al., 2021; Nathan et al., 2021) and suggest that vaccines that induce early Th17 responses will provide enhanced protection against TB. However, we have previously shown that Mtb actively limits the early generation of lung Th17 responses through the immunomodulatory functions of the Mtb serine protease Hip1 (Madan-Lala et al., 2014; Sia et al., 2017). Therefore, delineating the molecular basis for Th17 polarization following infection and the mechanisms employed by Mtb to limit Th17 generation will allow us to design efficacious TB vaccines that induce protective immunity.
Dendritic cells (DCs) are critical for initiating the activation, proliferation and polarization of naïve CD4 T cells by presenting pathogen-derived antigens, upregulating co-stimulatory molecules, and producing cytokines that contribute to polarization into Th1, Th17 and other Th subsets (Tascon et al., 2000; Wolf et al., 2007, 2008). However, Mtb is able to impede DC functions and impair DC-T cell crosstalk, leading to sub-optimal effector Th cell responses that fail to eliminate infection (Wolf et al., 2007; Madan-Lala et al., 2014; Sia et al., 2017). We recently demonstrated that the CD40−CD40L costimulatory pathway is necessary for generating Th17 responses and that Mtb limits Th17 responses by impairing interactions between CD40 on DCs and CD40L on CD4 T cells through Hip1 protease (Madan-Lala et al., 2014; Sia et al., 2017). Importantly, we showed that exogenously engaging CD40 on Mtb-infected DCs led to significant enhancement of mucosal Mtb-specific Th17 responses and improved control of Mtb lung burdens in mouse models (Sia et al., 2017). These studies demonstrate that targeting the CD40−CD40L pathway is an attractive approach for developing new Th17-inducing vaccine adjuvants and therapeutics.
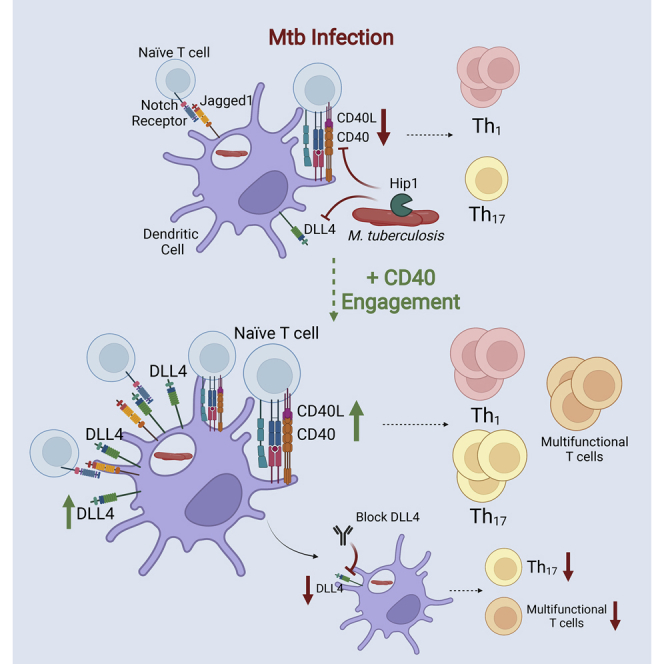
In order to better inform development of vaccine and therapeutic strategies that would augment DC responses and Th17 polarization, we sought to elucidate the molecular pathways downstream of CD40 signaling in DCs that preferentially induce Th17 polarization. Here we identify the Notch ligand delta-like canonical Notch ligand 4 (DLL4) as necessary for Th17 polarization and demonstrate that Mtb limits DLL4 signaling on DCs to prevent optimal Th17 responses. Notch signaling is initiated by ligand-receptor binding and requires binding of any of the five ligands: DLL1, DLL3, DLL4, Jagged1, or Jagged2 to any of the four Notch receptors, NOTCH1-4, on neighboring cells (Kopan and Ilagan, 2009). We found that while Mtb infection induced only low levels of Dll4 mRNA in DCs, exogenous CD40 engagement of Mtb-infected DCs substantially increased Dll4 expression and led to high frequencies of DLL4+ DCs. We hypothesized that CD40-dependent Th17 polarization is mediated by Notch ligands and tested this hypothesis by blocking Notch ligand signaling. Antibody blockade experiments showed that DLL4 is required for Th17 polarization during Mtb infection, both in vitro and in vivo, providing new evidence that links CD40, DLL4 and Th17 polarization. Moreover, CD40-engagement enabled DCs to induce multifunctional CXCR3+CCR6+ Th17 subsets in the lung, including double-positive (DP) IL-17+IFN-γ+ and IL-17+IL-22+ subsets and triple-positive (TP) IL-17+IFN-γ+IL-22+ subsets that have been associated with protective immunity against TB (Scriba et al., 2008; Arlehamn et al., 2014; Shanmugasundaram et al., 2020). IL-17 and IL-22 expression positively correlated with NOTCH2 receptor expression on Th subsets and these responses were abrogated upon blocking DLL4 on DCs. Moreover, DLL4-dependent IL-17 responses inversely correlated with Mtb lung burdens, suggesting that DLL4 signaling promotes Th17 responses and augments Mtb control. In addition, we found that the Hip1 protease impedes DLL4 expression on lung DCs through a CD40-dependent mechanism. To our knowledge, our studies are the first to demonstrate that Notch signaling downstream of CD40−CD40L interactions is necessary for generating Th17 responses during Mtb infection and that overcoming Mtb limitation of CD40 and Notch ligand signaling pathways can promote protective immunity. Our studies thus identify CD40 and DLL4 as targets for adjuvant-mediated immunomodulation during vaccination.
Results
Engaging CD40 on Mtb-infected dendritic cells augments pro-inflammatory cytokines and enhances expression of notch ligand Dll4
To identify the CD40-dependent mechanisms that enable DCs to polarize Th cells towards Th17 subsets, we employed our previously-described model in which we exogenously engaged CD40 using the multimeric CD40L reagent (CD40LT), which crosslinks CD40 and effectively simulates the membrane-assisted aggregation of CD40L (Sia et al., 2017). We infected bone marrow-derived dendritic cells (BMDCs) from C57BL/6 (B6) mice in the presence or absence of CD40LT for up to 72 h (Figure 1). At designated time points after infection, we used quantitative polymerase chain reaction (qPCR) to assess gene expression and harvested supernatants to measure IL-6 and IL-12p40 cytokine production by enzyme-linked immunosorbent assay (ELISA). IL-6 and IL-12p40 are two pro-inflammatory cytokines that are produced by activated DCs and contribute to Th17 and Th1 polarization, respectively. We confirmed that Mtb-infected BMDCs stimulated with CD40LT produced significantly higher levels of pro-inflammatory cytokines IL-6 and IL-12p40, as previously demonstrated (Sia et al., 2017) (Figure 1A), along with significant increase in Il6 and Il12b mRNA levels (Figure 1B). We next measured the expression of Notch ligands Dll4 and Jag1. The addition of CD40LT to Mtb-infected DCs increased mRNA corresponding to Dll4 by ∼100-fold compared to Mtb alone and led to a significant increase at 72H (Figure 1C). In contrast, Jag1 mRNA levels did not increase with the addition of CD40LT and were comparable to Mtb infection alone (Figure 1C). Although stimulating BMDCs with CD40LT alone induced Dll4 mRNA compared to uninfected (UI), addition of CD40LT in the context of Mtb infection induced substantially more expression of Dll4 than CD40LT or Mtb alone (Figure S1). We were unable to detect the expression of additional Notch ligands Dll1, Dll3 or Jag2 (A.B.E. and J.R., unpublished data). To compare our Mtb-DC infections to the responses induced by purified ligands that bind pattern recognition receptors (PRRs), we stimulated DCs with LPS (TLR4 agonist) or zymosan (TLR2/Dectin-1 agonist) (Figure S2). LPS and zymosan each led to significant induction of IL-6 and IL-12p40 protein and corresponding mRNA (Figures S2A–S2C). Importantly, both LPS and zymosan induced robust expression of Dll4 mRNA at levels that were comparable to Mtb + CD40LT conditions (Figure S2D). Consistent with Mtb infection results, neither LPS or zymosan induced robust expression of Jag1 (Figure S2D). We also found that addition of CD40LT significantly augmented Dll4 mRNA following stimulation with LPS, zymosan or the Th17-skewing fungal pathogen Candidia albicans (Hernández-Santos and Gaffen, 2012) (C. albicans; Figure S3). Taken together, our results suggest that the expression of Dll4 in DCs is limited during Mtb infection but can be augmented by engaging CD40 on DCs. Moreover, consistent with previous observations, Jag1 is constitutively expressed in DCs and is not further induced by stimulation with either Mtb or PRR ligands (Napolitani et al., 2005). These data show that engagement of CD40 on DCs augments Dll4 mRNA in the context of Mtb infection as well other PRR ligands and Th17- skewing pathogens.
Figure 1.

Engaging CD40 on Mtb-infected DCs augments pro-inflammatory cytokines and enhances expression of Notch ligand Dll4
BMDCs were infected with either media (UI) or Mtb H37Rv strain at an MOI of 1 in the presence or absence of 1 μg/mL of multimeric CD40L reagent (CD40LT). At designated time points, cellfree supernatants and RNA were collected to assay for cytokine secretion and mRNA transcript.
(A) ELISA measurements of IL-6 and IL-12p40 in supernatants.
(B and C) qPCR analysis of genes was standardized to the housekeeping gene GAPDH, analyzed using the ΔΔCt method, and presented as 2-ΔΔCt. Data are presented as mean ± SD (a) or mean ± SEM (B and C). Data are representative of 3 independent experiments. Data were analyzed in (A) using a one-way ANOVA with a correction for multiple comparisons and (B and C) using a two-way ANOVA with a correction for multiple comparisons. Statistical significance p value key is the following: ∗ = ≤ 0.05, ∗∗∗∗ = ≤ 0.0001. See also Figures S1–S4.
We next sought to test whether the induction of Dll4 observed following addition of CD40LT is dependent on Mtb viability. We stimulated BMDCs with heat-killed Mtb (HK Mtb) with or without CD40LT for up to 48 h and collected supernatant and RNA for ELISA and qPCR assays, respectively, as described in Figure 1. As with live Mtb infection, we observed a significant increase in IL-6 and IL-12p40 protein following the addition of CD40LT (Figure S4A) and increase in Il6 and Il12b mRNA levels (Figure S4B). Moreover, addition of CD40LT led to a significant increase in Dll4 compared to HK Mtb alone while Jag1 levels remained unaltered (Figure S4C). These results demonstrate that the lack of Dll4 upregulation following Mtb infection as well as the enhanced Dll4 expression upon CD40 engagement is not dependent on the presence of live bacteria.
Engaging CD40 on Mtb-stimulated DCs enhances surface expression of DLL4 and Jagged1
Notch ligands need to be present on the surface of cells in order to interact with Notch receptors on neighboring cells (Kopan and Ilagan, 2009). Therefore, having observed an increase in Dll4 mRNA following the addition of CD40LT, we next sought to test whether CD40 engagement also induces Notch ligand expression on the surface of Mtb-infected DCs. We stimulated BMDCs with HK Mtb and collected cells at different time points over a 24-h time course and assessed surface expression of DLL4 and Jagged1 using flow cytometry. The addition of CD40LT led to a significant increase in the populations of DCs expressing DLL4 or Jagged1 compared to Mtb alone as assessed by frequencies (Figures 2A and 2B) and MFI (Figure 2C). Notably, the majority of CD40-enagaged DCs expressed both DLL4 and Jagged1 simultaneously (Figures 2A and 2B). These data demonstrate that in addition to increasing Dll4 mRNA, CD40 engagement augments surface expression of DLL4 and that all DLL4-expressing DCs were also positive for Jagged1.
Figure 2.

Engaging CD40 on Mtb-stimulated DCs enhances surface expression of DLL4 and Jagged1
BMDCs were stimulated with either media (UI) or HK Mtb (MOI 30) with or without the addition of 1 μg/mL of CD40LT. At designated time points, cells were collected and stained for surface markers.
(A) Representative flow cytometry plot of DLL4+ and Jagged1+ frequencies.
(B) Frequency of DLL4+ and Jagged1+ and double-positive populations.
(C) MFI of DLL4 and Jagged1 expression and representative graphs. All populations are singlets/live cells/CD11c+MHCIIhi. UI presented is 0H UI. Data are presented as mean ± SD. Data are representative of 3 independent experiments. Data were analyzed in (B) using a two-way ANOVA with a correction for multiple comparisons and (C) using an unpaired Student’s t test. Statistical significance p value key is the following: ∗ = ≤ 0.05, ∗∗ = ≤ 0.01, ∗∗∗ = ≤ 0.001, ∗∗∗∗ = ≤ 0.0001.
