

Abstract
Gas phase hydrogen chloride (HCl) was measured at Pasadena and San Joaquin Valley (SJV) ground sites in California during May and June 2010 as part of the CalNex study. Observed mixing ratios were on average 0.83 ppbv at Pasadena, ranging from below detection limit (0.055 ppbv) to 5.95 ppbv, and were on average 0.084 ppbv at SJV with a maximum value of 0.776 ppbv. At both sites, HCl levels were highest during midday and shared similar diurnal variations with HNO3. Coupled phase partitioning behavior was found between HCl/Cl− and HNO3/NO3− using thermodynamic modelling and observations. Regional modeling of Cl− and HCl using CMAQ captures some of the observed relationships but underestimates measurements by a factor of 5 or more. Chloride in the 2.5-10 μm size range in Pasadena was sometimes higher than sea salt abundances, based on co-measured Na+, implying that sources other than sea salt are important. The acid-displacement of HCl/Cl− by HNO3/NO3− (phase partitioning of semi-volatile acids) observed at the SJV site can only be explained by aqueous phase reaction despite low RH conditions and suggests the temperature dependence of HCl phase partitioning behavior was strongly impacted by the activity coefficient changes under relevant aerosol conditions (e.g., high ionic strength). Despite the influence from activity coefficients, the gas-particle system was found to be well constrained by other stronger buffers and charge balance so that HCl and Cl− concentrations were reproduced well by thermodynamic models.
Plain language summary:
Despite the known importance of gas-particle partitioning in chlorine activation chemistry, the sources and atmospheric fate of HCl and particulate Cl− requires further study. HCl concentrations and the corresponding phase partitioning mechanisms with Cl− in PM2.5 were studied through field measurements and modelling as part of the CalNex field campaign. The measurements confirmed that gas phase HCl was the dominant component of atmospheric chlorine and suggested that even though the current modelling can explain the diurnal variation of HCl, the mixing ratios were severely underestimated. Temperature dependence of Cl− replacement reaction by HNO3 was observed at one site. This study suggests further model development and field observations of HCl, as well as more reliable understanding of HCl phase partitioning behavior are needed to better predict the atmospheric fate of HCl and Cl−.
I. Introduction
The Earth’s atmosphere is an acidic/oxidizing medium through which a range of trace chemical elements are cycled and transformed. Chlorine cycles through the lower atmosphere primarily as chloride (Cl−) on particles, and hydrogen chloride (HCl) in the gas phase, with some contribution from Cl−containing organics [Keene et al., 1999]. The conversion of these more inert forms of chlorine to active chlorine (i.e., chlorine atoms and associated radicals) can initiate photochemical oxidation. Observations have shown that reaction of N2O5 with Cl−-containing particles to form nitryl chloride (ClNO2) is a significant pathway for chlorine activation [Ahern et al., 2018; Mielke et al., 2011; Riedel et al., 2013; Tham et al., 2013; Xia et al., 2021; Zhou et al., 2018]. The most important sources of particulate Cl− to the lower atmosphere are sea salt and soil (wind-blown dust), which appear mostly in the super-micron size fraction [Jordan et al., 2015; Keene et al., 1990]. However, the N2O5-ClNO2 chemical system requires sufficient particle Cl− be present as precursor and enough aerosol surface area (considering the abundance of Cl− in super-micron particles, both sub- and super-micron particles are probably required) as a reaction medium to explain observations with known mechanisms and rates [Bertram and Thornton, 2009; Roberts et al., 2009; Staudt et al., 2019]. A relationship between super-micron particle Cl− and ClNO2 was observed at Pasadena during CalNex, suggesting that larger particles could contribute to the formation of ClNO2 [Mielke et al., 2013]. Gas phase HCl presents the means by which super-micron Cl− can be re-distributed to other particles. Hence, a correct description of HCl concentrations and dynamics (i.e., phase partitioning) is key to understanding and predicting chlorine activation in high-NOx environments [Haskins et al., 2019; Wang et al., 2019].
Gaseous HCl has been a known atmospheric constituent for a number of decades (see Eldering et al. [1991] for a discussion of the history of HCl in the atmosphere). It has also been clear for some time that displacement of Cl− from sea salt and to a lesser extent, soil-derived particles by the strong acids, sulfuric (H2SO4) and nitric (HNO3), is the major source of atmospheric HCl:
| R1 |
Additional sources include volcanic gases, biomass and trash burning, and coal-fired power plants [Ahern et al., 2018; Keene et al., 1999; Wang et al., 2019]. There have been only a few measurements of HCl in the SoCAB (California’s South Coast Air Basin) that encompasses the greater Los Angeles area; most of those studies employed sampling times on the order of a few hours to one day, and found levels as high as 4 ppbv [Appel et al., 1991; Eldering et al., 1991; Grosjean, 1990]. Measurements of HCl off the coast of Southern California, made during the CalNex 2010 study, had a median HCl mixing ratio of 1.3 ppbv and an interquartile range of 0.10 to 3.8 ppbv [Crisp et al., 2014]. They also suggested that point sources were potentially large in near-coastal Southern California and were underestimated by emission inventories, consistent with the suggestion of Riedel et al. [2012].
The ultimate removal of HCl occurs through wet and dry deposition. However, the exchange of HCl with Cl− and subsequent activation represents one of the most important roles that HCl/Cl− can play in the lower atmosphere. Some studies suggested that the uncertainties in the reported thermodynamic properties of HCl/Cl− could be responsible for the disagreement between aerosol pH revealed by HCl/Cl− phase partitioning and other thermodynamic methods (e.g., NH3/NH4+ and/or HNO3/NO3− phase partitioning) [Haskins et al., 2018; Keene et al., 2004; Pye et al., 2020; Sander, 2015; Sudheer and Rengarajan, 2015; Young et al., 2013]. In contrast, some other studies showed good agreement between the observed and the thermodynamically modelled HCl/Cl− phase partitioning [Dasgupta et al., 2007; Guo et al., 2017; Trebs, 2005]. There remains an opportunity to use ambient data to assess the implementation of laboratory measurements in a thermodynamic framework.
Activation chemistry occurs on particles; however, it is clear from simple mass balance consideration that rapid replacement of particle Cl− is required to produce the levels of active chlorine species that are observed. For example in modeling observed nitryl chloride (ClNO2) concentrations, Osthoff et al. [2008] found that in addition to sea salt, other continental Cl−-containing particles were required to explain the total ClNO2 produced. HCl provides that additional source and so is often viewed as the limiting quantity in the chlorine activation pathway [Brown et al., 2013; Roberts et al., 2009; Thornton et al., 2010; Wegner et al., 2012].
The measurements of HCl made at the Pasadena and SJV ground sites during the CalNex 2010 experiment are described herein along with measurements of particle Cl−. These observations provide context for the assessment of particle Cl− participation in chlorine activation chemistry. The measurements confirm that gas phase HCl is the key component in the budget of Cl− since it is often the most abundant inorganic chlorine species. The CalNex measurements provide benchmarks for chloride/chlorine inventories to be used in regional air quality models, and are compared here to Community Multiscale Air Quality (CMAQ) modeling results and associated emissions inventories.
II. Experimental and Modeling Approaches
Measurement sites
The measurements reported here were made between 18 May and 28 June, 2010, at two separate ground sites, one at Pasadena ground site, the other at SJV as part of the CalNex 2010 field project. Both sites have been described by previous studies [Liu et al., 2012; Ryerson et al., 2013; VandenBoer et al., 2014; Washenfelder et al., 2011]. Briefly, the Pasadena site was on the campus of the California Institute of Technology, approximately 16 km northeast of downtown Los Angeles, and the SJV site was located at the southern end of the San Joaquin Valley at the Kern County Cooperative Extension in Bakersfield. Pasadena is located downwind from direct sources and sea breeze in Los Angeles, while the SJV site is agricultural and rural with greater influence by upwind emissions that arrive as well-mixed air masses [Fast et al., 2014; Guzman-Morales et al., 2014; Kelly et al., 2014].
Measurements at Pasadena site
In Pasadena the measurements of gas phase acids were made by negative-ion proton transfer chemical ionization mass spectrometry (NI-PT-CIMS) using acetate as the reagent ion. This method is sensitive to a range of organic and inorganic acids [Roberts et al., 2010; Veres et al., 2008], which are detected as their corresponding anion. The NI-PT-CIMS instrument was installed atop one of the sampling trailers and an inlet (~1.3 m) was affixed that sampled air approximately 5 m above ground level. The inlet consisted of 1/8” OD PFA tubing that was thermostated at 75°C with the flow rate of 2 L min−1. Instrument backgrounds were measured by scrubbing ambient air with Na2CO3, or by overflowing the inlet with ultrapure zero air or nitrogen. The instrument was calibrated for response to HCl using a calibrated gas standard (Spectra Gases) and for HNO3 using a permeation tube calibrated by UV absorption spectrometry [Neuman et al., 2003]. The overall uncertainties in the atmospheric mixing ratios were ± (35% + 55 parts per trillion by volume, pptv) for HCl and ± (35% + 80 pptv) for HNO3. There exist potential interferences due to volatilization of HCl or HNO3 from NH4Cl or NH4NO3-containing particles. The possible magnitude and timing of these interferences is considered in the interpretation of the measurements and is discussed in the Results below.
Gas phase ammonia was measured by a quantum-cascade tunable infrared laser differential absorption spectrometer (QC-TILDAS) (Aerodyne Research Inc.) as described in detail by Ellis et al. [2010]. The instrument sampled air from an inlet that was mounted on the scaffold adjacent to the NI-PT-CIMS trailer at a height of approximately 8 m above ground level. The NH3 signal was zeroed by overflowing the inlet with NH3-free air from a zero-air generator and the instrument sensitivity was determined periodically using an NH3 permeation source that was independently calibrated by ion chromatography (IC). The overall uncertainties of the NH3 measurements were ± (10% + 0.42 ppbv).
