

Abstract
In our recent publication, we show for the first time that the fungal pathogen Cryptococcus neoformans is able to manipulate host cells by producing eicosanoids that mimic those found in the host. Using complementary in vivo zebrafish and in vitro macrophage cell culture models of Cryptococcus infection, we found that these eicosanoids manipulate host innate immune cells by activating the host receptor PPAR-gamma which is an important regulator of macrophage inflammatory phenotypes. We initially identified PGE2 as the eicosanoid species responsible for this effect; however, we later found that a derivative of PGE2—15-keto-PGE2—was ultimately responsible and that this eicosanoid acted as a partial agonist to PPAR-gamma. In this commentary, we will discuss some of the concepts and conclusions in our original publication and expand on their implications and future directions.
Keywords: Cryptococcus neoformans, fungal infection, host pathogen interactions, macrophages, zebrafish, eicosanoids, 15-keto-PGE2, PPAR-gamma
Comment on: Evans RJ, Pline K, Loynes CA, Needs S, Aldrovandi M, Tiefenbach J, Bielska E, Rubino RE, Nicol CJ, May RC, Krause HM, O’Donnell VB, Renshaw SA, Johnston SA. 15-keto-prostaglandin E2 activates host peroxisome proliferator-activated receptor gamma (PPAR-γ) to promote Cryptococcus neoformans growth during infection. PLoS Pathog. 2019 Mar;15(3):e1007597. doi: 10.1371/journal.ppat.1007597. eCollection 2019 Mar. PubMed PMID: 30921435; PubMed Central PMCID: PMC6438442.
Cryptococcus neoformans is a pathogenic fungus that is ubiquitous in our everyday environment, but only those with severe or unusual immune deficiencies, such as HIV AIDS, develop serious disease. 1 During infection Cryptococcus forms a close interaction with host macrophages—after phagocytosis by macrophages Cryptococcus is able survive and replicate within the phagosome, subverting macrophage function and turning the macrophage into a niche for the establishment of infection. 2 To kill Cryptococcus, macrophages must be activated by a Th1 CD4+ T-cell-mediated adaptive immune response (hence the prevalence in HIV AIDS patients) 3 ; failure to control intracellular infection can lead to dissemination from the lungs into the central nervous system and the development of fatal fungal meningitis.
Cryptococcus can influence the activation state of infected macrophages, shifting them from protective Th1 activation states to a nonprotective Th2 state although the biological mechanisms behind this are unclear. 4 Our hypothesis for this study was that this manipulation might be mediated by eicosanoid species produced by the fungus. 5 Cryptococcus can produce a number of eicosanoid species which closely resemble those found in the host but natural purpose of these lipids normally associated with cell to cell signaling in multicellular organisms is unknown. 6 Macrophages and other innate immune cells are highly responsive to eicosanoid species such as prostaglandins and leukotrienes so we reasoned that eicosanoids produced by the fungus during intracellular infection could interfere with normal host signaling pathways.