DLL4 is required for Th17 polarization but is dispensable for Th1 polarization
To investigate the role of DLL4 and Jagged1 in CD40-mediated Th17 polarization during Mtb infection, we set up DC-T cell co-culture assays as previously described (Sia et al., 2017). Because deletion of either DLL4 or Jagged1 in mice is embryonically lethal (Xue et al., 1999; Gale et al., 2004; Krebs et al., 2004; Duarte et al., 2004), we used blocking antibodies corresponding to each of the two Notch ligands. DCs exposed to Mtb in the presence or absence of CD40LT for 24 h were co-cultured with OT-II transgenic T cells and cognate OVA peptide (OVA323–339). At the time of co-culture, blocking antibodies to either DLL4, Jagged1, or a combination of DLL4 and Jagged1 antibodies were added to each well at increasing concentrations. Following 72 h of co-culture, cell-free supernatants were harvested and IFN-γ and IL-17 production were measured by ELISA to represent Th1 and Th17 cytokines, respectively (Figures 3A and 3B). As previously demonstrated (Sia et al., 2017), addition of CD40LT enhanced IL-17 levels in co-culture but did not augment IFN-γ levels (Figures 3A and 3B). CD40LT stimulation alone did not lead to an increase in Th17 polarization and was comparable to the uninfected condition (Figure S5). Antibody blockade of Jagged1 during co-culture led to a significant reduction in both IFN-γ and IL-17 compared to controls, whereas blocking DLL4 alone preferentially decreased IL-17 levels without significantly altering IFN-γ production (Figures 3A and 3B). We also measured IL-2 production (Figure 3C) and found that blockade of Jagged1 resulted in a significant reduction in IL-2. In contrast, addition of anti-DLL4 did not alter IL-2 production, suggesting that blockade of DLL4 did not impact the expansion of CD4 T cells. Isotype controls for each of the Notch ligand antibodies demonstrated the specificity of these results (Figure S6). Furthermore, providing Notch ligand blockade also led to a reduction in Th17 polarization following heat-killed C. albicans + CD40LT DC stimulation (Figure S7). These data indicate that CD40 engagement promotes Th17 polarization via Notch ligands. Importantly, DLL4 is specifically required for Th17 polarization but is not required for Th1 polarization.
Figure 3.

DLL4 is required for Th17 polarization but is dispensable for Th1 polarization
BMDCs were stimulated with either media alone (UI), or HK Mtb (MOI 30) with or without 1 μg/mL of CD40LT. Following 24H of stimulation, cells were pulsed with 10 μg/mL of cognate peptide (OVA323-339) for one hour and then co-cultured with purified naïve CD4 OT-II Tg Thy1.1 T cells at a ratio of 4:1 T cells to DCs. For blockade conditions, blocking antibodies to either DLL4, Jagged1, or both DLL4 and Jagged1 combined, were added at the time of co-culture at the following concentrations: 15, 30, or 60 μg/mL. After 72H of co-culture, cell-free supernatants were harvested and assayed for cytokines by ELISA.
(A) IFN-γ (Th1).
(B) IL-17 (Th17).
(C) IL-2. Data are presented as mean ± SD. Data are representative of 3 independent experiments. Data were analyzed using a one-way ANOVA with a correction for multiple comparisons (all data points were compared to Mtb + CD40LT). Statistical significance p value key is the following: ∗∗∗ = ≤ 0.001, ∗∗∗∗ = ≤ 0.0001. See also Figures S5–S7.
To further investigate the role of DLL4, either singly or in combination with Jagged1, in Th17 polarization in vivo, we used an intratracheal (IT) transfer model to transfer BMDCs directly into the lungs of mice (Sia et al., 2017). This model allows us to specifically dissect how CD40 and Notch ligands on DCs impact T cell polarization in vivo. Groups of BMDCs exposed to different experimental conditions (depicted in Figure 4A) were intratracheally transferred into mouse lungs in the presence or absence of blocking antibodies to DLL4 alone, or both DLL4 and Jagged1. One day before IT transfer of DCs, purified naïve CD4 T cells from ESAT1-20-Tg mice were adoptively transferred into mice via the intravenous (IV) route. Six days after intratracheal DC transfer, mice were euthanized and single-cell lung suspensions were stimulated with ESAT-61-20 peptide (to stimulate antigen-specific T cells), and responses were measured using flow cytometry. We observed a significant increase in the frequencies of IL-17+ CD4 T cells on transfer of CD40-engaged Mtb-DCs compared to controls, whereas IL-2+ and IFN-γ+ CD4 T cells were unaffected by CD40LT addition (Figures 4B and 4C). Providing DLL4 blocking antibodies led to significantly reduced frequencies of antigen-specific IL-17+ CD4 T cells but did not affect IFN-γ+ or IL-2+ CD4 T cell frequencies (Figures 4B and 4C). This supports our in vitro polarization assay data (Figure 3) and demonstrates that DLL4 blockade specifically affects Th17 polarization but does not impact Th1 polarization or overall T cell expansion. Blocking both DLL4 and Jagged1 significantly reduced IL-17+ and IL-2+ CD4 T cell frequencies but also resulted in lower IFN-γ+ CD4 T cells (Figures 4B and 4C). These data support a critical role for DLL4 alone or in combination with Jagged1 in Th17 polarization during Mtb stimulation in vivo.
Figure 4.

Blocking DLL4 singly or in combination with Jagged1 on DCs reduces antigen-specific IL-17+ CD4 T cell frequencies in vivo
(A) Experimental schema. BMDCs we stimulated with either media alone (UI), or HK Mtb (MOI 30) with or without 1 μg/mL of CD40LT for 24H. At this time in the CD40LT condition, either DLL4 or DLL4 and Jagged1 blocking antibodies (30 μg/mL) were added. One day before transfer, 1E6 ESAT-6 transgenic T cells were transferred into mice via the intravenous (IV) route. On the day of intratracheal (IT) transfer, 1E6 DCs were transferred. At 6 days after transfer, mice were euthanized and lung single cell suspensions were stimulated with 10 μg/mL ESAT-61-20 peptide to assess antigen-specific responses. Cells were then stained for flow cytometry.
(B) Representative flow cytometry plots of IFN-γ+ and IL-17+ frequencies.
(C) Cytokine-positive frequency of CD4 T cells. Populations shown are singlets/live cells/CD3+/CD4+. Experimental schema was made with BioRender.com. Data are presented as mean ± SD. Data are representative of 2 independent experiments. Data were analyzed using a one-way ANOVA with a correction for multiple comparisons. Statistical significance p value key is the following: ns = no significance, ∗ = ≤ 0.05, ∗∗ = ≤ 0.01, ∗∗∗ = ≤ 0.001, ∗∗∗∗ = ≤ 0.0001.
CD40-mediated Th17 polarization during Mtb infection is dependent on DLL4 in the lungs of mice
Our data demonstrates that Mtb limits DLL4 during infection and that CD40 engagement of Mtb-infected DCs leads to significant induction of DLL4 which is required for Th17 polarization (Figures 3 and 4). To further examine the role of DLL4 and CD40 in polarizing endogenous Th cells towards Th17 and other subsets in vivo, we transferred Mtb-infected DCs directly into the lungs of naïve B6 mice and carried out detailed phenotyping of the Th subsets in the presence or absence of DLL4 blocking antibodies (Figure 5A). BMDCs were infected in vitro for 48 h with Mtb in the presence or absence of CD40LT, with or without DLL4 blocking antibody. After the infection period, BMDCs were harvested and transferred into the lungs of mice as previously described (Sia et al., 2017) (Figure 5). At four weeks post-transfer, we euthanized mice and generated single cell lung suspensions to assess the ex vivo CD4 T cell cytokine responses induced by transferred DCs using flow cytometry. As with our previous data (Figure 4), CD40LT did not augment IL-2+ and IFN-γ+ CD4 T cell frequencies and DLL4 blockade did not impact these responses (Figure 5B). However, the transfer of CD40LT-treated DCs led to higher frequencies of IL-17+ CD4 T cells and providing DLL4 blocking antibody in this context significantly reduced Th17 responses (Figure 5B). In addition to IL-17, the cytokine IL-22 has also been reported to be produced by Th17 cells in mucosal settings (Liang et al., 2006). Notably, we found that CD40 engagement also led to significantly higher IL-22+ CD4 T cell frequencies and blocking DLL4 reversed this effect (Figure 5B). We extended our analyses to examine multiple-cytokine producing CD4 T cells, which have been implicated in protection against TB (Scriba et al., 2008; Arlehamn et al., 2014; Dijkman et al., 2019; Shanmugasundaram et al., 2020). Of interest, the Boolean analysis showed that CD40LT also augmented the frequencies of double-positive (DP) IL-17+IL-22+ and triple-positive (TP) IL-17+ IFN-γ+ IL-22+ CD4 T cells (Figure 5C). Moreover, the chemokine receptors CXCR3 and CCR6, which have been associated with protective Th17 and Th1/Th17 subsets (Arlehamn et al., 2014; Shanmugasundaram et al., 2020), were co-expressed on single-positive (SP) IL-17+ and DP IL-17+IL-22+ populations induced by CD40LT but these were abrogated upon DLL4 blockade (Figure 5D). We obtained similar results on assaying Mtb antigen-specific responses following ex vivo stimulation of lung cells with Mtb whole cell lysate (WCL) (Figure S8). Isotype controls for either antibody demonstrated the specificity of these results (Figure S9). Overall, these results demonstrate an essential role for DLL4 in generating both bulk and antigen-specific multifunctional Th17 responses during Mtb infection.
Figure 5.

Blocking DLL4 on Mtb-infected DCs reduces Th17 and multifunctional CD4 T cell responses in vivo
(A) Experimental schema. BMDCs were infected with Mtb at an MOI of 1, with or without CD40LT, for 48H. For antibody blockade studies, 60 μg/mL anti-DLL4 antibody was added during infection. DCs were then harvested and 1E6 were intratracheally (IT) transferred into the lungs of mice along with additional blocking antibody. At 4 weeks post transfer, mice were euthanized and lung suspensions were unstimulated to asses ex vivo responses using flow cytometry.
(B) Frequency of cytokine-positive CD4 T cells.
(C) Boolean analysis of frequency of multiple cytokine-positive CD4 T cells.
(D) Frequency of CXCR3+CCR6+ CD4 T cells and Boolean analysis of CXCR3+CCR6+ and cytokine-positive CD4 T cells. All populations are singlets/live cells/CD3+/CD4+. Experimental schema was made with BioRender.com. Data are presented as mean ± SD. Data are representative of 2 independent experiments. Data were analyzed using a one-way ANOVA with a correction for multiple comparisons. Statistical significance p value key is the following: ∗ = ≤ 0.05, ∗∗ = ≤ 0.01, ∗∗∗ = ≤ 0.001. SP = single-positive, DP = double-positive, TP = triple-positive. See also Figures S8 and S9.
The dependence on DLL4 for Th17 polarization demonstrates a critical role for Notch signaling during Mtb infection. The Notch signaling pathway is activated once a Notch ligand binds to a Notch receptor to initiate downstream effector functions (Kopan and Ilagan, 2009). Therefore, we next sought to test Notch receptor expression on T cells. Because murine CD4 T cells express NOTCH1 and NOTCH2 (Fiorini et al., 2009), we tested the presence of these two receptors in the same groups of mice. Interestingly, we found that transfer of Mtb-infected DCs plus CD40LT significantly increased the frequencies of NOTCH2+ and NOTCH1+NOTCH2+ CD4 T cells (Figure 6A). The median of values of NOTCH1+ appears to be greater in the Mtb + CD40LT condition, but this result is not significant (Figure 6A). DLL4 blockade led to a reduction in NOTCH2+ and NOTCH1+NOTCH2+ DP CD4 T cells (Figure 6A) and in IL-17+ cells expressing NOTCH1 or 2 (Figure 6B). Taken together, these data support the role of Notch ligand-Notch receptor signaling downstream of CD40 in generating Th17 responses during Mtb infection.
Figure 6.

Engaging CD40 on Mtb-infected DCs increases NOTCH2 expression on CD4 T cells in the lung
BMDCs were infected with Mtb at an MOI of 1, with or without CD40LT, for 48H. For antibody blockade studies, 60 μg/mL anti-DLL4 antibody was added during infection. DCs were then harvested and 1E6 were intratracheally (IT) transferred into the lungs of mice along with additional blocking antibody. At 4 weeks post transfer, mice were euthanized and lung suspensions were unstimulated to asses ex vivo responses using flow cytometry.
(A) Frequency of NOTCH receptor-positive CD4 T cells.
(B) Boolean analysis of NOTCH receptor-expressing and cytokine-positive CD4 T cells. All populations are singlets/live cells/CD3+/CD4+. Data are presented as mean ± SD. Data are representative of 2 independent experiments. Data were analyzed using a one-way ANOVA with a correction for multiple comparisons. Statistical significance p value key is the following: ∗ = ≤ 0.05, ∗∗ = ≤ 0.01, ∗∗∗ = ≤ 0.001, ∗∗∗∗ = ≤ 0.0001.
DLL4-mediated Th17 responses correlate with NOTCH2 expression and lower lung Mtb burden
To gain insights into the relationships between DC and T cell markers in the lung we next performed correlation analyses from our multiparameter flow cytometry data (Figure 7). We found that IL-17-producing CD4 T cells not only correlated with CD40L expression on CD4 T cells, consistent with our previous study (Sia et al., 2017), but also showed positive correlations with NOTCH1 and NOTCH2 expression (Figures 7A and 7B). IL-22 expression was positively correlated with CD40L and NOTCH2 but not with NOTCH1 (Figure 7B), whereas IFN-γ expression did not correlate with either of the two NOTCH receptors (Figure S10). We were curious to study the relationship between T cell markers in our panels and Mtb load (measured by plating for CFU) in the lungs of mice. We found that transfer of infected DCs treated with CD40LT led to significantly lower Mtb burdens in the lungs compared to infection alone, whereas Mtb burdens in mice that received DLL4 blocking antibody had significantly higher Mtb CFU, comparable to the CD40LT-untreated group (Figure 7C). Correlogram analyses showed that only two markers, IL-17 and NOTCH2, had significant negative correlations with CFU (Figure 7C). Taken together, these results suggest that CD40 engagement on DCs augments Mtb control through increased IL-17 responses and Notch signaling pathways.
Figure 7.