Concentrations of particulate matter with aerodynamic diameter less than 2.5 μm (PM2.5) ionic species were measured online with a Particle-Into-Liquid-Sampler coupled to ion chromatographs (PILS-IC) [Orsini et al., 2003; Weber et al., 2001]. Prior to the PILS-IC, sample air was aspirated at 16.7 ± 0.7 L/min through a URG PM2.5 cyclone, a parallel plate carbon denuder [Eatough et al., 1993] and honeycomb acid (citric acid) and base (sodium carbonate) coated denuders. Liquid samples from the PILS were analyzed via two different ICs. Anions were measured with a Dionex Ion Chromatograph (UTAC-ULP1 concentrator, AG11 guard and AS11 anion columns) using a gradient NaOH eluent procedure lasting 20 minutes. Cations were determined only during the latter part of the study from 8-14 June with a Metrohm compact 761 IC, equipped with a Metrosep C4 cation column. Time resolution of cation measurement was 15-minutes. Systematic blank measurements were made on a daily basis throughout the study period by diverting sample flow through a HEPA filter downstream of the denuder and upstream of the PILS. A linear interpolation of blank data was performed and subtracted from the ambient data. Random measurement uncertainty based on a quadrature sum of squares that included the precision of standards, variability in sample air flow rate, liquid flow rate and blanks (all one standard deviation), was estimated at ± 15% for anions and ± 8% for cations. The coarse mode (2.5-10 μm) elemental Na and Cl were sampled by an 8-stage rotating drum impactor and measured by synchrotron X-ray fluorescence with time-resolution of 3 hours. The description of the apparatus and analytical technique is presented in Perry et al. [2004].
Measurements at SJV site
At the SJV site, the gas phase and PM2.5 water-soluble ionic components were sampled and measured using a modified AIM-IC (Ambient Ion Monitor- Ion Chromatography) system (URG Corp., Chapel Hill, NC), which provided the hourly time-resolved chemical composition data in both gas phase (NH3, HNO3, HCl and HONO) and particle phase (NH4+, SO42−, NO3−, NO2−, Na+, Cl−, Mg2+ and Ca2+). A detailed description of the performance of this sampling system for these species and at the field site can be found in Markovic et al. [2012], VandenBoer et al. [2014] and Markovic et al. [2014]. Combined gas and PM2.5 samples were collected into a modified inlet housing, which was mounted to a scaffold tower at 4.5 m above ground level, with PM2.5 size selection governed by an inertial impactor, at a flow of 3 L min−1. Collection of each fraction was made into aqueous solution, transported to cation and anion ICs, and analyzed every 30 minutes, yielding hourly time resolution for both phases. Background determinations were made by overflowing the inlet with zero air before, during and following the field observations. Linear interpolations between each assessment were subtracted from ambient data. Blanks were paired with calibration by mixed ion primary standards for both anion and cation suites (e.g., Dionex Combined Seven Anion Standard, P/N 056933). Time response and sampling efficiencies for NH3 and HNO3 were performed by standard addition, and intercomparison with CIMS observations, respectively [Crounse et al., 2006; Markovic et al., 2014]. The overall uncertainties for HCl and Cl− were ± (≤ 15% + 19 pptv) and ± (≤ 15% + 27 ng m−3), HNO3 and NO3− were ± (≤ 15% + 65 pptv) and ± (≤ 15% + 45 ng m−3), and NH3 and NH4+ were ± (≤ 15% + 41 pptv) and ± (≤ 15% + 29 ng m−3).
HCl and Cl− modelling
The phase partitioning of HCl and Cl− was modelled in this study using thermodynamic model calculations. Apart from the commonly used thermodynamic Extended Aerosol Inorganic Model (E-AIM) [Wexler, 2002] and ISORROPIA (Greek for “equilibrium”) II [Fountoukis and Nenes, 2007], the observed gas-particle phase partitioning behavior of HCl coupled with HNO3 was also calculated and assessed in this study to better understand the potential influence of uncertainties in thermodynamic parameters and activity coefficients that remain open questions in the community (e.g., [[Pye et al., 2020]]). In principle, if the reaction takes place in the solid phase:
| R2 |
The thermodynamic equilibrium of this reaction can be described as the theoretical partial pressure ratio of gaseous HCl and HNO3 with its temperature dependence [Seinfeld and Pandis, 2006]:
| E1 |
However, if the reaction is mainly controlled by aqueous phase acids exchange, the equilibrium expression is altered to be:
| R3 |
where the aqueous phase activities of Cl− and NO3− will also influence the p(HCl)/p(HNO3) ratio, the following relationships for the aqueous species must also be considered:
| R4 |
| E2 |
| R5 |
| E3 |
Where K(HCl) and K(HNO3) correspond to the effective Henry’s Law Constant (defined as the resulting equilibrium constants when R4 and R5 are combined, which includes both gas-liquid phase partitioning equilibrium and acid dissociation) of HCl or HNO3 and a(H+), a(NO3−) and a(Cl−) correspond to the liquid phase activities of H+, NO3− and Cl−. Taking the ratio of these two expressions, it can be derived that:
| E4 |
Since both K(HCl) and K(HNO3) are a function of temperature with the relationship:
| E5 |
where ΔrH and ΔrS are the standard enthalpy (J mol−1) and entropy (J mol−1 K−1) change of the corresponding equilibrium, such that:
| E6 |
Combining E4 and E6, there should exist a linear relationship between ln[p(HNO3)a(Cl−)/p(HCl)a(NO3−)] against 1/T with the resulting slope equal to −[ΔrH(HCl) − ΔrH(HNO3)]/R. Ion activities cannot be directly measured so that values are usually predicted using thermodynamic computation.
Considering that for each chemical species, X:
| E7 |
where n represents the ambient mole concentrations and γ represents the activity coefficient. E4 and E7 can be combined to give:
| E8 |
| E9 |
The left side of E9 can be directly obtained from ambient concentration measurements, which is influenced by the ratio of the thermodynamic equilibrium constants and the activity coefficients. The ambient concentration ratio of n(Cl−)/n(NO3−) used in E9 does not require knowledge of aerosol liquid water content or the need to apply mole-fraction-based concentrations. Using E9, the slope of observed ln[n(HNO3)n(Cl−)/n(HCl)n(NO3−)] against the corresponding 1/T values is jointly contributed to by the enthalpy differences and the temperature dependence of the activity coefficients.
The temporal variations in the mass concentrations of HCl and Cl− were also simulated during the measurement period using the Community Multiscale Air Quality (CMAQ; www.epa.gov/cmaq) model. The CMAQ model used here is based on version 5.0.2 but with updates to the sea-spray emission parameterization that were included in version 5.1, as described by Gantt et al. [2015]. Sea-spray emissions are calculated online in CMAQ as a function of the modeled wind speed and sea-surface temperature and are enhanced in coastal grid cells to represent the effect of wave breaking on emissions at the coast [Gantt et al., 2015; Kelly et al., 2010]. Since underpredictions of sodium concentrations were identified previously during CalNex [Kelly et al., 2014], a sensitivity simulation was also conducted where sea-spray emissions were increased by a factor of three. This simulation is referred to as “SSx3” hereafter to distinguish it from the “Base” simulation. CMAQ represents atmospheric particles with three lognormal modes [Binkowski and Roselle, 2003]. Inorganic components of the two fine particle modes (Aitken and accumulation) are equilibrated with their gas-phase counterparts using ISORROPIA II [Fountoukis and Nenes, 2007], and diffusive mass transfer is simulated explicitly for the coarse particle mode [Kelly et al., 2010]. In addition to primary emissions, HCl is generated in CMAQ from evaporation of particle Cl− that often occurs due to particle acidification from H2SO4 and HNO3 condensation.
The CMAQ simulations were conducted for the 4 May – 30 June 2010 period using a domain that covers nearly all of California and parts of surrounding areas with 4-km horizontal resolution and 34 vertical layers. Point source emissions were based on state-submitted 2010 emission totals, and other anthropogenic, nonmobile source emissions were based on the 2008 National Emissions Inventory version 2. Mobile source emissions were based on total amounts provided by the California Air Resources Board with temporal and spatial allocations developed using the SMOKE-MOVES model (www.cmascenter.org/smoke). Meteorology used to drive the air quality simulations was generated with WRFv3.4 and was found to capture the major transport patterns in SoCAB [Baker et al., 2013]. Additional details on the model configuration are available in Kelly et al. [2014].
III. Results and discussion
HCl concentration in Pasadena and SJV sites
The gas phase HCl concentrations measured at the two ground sites during CalNex are shown in Figure 1a and b, respectively. At the Pasadena site, along with the PM2.5 Cl− mass loadings, coarse-mode (2.5-10 μm, PM2.5-10) Cl− mass loading is also presented. The loadings of HCl reached slightly above 8 μg/m3 (6 ppbv mixing ratio), while PM2.5 Cl− ranged up to almost 4 μg/m3, and coarse-mode Cl− 8 μg/m3. The Cl− quantities were generally much lower than HCl and had a less pronounced diurnal dependence. From 26 May to 9 June, it is worth noting that the particulate Cl− was depleted in both PM2.5 and PM2.5-10 while there was still strong diurnal variation of gaseous HCl, indicating the existence of other primary or secondary sources of HCl. The additional HCl could form from other Cl− salts (e.g., CaCl2 and NH4Cl) that could be responsible for the relative abundance of Cl− to Na+ observed during some periods (e.g., May 20-26 and June 11-13). Dust salts can act as temporal reservoir for Cl− [Sullivan et al., 2007] and Royer et al. [2021] also suggested that dust components could promote the reactive halogen formation. The meteorological conditions at both sites are illustrated in Figure S1, which shows the drier conditions at the SJV site compared to Pasadena. The dominant wind direction was mainly southwest for Pasadena (sea breeze) and northwest for SJV (transport through the Central Valley).
Figure 1.
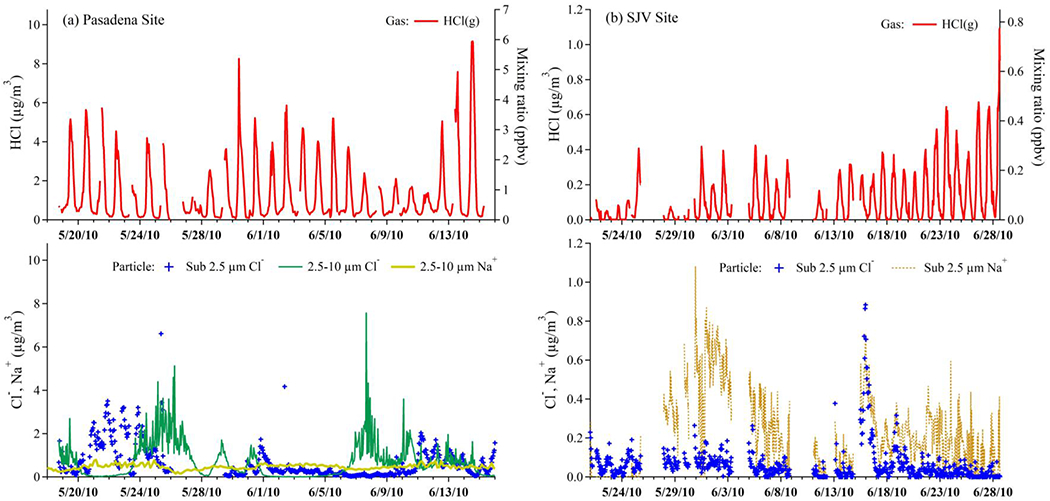
(a) Timeseries of NI-PI-CIMS HCl (red line) and PILS-IC PM2.5 Cl− (blue crosses) quantities as mass loadings (left) and mixing ratios (right) and XRF PM2.5-10 measurements of Na+ and Cl− (lower panel) measured at the Pasadena ground site. (b) The AIM-IC SJV quantities of HCl, and PM2.5 Cl− and Na+. Gaps in datasets indicate periods of instrument maintenance, calibration, backgrounds, or where data quality was not validated.
The possibility that volatilization of Cl− could represent an interference in the HCl measurement in Pasadena can be explored by simple mass balance considerations. First of all, daytime HCl is much larger than particulate Cl−, on average one order of magnitude higher from 10:00 to 18:00 local time. Thus, there is no possibility of significant interference during those periods. Nighttime levels of the two quantities were usually low (<0.5 μg/m3) and elevated nighttime Cl− did not result in observations of elevated nighttime HCl. The few nights during which Cl− approached 4 μg/m3 can be used to put limits on how much of an interference might be occurring. For example, on the night of 23-24 May, HCl was only 5% of Cl−. On the night of 11-12 June, HCl was at most 10% of Cl−. Therefore, the conservative implication is that volatilization was at most a 5-10% effect positive bias and this process on the CIMS inlet is not a significant interference. As a result, the background measurement frequency is expected to be sufficient to account for the majority of HCl released from particles deposited on inlet surfaces. In general, there is usually sufficient soluble chloride (HCl + Cl−) mass loadings in the ambient to sustain the nighttime production of ppbv levels of ClNO2 from N2O5 condensed phase chemistry in Pasadena reported by Mielke et al. [2013] and Young et al. [2012].
By comparison, the time series of HCl, particulate Cl−, and Na+ concentrations in PM2.5 are plotted in Figure 1b, where it shows that at the SJV site, the levels of HCl were significantly lower than those measured at the Pasadena site, ranging from below the 0.019 ppbv (0.028 μg m−3) detection limit to 0.78 ppbv (1.09 μg m−3) with a mean value of 0.085 ppbv (0.12 μg m−3) and standard deviation of 0.11 ppbv (0.15 μg m−3). Data for PM2.5-10 Cl− were not available at the SJV site. Using the measured PM2.5 Na+ and Cl− loadings as a proxy for coarse particle composition, particulate NaCl was frequently observed to be aged by acid-displacement reactions as the particles were depleted in Cl−, which would release a significant quantity of HCl [Abbatt and Waschewsky, 1998; Angelucci et al., 2021]. Other direct sources of HCl, if they exist, were much less important toward explaining the observations compared to the Pasadena site. This conclusion is supported by the PM2.5 Na+-Cl− charge balance relationship where a large molar equivalent excess of Na+ compared to Cl− suggests that sea salt dominated over other potential Cl− sources.
The HCl observed during CalNex at the two sites can be compared with previous HCl measurements in the SoCAB. Figure 2 shows the statistical distribution of the CalNex observations at both sites compared to two prior data sets from measurements made in the 1980s. Grosjean [1990] reported HCl measurements for 9 days, 12 to 21 August 1986, at Glendora, CA (~30 km east of Pasadena) from integrated samples taken at 5 different sampling intervals throughout any 24 hour period. The measurements reported by Eldering et al. [1991] were 24 hour samples taken every 6 days for an entire year at sites throughout the SoCAB in 1986. The 15 May to 30 June observations from the 3 sites closest to Pasadena: Downtown, Burbank and Upland, were selected for use in Figure 2. Several publications using data from the CalNex 2010 dataset have concluded that there have been large decreases (7-8%/yr) in VOCs, NOx and NOx-product compounds over the past 40 years in the SoCAB [Pollack et al., 2013; Warneke et al., 2012]. Although there is not nearly the same degree of data coverage, HCl measurements over the 25 years spanning 1985-2010, do not appear to show any significant decrease. This is perhaps not surprising considering that HCl volatilization from sea salt, and transported dust and soil, even after dry deposition, are probably the largest sources in this air basin.
Figure 2.
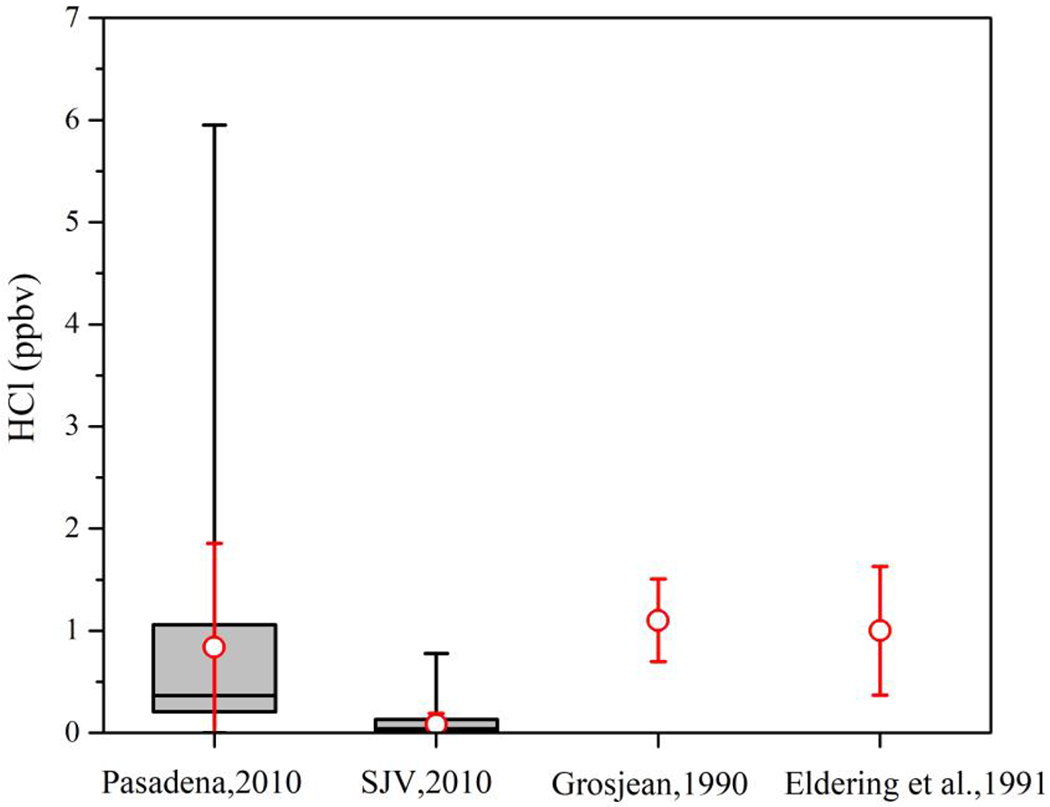
Distributions of HCl measured at or near the CalNex 2010 Pasadena and SJV site, the May 15 to June 30 observations (n=7) of Eldering et al. [1991], August 12-21 data (n=43) of Grosjean [1990]. The prior studies were both conducted in the SoCAB, with comparison sites selected for proximity to Pasadena. The black box and whiskers represent the central 50% (box) and max and min of the CalNex observation, while the red circle and error bars are the observation averages ± 1σ.
The variation of the HCl measured in Pasadena with time of day is shown in Figure 3, in addition to data from other studies conducted in the SoCAB. The CalNex data show a clear diurnal pattern characteristic of species such as O3 or HNO3 that have a photochemical daytime source, and are deposited to the surface at night. Other HCl data sets from the SoCAB have similar diurnal patterns, but with limited temporal resolution. Mid-day levels of HCl observed at the surface average slightly more than 2 ppbv, and are probably characteristic of the HCl mixed throughout the planetary boundary layer (PBL) at this location because vertical mixing is at a maximum during this period. The relationship between HCl and HNO3 at the two sites are plotted and compared in Figure 4, with points colored by date. In Pasadena, it is clearly illustrated that HCl and HNO3 are closely correlated on individual days, but the dataset did not exhibit a strong correlation between the two acids overall (R2= 0.30). This is consistent with the results of Appel et al. [1991] who noted that there was only weak correlation between HCl and HNO3 measured during their study (R2 = 0.36). The slope of HCl against HNO3 ranged from less than 1:10 up to 2:1. In contrast, there is a consistent linear relationship between HNO3 and HCl in the gas phase throughout the whole campaign at the SJV site (Figure 4b). The strong linear correlations between increasing HNO3 and HCl in the gas phase further suggest that the phase partitioning equilibria of HCl/Cl− and HNO3/NO3− are probably highly coupled with each other at the SJV site. It is also reasonable to suggest that the day-to-day variation of HCl-HNO3 relationship in Pasadena indicates there are other important sources for HCl (e.g., acidification of soil particles or chloride-containing urban surfaces) or HNO3 (photochemistry production). In either case, the outcome is that the two constituents have not been in the same airmass long enough to equilibrate.
Figure 3.
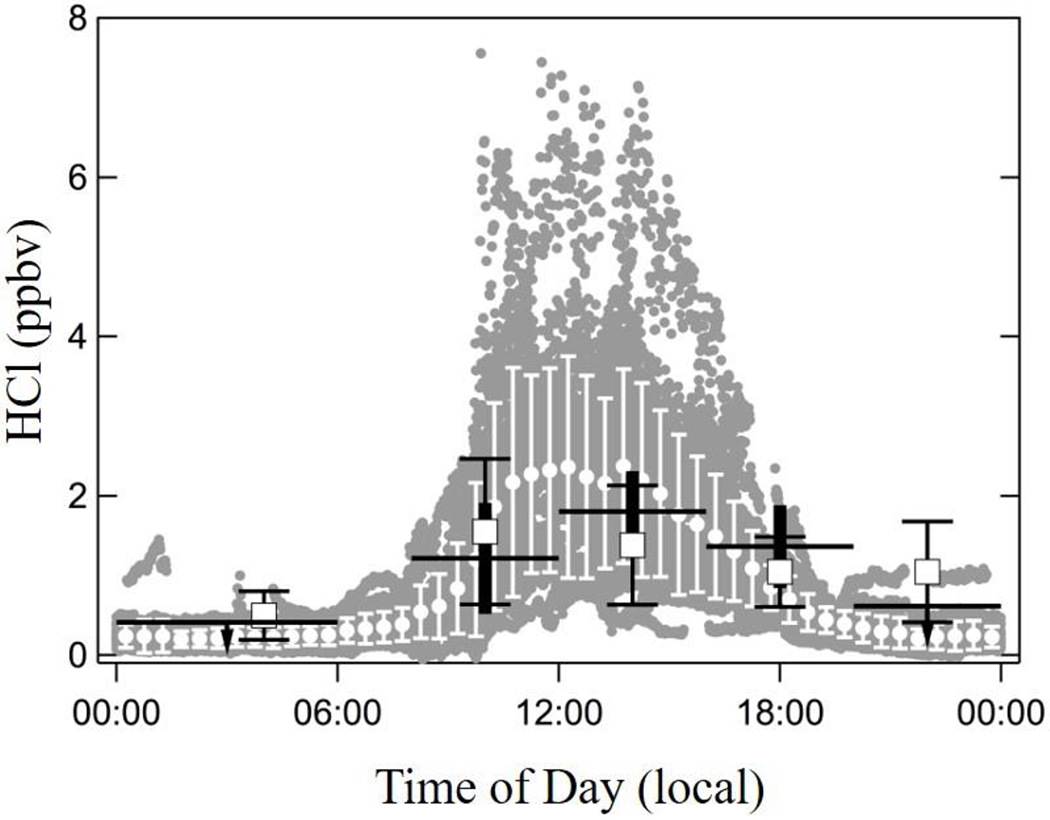
Measurements of HCl versus time of day. The one-minute averages from Pasadena are shown as grey circles and the half hour averages are shown as white points with the error bars indicating ±1σ. Data from 11-17 September 1985 - taken at Claremont CA - are shown as horizontal lines with thick vertical bars that indicate ±1σ. Note that the 00:00-06:00 and 20:00-24:00 numbers are upper limits [Appel et al., 1991]. Data from 12-21 August 1986 taken at Glendora, CA, are shown as open squares with narrow capped error bars that indicate ±1σ [Grosjean, 1990].