Th17 responses correlate with NOTCH2 expression and lower lung CFU
BMDCs were infected with Mtb at an MOI of 1, with or without CD40LT, for 48H. For antibody blockade studies, 60 μg/mL anti-DLL4 antibody was added during infection. DCs were then harvested and 1E6 were intratracheally (IT) transferred into the lungs of mice along with additional blocking antibody. At 4 weeks post transfer, mice were euthanized and lung suspensions were unstimulated to asses ex vivo responses using flow cytometry and lung homogenates were plated to enumerate Mtb burdens.
(A) Correlogram using R package “corrplot” showing correlation between the frequency of marker-positive CD4 T cells and frequency of cytokine-positive CD4 T cells.
(B) Correlations in R using “ggscatter” between the frequency of marker-positive CD4 T cells and frequency of cytokine-positive CD4 T cells.
(C) Mtb lung colony-forming unit (CFU) and correlations between CFU and frequencies of marker-positive or cytokine-positive CD4 T cells using “ggscatter” in R. All correlations presented are Pearson’s correlations. Data in C) are presented as mean ± SD and were analyzed using an unpaired Student’s t test. Data are representative of 2 independent experiments. Statistical significance p value key is the following: ∗ = ≤ 0.05, ∗∗ = ≤ 0.01, ∗∗∗ = ≤ 0.001, ∗∗∗∗ = ≤ 0.0001. See also Figure S10.
Mtb restriction of CD40 and DLL4 signaling in lung DCs is mediated by the Hip1 serine protease
We previously showed that Mtb prevents CD40 expression on infected DCs through the immunomodulatory serine protease Hip1 (Madan-Lala et al., 2014; Sia et al., 2017; Georgieva et al., 2018). A hip1mutant induced robust CD40 expression and higher Th17 responses compared to wild type Mtb (Madan-Lala et al., 2014; Sia et al., 2017; Georgieva et al., 2018). We also showed that Mtb Hip1 restricts optimal Th17 polarization by dampening the CD40 costimulatory pathway and that engaging CD40 on DCs enhances Th17 responses to levels comparable to that induced by the hip1mutant (Sia et al., 2017; Georgieva et al., 2018). Thus, the hip1mutant provides us with a unique tool to further probe the relationship between CD40 and DLL4 pathways during in vivo Mtb infection. We infected C57BL/6 mice with either wild type Mtb or the hip1 mutant via the aerosol route and euthanized mice two weeks post-infection to assess early immune responses (Figure 8A). We harvested the lungs and stained single cell suspensions for cell surface and intracellular markers to analyze by flow cytometry. We observed that the hip1 mutant induced robust CD40 expression on DCs in the lung compared to wild type Mtb (Figure 8B). We next examined the expression of DLL4 and Jagged1 in two populations of lung DCs: CD11b+ and CD103+ DCs. These are the two main populations of classical DCs in the lungs and have been implicated in Th polarization (Furuhashi et al., 2012; Zelante et al., 2015; Lai et al., 2018). Infection with the hip1 mutant, but not wild type Mtb, led to significantly higher frequencies of DLL4+ and DLL4+Jagged1+ DCs of both types in the lungs of infected mice (Figures 8C and 8D). Frequencies of Jagged1+ DCs in the lung were higher in both infected groups compared to uninfected mice, but indistinguishable between wild type and hip1 mutant groups (Figures 8C and 8D). We next assessed ex vivo T cell responses in these same groups of mice at this early time point. Consistent with previous data, infection with hip1mutant resulted in significantly higher frequencies of IL-17+ CD4 T cells (Madan-Lala et al., 2014; Sia et al., 2017; Georgieva et al., 2018) as well as significantly higher frequencies of IL-2+, IFN-γ+, and IL-22+ CD4 T cells in the lung compared to wild type Mtb infection (Figure 8E). Interestingly, the hip1mutant also induced higher frequencies of Notch receptor-expressing T cells compared to Mtb infection (Figure 8E). Furthermore, correlogram analyses showed that DLL4 expression on CD103+ DCs positively correlated with IL-17 and NOTCH2 expression on T cells (Figure 8F) but did not show any association with IFN-γ, IL-22, or NOTCH1 (Figure S11). These data suggest that DLL4-NOTCH2 interactions likely mediate Th17 polarization during Mtb infection. Thus, the absence of Hip1 augments CD40 and DLL4 signaling and subsequent Th17 responses whereas the presence of Hip1 impairs CD40-DLL4 signaling to limit Th17 responses.
Figure 8.

Mtb restricts DLL4 expression and early CD4 T cells responses in the lung through the Hip1 serine protease
(A) Experimental schema. C57BL/6 mice were infected via the aerosol route with a low-dose of Mtb or hip1mutant. Following 2 weeks post-infection, mice were euthanized and lung ex vivo responses were measured using flow cytometry.
(B–D) (B) Frequency of CD40-expressing CD11b+ DCs and CD40-expressing CD103+ DCs. Representative flow plots and frequency of DLL4+, Jagged1+, and DLL4+Jagged1+ for (C) CD11b+ DCs and (D) CD103+ DCs.
(E) Frequency of cytokine-positive and NOTCH receptor-positive CD4 T cells.
(F) Correlations between different CD4 T cell and innate immune population markers using “ggscatter” in R. All CD11b+ DC populations are singlets/live cells/CD45+/CD3-/CD64-F4/80-/MHCII+CD11c+/CD11b+CD103-. All CD103+ DC populations are singlets/live cells/CD45+/CD3-/CD64-F4/80-/MHCII+CD11c+/CD11b−CD103+. All T cell populations are singlets/live cells/CD3+/CD4+. All correlations presented are Pearson’s correlations. Experimental schema was made with BioRender.com. Data in B-E are presented as mean ± SD. Data were analyzed in B–E using unpaired Student’s t-tests. Data are representative of 2 independent experiments. Statistical significance p value key is the following: ∗ = ≤ 0.05, ∗∗ = ≤ 0.01. See also Figure S11.
The similarity between the responses elicited by exogenous engagement of CD40 in the context of wild type Mtb infection and infection with the hip1mutant led us to hypothesize that the hip1mutant increases DLL4 expression on lung DCs through CD40 engagement. To test the requirement for CD40 in Notch ligand expression, we infected mice lacking CD40 (CD40−/−) or C57BL/6 mice with intact CD40 (CD40+/+) with either wild type Mtb or the hip1mutantvia the aerosol route. We assessed DC and ex vivoT cell profiles two weeks post-infection using flow cytometry (Figure 9). Figure 9A shows that DLL4+ and DLL4+Jagged1+ lung DC populations induced by the hip1mutant are dependent on CD40. We observed a significant reduction in the frequencies of IL-2+, IFN-γ+ and IL-22+ CD4 T cells and a reduction in IL-17+ CD4 T cell frequencies in the CD40−/− group relative to the CD40+/+ group (Figure 9B). In addition, we observed reduced levels of NOTCH2+ and NOTCH1+NOTCH2+ co-expressing cells and significantly reduced levels of NOTCH1+ (Figure 9C). Taken together, these data demonstrate that in vivo induction of DLL4 signaling is dependent on CD40 expression on lung DCs and suggests that DLL4-NOTCH2 interactions in turn promote optimal polarization of Th17 responses. Importantly, these data implicate the Hip1 serine protease in impeding CD40-dependent Notch ligand signaling and highlight limitation of CD40 and DLL4 signaling as an immune evasion mechanism that dampens Th17 polarization during Mtb infection.
Figure 9.

Hip1 impedes DLL4 expression on lung DCs via a CD40-dependent mechanism
C57BL/6 (CD40+/+) or CD40−/− mice were infected via the aerosol route with a low-dose of Mtb or hip1mutant. Following 2 weeks post-infection, mice were euthanized and lung ex vivo responses were measured using flow cytometry.
(A) Fold of infected mice over uninfected mice (from the same mouse strain) for DLL4+ and DLL4+Jagged1+ in the CD103+ DC population.
(B) Fold over uninfected for cytokine-positive CD4 T cells.
(C) Fold over uninfected for NOTCH receptor-positive CD4 T cells. All CD11b+ DC populations are singlets/live cells/CD45+/CD3-/CD64-F4/80-/MHCII+CD11c+/CD11b+CD103-. All CD103+ DC populations are singlets/live cells/CD45+/CD3-/CD64-F4/80-/MHCII+CD11c+/CD11b−CD103+. All T cell populations are singlets/live cells/CD3+/CD4+. Data were analyzed using a one-way ANOVA with a correction for multiple comparisons. Data are presented as fold over uninfected mean ± SD. Data are representative of 2 independent experiments. Statistical significance p value key is the following: ∗ = ≤ 0.05, ∗∗ = ≤ 0.01, ∗∗∗ = ≤ 0.001.
Discussion
In this study, we identify Notch ligand signaling on DCs as a critical mechanism for Th17 polarization during Mtb infection. We demonstrate that induction of the Notch ligand DLL4 downstream of CD40 signaling augments Th17 responses, which correlated with lower Mtb lung burdens. DLL4 is required for inducing multifunctional CXCR3+CCR6+-expressing DP IL-17+IFN-γ+, IL-17+IL-22+ and TP IL-17+IFN-γ+IL-22+ subsets in the lung. In addition, we provide evidence that Mtb limits CD40-dependent Notch ligand signaling and dampens Th17 responses through the immunomodulatory Hip1 serine protease. Overall, our studies provide new molecular insights into Th17 responses during Mtb infection and reveal key innate pathways that can be targeted to enhance protective CD4 T cell responses and improve pulmonary control of Mtb.
DCs are critical for shaping adaptive immunity and are necessary for initiating T cell responses in the lung following Mtb infection. However, it is now well established that Mtb impedes DC functions to subvert early protective T cell responses (Wolf et al., 2007; Madan-Lala et al., 2014; Sia et al., 2017; Georgieva et al., 2018) and restrict Th17 polarization (Madan-Lala et al., 2014; Sia et al., 2017; Georgieva et al., 2018). Our group reported that Mtb prevents optimal crosstalk between DCs and CD4 T cells by impairing the CD40 costimulatory pathway (Sia et al., 2017). Specifically, we showed that interactions between CD40 on DCs and CD40L on T cells are necessary for Th17 polarization during Mtb infection, even when Th17-polarizing cytokines such as IL-6, IL-1b and IL-23 are present, and that exogenously triggering CD40 signaling on DCs enhances Th17 responses and improves control of pathogen burdens in the lungs (Sia et al., 2017). However, the mechanism by which CD40 orchestrates Th17 polarization in response to Mtb or other stimuli was not well understood. We now demonstrate that engaging CD40 on DCs during Mtb infection leads to upregulation of the Notch ligand Dll4 (Figures 1 and S4) and increases cell surface expression of DLL4 and Jagged1 on DCs (Figure 2). In contrast, Jag1 is constitutively expressed and is not further induced by infection (Figures 1 and S4). Notably all DLL4+ DCs were also positive for Jagged1, leading to high frequencies of double-positive DLL4+Jagged1+ DC populations (Figure 2). We also show that blocking DLL4 abrogates CD40-dependent Th17 polarization in vitro (Figure 3) and in vivo (Figures 4 and 5) but does not significantly impact Th1 polarization, highlighting a critical role for DLL4 in Th17 polarization. These findings reveal important insights into how DC responses mediate Th17 polarization during Mtb infection. In addition, our results on C. albicans and other PRR ligands (Figures S3 and S7) suggests that these insights are likely to also be important for understanding Th17 polarization beyond Mtb infection.
Although collaboration between CD40 and DLL4 in Th17 polarization has not been previously reported, expression of the Notch ligands Dll4 and Jag1 has largely been studied in the context of Th1/Th2 differentiation in response to TLR stimulation and anti-CD40 antibody (Sauma et al., 2011). Some reports have linked DLL4 with Th17 polarization in response to stimulating with TLR ligands in vitro or following BCG infection, but these studies did not demonstrate a clear requirement for DLL4 or CD40-DLL4 collaboration in generating Th17 responses (Ito et al., 2009; Mukherjee et al., 2009; Meng et al., 2016). Thus, to our knowledge, this is the first study to link CD40 and DLL4 signaling on DCs to Th17 polarization during Mtb infection and provides key insights that can be applied to other experimental models where immunomodulation of Th17 responses is of interest. In addition to their roles in Th17 polarization, our study also identifies a role for CD40 and Notch ligands in promoting multifunctional lung Th responses in vivo (Figure 5), including induction of DP IL-17+IL-22+ and TP IL-17+ IFN-γ+ IL-22+ CD4 T cells. Studies in humans and non-human primate models of TB have suggested that CD4 T cells that simultaneously expressing multiple cytokines are protective against disease (Scriba et al., 2008; Dijkman et al., 2019; Shanmugasundaram et al., 2020; Arlehamn et al., 2014). IL-22, an IL-10 family member, is often co-expressed by Th17 cells (Liang et al., 2006) and has been implicated in promoting protective immunity to Mtb (Scriba et al., 2008; Treerat et al., 2017). Additional Th subsets that are thought to promote protective functions in TB include CXCR3+CCR6+T cells that co-express IFN-γ and IL-17, (sometimes referred to as Th1∗ subsets), which were identified in the peripheral blood of latently infected individuals (Arlehamn et al., 2014). Interestingly, our group recently showed that CXCR3+CCR6+ dual Th1/Th17 cells are also present in lung compartments of asymptomatic Mtb-infected rhesus macaques, where they were associated with pulmonary control of Mtb infection (Shanmugasundaram et al., 2020). We now show that CXCR3+CCR6+ CD4 T cells that express both IL-17 and IFN-γ are induced via CD40 and DLL4 signaling, highlighting an essential role for crosstalk between DC costimulatory and Notch ligand pathways for generating these responses (Figure 6). Moreover, our data showing that triggering CD40-DLL4 signaling can overcome Mtb restriction of IL-17, IL-22 and IFN-γ producing multifunctional responses in the lung, provides new mechanistic insights that can be leveraged for inducing protective Th17 subsets via vaccination.