Figure 4.
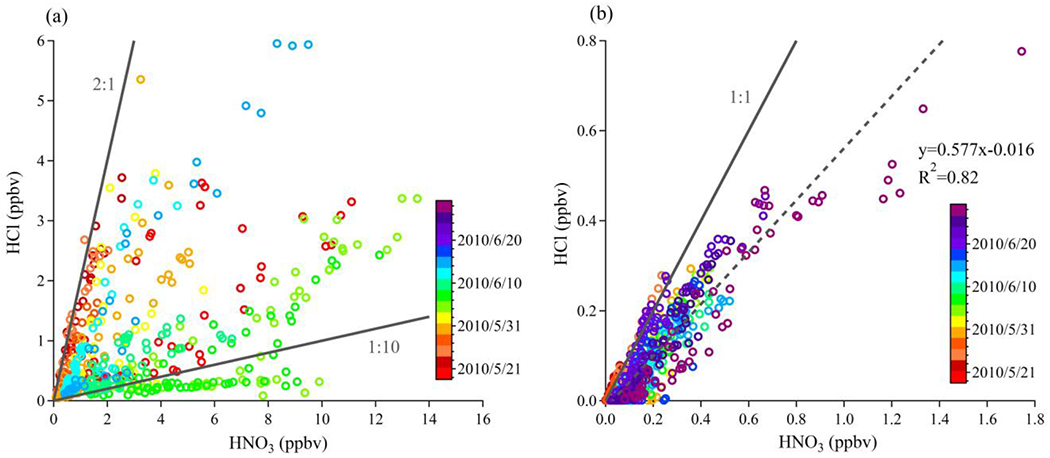
Correlations of HCl with HNO3 colored by observation date for the CalNex 2010 datasets at: (a) the Pasadena site and (b) the SJV site. Note that observations at Pasadena ended before those at SJV (see Fig.1). Due to the wide variance in the relationship between these species in Pasadena, the range of ratios from 2:1 to 1:10 is denoted using solid black lines. At the SJV site, the 1:1 ratio is denoted by the solid black line, while the orthogonal least-squares linear regression fit is shown with the dashed grey line.
HCl concentration and phase partitioning
The two sites explored here vary greatly in several respects, including RH and T (Figure S1), which will affect phase partitioning. SJV was characterized by average (± standard deviation) RH of 38 ± 17 % and T of 23.5 ± 6.7 °C. Pasadena was generally wetter and cooler, with RH of 69 ± 16 % and T of 17.8 ± 4.3 °C. To further study the relationship between HCl and HNO3 phase partitioning, the temperature dependence of thermodynamic equilibrium constants under the assumption of solid phase or aqueous phase reaction for SJV is shown in Figure 5a and b, respectively. The data points with Cl− concentrations lower than 1 nmol m−3 were not included in the comparison (grey points, Figures 5a–b) to ensure that NaCl-containing particles were abundant enough for the HCl and HNO3 phase partitioning to mostly corresponding to the aerosol ageing process. For the assumption of solid phase reaction (i.e., R2), it can be seen in Figure 5a that the theoretical calculation always overestimates the ln[p(HCl)/p(HNO3)] values. Generally, the solid phase equilibrium predicts the HCl to be more concentrated than HNO3, which does not agree with the observation (Figure 4b suggests that p(HCl)<p(HNO3) almost throughout the whole campaign). Therefore, even though the SJV site had low RH (36.6 ± 16.5 %), observed HCl/Cl− cannot be explained by a purely solid-phase reaction so that some liquid partitioning in metastable or fully deliquesced particles must be occurring.
Figure 5.
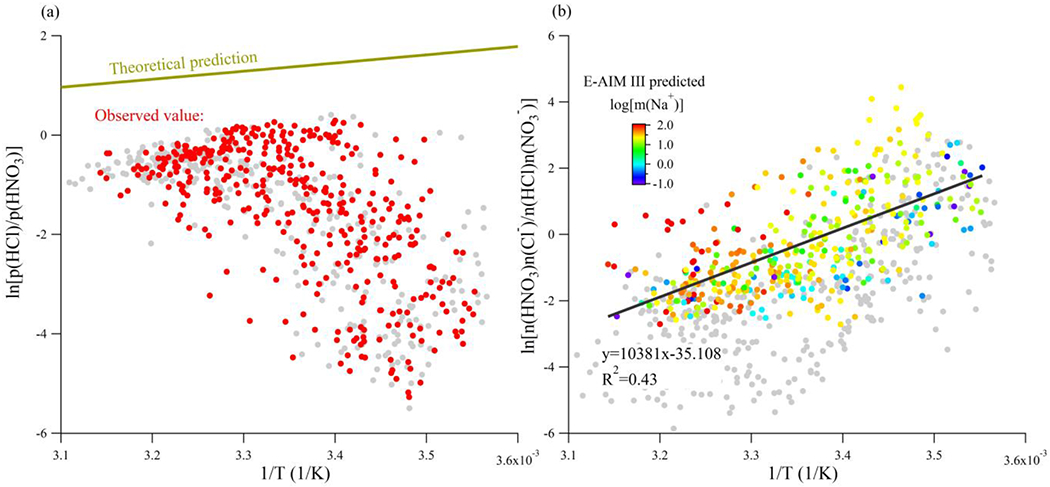
(a) The theoretical prediction and observed temperature dependence of ln[p(HCl)/p(HNO3)] against 1/T in (1/K) for the SJV site under the assumption of solid phase reaction. (b) The linear regression results of ln[n(HNO3)n(Cl−)/n(HCl)n(NO3−)] against 1/T in (1/K) colored by the molality of Na+ calculated by E-AIM III for the SJV site under the assumption of aqueous phase reaction. The data with Cl− concentrations less than 1 nmol m−3 (grey points) representing 22 % of the measured data, have been excluded from each regression analysis.
The efflorescence relative humidity for NaCl is around 43% (higher than the average RH in SJV), while the efflorescence relative humidity for NaNO3 is not observed [Seinfeld and Pandis, 2006] or the time scale is too long to be atmospherically relevant [Kim et al., 2012]. Therefore, the deliquesced sea salt aerosol prior to transport and the efficient formation of nitrate salt can sustain particles in metastable state. Other than significantly lowering the efflorescence RH, the formation of NaNO3 can also substantially increase the ionic strength of NaCl-containing particles (see Figure S2 for the E-AIM III-calculated Na+ molality under different NaNO3:NaCl molar ratios). This could be important to evaluate the ClNO2 yields on particle surfaces. For example, Mitroo et al. [2019] found that deliquesced chloride salts can have high ClNO2 yields. The existence of other hygroscopic salts such as CaCl2 and MgCl2 can also lower the deliquescence point of particles [Tang and Munkelwitz, 1993] and promote ClNO2 formation [Royer et al., 2021].
In Figure 5b the ln[n(HNO3)n(Cl−)/n(HCl)n(NO3−)] value (see Methods for detail) was explored as a function of 1/T. The resulting linear relationship of the SJV site shows a moderate correlation (R2=0.43, Figure 5). A similar relationship can also be computed between HCl/Cl− and HONO/NO2− phase partitioning behavior at the SJV site (Figure S3), indicating that these two gas-particle partitioning equilibria are also likely coupled. The temperature dependence observed is derived from both thermodynamic parameters and activity coefficients. Several thermodynamic modelling results suggest that ΔrH for HCl phase partitioning is only several kJ/mol lower than that of HNO3 phase partitioning [Carslaw et al., 1995; Fountoukis and Nenes, 2007; Seinfeld and Pandis, 2006] (also listed in Table S1). Therefore, the strong temperature dependence observed in Figure 5b can only be explained by the effect of activity coefficients. If the contribution from activity coefficients is neglected, it would require −[ΔrH(HCl)- ΔrH(HNO3)] to reach 86.3 kJ/mol to explain the observed temperature dependence. To assess whether this assumption is reasonable, the molality of Na+ was predicted by E-AIM III under the assumption of metastable state. Current versions of E-AIM cannot calculate the temperature dependence of NaCl activity coefficients under RH<60%, so E-AIM III is used here with fixed temperature at 25 °C to determine the RH dependence of activity coefficients as the reference. Molality of Na+ was generally on the order of 101~2 M, suggesting that the aerosol likely consisted of deliquesced salts with high ionic strength. The influence of activity coefficients may be significant under this situation.
In contrast, there is no apparent temperature dependence of ln[n(HNO3)n(Cl−)/n(HCl)n(NO3−)] at the Pasadena site where RH was 69.3 ± 15.3 %, significantly higher than that at the SJV site (Figure S4). One of the reasons could be that, even though the Pasadena site was more likely to have fully deliquesced aerosol to go through aqueous phase HCl-HNO3 exchange reaction, there were simultaneous direct sources for HNO3 and sea salt arriving at the measurement location such that the particles were not fully internally mixed as revealed from the interpretation of Figure 4. To explore this, we performed analysis on a day by day basis (Figure S5), finding that for most days there were obvious diurnal cycles in the temperature dependence of ln[n(HNO3)n(Cl−)/n(HCl)n(NO3−)], consistent with the frequent sea-land breeze transporting externally mixed aerosol to this site [Washenfelder et al., 2011]. Furthermore, it is found that in SJV site Cl− activity coefficients spanned two orders of magnitudes (1~100) while they remained close to unity in Pasadena site; by comparison, the range of NO3− activity coefficients at both sites showed a much smaller range of variation and were around 0.3 (shown in detail in Figure S6) so that the influence of activity coefficients was not strong enough to be observed at the Pasadena site.
Apart from the strong influence from activity coefficient changes, another possibility is that the enthalpy of HCl phase partitioning was underestimated by the previous studies. Several literature values for standard effective Henry’s Law Constants (at 298 K), either determined through experiments, field observation or computation, exhibit large differences (listed in Table S1) while experimentally determined parameters should have higher reliability. Some of these values have been proven to be unreliable or have some flaws in the methodology [Clegg and Brimblecombe, 1988; Keene and Savoie, 1999; Marsh and McElroy, 1985; Sander, 2015]. Among the values for KH of HCl listed by Sander [2015], the one provided by Marsh and McElroy [1985] was derived from an incorrect equation, and the sources of Brimblecombe and Clegg [1988] and Pandis and Seinfeld [1989] were also incorrect or questionable as suggested by the notes in Sander [2015] stating that this compiled paper did not list the temperature dependence parameters with reliable sources. While beyond the scope of this work, a complete evaluation of the existing thermodynamic parameters would be beneficial to improve our understanding of the dominant factors influencing HCl/Cl− phase partitioning and model-observation disagreement found in some studies.
For activity coefficients, different calculation methods also can have large discrepancies. Kim et al. [1993] illustrated in detail that for a NaCl solution with ionic strength of 40 mol/kg, the difference of activity coefficients calculated by three common methods K-M method [Kusik and Meissner, 1978], Pitzer method [Pitzer and Mayorga, 2002] and Bromley method [Bromley, 1973] can reach two orders of magnitudes. In comparison, the activity coefficients of NaNO3 solution calculated by the above-mentioned three methods vary in the relatively narrow range of 0.6~3. The activity coefficients of NaCl solution under high ionic strength conditions tremendously depends on the chosen calculation method. The activity calculation methods used in E-AIM III are introduced in detail in Clegg et al. [1992] and [Clegg et al., 1997], which are mainly validated at 298.15 K and RH > 40%.
The disagreement between the observed and modelled phase partitioning of HCl/Cl− is usually attributed to factors such as measurement uncertainties, other unconsidered chemical components or externally mixed particles [Shon et al., 2012; Sudheer and Rengarajan, 2015; Trebs, 2005]. Here, we suggest another potential factor is the uncertainties of their thermodynamic properties which complements the discussion raised by Young et al. [2013] and Keene et al. [2004]. Interestingly, the −[ΔrH(HONO)- ΔrH(HNO3)] calculated based on the slopes of Figure 5b and Figure S3 (neglecting the contribution from activity coefficients) yields a value of 24.8 kJ/mol at SJV, falls within 60% difference of prior reports (listed in Table S1). Unlike Cl−, the logarithm of activity coefficient of NO2− in NaNO2 solution does not show the obvious dependence on molality when its molality is higher than 4 mol kg−1 [Staples, 1981]. It is reasonable to assume when activity coefficients show no strong dependence on RH, the observed coupled phase partitioning behavior is mainly controlled by the temperature dependent effective Henry’s Law constants; in contrast, when activity coefficients are strongly affected by RH, the phase partitioning behavior can be dominated by RH-related activity coefficients changes. Considering the wide ranges of HCl Henry’s Law constants from different studies, a complete evaluation of those values will help distinguish the contribution from both terms when interpreting HCl phase partitioning behavior.
Application of aerosol thermodynamic models to the Pasadena dataset can also help explain the factors controlling HCl and HCl/Cl− distribution in the SoCAB environment. Semi-volatile NH4Cl formation is not favored to form at either site (described in Supplementary Information: section 1). Therefore, two models that take account of all the major inorganic ionic components were used in this work, the E-AIM described by Wexler [2002], and ISORROPIA described by Fountoukis and Nenes [2007]. Attempts to model the gas/particle system using only PM1.0 data i.e., AMS data, or PM2.5 data without explicit cation measurements were not very successful. The final period of the Pasadena observation had cation measurements for the PM2.5 size range and showed good agreement between measured and modeled HCl and Cl− for both models (shown in Figure 6). The good fit between the modelled and observed concentration indicates that the gas-particle system is adequately constrained by thermodynamic processes such as charge balance, or the existence of stronger buffer in the system (e.g., NH3/NH4+ and HNO3/NO3− phase partitioning) [Zheng et al., 2020], when needing to describe HCl/Cl− phase partitioning. However, considering that the disagreement between observed and modelled phase partitioning of HCl/Cl− is frequently reported [Shon et al., 2012; Sudheer and Rengarajan, 2015; Trebs, 2005], caution should be taken in applying this conclusion more generally.
Figure 6.
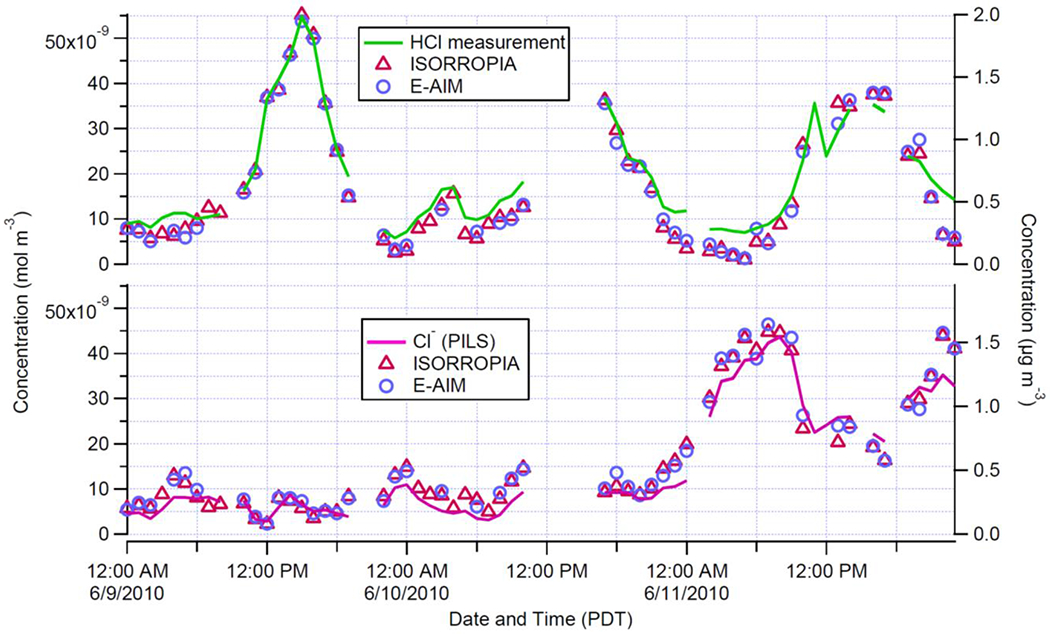
A comparison of aerosol thermodynamic models, ISORROPIA (red triangles), E-AIM (blue circles), with observations: HCl (green line) top panel and Cl− (magenta line) bottom panel for a three-day period during CalNex-LA.
Regional modelling of HCl
Hourly average observed and predicted HCl mixing ratios at the Pasadena site are compared in Figure 7a, and hourly average observed PM2.5 Cl− concentrations are compared with the sum of the predicted Aitken and accumulation mode Cl− in Figure 7b. Large underestimates of HCl mixing ratio in both model simulations are evident. For instance, the observed mean HCl mixing ratio across all hours is underestimated by a factor of 6.6 in the Base simulation and 4.8 in the SSx3 simulation. This underestimation is most pronounced during daytime when observed HCl mixing ratios are greatly enhanced compared to nighttime. From 11-18 PST, Cl− is almost entirely depleted from fine mode particles in the model while the median observed PM2.5 Cl− is 0.36 μg m−3 (Figure 7b). To the extent that PM2.5 Cl− is the source of HCl, the modeled amounts are insufficient to maintain the levels of HCl observed. Furthermore, as indicated by Figure 1a, the coarse mode particles also cannot explain the high HCl concentrations observed in the mid-day.
Figure 7.
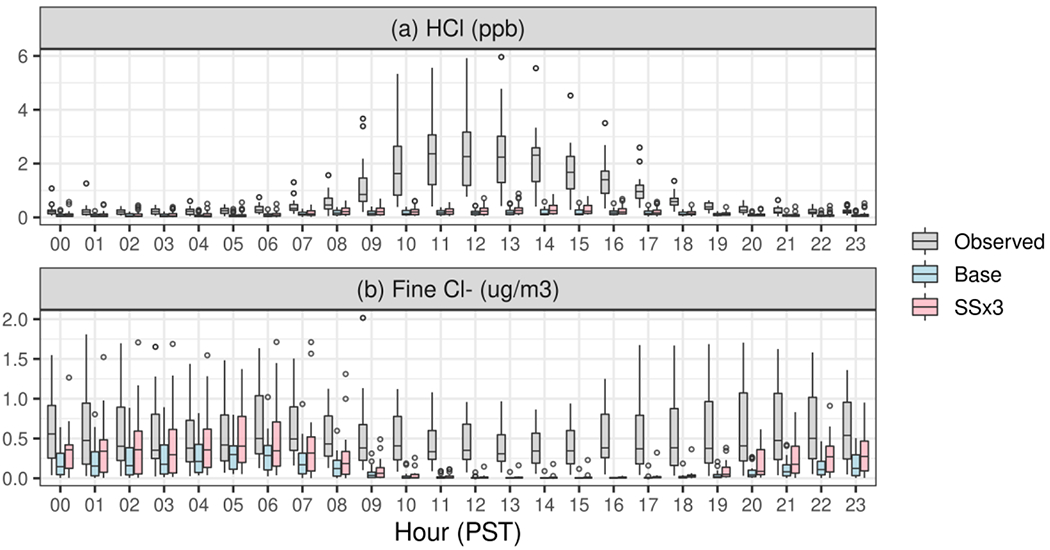
Modeled and measured HCl and PM2.5 chloride at the Pasadena site during 15 May and 15 June 2010.