One of the ways by which Mtb evades host immunity is through expression of immunomodulatory proteins that interfere with DC-T cell crosstalk. Our lab has previously shown that Mtb prevents CD40 expression on DCs and restricts Th17 polarization through the Hip1 serine protease (Madan-Lala et al., 2014; Sia et al., 2017; Georgieva et al., 2018). A hip1mutant strain of Mtb induces robust CD40 expression and higher Th17 responses relative to wild type Mtb (Madan-Lala et al., 2014; Sia et al., 2017). We now show that in contrast to wild type Mtb, which did not induce DLL4 on the cell surface of lung DCs, mice infected with a hip1mutant significantly increased DLL4+ and DLL4+Jagged1+ DC populations in the lung (Figure 8). This was accompanied by higher frequencies of IL-17- and IL-22-producing Th subsets along with increased expression of NOTCH1 and NOTCH2 receptors on T cells early in infection relative to wild type Mtb (Figure 8). Moreover, levels of DLL4 on DCs positively correlated with IL-17 and NOTCH2 expression on Th subsets suggesting that DLL4-NOTCH2 interactions likely mediate Th17 polarization (Figure 8). The induction of DLL4+Jagged1+ DC and NOTCH1+NOTCH2+ CD4 T cells following CD40 engagement or hip1mutant infection suggests that combinatorial Notch-ligand-Notch receptor interactions may promote balanced Th1/Th17 responses. Infection of CD40−/− mice demonstrated that induction of DLL4+ and DLL4+Jagged1+ DCs by the hip1mutant was abrogated in the absence of CD40 (Figure 9), demonstrating that Notch ligand expression is dependent on CD40 signaling. These results provide evidence that Mtb limits DLL4-Notch receptor interactions during infection via a mechanism that involves Hip1. We have previously shown that Hip1 prevents optimal CD40 expression through proteolysis of its substrate GroEL2 (Naffin-Olivos et al., 2014; Georgieva et al., 2018). We showed that full-length recombinant GroEL2 protein induces robust CD40 expression on DCs through a TLR2-dependent mechanism (Madan-Lala et al., 2014). However, the cleaved form of GroEL2, which is the form that predominates in wild type Mtb, and which is present in both live and killed cultures, is unable to induce CD40 (Georgieva et al., 2018). Therefore, we posit that the ability of the hip1mutant to induce Notch ligands on lung DCs is mediated by the presence of full length GroEL2, as this protein remains uncleaved in the absence of the Hip1 protease (Rengarajan et al., 2008; Naffin-Olivos et al., 2014; Georgieva et al., 2018). We posit that full length GroEL2 specifically limits CD40 signaling on DCs (and not general DC activation or other co-stimulatory markers such as CD80/CD86) as we have demonstrated that Hip1 limits Notch ligand expression through a CD40-dependent mechanism (Figure 9). Thus, by impeding the CD40 costimulatory pathway, wild type Mtb limits DLL4-NOTCH receptor signaling, leading to delayed and sub-optimal Th17 responses. In addition, our data on C. albicans (Figures S3 and S7) support the idea that modulating Notch signaling may be a strategy employed by Th17-polarizing bacteria to promote Th17 responses as well as by other pathogens that manipulate DC-T cell crosstalk and promote disease, thereby extending our insights beyond TB.
Our results also suggest that Notch signaling plays a role in generating protective immune responses that help control mycobacterial burdens. Correlation analysis revealed that IL-17 and NOTCH2 inversely correlate with Mtb CFU (Figure 7). In our DC IT transfer experiment, we observed induction of NOTCH2+ lung CD4 T cells following CD40-engagement was reduced after DLL4 blockade (Figure 6). Additionally, infection with the hip1mutant, which naturally engages the CD40−CD40L pathway, led to induction of DLL4 on lung DCs and NOTCH2 on T cells (Figure 8). These results suggest that wild type Mtb actively dampens Notch ligand and Notch receptor expression to impede DC-T cell crosstalk in order to promote disease and maintain bacterial burdens in the lung during infection. Previous work from our lab showed that infection with the hip1mutant significantly prolonged survival of mice and resulted in dramatically lower lung immunopathology (Rengarajan et al., 2008). Our data suggest that early induction of CD40 and DLL4 in the absence of Hip1 leads to higher IL-17+NOTCH2+ Th subsets and more protective immune responses. Our results that suggest Notch signaling is important for protection aligns with recent studies in other experimental models (Tu et al., 2005; Ito et al., 2009; Webb et al., 2019). Of interest, a recent study on SARS-CoV-2 infection found that Notch signaling is upregulated in juvenile compared to older macaques and suggests that lack of Notch signaling could be a risk factor for the increased susceptibility of older individuals to COVID-19 (Rosa et al., 2021). Our results on Notch signaling in Mtb-infected mice highlight the need to better understand this pathway in human TB, where there is limited data. A recent study measured the expression of Notch ligands and receptors in human PBMC samples and found that individuals with active TB exhibited higher expression of DLL4 on monocytes and NOTCH1 on T cells compared to healthy individuals (Castro et al., 2020). Another study found that DLL4 expression on monocytes in individuals with TB was reduced following anti-TB treatment (Schaller et al., 2016). However, these studies focus on chronic stages of TB disease and do not study Notch ligand expression during early events following Mtb infection. Therefore, additional studies in humans and non-human primate models of latent and active TB are needed to dissect the role of Notch ligand signaling in initiating Th17 responses in lung compartments following Mtb infection as well as within granulomas in chronic stages of TB disease. The timing and location of DLL4 and Jagged1 signaling during Mtb infection will likely dictate Th1/Th17 balance and protective versus pathogenic outcomes, along with other suppressive pathways that have been identified to function during Mtb infection. Moreover, because we know that aberrant IL-17 or DLL4 expression is not beneficial for the host (Cruz et al., 2006, 2010; Pollara et al., 2021; Tran et al., 2013; Mochizuki et al., 2013; Chung et al., 2017), inducing balanced Th1/Th17 immunity along with their temporal and spatial context will be important considerations in designing vaccines and host-directed therapies for TB. DLL4 has also been shown to be important for reducing inflammation in non-TB contexts (Huang et al., 2013, 2017), where it has been implicated in activating T cells and other responses that mediate graft-versus-host disease and autoimmunity (Tran et al., 2013; Mochizuki et al., 2013; Chung et al., 2017). Moreover, excessive IL-17 is well established as a mechanism of autoimmune-driven pathology and can also be detrimental in TB disease depending on the timing and location (Cruz et al., 2006, 2010; Pollara et al., 2021). Furthermore, studies on immunomodulation of DLL4, either by targeting CD40 signaling or via small molecules that directly target Notch ligands on DCs, will be necessary for determining the utility of modulating the CD40-DLL4 axis for host directed therapies that prevent excessive TB pathology.
In addition to providing new insights into Mtb immune evasion strategies, identification of the CD40-DLL4 axis in Th17 polarization has implications for improving vaccine and adjuvant design. Studies on Mtb immunomodulatory proteins that subvert host protective immunity, such as those presented here, are vital for elucidating pathways that should be overcome in the context of live-attenuated vaccines. Several studies have demonstrated that deletion of immune evasion proteins in either Mtb or the BCG vaccine strain is a viable strategy for developing more efficacious vaccines against TB (Grode et al., 2005; Festjens et al., 2011; Kaushal et al., 2015; Martinot et al., 2020). BCG has previously been shown to affect DC expression of Notch ligands (Ito et al., 2009; Schaller et al., 2016) and TLR9 was reported to regulate granulomas induced by BCG via DLL4 (Ito et al., 2009). Our lab has developed a knockout of hip1 in BCG (BCGΔhip1) and found it to induce higher levels of IL-17 than wild type BCG (Bizzell et al., 2018). Thus, it would be interesting to test whether DLL4 signaling is operant in the context of BCGΔhip1 vaccination and whether engaging the CD40-DLL4 axis has an adjuvant effect on BCGΔhip1 vaccination. Furthermore, designing adjuvants that crosslink CD40 and/or DLL4 on DCs during subunit vaccination can induce beneficial early Th17 polarization and a more balanced Th1/Th17 response to vaccination.
In summary, our study demonstrates that engaging CD40 during Mtb infection is critical for inducing the Notch ligand DLL4, which is necessary for Th17 polarization during infection. By delineating the DLL4-CD40-Th17 axis in TB, our work provides clear targets that can be harnessed for new adjuvant and vaccination approaches not only in TB, but also in other pathogenic infections and autoimmune disease states where therapeutically manipulating Th17 response is desirable.
Limitations of the study
We identify an important role for CD40-dependent DLL4 signaling during Th17 polarization in Mtb infection, but recognize there are limitations to our study. Although we have shown that Mtb limits CD40 and Notch signaling through the Hip1 protease and that infection with a hip1mutant strain results in higher Th17 responses, we have not demonstrated that blockade of DLL4 during hip1mutant infection abrogates Th17 polarization and/or worsens disease outcomes, pathology or CFU. Because DLL4 knockout mice are embryonic lethal, we used blocking antibodies against DLL4 for our studies. However, blockade of DLL4 in the context of examining disease progression and pathology following hip1mutant infection, necessitates using multiple doses of antibody over long periods of time. Prolonged use of blocking antibodies in vivo can lead to toxicity, which would complicate the interpretation of the results. Furthermore, work will be necessary to elucidate the role of DLL4/CD40 signaling in long-term disease outcomes for TB. We have also not determined whether the effect of DLL4 blockade is specific to Th17 polarization or expansion, although we have demonstrated that Th1 and other cytokine production (apart from those associated with Th17) remains intact. A future experiment measuring proliferation can better differentiate between these two possibilities in vivo.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Murine DLL4 blocking antibody (Clone: HMD4; BioXCell) | BioXCell | Catalog #: BE0127; RRID: AB_10950366 |
| InVivoMAb polyclonal Armenian hamster IgG [DLL4 isotype] (BioXCell) | BioXCell | Catalog #: BE0091; RRID: AB_1107773 |
| Jagged1 blocking antibody (Clone: HMJ1-29; Biolegend) | Biolegend | Catalog #: 130902; RRID: AB_2561301 |
| Purified Armenian hamster IgG Isotype [Jagged1 isotype] (Biolegend) | Biolegend | Catalog #: 400902 |
| FITC anti-mouse I-A/I-E (Clone: M5/114.15.2) | Biolegend | Catalog #: 107606; RRID: AB_313321 |
| PE anti-mouse DLL4 (Clone: HMD4-1) | Biolegend | Catalog #: 130807; RRID: AB_1227634 |
| PE-Cy5 anti-mouse CD40 (Clone: 1C10) | Invitrogen | Catalog #: 15-0401-82; RRID: AB_468747 |
| PE-Cy7 anti-mouse CD11c (Clone: N418) | Invitrogen | Catalog #: 25-0114-81; RRID: AB_469589 |
| APC anti-mouse CD339 (Jagged1) (Clone: HMJ1-29) | Biolegend | Catalog #: 130914; RRID: AB_2561305 |
| Alexa700 anti-mouse CD86 (Clone: GL-1) | Biolegend | Catalog #: 105024; RRID: AB_493721 |
| APC-Cy7 anti-mouse CD11b (Clone: M1/70) | Biolegend | Catalog #: 101225; RRID: AB_830641 |
| FITC anti-mouse CD14 (Clone: Sa14-2) | Biolegend | Catalog #: 123308; RRID: AB_940580 |
| FITC anti-mouse NK-1.1 (Clone: PK136) | Biolegend | Catalog #: 108706; RRID: AB_313393 |
| FITC anti-mouse TER-119/Erythroid (Clone: TER-119) | Biolegend | Catalog #: 116206; RRID: AB_313707 |