In Figure 8, distributions of modeled mixing ratios of HCl and PM2.5 Na+ and Cl− are shown for Pasadena. The mean PM2.5 Na+ mixing ratio in the SSx3 simulation is 2.6 times that in the Base simulation and indicates that Na+ concentrations in Pasadena increase roughly linearly with sea-spray emissions. The mean PM2.5 Cl− is 64% greater in the SSx3 than Base simulation at Pasadena. The sub-linear increase in PM2.5 Cl− in Pasadena with increasing sea spray emissions is likely due to the displacement of Cl− from the particles as they are transported inland, consistent with the daily plots of the thermodynamic system (Figure S5). However, the increase in HCl mixing ratio that would result from this Cl− displacement is insufficient to resolve the model HCl underpredictions. Much greater fine-mode sea-spray emissions or other temporally variable surface reservoirs of Cl− would be needed to explain the HCl underpredictions via this pathway. The combination of too rapid removal of coarse sea spray and underestimation of coarse particle Cl− displacement could in part explain the HCl underpredictions. However, other sources of HCl in the basin that were uncharacterized or underestimated [Crisp et al., 2014] also likely contribute to the underpredictions. Additional potential sources could be a persistent surface reservoir of Cl− salts, including dust, grime or surface soil, that release HCl coupled with HNO3 dry deposition [Baergen et al., 2015; McNamara et al., 2020]. HCl and Cl− concentrations were also underpredicted by the model at the SJV site (shown in Figure S7) and have been reported previously in Colorado [Kelly et al., 2016] and Florida [Kelly et al., 2010], and so improved understanding of HCl and Cl− sources would broadly benefit model performance.
Figure 8.
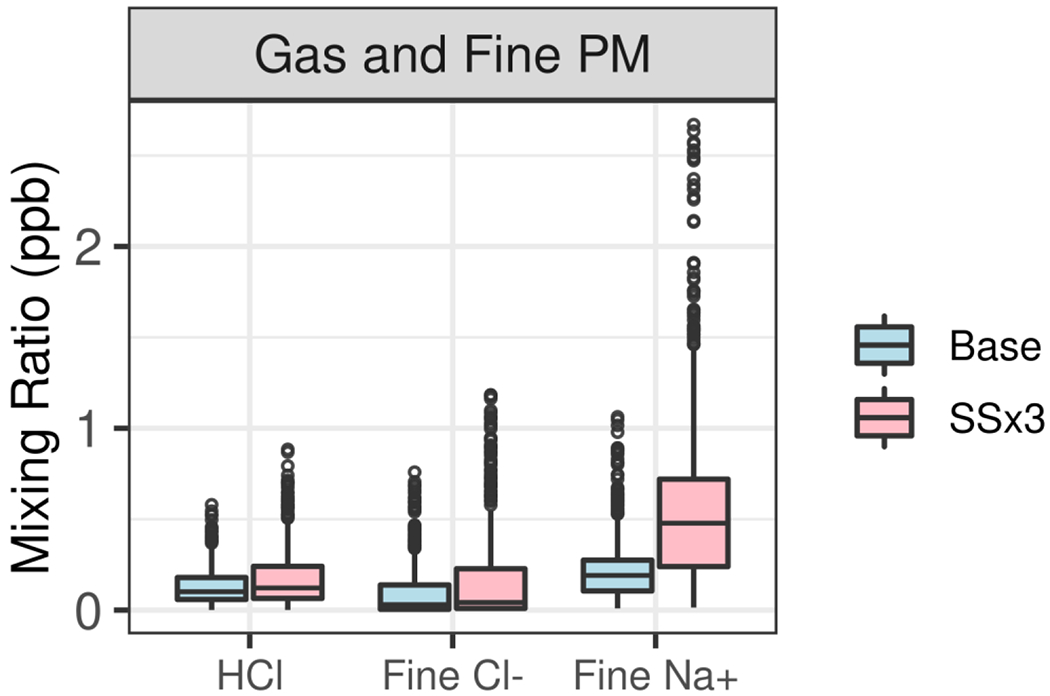
Modeled distributions of HCl and fine particle sodium and chloride and at the Pasadena site during 15 May to 15 June 2010. Circles represent values greater than 1.5 times the interquartile range from either end of the box.
IV. Conclusions
HCl was measured from May 15 to June 15, 2010 during the CalNex experiment. Observed mixing ratios ranged from below detection limit (55pptv) up to 5.95 ppbv with a mean of 0.83 ppbv in Pasadena and was on average 0.084 ppbv at SJV with a maximum value of 0.78 ppbv. At both sites, HCl levels were highest during midday and were well-correlated with HNO3 on a given day. However, in the Pasadena site the slope of the HCl: HNO3 correlation varied a great deal (2:1 to 1:10) from day-to-day. At the SJV site, the HCl was well correlated with HNO3 throughout the whole campaign. Lowest HCl levels were observed at night and were consistent with strong deposition of this highly soluble compound. In general, there was no correlation between HCl and episodically high particle Cl− levels. HCl was more likely influenced by other factors such as the reaction of chlorine atoms (generated by photolysis of photolabile compounds such as ClNO2) [Angelucci et al., 2021; Mielke et al., 2013; Young et al., 2014].
The gas-particle exchange of HCl can be governed by NH3 during periods of lower temperature and high (>90%) humidity. The observed levels of HCl and Cl− are in agreement with aerosol thermodynamic models using PM2.5 composition, NH3, HNO3, and constrained by base cation measurements. At the SJV site, the phase partitioning of HCl/Cl− was found coupled with HNO3/NO3− and HONO/NO2− phase partitioning that was likely governed by activity coefficient changes. Considering that the Henry’s Law constants for HCl reported by different literature have order-of-magnitude differences, its temperature dependence should also have a more reliable assessment as well as an evaluation of the influence that aerosol components have on overall phase partitioning behavior. It is possible that ambient measurement could provide this information. The ideal situation to perform such observation-based calculations would be in consistently high RH (e.g., >90%) scenario where the influence from activity coefficient changes is minimized. However, such conditions did not exist during this campaign.
Measured HCl mixing ratios were underestimated by factors of 5 or more in regional air quality modeling. A sensitivity simulation with increased sea-spray emissions suggests that underestimates of these emissions are not the sole cause of the HCl underpredictions. Other possible contributors to model underpredictions include too rapid deposition of coarse particle chloride, underestimates of Cl− displacement from coarse particles, and other uncharacterized or underestimated HCl sources in the basin such as soil or wind-blown dust. However, these are areas of future studies. Model development [Appel et al., 2020] and high time-resolution measurement will be necessary to fully understand the causes of the model underestimates.
Supplementary Material
V. Acknowledgments and Disclaimer –
We thank California Air Resources Board for funding both CalNex sites, California Institute of Technology for supporting the Pasadena site, and John Karlik, University of California Agricultural Cooperative Extension Staff and Kern County Staff, for logistical support at the SJC site. We acknowledge financial support for the field measurements from the NOAA Climate Change and Health of the Atmosphere programs and support for the analysis from the Natural Science and Engineering Research Council. The authors also thank Brett Gantt for providing an updated version of CMAQ and Chris Allen and Allan Beidler for emissions inventory development. The views expressed in this manuscript are those of the authors alone and do not necessarily reflect the views and policies of the US Environmental Protection Agency.
Footnotes
The data used in this paper are available at: http://csl.noaa.gov/groups/cs17/measurements/2010calnex/Ground/DataDownload/.
VII. References
- Abbatt JPD, and Waschewsky GCG (1998), Heterogeneous interactions of HOBr, HNO3, O3, and NO2 with deliquescent NaCl aerosols at room temperature, The Journal of Physical Chemistry A, 102(21), 3719–3725. [Google Scholar]
- Ahern AT, Goldberger L, Jahl L, Thornton J, and Sullivan RC (2018), Production of N2O5 and ClNO2 through nocturnal processing of biomass-burning aerosol, Environ Sci Technol, 52(2), 550–559. [DOI] [PubMed] [Google Scholar]
- Angelucci AA, Furlani TC, Wang X, Jacob DJ, VandenBoer TC, and Young CJ (2021), Understanding Sources of Atmospheric Hydrogen Chloride in Coastal Spring and Continental Winter, ACS Earth and Space Chemistry, 5(9), 2507–2516. [Google Scholar]
- Appel BR, Tokiwa Y, Povard V, and Kothny EL (1991), The measurement of atmospheric hydrochloric acid in southern California, Atmospheric Environment. Part A. General Topics, 25(2), 525–527. [Google Scholar]
- Appel KW, et al. (2020), The community multiscale air quality (CMAQ) model versions 5.3 and 5.3.1: system updates and evaluation, Geosci. Model Dev. Discuss, 2020, 1–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baergen AM, Styler SA, van Pinxteren D, Muller K, Herrmann H, and Donaldson DJ (2015), Chemistry of urban grime: inorganic ion composition of grime vs particles in Leipzig, Germany, Environ Sci Technol, 49(21), 12688–12696. [DOI] [PubMed] [Google Scholar]
- Baker KR, Misenis C, Obland MD, Ferrare RA, Scarino AJ, and Kelly JT (2013), Evaluation of surface and upper air fine scale WRF meteorological modeling of the May and June 2010 CalNex period in California, Atmospheric Environment, 80, 299–309. [Google Scholar]
- Becker KH, Kleffmann J, Martin Negri R, and Wiesen P (1998), Solubility of nitrous acid (HONO) in ammonium sulfate solutions, Journal of the Chemical Society, Faraday Transactions, 94(11), 1583–1586. [Google Scholar]
- Bertram TH, and Thornton JA (2009), Toward a general parameterization of N2O5 reactivity on aqueous particles: the competing effects of particle liquid water, nitrate and chloride, Atmospheric Chemistry and Physics, 9(21), 8351–8363. [Google Scholar]
- Binkowski FS, and Roselle SJ (2003), Models - 3 Community Multiscale Air Quality (CMAQ) model aerosol component 1. Model description, Journal of Geophysical Research: Atmospheres, 108(D6). [Google Scholar]
- Brimblecombe P, and Clegg SL (1988), The solubility and behaviour of acid gases in the marine aerosol, J Atmos Chem, 7(1), 1–18. [Google Scholar]
- Bromley LA (1973), Thermodynamic properties of strong electrolytes in aqueous solutions, AIChE Journal, 19(2), 313–320. [Google Scholar]
- Brown SS, et al. (2013), Nitrogen, Aerosol Composition, and Halogens on a Tall Tower (NACHTT): Overview of a wintertime air chemistry field study in the front range urban corridor of Colorado, Journal of Geophysical Research: Atmospheres, 118(14), 8067–8085. [Google Scholar]
- Carslaw KS, Clegg SL, and Brimblecombe P (1995), A Thermodynamic Model of the System HCl-HNO3-H2SO4-H2O, Including Solubilities of HBr, from <200 to 328 K, The Journal of Physical Chemistry, 99(29), 11557–11574. [Google Scholar]
- Clegg SL, and Brimblecombe P (1988), Equilibrium partial pressures of strong acids over concentrated saline solutions—I. HNO3, Atmospheric Environment (1967), 22(1), 91–100. [Google Scholar]
- Clegg SL, and Brimblecombe P (1990), Equilibrium partial pressures and mean activity and osmotic coefficients of 0-100% nitric acid as a function of temperature, The Journal of Physical Chemistry, 94(13), 5369–5380. [Google Scholar]
- Clegg SL, Pitzer KS, and Brimblecombe P (1992), Thermodynamics of multicomponent, miscible, ionic solutions. Mixtures including unsymmetrical electrolytes, The Journal of Physical Chemistry, 96(23), 9470–9479. [Google Scholar]
- Clegg SL, Brimblecombe P, Liang Z, and Chan CK (1997), Thermodynamic Properties of Aqueous Aerosols to High Supersaturation: II—A Model of the System Na+–Cl−–NO3−–SO42−–H2O at 298.15 K, Aerosol Science and Technology, 27(3), 345–366. [Google Scholar]
- Crisp TA, Lerner BM, Williams EJ, Quinn PK, Bates TS, and Bertram TH (2014), Observations of gas phase hydrochloric acid in the polluted marine boundary layer, Journal of Geophysical Research: Atmospheres, 119(11), 6897–6915. [Google Scholar]
- Crounse JD, McKinney KA, Kwan AJ, and Wennberg PO (2006), Measurement of gas-phase hydroperoxides by chemical ionization mass spectrometry, Anal Chem, 78(19), 6726–6732. [DOI] [PubMed] [Google Scholar]
- Dasgupta PK, Campbell SW, Al-Horr RS, Ullah SMR, Li J, Amalfitano C, and Poor ND (2007), Conversion of sea salt aerosol to NaNO3 and the production of HCl: Analysis of temporal behavior of aerosol chloride/nitrate and gaseous HCl/HNO3 concentrations with AIM, Atmospheric Environment, 41(20), 4242–4257. [Google Scholar]
- Eatough DJ, Wadsworth A, Eatough DA, Crawford JW, Hansen LD, and Lewis EA (1993), A multiple-system, multi-channel diffusion denuder sampler for the determination of fine-particulate organic material in the atmosphere, Atmospheric Environment. Part A. General Topics, 27(8), 1213–1219. [Google Scholar]
- Eldering A, Solomon PA, Salmon LG, Fall T, and Cass GR (1991), Hydrochloric acid: A regional perspective on concentrations and formation in the atmosphere of Southern California, Atmospheric Environment. Part A. General Topics, 25(10), 2091–2102. [Google Scholar]
- Ellis RA, Murphy JG, Pattey E, van Haarlem R, O’Brien JM, and Herndon SC (2010), Characterizing a Quantum Cascade Tunable Infrared Laser Differential Absorption Spectrometer (QC-TILDAS) for measurements of atmospheric ammonia, Atmospheric Measurement Techniques, 3(2), 397–406. [Google Scholar]
- Fast JD, et al. (2014), Modeling regional aerosol and aerosol precursor variability over California and its sensitivity to emissions and long-range transport during the 2010 CalNex and CARES campaigns, Atmospheric Chemistry and Physics, 14(18), 10013–10060. [Google Scholar]
- Fountoukis C, and Nenes A (2007), ISORROPIA II: a computationally efficient thermodynamic equilibrium model for K+–Ca2+–Mg2+–NH4+–Na+–SO42−–NO3−–Cl−–H2O aerosols, Atmospheric Chemistry and Physics, 7(17), 4639–4659. [Google Scholar]
- Gantt B, Kelly JT, and Bash JO (2015), Updating sea spray aerosol emissions in the Community Multiscale Air Quality (CMAQ) model version 5.0.2, Geoscientific Model Development, 8(11), 3733–3746. [Google Scholar]
- Grosjean D (1990), Liquid-chromatography analysis of chloride and nitrate with “negative” ultraviolet detection: ambient levels and relative abundance of gas-phase inorganic and organic acids in southern California, Environmental Science & Technology, 24(1), 77–81. [Google Scholar]
- Guo H, Liu J, Froyd KD, Roberts JM, Veres PR, Hayes PL, Jimenez JL, Nenes A, and Weber RJ (2017), Fine particle pH and gas–particle phase partitioning of inorganic species in Pasadena, California, during the 2010 CalNex campaign, Atmospheric Chemistry and Physics, 17(9), 5703–5719. [Google Scholar]
- Guzman-Morales J, et al. (2014), Estimated contributions of primary and secondary organic aerosol from fossil fuel combustion during the CalNex and Cal-Mex campaigns, Atmospheric Environment, 88, 330–340. [Google Scholar]
- Haskins JD, et al. (2019), Observational Constraints on the Formation of Cl2 From the Reactive Uptake of ClNO2 on Aerosols in the Polluted Marine Boundary Layer, Journal of Geophysical Research: Atmospheres, 124(15), 8851–8869. [Google Scholar]
- Haskins JD, et al. (2018), Wintertime gas - particle partitioning and speciation of inorganic chlorine in the lower troposphere over the Northeast United States and coastal ocean, Journal of Geophysical Research: Atmospheres, 123(22). [Google Scholar]
- Jordan CE, Pszenny AAP, Keene WC, Cooper OR, Deegan B, Maben J, Routhier M, Sander R, and Young AH (2015), Origins of aerosol chlorine during winter over north central Colorado, USA, Journal of Geophysical Research: Atmospheres, 120(2), 678–694. [Google Scholar]
- Keene WC, and Savoie DL (1999), Correction to “The pH of deliquesced sea-salt aerosol in polluted marine air”, Geophysical Research Letters, 26(9), 1315–1316. [Google Scholar]
- Keene WC, Pszenny AAP, Maben JR, Stevenson E, and Wall A (2004), Closure evaluation of size-resolved aerosol pH in the New England coastal atmosphere during summer, Journal of Geophysical Research: Atmospheres, 109(D23). [Google Scholar]
- Keene WC, Pszenny AAP, Jacob DJ, Duce RA, Galloway JN, Schultz-Tokos JJ, Sievering H, and Boatman JF (1990), The geochemical cycling of reactive chlorine through the marine troposphere, Global Biogeochemical Cycles, 4(4), 407–430. [Google Scholar]
- Keene WC, et al. (1999), Composite global emissions of reactive chlorine from anthropogenic and natural sources: Reactive Chlorine Emissions Inventory, Journal of Geophysical Research: Atmospheres, 104(D7), 8429–8440. [Google Scholar]
- Kelly JT, Bhave PV, Nolte CG, Shankar U, and Foley KM (2010), Simulating emission and chemical evolution of coarse sea-salt particles in the Community Multiscale Air Quality (CMAQ) model, Geoscientific Model Development, 3(1), 257–273. [Google Scholar]
- Kelly JT, Baker KR, Nolte CG, Napelenok SL, Keene WC, and Pszenny AAP (2016), Simulating the phase partitioning of NH3, HNO3, and HCl with size-resolved particles over northern Colorado in winter, Atmospheric Environment, 131, 67–77. [Google Scholar]
- Kelly JT, et al. (2014), Fine-scale simulation of ammonium and nitrate over the South Coast Air Basin and San Joaquin Valley of California during CalNex-2010, J Geophys Res-Atmos, 119(6), 3600–3614. [Google Scholar]
- Kim H, Lee M-J, Jung H-J, Eom H-J, Maskey S, Ahn K-H, and Ro C-U (2012), Hygroscopic behavior of wet dispersed and dry deposited NaNO3 particles, Atmospheric Environment, 60, 68–75. [Google Scholar]
- Kim YP, Seinfeld JH, and Saxena P (1993), Atmospheric Gas-Aerosol Equilibrium I. Thermodynamic Model, Aerosol Science and Technology, 19(2), 157–181. [Google Scholar]
- Kusik CL, and Meissner HP (1978), Electrolyte activity coefficients in inorganic processing, A.I.Ch.E. Journal Symposium Series, 173(1978), 14–20. [Google Scholar]
- Liu S, et al. (2012), Secondary organic aerosol formation from fossil fuel sources contribute majority of summertime organic mass at Bakersfield, Journal of Geophysical Research: Atmospheres, 117(D24). [Google Scholar]
- Markovic MZ, VandenBoer TC, and Murphy JG (2012), Characterization and optimization of an online system for the simultaneous measurement of atmospheric water-soluble constituents in the gas and particle phases, J Environ Monit, 14(7), 1872–1884. [DOI] [PubMed] [Google Scholar]
- Markovic MZ, VandenBoer TC, Baker KR, Kelly JT, and Murphy JG (2014), Measurements and modeling of the inorganic chemical composition of fine particulate matter and associated precursor gases in California’s San Joaquin Valley during CalNex 2010, Journal of Geophysical Research: Atmospheres, 119(11), 6853–6866. [Google Scholar]
- Marsh ARW, and McElroy WJ (1985), The dissociation constant and Henry’s law constant of HCl in aqueous solution, Atmospheric Environment (1967), 19(7), 1075–1080. [Google Scholar]
- McGrath MJ, Kuo IF, Ngouana WB, Ghogomu JN, Mundy CJ, Marenich AV, Cramer CJ, Truhlar DG, and Siepmann JI (2013), Calculation of the Gibbs free energy of solvation and dissociation of HCl in water via Monte Carlo simulations and continuum solvation models, Phys Chem Chem Phys, 15(32), 13578–13585. [DOI] [PubMed] [Google Scholar]
- McNamara SM, et al. (2020), Observation of road salt aerosol driving inland wintertime atmospheric chlorine chemistry, ACS Cent Sci, 6(5), 684–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Z, and Seinfeld JH (1996), Time scales to achieve atmospheric gas-aerosol equilibrium for volatile species, Atmospheric Environment, 30(16), 2889–2900. [Google Scholar]
- Mielke LH, Furgeson A, and Osthoff HD (2011), Observation of ClNO2 in a mid-continental urban environment, Environ Sci Technol, 45(20), 8889–8896. [DOI] [PubMed] [Google Scholar]
- Mielke LH, et al. (2013), Heterogeneous formation of nitryl chloride and its role as a nocturnal NOx reservoir species during CalNex - LA 2010, Journal of Geophysical Research: Atmospheres, 118(18). [Google Scholar]
- Mitroo D, Gill TE, Haas S, Pratt KA, and Gaston CJ (2019), ClNO2 Production from N2O5 Uptake on Saline Playa Dusts: New Insights into Potential Inland Sources of ClNO2, Environ Sci Technol, 53(13), 7442–7452. [DOI] [PubMed] [Google Scholar]
- Neuman JA, Ryerson TB, Huey LG, Jakoubek R, Nowak JB, Simons C, and Fehsenfeld FC (2003), Calibration and evaluation of nitric acid and ammonia permeation tubes by UV optical absorption, Environ Sci Technol, 37(13), 2975–2981. [DOI] [PubMed] [Google Scholar]