| FITC anti-mouse CD19 (Clone: 1D3/CD19) | Biolegend | Catalog #: 152404; RRID: AB_2629813 |
| FITC Rat anti-mouse IL-2 (Clone: JES6-5H4) | BD | Catalog #: 554427; RRID: AB_395385 |
| PE Rat anti-mouse Vβ6 T-Cell (Clone: RR4-7) | BD | Catalog #: 553194; RRID: AB_394701 |
| PerCP anti-mouse CD45 (Clone: 30-F11) | Biolegend | Catalog #: 103130; RRID: AB_893339 |
| Alexa 700, Hamster anti-mouse CD3ε (Clone: 500A2) | BD | Catalog #: 557984; RRID: AB_396972 |
| APC-Cy7 anti-mouse CD8α (Clone: 53-6.7) | Biolegend | Catalog #: 100714; RRID: AB_312753 |
| BV650 anti-mouse/human CD44 (Clone: IM7) | Biolegend | Catalog #: 103049; RRID: AB_2562600 |
| BV650 anti-mouse CD183 (CXCR3) (Clone: CXCR3-173) | Biolegend | Catalog #: 126531; RRID: AB_2563160 |
| BV711 anti-mouse CD8α (Clone: 53-6.7) | Biolegend | Catalog #: 100748; RRID: AB_2562100 |
| BV785 anti-mouse CD196 (CCR6) (Clone: 29-2L17) | Biolegend | Catalog #: 129823; RRID: AB_2715923 |
| APC-Cy7 Rat anti-mouse CD44 (Clone: IM7) | BD | Catalog #: 560568: AB_1727481 |
| BUV395 Rat Anti-Mouse CD84 (Clone: 1D3/CD84) | BD | Catalog #: 749570; RRID: AB_2873895 |
| BUV496 Rat Anti-Mouse I-A/I-E (Clone: 2G9) | BD | Catalog #: 750171; RRID: AB_2874376 |
| BUV563 Hamster Anti-Mouse CD80 (Clone: 16-10A1) | BD | Catalog #: 741272; RRID: AB_2870813 |
| BUV661 Rat Anti-Mouse CD115 (Clone: T38-320) | BD | Catalog #: 749973; RRID: AB_2874200 |
| BUV737 Rat Anti-Mouse DLL4 (Clone: 9A1.5) | BD | Catalog #: 748394; RRID: AB_2872813 |
| BUV805 Rat Anti-Mouse F4/80 (Clone: T45-2342) | BD | Catalog #: 749282; RRID: AB_2873657 |
| BV421 Rat Anti-Mouse CD172a (Clone: P84) | BD | Catalog #: 740071; RRID: AB_2739835 |
| BV421 anti-mouse CD169 (Clone: 3D6.112) | Biolegend | Catalog #: 142421; RRID: AB_2734202 |
| BV480 Hamster Anti-Mouse CD103 (Clone: 2E7) | BD | Catalog #: 748252; RRID: AB_2872682 |
| BV570 anti-mouse CD3 (Clone: 17A2) | Biolegend | Catalog #: 100225; RRID: AB_10900444 |
| BV570 anti-mouse CD19 (Clone: 6D5) | Biolegend | Catalog #: 115535; RRID: AB_10933260 |
| BV650 anti-mouse/rat XCR1 (Clone: ZET) | Biolegend | Catalog #: 148220; RRID: AB_2566410 |
| BV711 anti-mouse CD11c (Clone: N418) | Biolegend | Catalog #: 117349; RRID: AB_2563905 |
| BV750 anti-mouse CD45 (Clone: 30-F11) | Biolegend | Catalog #: 103157; RRID: AB_2734155 |
| BV786 Mouse Anti-Mouse CD64 a/b (Clone: X54-5/7.1) | Biolegend | Catalog #: 741024; RRID: AB_2740644 |
| FITC anti-mouse Ly-6G (Clone: 1A8) | Biolegend | Catalog #: 127606; RRID: AB_1236494 |
| BB700 Rat Anti-Mouse CD124 (Clone: mIL4R-M1) | BD | Catalog #: 742172; RRID: AB_2871410 |
| PE anti-mouse Jagged1 (Clone: HMJ1-29) | Biolegend | Catalog #: 130908; RRID: AB_2561303 |
| PE/Cy5 anti-mouse CD3ε (Clone: 145-2C11) | Biolegend | Catalog #: 100310; RRID: AB_312675 |
| PE-Cy7 anti-mouse/human CD11b (Clone: M1/70) | Biolegend | Catalog #: 101215; RRID: AB_312798 |
| PE/Dazzle 594 anti-mouse Ly-6C (Clone: HK1.4) | Biolegend | Catalog #: 128043; RRID: AB_2566576 |
| PE-Cy5 anti-mouse CD24 (Clone: M1/69) | Biolegend | Catalog #: 101812; RRID: AB_439714 |
| PE-Cy7 anti-mouse JAML (Clone: 4/E10) | Novus Biologicals | Catalog #: NBP1-43309PECY7 |
| Alexa 647 Rat Anti-Mouse S100A9 (Clone: 2B10) | BD | Catalog #: 565833; RRID: AB_2739373 |
| Alexa 700 anti-mouse/human CD11b (Clone: M1/70) | Biolegend | Catalog #: 101222; RRID: AB_493705 |
| PE-CF594 Rat anti-mouse IL-17A (Clone: TC11-18H10) | BD | Catalog #: 562542; RRID: AB_2737643 |
| PE-Cy7 Rat anti-mouse TNF (Clone: MP6-XT22) | BD | Catalog #: 557644; RRID: AB_396761 |
| BV786 Rat anti-mouse CD4 (Clone: RM4-5) | BD | Catalog #: 563727; RRID: AB_2728707 |
| APC anti-mouse IFN-γ (Clone: XMG1.2) | Invitrogen | Catalog #: 17-7311-81; RRID: AB_469503 |
| BV421 Rat anti-mouse IL-2 (Clone: JES6-5H4) | BD | Catalog #: 562969; RRID: AB_2737923 |
| PE Rat anti-mouse NOTCH1 (Clone: 22E5.5) | BD | Catalog #: 562754; RRID: AB_2737770 |
| PerCP-Cy5.5 anti-mouse IL-22 (Clone: Poly5164) | Biolegend | Catalog #: 516411; RRID: AB_2563373 |
| PE-Cy7 anti-mouse CD40L (Clone: SA047C3) | Biolegend | Catalog #: 157008; RRID: AB_2832545 |
| V450 Hamster anti-mouse CD3ε (Clone: 500A2) | BD | Catalog #: 560801; RRID: AB_2034005 |
| BV605 Rat anti-mouse NOTCH2 (Clone: 16F11) | BD | Catalog #: 745122; RRID: AB_2742726 |
| Alexa700 Rat anti-mouse CD4 (Clone: RM4-5) | BD | Catalog #: 557956; RRID: AB_396956 |
| Bacterial and virus strains | ||
| M. tuberculosis H37Rv | BEI Resources | Catalog #: NR-123 |
| M. tuberculosis H37Rv hip1mutant | Rengarajan et al. (2008) | |
| Chemicals, peptides, and recombinant proteins | ||
| Red Blood Lysis Buffer | Sigma | Catalog #: R7757-100ML |
| RMPI-1640 | Lonza | Catalog #: 12-702F |
| L-Glutamine | Lonza | Catalog #: 17-605E |
| MEM NEAA | Gibco | Catalog #: 11140-050 |
| HEPES Buffer | Corning | Catalog #: 25-060-CI |
| Sodium Pyruvate | Lonza | Catalog #: 13-115E |
| Fetal Bovine Serum (FBS) | Gemini | Catalog #: 900-108 |
| Penicillin/Streptomycin | Lonza | Catalog #: 17-602E |
| 2-Mercaptoethanol | Gibco | Catalog #: 21985-023 |
| Murine rGM-CSF | R&D Systems | Catalog #: 415-ML-005/CF |
| Trypan blue stain | Invitrogen | Catalog #: T10282 |
| Hanks’ Balanced Salt Solution (HBSS) | Corning | Catalog #: 21-021-CV |
| Collagenase, Type IV | Worthington | Catalog #: LS004210 |
| DNAse I | Worthington | Catalog #: LS002058 |
| Amikacin sulfate salt | Sigma | Catalog #: A2324-5G |
| Triton X-100 | Fisher Scientific/ACROS Organics | Catalog #: AC215682500 |
| ESAT-61-20 peptide | Genemed Synthesis Inc. | Sequence: MTEQQWNFAGIEAAASAIQG |
| OVA323-339 Peptide | Invivogen | Catalog #: vac-isq |
| Whole Cell Lysate (WCL) | BEI Resources | Catalog #: NR-14822 |
| Mouse CD40 Ligand Trimer (CD40LT) | Adipogen | Catalog #: AG-40B-0020 |
| LPS-EB | Invivogen | Catalog #: tlrl-3pelps |
| Zymosan | Invivogen | Catalog #: tlrl-zyn |
| Heat-Killed C. albicans | Invivogen | Catalog #: tlrl-hkca |
| CD11c MicroBeads UltraPure, mouse | Miltenyi Biotec | Catalog #: 130-108-338 |
| CD4 (L3T4) MicroBeads, mouse | Miltenyi Biotec | Catalog #: 130-117-043 |
| Middlebrook 7H9 | BD | Catalog #: 271310 |
| Glycerol | Sigma | Catalog #: G6279-1L |
| OADC | BD | Catalog #: 212351 |
| Tween80 | VWR | Catalog #: 97061-674 |
| Kanamycin solution from Streptomyces kanamyceticus | Sigma | Catalog #: K0254-20ML |
| Middlebrook 7H10 | BD | Catalog #: 262710 |
| Cycloheximide | Sigma | Catalog #: C7698-5G |
| Brefeldin A | Sigma | Catalog #: B7651-5MG |
| GolgiStop Protein Transport Inhibitor (GolgiStop) | BD | Catalog #: 51-2092KZ |
| Fixable Aqua Dead Cell Stain Kit | Invitrogen | Catalog #: L34957 |
| Fixable Near-IR Dead Cell Stain Kit | Invitrogen | Catalog #: L34976 |
| Purified Rat Anti-Mouse CD16/CD32 Fc Block | BD | Catalog #: 553141 |
| Dulbecco’s Phosphate Buffered Saline (PBS) | Sigma | Catalog #: D8537-500ML |
| EDTA | Corning | Catalog #: 46-034-CI |
| Cytofix/Cytoperm Fixation/Permeabilization Kit | BD | Catalog #: 554714 |
| 4% Paraformaldehyde | Electron Microscopy Sciences | Catalog #: 157-4-100 |
| Anti-Rat and Anti-Hamster Ig κ /Negative Control Compensation Particles | BD | Catalog #: 552845 |
| UltraComp eBeads Compensation Beads | Invitrogen | Catalog #: 01-2222-42 |
| ArC Amine Reactive Compensation Bead Kit | Invitrogen | Catalog #: A10346 |
| SYBR Green PCR Master Mix | Applied Biosystems | Catalog #: 4309155 |
| Water, Molecular Biology | Quality Biological | Catalog #: 351-029-721 |
| Critical commercial assays | ||
| Mouse Naïve CD4+ T Cell Isolation Kit | StemCell | Catalog #: 19765 |
| Mouse IL-6 ELISA | BD | Catalog #: 555240 |
| Mouse IL-12p40 ELISA | BD | Catalog #: 555165 |
| Mouse IFN-γ ELISA | Mabtech | Catalog #: 3321-1H-6 |
| Mouse IL-2 ELISA | BD | Catalog #: 555148 |
| Mouse IL-17A ELISA | Invitrogen | Catalog #: 88-7371-88 |
| Mouse IL-22 ELISA | R&D Systems | Catalog #: DY582-05 |
| Quick-RNA Miniprep Kit | Zymo | Catalog #: R1055 |
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Catalog #: 4368814 |
| Experimental models: Cell lines | ||
| Bone marrow-derived dendritic cells (BMDCs) | C57BL/6 mice; this study. | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6 | The Jackson Laboratory | Catalog #: 000664 |
| Mouse: C57BL/6 CD40−/− (B6.129P2-Cd40tm1Kik/J) | The Jackson Laboratory | Catalog #: 002928 |
| Mouse: C57BL/6 OT-II OVA323–339 Thy1.1+ | Provided by: Bali Pulendran, Stanford University (formerly Emory University) Developed by: Francis Carbone, University of Melbourne) |
N/A |
| Mouse: C57BL/6 ESAT-61-20/I-Ab | Andrea Cooper, University of Leicester (formerly Trudeau Institute) | N/A |
| Oligonucleotides | ||
| KiCqStart SYBR Primer: Murine Il6 | Sigma | M_Il6_1 |
| KiCqStart SYBR Primer: Murine Il12b | Sigma | M_Il12b_1 |
| KiCqStart SYBR Primer: Murine Dll4 | Sigma | M_Dll4_3 |
| KiCqStart SYBR Primer: Murine Jag1 | Sigma | M_Jag1_1 |
| GAPDH qPCR Primer | Eurofins | F: TGGCCTTCCGTGTTCCTAC R: GAGTTGCTGTTGAAGTCGCA |
| Software and algorithms | ||
| GraphPad Prism v9 | GraphPad | www.graphpad.com |
| FlowJo v10 | BD | www.flowjo.com |
| R Studio | R | www.rstudio.com |
| BioRender | BioRender | www.biorender.com |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by Jyothi Rengarajan (jrengar@emory.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Mice
C57BL/6 wild type (WT) and CD40−/− (B6.129P2-Cd40tm1Kik) female mice were purchased from The Jackson Laboratory. C57BL/6 OT-II OVA323–339 Thy1.1+ mice (originally developed by Dr. Francis Carbone, University of Melbourne) were kindly provided by Dr. Bali Pulendran (Stanford University, formerly Emory University) and bred in the Yerkes vivarium. C57BL/6 ESAT-61-20/I-Ab transgenic mice were kindly provided by Dr. Andrea Cooper (University of Leicester, formerly Trudeau Institute) and bred in the Yerkes vivarium. All WT mice used for experiments were eight-to-twelve weeks of age and all transgenic mice used were eight-to-eighteen weeks of age. Mice were housed in either the Yerkes National Primate Center animal BSL-3 or BSL-1 vivarium under sterile conditions with food and water provided ad libitum. All animals were handled according to the regulations formulated by the Emory University Institutional Animal Care and Use Committee (IACUC).
Bone marrow-derived dendritic cells
For most experiments in this study, primary cell culture bone marrow-derived dendritic cells (BMDCs) were used. BMDCs were generated from female C57BL/6 WT mice purchased from The Jackson Laboratory as previously described (Sia et al., 2017). Briefly, the femur and tibia of mice were extracted and flushed using cold RPMI-1640 (Lonza). Following red blood cell (RBC) lysis using RBC Lysis Buffer (Sigma), progenitor cells were spun down and plated at a concentration of 1E6 cells/mL in R10 media (RPMI-1640 [Lonza] with 2mM L-glutamine [Lonza], 0.1 mM NEAA [Gibco], 10 mM HEPES [Corning], 1mM Sodium Pyruvate [Lonza], 10% heat-inactivated FBS [Gemini]) supplemented with 1:100 Penicillin/Streptomycin (Lonza), 1:1000 2-mercaptoethanol (BME; Gibco) and 20 ng/mL murine rGM-CSF (R&D Systems). Cells were grown at 37°C with 5% CO2. On Day 3 and 6 following plating, cells were fed using R10 media supplemented with 2-mercaptoethanol and rGM-CSF. On day 8 following plating, cells were harvested and purified using mouse CD11c+ beads (MiltenyiBiotec) according to manufacturer recommendations. Purity of BMDCs was confirmed using flow cytometry. For BMDC stimulations and infections, purified BMDCs were plated in 24-well tissue culture plates at a concentration of 6E5 cells/mL in R10 media supplemented with 1:1000 BME. For intratracheal assay BMDC preparations, cells were plated in tissue culture plates at a concentration of 1E6 cells/mL. Cells were allowed to adhere to the plate (4 h post-plating to overnight) before use. All cells throughout the study were counted using trypan blue stain (Thermo-Fisher Scientific) on a Countess Automated Cell Counter (Invitrogen).
Bacterial strains
Mycobacterium tuberculosis (Mtb) strains H37Rv and H37Rv hip1mutant (Rengarajan et al., 2008) were used. As previously described (Madan-Lala et al., 2014; Naffin-Olivos et al., 2014; Sia et al., 2017), Mtb strains were grown in liquid media Middlebrook 7H9 (BD Difco) supplemented with 0.5% glycerol (Sigma), 10% oleic acid-albumin-dextrose-catalase (OADC) (BD) and 0.05% Tween 80 (VWR) at 37°C and shaking at 75 rpm. Additionally, 20 μg/mL kanamycin (Sigma) was included for growing the hip1mutant. Stocks were prepared by growing cultures to an OD600 of 0.4–0.6, then filtered and resuspended in 7H9 media with 25% glycerol (Sigma) and stored at −80°C. Before use, stocks were titered to determine CFU. Heat-killed stocks were prepared as previously described (Sia et al., 2017).
Method details
Mtb in vitro infection and stimulation of DCs
For in vitro infections with live Mtb, purified BMDCs were infected with H37Rv at an MOI of 1.0. Briefly, bacterial cultures were centrifuged and resuspended in R10 media supplemented with 1:1000 BME (and 1 μg/mL CD40LT (Adipogen) for relevant conditions). Plates were then placed in the 37°C incubator (with 5% CO2) for 6 h. Following infection, a 200 μg/mL Amikacin (Sigma) solution (in R10 supplemented with 1:1000 BME) was added to cells for 30 min to kill extracellular bacteria. Afterwards, wells were washed 3× PBS (Sigma) and resuspended in R10 media supplemented with 2-mercaptoethanol (and 1 μg/mL CD40LT for relevant conditions) and the plates were placed in the 37°C incubator until designated time point. For certain wells, cells were lysed using PBS +0.5% Triton X-(Fisher Scientific) and plated to determine intracellular CFU on Middlebrook 7H10 agar plates (supplemented with 0.5% glycerol [Sigma], 10% OADC [BD] and 0.1 mg/mL cycloheximide solution [Sigma]). For BMDC stimulations, cells were allowed to adhere until stimulation, and existing supernatant was removed and replaced with R10 media with 1:1000 BME containing stimuli. Cells were then placed in the incubator until designated time points. Heat-killed bacteria were used at an MOI of 30. Stimulations with CD40LT alone used 1 μg/mL CD40LT. For PRR/TLR stimulations, 0.1 μg/mL LPS (Invivogen), 10 μg/mL Zymosan (Invivogen), or heat-killed C. albicans (Invivogen) at an MOI of 17 were used. Following stimulation, cell-free supernatants were removed from each well and stored for protein quantification (for live infection, supernatants were filtered using a 0.23 μm filter and removed from the BSL-3). To collect samples for RNA purification, wells were washed ×1 using PBS (Sigma) and 300 μL of RNA Lysis Buffer (Zymo) was added to each well. Samples were then flash-frozen using 70% ethanol and dry ice and stored in the −80°C until RNA purification.
RNA extraction, cDNA generation, and qPCR
RNA samples were purified using the Quick-RNA Miniprep Kit (Zymo) according to manufacturer’s instructions. For BSL-3 samples, RNA was purified in the BSL-3 and removed at the elution step. Following purification, RNA was quantified using a spectrophotometer (NanoDrop ND-1000 or NanoDrop One [Fisher Scientific]). cDNA was made from each sample using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) using 100 ng of RNA and carried out in a C1000 Thermal Cycler (BioRad) according to manufacturer’s recommendation. Quantitative Polymerase Chain Reactions (qPCR) were carried out in a 384-well plate format on a QuantStudio 5 Smart Start (Thermo-Fisher Scientific) machine for “SYBR” reactions. SYBR Green PCR Master Mix (Applied Biosystems) was combined with molecular-biology grade water (Quality Biological), cDNA, and primers (10 μM) and pipetted into each well. Murine KiCqStart SYBR Primers used for this study (mouse m_Il6_1, m_Il12b_1, m_Dll4_3, m_Jag1_1) were purchased from Sigma. GAPDH primer was purchased from Eurofins (sequence: F: TGGCCTTCCGTGTTCCTAC R: GAGTTGCTGTTGAAGTCGCA). Prior to use, primer efficiency curves were generated for each primer. Each sample for qPCR was run in triplicate. All qPCR data were analyzed using the ΔΔCt method and expression of genes was standardized to 0H uninfected (UI) sample GAPDH. All qPCR Data are presented as 2−ΔΔCt.
Enzyme-Linked Immunosorbent Assay
Cell-free supernatants were used to enumerate cytokine protein levels using Enzyme-Linked Immunosorbent Assay (ELISA) assay. All the ELISAs were run according to manufacturer instructions: murine IL-6 (BD), murine IL-12p40 (BD), murine IFN-γ (Mabtech), murine IL-2 (BD), murine IL-17A (Invitrogen), and murine IL-22 (R&D Systems). Plates were washed using a Biotek ELx405 machine and measured using a BioTek ELx808 reader.
DC-T cell co-culture assays
Purified BMDCs were stimulated with Mtb (as described above) with different conditions for 24 h. Following this time period, supernatant was removed and wells were washed 1× with PBS (Sigma). A mixture of R10 (supplemented with 1:1000 BME) with 10 μg/mL cognate peptide (OVA323-339, Invivogen) was then added and the BMDCs were “pulsed” for 1 h. Afterwards, OT-II OVA323–339-specific Thy1.1+ CD4 T cells were added at a ratio of 4:1 T cell: DCs to each well. For conditions that required antibody blockade, the following antibodies (15 μg/mL - 60 μg/mL) were added at the co-culture step: murine DLL4 blocking antibody (Clone: HMD4; BioXCell), InVivoMAb polyclonal Armenian hamster IgG [DLL4 isotype] (BioXCell), Jagged1 blocking antibody (Clone: HMJ1-29; Biolegend), purified Armenian hamster IgG Isotype [Jagged1 isotype] (Biolegend). The cell culture plate was placed in a 37°C incubator (with 5% CO2) for 72 h. Supernatants were harvested and then briefly spun to ensure a cell-free mixture. Naive CD4 T cells were purified from the spleens of female and male C57BL/6 OT-II OVA323–339 Thy1.1+ mice using the mouse naive CD4+T cell isolation kit (StemCell) according to manufacturers’ instructions [isolated cells were >95% viable].
IT instillation of DCs and mouse tissue harvest
Purified BMDCs were stimulated or infected (as outlined above) in tissue culture plates for intratracheal (IT) transfer. BMDCs were harvested either at 24 h (stimulated BMDCs) or at 48 h (infected BMDCs). For conditions using CD40LT and antibody blockade, relevant blocking antibodies were provided during infection/stimulation. Afterwards, cells were harvested from the plates, washed 1× using PBS (Sigma), and then spun down followed by counting. Cells were then resuspended at 20E6/mL in PBS. For experiments in which Mtb-infected BMDC were transferred IT in the presence of blocking antibodies (anti-DLL4 or isotype controls), cells were resuspended in PBS and antibodies were added at 60 μg/mL. For experiments in which Mtb-stimulated BMDCs were transferred IT in the presence of blocking antibodies (anti-DLL4, anti-Jagged1, or isotype controls), cells were resuspended in PBS and antibodies were added at 30 μg/mL. For IT transfer, mice were anesthetized using isoflurane in a closed isoflurane chamber (Med-Vet) and 1 × 106 infected BMDCs (in 50 μL volume) were instilled into the trachea of mice as previously described (Sia et al., 2017). For Mtb-stimulated BMDC IT experiments, C57BL/6 ESAT-61-20/I-Ab transgenic naive CD4 T cells were transferred into mice one day before IT transfer. Briefly, spleens from female and male ESAT-6 transgenic mice were harvested and made into a single-cell suspension. CD4 T cells were purified using mouse CD4 (L3T4) Micro-Beads (MiltenyiBiotec) according to manufacturer’s instructions. The resulting purified CD4 T cells were resuspended at a concentration of 1E7 cells/mL and 1E6 cells were transferred to mice via the intravenous (IV) route one day before IT transfer. Mice were euthanized at either six days or four weeks post-IT, depending on experiment, using isoflurane overdose. Lungs were placed into lung C-Tubes (MiltenyiBiotec) containing HBSS (Corning) supplemented with 2% heat-inactivated FBS (Gemini) and 10mM HEPES (Corning). A mixture of 0.1% collagenase, type IV (Worthington) and 0.01% DNAse I (Worthington) was added into each tube. Lung tissue was homogenized using an automated gentleMACS Dissociator (MiltenyiBiotec) using the manufacturer’s murine lung processing program. After addition of collagenase/DNAse mixture, lungs were dissociated and placed in a 37°C (with 5% CO2) incubator for 30 min. Following this time, lungs were dissociated again using a murine lung processing program. The tubes were then spun down and red blood cells were lysed from the mixture using RBC lysis buffer (Sigma). Lungs were resuspended at 10 × 106E6/mL in R10 supplemented with 1:1000 BME and 1E6 cells were plated per well for ex vivo or ESAT-61-20 stimulation in 96-well U-bottom propylene plates and placed in the 37°C incubator (with 5% CO2). For enumeration of bacteria, a portion of the lungs was harvested in sterile 2 mL tubes (Sarstedt) containing stainless steel beads (Next Advance) and PBS +0.02% Tween80. The lungs were then homogenized in a Bullet Blender (Next Advance). Serial dilutions were plated onto Middlebrook 7H10 plates (with or without 20 μg/mL Kanamycin for hip1mutant) to determine the CFU.
Aerogenic infection of mice with Mtb
Mtb cultures for aerosol infection were prepared as previously described (Sia et al., 2017). Briefly, mice were infected via the aerosol route (∼100 CFU) using a nose only exposure chamber (In-Tox Products). A day following aerosol infection, mice were euthanized to determine bacterial burdens as described above.