- Orsini DA, Ma Y, Sullivan A, Sierau B, Baumann K, and Weber RJ (2003), Refinements to the particle-into-liquid sampler (PILS) for ground and airborne measurements of water soluble aerosol composition, Atmospheric Environment, 37(9–10), 1243–1259. [Google Scholar]
- Osthoff HD, et al. (2008), High levels of nitryl chloride in the polluted subtropical marine boundary layer, Nature Geoscience, 1(5), 324–328. [Google Scholar]
- Pandis SN, and Seinfeld JH (1989), Sensitivity analysis of a chemical mechanism for aqueous-phase atmospheric chemistry, Journal of Geophysical Research, 94(D1). [Google Scholar]
- Park JY, and Lee YN (1988), Solubility and decomposition kinetics of nitrous acid in aqueous solution, The Journal of Physical Chemistry, 92(22), 6294–6302. [Google Scholar]
- Perry KD, Cliff SS, and Jimenez-Cruz MP (2004), Evidence for hygroscopic mineral dust particles from the Intercontinental Transport and Chemical Transformation Experiment, Journal of Geophysical Research: Atmospheres, 109(D23). [Google Scholar]
- Pio CA, and Harrison RM (1987), Vapour pressure of ammonium chloride aerosol: Effect of temperature and humidity, Atmospheric Environment (1967), 21(12), 2711–2715. [Google Scholar]
- Pitzer KS, and Mayorga G (2002), Thermodynamics of electrolytes. II. Activity and osmotic coefficients for strong electrolytes with one or both ions univalent, The Journal of Physical Chemistry, 77(19), 2300–2308. [Google Scholar]
- Pollack IB, Ryerson TB, Trainer M, Neuman JA, Roberts JM, and Parrish DD (2013), Trends in ozone, its precursors, and related secondary oxidation products in Los Angeles, California: A synthesis of measurements from 1960 to 2010, Journal of Geophysical Research: Atmospheres, 118(11), 5893–5911. [Google Scholar]
- Pye HOT, et al. (2020), The acidity of atmospheric particles and clouds, Atmospheric Chemistry and Physics, 20(8), 4809–4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedel TP, et al. (2012), Nitryl chloride and molecular chlorine in the coastal marine boundary layer, Environ Sci Technol, 46(19), 10463–10470. [DOI] [PubMed] [Google Scholar]
- Riedel TP, et al. (2013), Chlorine activation within urban or power plant plumes: Vertically resolved ClNO2 and Cl2 measurements from a tall tower in a polluted continental setting, Journal of Geophysical Research: Atmospheres, 118(15), 8702–8715. [Google Scholar]
- Roberts JM, Osthoff HD, Brown SS, Ravishankara AR, Coffman D, Quinn P, and Bates T (2009), Laboratory studies of products of N2O5 uptake on Cl–containing substrates, Geophysical Research Letters, 36(20). [Google Scholar]
- Roberts JM, et al. (2010), Measurement of HONO, HNCO, and other inorganic acids by negative-ion proton-transfer chemical-ionization mass spectrometry (NI-PT-CIMS): application to biomass burning emissions, Atmospheric Measurement Techniques, 3(4), 981–990. [Google Scholar]
- Royer HM, Mitroo D, Hayes SM, Haas SM, Pratt KA, Blackwelder PL, Gill TE, and Gaston CJ (2021), The Role of Hydrates, Competing Chemical Constituents, and Surface Composition on ClNO2 Formation, Environ Sci Technol, 55(5), 2869–2877. [DOI] [PubMed] [Google Scholar]
- Rubio MA, Lissi E, Villena G, Elshorbany YF, Kleffmann J, Kurtenbach R, and Wiesen P (2009), Simultaneous measurements of formaldehyde and nitrous acid in dews and gas phase in the atmosphere of Santiago, Chile, Atmospheric Environment, 43(38), 6106–6109. [Google Scholar]
- Ryerson TB, et al. (2013), The 2010 California Research at the Nexus of air quality and climate change (CalNex) field study, Journal of Geophysical Research: Atmospheres, 118(11), 5830–5866. [Google Scholar]
- Sander R (2015), Compilation of Henry’s law constants (version 4.0) for water as solvent, 15(8), 4399–4981. [Google Scholar]
- Seinfeld JH, and Pandis SN (2006), Atmospheric chemistry and physics: from air pollution to climate change, edited, John Wiley & Sons, Inc., New Jersey, USA. [Google Scholar]
- Shon Z-H, Kim K-H, Song S-K, Jung K, Kim N-J, and Lee J-B (2012), Relationship between water-soluble ions in PM2.5 and their precursor gases in Seoul megacity, Atmospheric Environment, 59, 540–550. [Google Scholar]
- Staples BR (1981), Activity and osmotic coefficients of aqueous alkali metal nitrites, Journal of Physical and Chemical Reference Data, 10(3), 765–778. [Google Scholar]
- Staudt S, Gord JR, Karimova NV, McDuffie EE, Brown SS, Gerber RB, Nathanson GM, and Bertram TH (2019), Sulfate and Carboxylate Suppress the Formation of ClNO2 at Atmospheric Interfaces, ACS Earth and Space Chemistry, 3(9), 1987–1997. [Google Scholar]
- Sudheer AK, and Rengarajan R (2015), Time-resolved inorganic chemical composition of fine aerosol and associated precursor gases over an urban environment in western India: Gas-aerosol equilibrium characteristics, Atmospheric Environment, 109, 217–227. [Google Scholar]
- Sullivan RC, Guazzotti SA, Sodeman DA, and Prather KA (2007), Direct observations of the atmospheric processing of Asian mineral dust, Atmospheric Chemistry and Physics, 7(5), 1213–1236. [Google Scholar]
- Tang IN, and Munkelwitz HR (1993), Composition and temperature dependence of the deliquescence properties of hygroscopic aerosols, Atmospheric Environment. Part A. General Topics, 27(4), 467–473. [Google Scholar]
- Tham YJ, Yan C, Xue L, Zha Q, Wang X, and Wang T (2013), Presence of high nitryl chloride in Asian coastal environment and its impact on atmospheric photochemistry, Chinese Science Bulletin, 59(4), 356–359. [Google Scholar]
- Thornton JA, et al. (2010), A large atomic chlorine source inferred from mid-continental reactive nitrogen chemistry, Nature, 464(7286), 271–274. [DOI] [PubMed] [Google Scholar]
- Trebs I (2005), The NH4+-NO3−-Cl−-SO42−-H2O aerosol system and its gas phase precursors at a pasture site in the Amazon Basin: How relevant are mineral cations and soluble organic acids?, Journal of Geophysical Research, 110(D7). [Google Scholar]
- VandenBoer TC, et al. (2014), Evidence for a nitrous acid (HONO) reservoir at the ground surface in Bakersfield, CA, during CalNex 2010, Journal of Geophysical Research: Atmospheres, 119(14), 9093–9106. [Google Scholar]
- Veres P, Roberts JM, Warneke C, Welsh-Bon D, Zahniser M, Herndon S, Fall R, and de Gouw J (2008), Development of negative-ion proton-transfer chemical-ionization mass spectrometry (NI-PT-CIMS) for the measurement of gas-phase organic acids in the atmosphere, International Journal of Mass Spectrometry, 274(1-3), 48–55. [Google Scholar]
- Wang X, et al. (2019), The role of chlorine in global tropospheric chemistry, Atmospheric Chemistry and Physics, 19(6), 3981–4003. [Google Scholar]
- Warneke C, de Gouw JA, Holloway JS, Peischl J, Ryerson TB, Atlas E, Blake D, Trainer M, and Parrish DD (2012), Multiyear trends in volatile organic compounds in Los Angeles, California: Five decades of decreasing emissions, Journal of Geophysical Research: Atmospheres, 117(D21). [Google Scholar]
- Washenfelder RA, et al. (2011), The glyoxal budget and its contribution to organic aerosol for Los Angeles, California, during CalNex 2010, Journal of Geophysical Research: Atmospheres, 116(D21). [Google Scholar]
- Weber RJ, Orsini D, Daun Y, Lee YN, Klotz PJ, and Brechtel F (2001), A Particle-into-Liquid Collector for rapid measurement of aerosol bulk chemical composition, Aerosol Science and Technology, 35(3), 718–727. [Google Scholar]
- Wegner T, Grooß JU, von Hobe M, Stroh F, Sumińska-Ebersoldt O, Volk CM, Hösen E, Mitev V, Shur G, and Müller R (2012), Heterogeneous chlorine activation on stratospheric aerosols and clouds in the Arctic polar vortex, Atmospheric Chemistry and Physics, 12(22), 11095–11106. [Google Scholar]
- Wexler AS (2002), Atmospheric aerosol models for systems including the ions H+, NH4+, Na+, SO42−, NO3−, Cl−, Br−, and H2O, Journal of Geophysical Research, 107(D14). [Google Scholar]
- Xia M, et al. (2021), Winter ClNO2 formation in the region of fresh anthropogenic emissions: seasonal variability and insights into daytime peaks in northern China, Atmospheric Chemistry and Physics, 21(20), 15985–16000. [Google Scholar]
- Young AH, Keene WC, Pszenny AAP, Sander R, Thornton JA, Riedel TP, and Maben JR (2013), Phase partitioning of soluble trace gases with size-resolved aerosols in near-surface continental air over northern Colorado, USA, during winter, Journal of Geophysical Research: Atmospheres, 118(16), 9414–9427. [Google Scholar]
- Young CJ, et al. (2012), Vertically resolved measurements of nighttime radical reservoirs in Los Angeles and their contribution to the urban radical budget, Environ Sci Technol, 46(20), 10965–10973. [DOI] [PubMed] [Google Scholar]
- Young CJ, et al. (2014), Chlorine as a primary radical: evaluation of methods to understand its role in initiation of oxidative cycles, Atmospheric Chemistry and Physics, 14(7), 3427–3440. [Google Scholar]
- Zheng G, Su H, Wang S, Andreae MO, Poschl U, and Cheng Y (2020), Multiphase buffer theory explains contrasts in atmospheric aerosol acidity, Science, 369(6509), 1374–1377. [DOI] [PubMed] [Google Scholar]
- Zhou W, et al. (2018), Production of N2O5 and ClNO2 in summer in urban Beijing, China, Atmospheric Chemistry and Physics, 18(16), 11581–11597. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.