Flow cytometry
For lung suspensions, cells were either left unstimulated (ex vivo) or stimulated with 10 μg/mL ESAT-61-20 peptide (Genemed Synthesis, Inc) or 10 μg/mLwhole cell lysate (WCL) [BEI]. Plates were then placed in the 37°C incubator (with 5% CO2) and after 2 h, a mixture of 5 μg/mL Brefeldin A (Sigma) and 1:1500 GolgiStop (BD) in R10 supplemented with 1:1000 BME was added. The plate was then returned to the incubator for 4H (6H total) for ESAT-6 stimulations or overnight for WCL stimulations and the cells were stained the next day. For staining BMDCs, cells were harvested from plates at designated time points and stained directly. To distinguish between live and dead cells, all cells were stained with Fixable Aqua Dead Cell Stain Kit (Molecular Probes) or Fixable Near-IR Dead Cell Stain Kit (Invitrogen). Additionally, mouse Fc block (BD) was used before staining. To stain BMDCs, the following antibodies were used (all surface): FITC anti-mouse I-A/I-E (clone: M5/114.15.2, Biolegend), PE anti-mouse DLL4 (clone: HMD4-1, Biolegend), PE-Cy5 anti-CD40 (clone: 1C10, eBioscience), PE-Cy7 anti-mouse CD11c (clone: N418, eBioscience), APC anti-mouse CD339 (Jagged1) (clone: HMJ1-29, Biolegend), Alexa700 anti-mouse CD86 (clone: GL-1, Biolegend), APC-Cy7 anti-mouse CD11b (clone: M1/70, Biolegend). To stain lung T cells, the following surface stain antibodies were used: FITC anti-mouse CD14 (clone: Sa14-2, Biolegend), FITC anti-mouse NK-1.1 (clone: PK136, Biolegend), FITC anti-mouse TER-119/Erythroid (clone: TER-119, Biolegend), FITC anti-mouse CD19 (clone: 1D3/CD19, Biolegend), FITC Rat anti-mouse IL-2 (clone: JES6-5H4, BD), PE Rat anti-mouse Vβ6 T-Cell (clone: RR4-7, BD), PerCP anti-mouse CD45 (clone: 30-F11, Biolegend), Alexa 700, Hamster anti-mouse CD3ε (clone: 500A2, BD), APC-Cy7 anti-mouse CD8α (clone: 53-6.7, Biolegend), BV650 anti-mouse/human CD44 (clone: IM7, Biolegend), BV650 anti-mouse CD183 (CXCR3) (clone: CXCR3-173, Biolegend), BV711 anti-mouse CD8α (clone: 53-6.7, Biolegend), BV785 anti-mouse CD196 (CCR6) (clone: 29-2L17, Biolegend), APC-Cy7 Rat anti-mouse CD44 (clone: IM7, BD). To stain lung innate cells, the following surface stain antibodies were used: BUV395 Rat Anti-Mouse CD84 (clone: 1D3/CD84, BD), BUV496 Rat Anti-Mouse I-A/I-E (clone: 2G9, BD), BUV563 Hamster Anti-Mouse CD80 (clone: 16-10A1, BD), BUV661 Rat Anti-Mouse CD115 (clone: T38-320, BD), BUV737 Rat Anti-Mouse DLL4 (clone: 9A1.5, BD), BUV805 Rat Anti-Mouse F4/80 (clone: T45-2342, BD), BV421 Rat Anti-Mouse CD172a (clone: P84, BD), BV421 anti-mouse CD169 (clone: 3D6.112, Biolegend), BV480 Hamster Anti-Mouse CD103 (clone: 2E7, BD), BV570 anti-mouse CD3 (clone: 17A2, Biolegend), BV570 anti-mouse CD19 (clone: 6D5, Biolegend), BV650 anti-mouse/rat XCR1 (clone: ZET, Biolegend), BV711 anti-mouse CD11c (clone: N418, Biolegend), BV750 anti-mouse CD45 (clone: 30-F11, Biolegend), BV786 Mouse Anti-Mouse CD64 a/b (clone: ×54-5/7.1, BD), FITC anti-mouse Ly-6G (clone: 1A8, Biolegend), BB700 Rat Anti-Mouse CD124 (clone: mIL4R-M1, BD), PE anti-mouse Jagged1 (clone: HMJ1-29, Biolegend), PE/Cy5 anti-mouse CD3ε (clone: 145-2C11, Biolegend), PE-Cy7 anti-mouse/human CD11b (clone: M1/70, Biolegend), PE/Dazzle 594 anti-mouse Ly-6C (clone: HK1.4, Biolegend), PE-Cy5 anti-mouse CD24 (clone: M1/69, Biolegend), PE-Cy7 anti-mouse JAML (clone: 4/E10, Novus Biologicals), Alexa 647 Rat Anti-Mouse S100A9 (clone: 2B10, BD), and Alexa 700 anti-mouse/human CD11b (clone: M1/70, Biolegend). To stain lung T cells, the following intracellular stain antibodies were used: PE-CF594 Rat anti-mouse IL-17A (clone: TC11-18H10, BD), PE-Cy7 Rat anti-mouse TNF (clone: MP6-XT22, BD), BV786 Rat anti-mouse CD4 (clone: RM4-5, BD), APC anti-mouse IFN-γ (clone: XMG1.2, eBioscience), BV421 Rat anti-mouse IL-2 (clone: JES6-5H4, BD), PE Rat anti-mouse NOTCH1 (clone: 22E5.5, BD), PerCP-Cy5.5 anti-mouse IL-22 (clone: Poly5164, Biolegend), PE-Cy7 anti-mouse CD40L (clone: SA047C3, Biolegend), V450 Hamster anti-mouse CD3ε (clone: 500A2, BD), BV605 Rat anti-mouse NOTCH2 (clone: 16F11, BD), Alexa700 Rat anti-mouse CD4 (clone: RM4-5, BD). Prior to use, all antibodies were titrated for optimal concentration. The live/dead stain (1:500) and Fc block (1:50) mixture was diluted in PBS (Sigma). The surface stain antibodies were diluted in FACS buffer (PBS [Sigma], 2% heat-inactivated FBS [Gemini], and 2 mM EDTA [Corning]). For intracellular staining, the BD Cytofix/Cytoperm kit and buffers were used according to manufacturer’s instructions. Following staining, cells were fixed in 1:1 FACS buffer to 4% paraformaldehyde (PFA) (Electron Microscopy Sciences) for BSL-3 samples, or 2% PFA for non-BSL-3 samples and placed in the 4°C until acquisition (up to 24 h post-staining). For compensation, Anti-Rat and Anti-Hamster Ig κ/Negative Control Compensation Particles (BD), Ultra-Comp eBeads compensation beads (Invitrogen), or ArC Amine Reactive Compensation Bead Kit (Invitrogen) were used. All samples were acquired using an LSR-II machine (BD) or an A5 Symphony (BD) using FACSDiva (BD) software. All Data were analyzed using FlowJo software (FlowJo LLC). For certain experiments, FlowAI plugin was used to select for optimal events (Monaco et al., 2016). For DC flow cytometry data, cells were gated: singlets/live cells/CD11c+MHCIIhi (for in vitro experiments) or singlets/live cells/CD45+/CD3-/CD64-F4/80-/MHCII+CD11c+/CD11b+CD103- (for in vivo CD11b+ DC populations) or singlets/live cells/CD45+/CD3-/CD64-F4/80-/MHCII+CD11c+/CD11b−CD103+ (for in vivo CD103+ DC populations). For T cell flow cytometry data, cells were gated: singlets/live cells/CD3+/CD4+.
Quantification and statistical analysis
Statistical analyses of data and graphs were mostly conducted using Prism (GraphPad). For correlations, R statistical software was used, in particular the corrplot (Murdoch and Chow, 1996; Friendly, 2012) and “ggscatter” packages (ggscatter is part of “ggpubr” base package developed by Alboukadel Kassambara, PhD). The experimental schema figures and graphical abstract were generated using BioRender.com. All data presented are representative of 2 to 4 independent experiments and are presented as mean ±standard deviation (SD) or mean ± (standard error of the mean) SEM [indicated in figure legend]. Statistical significance pvalue key is the following: ns = no significance, ∗ = ≤ 0.05, ∗∗ = ≤ 0.01, ∗∗∗ = ≤ 0.001, ∗∗∗∗ = ≤ 0.0001. Statistical tests performed for each figure are noted in individual figure key. All correlations presented are Pearson’s correlations.
Acknowledgments
We thank members of the Rengarajan lab for helpful discussions and for input on the manuscript. This work was supported by R56AI083366 and R01AI134244 (to J.R.), an Administrative Supplement to R01AI134244 to Promote Diversity in Health-Related Research (to A.B.E.), an ARCS Foundation Herz Global Impact Award (to A.B.E), and a Laney Graduate School Fellowship (to A.B.E.).
Author contributions
Conceptualization, A.B.E., J.K.S., J.R.; Methodology, A.B.E., J.K.S., J.R.; Investigation, A.B.E., J.K.S., H.K.D., SL.G., M.Q., K.L.S., J.R.; Validation, A.B.E., J.R.; Formal Analysis, A.B.E., J.R.; Data Curation A.B.E.; Resources J.R.; Writing-original paper, A.B.E., J.R.; Writing – Review & Editing, A.B.E., J.K.S., H.K.D., SL.G., M.Q., J.R.; Visualization, A.B.E.; Supervision, J.R.; Funding Acquisition, A.B.E., J.R.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper received support from a program designed to increase minority representation in science. The author list of this paper includes contributors from the location where the research was conducted who participated in the data collection, design, analysis, and/or interpretation of the work.
Published: May 20, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.104305.
Supplemental information
Data and code availability
-
•
This study did not generate new sequencing data. All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Arlehamn C., Seumois G., Gerasimova A., Huang C., Fu Z., Yue X., Sette A., Vijayanand P., Peters B. Transcriptional profile of tuberculosis antigen-specific T cells reveals novel multifunctional features. J. Immunol. 2014;193:2931–2940. doi: 10.4049/jimmunol.1401151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bizzell E., Sia J.K., Quezada M., Enriquez A., Georgieva M., Rengarajan J. Deletion of BCG Hip1 protease enhances dendritic cell and CD4 T cell responses. J. Leukoc. Biol. 2018;103:739–748. doi: 10.1002/jlb.4a0917-363rr. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro R., Zambuzi F., Fontanari C., De Morais F., Bollela V., Kunkel S., Schaller M., Frantz F. NOTCH1 and DLL4 are involved in the human tuberculosis progression and immune response activation. Tuberculosis (Edinb.) 2020;124:101980. doi: 10.1016/j.tube.2020.101980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung J., Ebens C.L., Perkey E., Radojcic V., Koch U., Scarpellino L., Tong A., Allen F., Wood S., Feng J., et al. Fibroblastic niches prime T cell alloimmunity through Delta-like Notch ligands. J. Clin. Invest. 2017;127:1574–1588. doi: 10.1172/jci89535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper A.M. Cell-mediated immune responses in tuberculosis. Annu. Rev. Immunol. 2009;27:393–422. doi: 10.1146/annurev.immunol.021908.132703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper A.M., Dalton D.K., Stewart T.A., Griffin J.P., Russell D.G., Orme I.M. Disseminated tuberculosis in interferon gamma gene-disrupted mice. J. Exp. Med. 1993;178:2243–2247. doi: 10.1084/jem.178.6.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz A., Fraga A.G., Fraga A., Fountain J.J., Fountain J., Rangel-Moreno J., Torrado E., Saraiva M., Pereira D., Randall T.D., et al. Pathological role of interleukin 17 in mice subjected to repeated BCG vaccination after infection with Mycobacterium tuberculosis. J. Exp. Med. 2010;207:1609–1616. doi: 10.1084/jem.20100265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz A., Khader S., Torrado E., Fraga A., Pearl J., Pedrosa J., Cooper A., Castro A. Cutting edge: IFN-γamma regulates the induction and expansion of IL-17-producing CD4 T cells during mycobacterial infection. J. Immunol. 2006;177:1416–1420. doi: 10.4049/jimmunol.177.3.1416. [DOI] [PubMed] [Google Scholar]
- Dijkman K., Sombroek C.C., Vervenne R.A.W., Hofman S.O., Boot C., Remarque E.J., Kocken C.H.M., Ottenhoff T.H.M., Kondova I., Khayum M.A., et al. Prevention of tuberculosis infection and disease by local BCG in repeatedly exposed rhesus macaques. Nat. Med. 2019;25:255–262. doi: 10.1038/s41591-018-0319-9. [DOI] [PubMed] [Google Scholar]
- Duarte A., Hirashima M., Benedito R., Trindade A., Diniz P., Bekman E., Costa L., Henrique D., Rossant J. Dosage-sensitive requirement for mouse Dll4 in artery development. Genes Dev. 2004;18:2474–2478. doi: 10.1101/gad.1239004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Festjens N., Bogaert P., Batni A., Houthuys E., Plets E., Vanderschaeghe D., Laukens B., Asselbergh B., Parthoens E., De Rycke R., et al. Disruption of the SapM locus in Mycobacterium bovis BCG improves its protective efficacy as a vaccine against M. tuberculosis. EMBO Mol. Med. 2011;3:222–234. doi: 10.1002/emmm.201000125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorini E., Merck E., Wilson A., Ferrero I., Jiang W., Koch U., Auderset F., Laurenti E., Tacchini-Cottier F., Pierres M., et al. Dynamic regulation of notch 1 and notch 2 surface expression during T cell development and activation revealed by novel monoclonal antibodies. J. Immunol. 2009;183:7212–7222. doi: 10.4049/jimmunol.0902432. [DOI] [PubMed] [Google Scholar]
- Flynn J.L., Chan J., Triebold K.J., Dalton D.K., Stewart T.A., Bloom B.R. An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J. Exp. Med. 1993;178:2249–2254. doi: 10.1084/jem.178.6.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friendly M. Corrgrams: exploratory displays for correlation matrices. Am. Stat. 2012;56:316–324. doi: 10.1198/000313002533. [DOI] [Google Scholar]
- Furuhashi K., Suda T., Hasegawa H., Suzuki Y., Hashimoto D., Enomoto N., Fujisawa T., Nakamura Y., Inui N., Shibata K., et al. Mouse lung CD103+ and CD11bhigh dendritic cells preferentially induce distinct CD4+ T-cell responses. Am. J. Respir. Cell Mol. Biol. 2012;46:165–172. doi: 10.1165/rcmb.2011-0070oc. [DOI] [PubMed] [Google Scholar]
- Gale N.W., Dominguez M.G., Noguera I., Pan L., Hughes V., Valenzuela D.M., Murphy A.J., Adams N.C., Lin H.C., Holash J., et al. Haploinsufficiency of delta-like 4 ligand results in embryonic lethality due to major defects in arterial and vascular development. Proc. Natl. Acad. Sci. U S A. 2004;101:15949–15954. doi: 10.1073/pnas.0407290101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgieva M., Sia J.K., Bizzell E., Madan-Lala R., Rengarajan J. Mycobacterium tuberculosis GroEL2 Modulates Dendritic Cell Responses. Infect. Immun. 2018;86 doi: 10.1128/iai.00387-17. e00387-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopal R., Monin L., Slight S., Uche U., Blanchard E., A Fallert Junecko B., Ramos-Payan R., Stallings C.L., Reinhart T.A., Kolls J.K., et al. Unexpected role for IL-17 in protective immunity against hypervirulent Mycobacterium tuberculosis HN878 infection. PLoS Pathog. 2014;10 doi: 10.1371/journal.ppat.1004099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grode L., Seiler P., Baumann S., Hess J., Brinkmann V., Nasser Eddine A., Mann P., Goosmann C., Bandermann S., Smith D., et al. Increased vaccine efficacy against tuberculosis of recombinant Mycobacterium bovis bacille Calmette-Guérin mutants that secrete listeriolysin. J. Clin. Invest. 2005;115:2472–2479. doi: 10.1172/jci24617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández-Santos N., Gaffen S.L. Th17 cells in immunity to Candida albicans. Cell Host Microbe. 2012;11:425–435. doi: 10.1016/j.chom.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H.M., Huang H., Hsiao G., Fan C.K., Fan C., Lin C., Leu S.J., Leu S., Chiang B., Lee Y.L., Lee Y. Notch ligand delta-like 4-pretreated dendritic cells alleviate allergic airway responses by enhancing IL-10 production. PLoS One. 2013;8:e63613. doi: 10.1371/journal.pone.0063613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M.T., Huang M., Chen Y., Lien C.I., Lien C., Liu W.L., Liu W., Hsu L.C., Hsu L., Yagita H., Chiang B.L., Chiang B. Notch ligand DLL4 alleviates allergic airway inflammation via induction of a homeostatic regulatory pathway. Sci. Rep. 2017;7:43535. doi: 10.1038/srep43535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T., Schaller M., Hogaboam C.M., Standiford T.J., Sandor M., Lukacs N.W., Chensue S.W., Kunkel S.L. TLR9 regulates the mycobacteria-elicited pulmonary granulomatous immune response in mice through DC-derived Notch ligand delta-like 4. J. Clin. Invest. 2009;119:33–46. doi: 10.1172/JCI35647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushal D., Foreman T.W., Gautam U.S., Alvarez X., Adekambi T., Rangel-Moreno J., Golden N.A., Johnson A.M.F., Phillips B.L., Ahsan M.H., et al. Mucosal vaccination with attenuated Mycobacterium tuberculosis induces strong central memory responses and protects against tuberculosis. Nat. Commun. 2015;6:8533. doi: 10.1038/ncomms9533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopan R., Ilagan M.X.G. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216–233. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs L.T., Shutter J.R., Tanigaki K., Honjo T., Stark K.L., Gridley T. Haploinsufficient lethality and formation of arteriovenous malformations in Notch pathway mutants. Genes Dev. 2004;18:2469–2473. doi: 10.1101/gad.1239204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai R., Jeyanathan M., Afkhami S., Zganiacz A., Hammill J., Yao Y., Kaushic C., Xing Z. CD11b + dendritic cell-mediated anti- Mycobacterium tuberculosis Th1 activation is counterregulated by CD103 + dendritic cells via IL-10. J. Immunol. 2018;200:1746–1760. doi: 10.4049/jimmunol.1701109. [DOI] [PubMed] [Google Scholar]
- Liang S.C., Tan X.Y., Luxenberg D.P., Karim R., Dunussi-Joannopoulos K., Collins M., Fouser L.A. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 2006;203:2271–2279. doi: 10.1084/jem.20061308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macmicking J., Xie Q.w., Nathan C. Nitric oxide and macrophage function. Annu. Rev. Immunol. 1997;15:323–350. doi: 10.1146/annurev.immunol.15.1.323. [DOI] [PubMed] [Google Scholar]
- Madan-Lala R., Sia J.K., King R., Adekambi T., Monin L., Khader S.A., Pulendran B., Rengarajan J. Mycobacterium tuberculosis impairs dendritic cell functions through the serine hydrolase Hip1. J. Immunol. 2014;192:4263–4272. doi: 10.4049/jimmunol.1303185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangtani P., Abubakar I., Ariti C., Beynon R., Pimpin L., Fine P.E.M., Rodrigues L.C., Smith P.G., Lipman M., Whiting P.F., Sterne J.A. Protection by BCG vaccine against tuberculosis: a systematic review of randomized controlled trials. Clin. Infect. Dis. 2014;58:470–480. doi: 10.1093/cid/cit790. [DOI] [PubMed] [Google Scholar]
- Martinot A.J., Blass E., Yu J., Aid M., Mahrokhian S.H., Cohen S.B., Plumlee C.R., Larocca R.A., Siddiqi N., Wakabayashi S., et al. Protective efficacy of an attenuated Mtb ΔLprG vaccine in mice. PLoS Pathog. 2020;16:e1009096. doi: 10.1371/journal.ppat.1009096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng L., Bai Z., He S., Mochizuki K., Liu Y., Purushe J., Sun H., Wang J., Yagita H., Mineishi S., et al. The notch ligand DLL4 defines a capability of human dendritic cells in regulating Th1 and Th17 differentiation. J. Immunol. 2016;196:1070–1080. doi: 10.4049/jimmunol.1501310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki K., Xie F., He S., Tong Q., Liu Y., Mochizuki I., Guo Y., Kato K., Yagita H., Mineishi S., Zhang Y. Delta-like ligand 4 Identifies a previously uncharacterized population of inflammatory dendritic cells that plays important roles in eliciting allogeneic T cell responses in mice. J. Immunol. 2013;190:3772–3782. doi: 10.4049/jimmunol.1202820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaco G., Chen H., Poidinger M., Chen J., De Magalhães J.P., Larbi A. flowAI: automatic and interactive anomaly discerning tools for flow cytometry data. Bioinformatics. 2016;32:2473–2480. doi: 10.1093/bioinformatics/btw191. [DOI] [PubMed] [Google Scholar]
- Mukherjee S., Schaller M.A., Neupane R., Kunkel S.L., Lukacs N.W. Regulation of T cell activation by Notch ligand, DLL4, promotes IL-17 production and Rorc activation. J. Immunol. 2009;182:7381–7388. doi: 10.4049/jimmunol.0804322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murdoch D.J., Chow E.D. A graphical display of large correlation matrices. Am. Stat. 1996;50:178–180. doi: 10.1080/00031305.1996.10474371. [DOI] [Google Scholar]
- Naffin-Olivos J.L., Georgieva M., Goldfarb N., Madan-Lala R., Dong L., Bizzell E., Valinetz E., Brandt G.S., Yu S., Shabashvili D.E., et al. Mycobacterium tuberculosis Hip1 modulates macrophage responses through proteolysis of GroEL2. PLoS Pathog. 2014;10 doi: 10.1371/journal.ppat.1004132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitani G., Rinaldi A., Bertoni F., Sallusto F., Lanzavecchia A. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat. Immunol. 2005;6:769–776. doi: 10.1038/ni1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan A., Beynor J.I., Beynor J., Baglaenko Y., Suliman S., Ishigaki K., Asgari S., Huang C.C., Huang C., Luo Y., et al. Multimodally profiling memory T cells from a tuberculosis cohort identifies cell state associations with demographics, environment and disease. Nat. Immunol. 2021;22:781–793. doi: 10.1038/s41590-021-00933-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newport M.J., Huxley C.M., Huston S., Hawrylowicz C.M., Oostra B.A., Williamson R., Levin M. A mutation in the interferon-gamma-receptor gene and susceptibility to mycobacterial infection. N. Engl. J. Med. 1996;335:1941–1949. doi: 10.1056/nejm199612263352602. [DOI] [PubMed] [Google Scholar]
- Ogongo P., Tezera L., Ardain A., Nhamoyebonde S., Ramsuran D., Singh A., Ngoepe A., Karim F., Naidoo T., Khan K., et al. Tissue resident-like CD4+ T cells secreting IL-17 control Mycobacteria tuberculosis in the human lung. J. Clin. Invest. 2021;131:e142014. doi: 10.1172/JCI142014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada S., Markle J.G., Deenick E.K., Mele F., Averbuch D., Lagos M., Alzahrani M., Al-Muhsen S., Halwani R., Ma C.S., et al. Impairment of immunity to Candida and Mycobacterium in humans with bi-allelic RORC mutations. Science. 2015;349:606–613. doi: 10.1126/science.aaa4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Organization W.H. WHO; 2021. Global Tuberculosis Report 2021. [Google Scholar]
- Perreau M., Rozot V., Welles H.C., Belluti-Enders F., Vigano S., Maillard M., Dorta G., Mazza-Stalder J., Bart P.A., Roger T., et al. Lack of Mycobacterium tuberculosis-specific interleukin-17A-producing CD4+ T cells in active disease. Eur. J. Immunol. 2013;43:939–948. doi: 10.1002/eji.201243090. [DOI] [PubMed] [Google Scholar]
- Pollara G., Turner C.T., Turner C., Rosenheim J., Chandran A., Bell L.C.K., Bell L., Khan A., Patel A., Peralta L., et al. Exaggerated IL-17A activity in human in vivo recall responses discriminates active tuberculosis from latent infection and cured disease. Sci. Transl. Med. 2021;13:eabg7673. doi: 10.1126/scitranslmed.abg7673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rengarajan J., Murphy E., Park A., Krone C.L., Hett E.C., Bloom B.R., Glimcher L.H., Rubin E.J. Mycobacterium tuberculosis Rv2224c modulates innate immune responses. Proc. Natl. Acad. Sci. U S A. 2008;105:264–269. doi: 10.1073/pnas.0710601105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues L., Diwan V., Wheeler J. Protective effect of BCG against tuberculous meningitis and miliary tuberculosis: a meta-analysis. Int. J. Epidemiol. 1993;22:1154–1158. doi: 10.1093/ije/22.6.1154. [DOI] [PubMed] [Google Scholar]
- Rosa B., Ahmed M., Singh D.K., Singh D., Choreño-Parra J.A., Choreño-Parra J., Cole J., Jiménez-Álvarez L.A., Jiménez-Álvarez L., Rodríguez-Reyna T., et al. IFN signaling and neutrophil degranulation transcriptional signatures are induced during SARS-CoV-2 infection. Commun. Biol. 2021;4:290. doi: 10.1038/s42003-021-01829-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauma D., Espejo P., Ramirez A., Fierro A., Rosemblatt M., Bono M.R. Differential regulation of Notch ligands in dendritic cells upon interaction with T helper cells. Scand. J. Immunol. 2011;74:62–70. doi: 10.1111/j.1365-3083.2011.02541.x. [DOI] [PubMed] [Google Scholar]
- Schaller M.A., Allen R.M., Kimura S., Day C.L., Kunkel S.L. Systemic expression of notch ligand delta-like 4 during mycobacterial infection alters the T cell immune response. Front. Immunol. 2016;7:527. doi: 10.3389/fimmu.2016.00527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scriba T., Kalsdorf B., Abrahams D., Isaacs F., Hofmeister J., Black G., Hassan H., Wilkinson R., Walzl G., Gelderbloem S., et al. Distinct, specific IL-17- and IL-22-producing CD4+ T cell subsets contribute to the human anti-mycobacterial immune response. J. Immunol. 2008;180:1962–1970. doi: 10.4049/jimmunol.180.3.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanmugasundaram U., Bucsan A.N., Ganatra S.R., Ibegbu C., Quezada M., Blair R.V., Alvarez X., Velu V., Kaushal D., Rengarajan J. Pulmonary Mycobacterium tuberculosis control associates with CXCR3- and CCR6-expressing antigen-specific Th1 and Th17 cell recruitment. JCI insight. 2020;5:e137858. doi: 10.1172/jci.insight.137858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sia J.K., Bizzell E., Madan-Lala R., Rengarajan J. Engaging the CD40-CD40L pathway augments T-helper cell responses and improves control of Mycobacterium tuberculosis infection. PLoS Pathog. 2017;13 doi: 10.1371/journal.ppat.1006530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tascon R.E., Soares C.S., Ragno S., Stavropoulos E., Hirst E.M.A., Colston M.J. Mycobacterium tuberculosis-activated dendritic cells induce protective immunity in mice. Immunology. 2000;99:473–480. doi: 10.1046/j.1365-2567.2000.00963.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran I.T., Sandy A.R., Carulli A.J., Ebens C., Chung J., Shan G.T., Radojcic V., Friedman A., Gridley T., Shelton A., et al. Blockade of individual Notch ligands and receptors controls graft-versus-host disease. J. Clin. Invest. 2013;123:1590–1604. doi: 10.1172/jci65477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treerat P., Prince O., Cruz-Lagunas A., Munoz-Torrico M., Salazar-Lezama M.A., Selman M., Fallert-Junecko B., Reinhardt T.A., Alcorn J.F., Kaushal D., et al. Novel role for IL-22 in protection during chronic Mycobacterium tuberculosis HN878 infection. Mucosal Immunol. 2017;10:1069–1081. doi: 10.1038/mi.2017.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu L., Fang T.C., Artis D., Shestova O., Pross S.E., Maillard I., Pear W.S. Notch signaling is an important regulator of type 2 immunity. J. Exp. Med. 2005;202:1037–1042. doi: 10.1084/jem.20050923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb L.M., Oyesola O.O., Früh S.P., Kamynina E., Still K.M., Patel R.K., Peng S.A., Cubitt R.L., Grimson A., Grenier J.K., et al. The Notch signaling pathway promotes basophil responses during helminth-induced type 2 inflammation. J. Exp. Med. 2019;216:1268–1279. doi: 10.1084/jem.20180131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf A.J., Desvignes L., Linas B., Banaiee N., Tamura T., Takatsu K., Ernst J.D. Initiation of the adaptive immune response to Mycobacterium tuberculosis depends on antigen production in the local lymph node, not the lungs. J. Exp. Med. 2008;205:105–115. doi: 10.1084/jem.20071367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf A.J., Linas B., Trevejo-Nunez G.J., Kincaid E., Tamura T., Takatsu K., Ernst J.D. Mycobacterium tuberculosis infects dendritic cells with high frequency and impairs their function in vivo. J. Immunol. 2007;179:2509–2519. doi: 10.4049/jimmunol.179.4.2509. [DOI] [PubMed] [Google Scholar]
- Xue Y., Gao X., Lindsell C.E., Norton C.R., Chang B., Hicks C., Gendron-Maguire M., Rand E.B., Weinmaster G., Gridley T. Embryonic lethality and vascular defects in mice lacking the Notch ligand Jagged1. Hum. Mol. Genet. 1999;8:723–730. doi: 10.1093/hmg/8.5.723. [DOI] [PubMed] [Google Scholar]
- Zelante T., Wong A., Ping T., Chen J., Sumatoh H., Viganò E., Hong Bing Y., Lee B., Zolezzi F., Fric J., et al. CD103(+) dendritic cells control Th17 cell function in the lung. Cell Rep. 2015;12:1789–1801. doi: 10.1016/j.celrep.2015.08.030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
This study did not generate new sequencing data. All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.