Quantifying Eicosanoids During Cryptococcus Infection and Determining Their Source
Very little is known about eicosanoid synthesis pathways in Cryptococcus; only two Cryptococcus enzymes—phospholipase B1 (PLB1) and laccase (LAC1)—have been linked to eicosanoid synthesis in the fungus.7,8 The lack of homologs to eicosanoid synthesis enzymes found in higher organisms suggests that the pathway is distinct from anything previously described. Deletion mutants for PLB1 8 and LAC1 9 have been characterized in Cryptococcus. The PLB1 mutant (Δplb1) shows a profound decrease in all eicosanoids produced by C neoformans suggesting this enzyme is central to eicosanoid synthesis—possibly fulfilling a role analogous to phospholipase A2 in mammalian cells. The LAC1 mutant (Δlac1) is deficient in only PGE2 and its derivative 15-keto-PGE2, suggesting this enzyme might fulfill a role analogous to prostaglandin E2 synthase in mammalian cells. Both of these strains were used in our study to differentiate between host- and pathogen-derived eicosanoids; to aid the study of these strains in our zebrafish model, we produced green fluorescent protein–tagged versions of each strain. Δplb1 is known to have a growth defect in macrophages, 10 whereas laccase activity has been found to positively correlate with increased mortality in patients with HIV-associated cryptococcosis 11 —although how much this is due to PGE2 synthesis as opposed to the role of laccase in the production of another cryptococcal virulence factor melanin. In our study, we were able to rescue the in vitro intracellular proliferation defect of Δplb1 with exogenous PGE2; we also found that both Δplb1 and Δlac1 had reduced in vivo growth in our zebrafish larvae cryptococcosis model; however, only Δplb1-infected fish responded to exogenous PGE2 or 15-keto-PGE2. 5 We attribute Δlac1’s unresponsiveness to exogenous prostaglandin treatment to the fact that laccase is also responsible for aforementioned melanin synthesis—thus, it is possible that for this strain both melanin and PGE2 are required for wild-type levels of growth—or an unknown defect that was responsible for it being much more attenuated in animal infection than the Δplb mutant. We attempted to circumvent this difference by disrupting the macrophage immune response but found that any immunocompromise of this response was critical to survival and confounded any differences. 12
A major challenge we faced in our study was measuring eicosanoid levels during host-pathogen interactions and determining whether the eicosanoids measured were host or pathogen derived. A previous study by Shen and Liu 13 found that pulmonary levels of PGE2 increased in mice infected with C neoformans; however, they were unable to attribute this to host or pathogen production. In our study, we performed experiments to measure differences in PGE2 content between wild-type H99 and Δplb1-infected macrophages using 2 different methods—ELISA (enzyme-linked immunosorbent assay) and LC MS/MS (liquid chromatography-tandem mass spectrometry). We found that J774 macrophages did produce detectable levels of PGE2; however, we did not see any significant difference between infected or uninfected macrophages or between H99, Δplb1 and Δplb1:PLB1-infected cells (although the concentrations detected with ELISA and LC MS/MS were very similar). 5 This suggested that Cryptococcus-derived eicosanoids present during infection were likely to be contained within the macrophage. Measurement of these small, localized eicosanoid levels within infected macrophages proved very difficult with current analytical methods—to our knowledge, intracellular levels of pathogen-derived eicosanoids have never been quantitatively measured before. To overcome this difficulty, we took a different approach; we used a co-infection assay which has previously been used to investigate the interaction of different Cryptococcus gattii strains within the same macrophage. 14 We predicted that the parental cryptococcal strain produced growth-promoting eicosanoids but Δplb1 could not; the Δplb1 strain should display improved intracellular replication when H99 is also present within the same macrophage. Indeed, we found that Δplb1 proliferated better when accompanied by 2 wild-type yeast cells in the same macrophage as opposed to when 2 Δplb1 yeast cells were accompanied by 1 wild-type yeast cell. These experiments confirmed to us that Cryptococcus produced eicosanoids during macrophage infection and suggested that they did remain contained within the macrophage—important because it indicated that any host receptor targeted was likely to be intracellular.
Identifying a Mechanism
Our initial experiments indicated that PGE2 was the eicosanoid species required for Cryptococcus growth because exogenous addition of this species was sufficient to rescue the growth defects of Δplb1 in J774 macrophages and zebrafish larvae. Intending to boost the observed effects of PGE2, we used a chemically altered version of PGE2 called 16,16 dimethyl PGE2 that cannot be metabolized. 15 To our surprise, the opposite outcome occurred—16,16-dimethyl PGE2 could no longer rescue the growth of Δplb1. Under physiological conditions, PGE2 can be further converted to 15-keto-PGE2 by the enzyme 15-hydroxy prostaglandin dehydrogenase (PGDH; Figure 1). 16 We assumed that conversion from PGE2 to 15-keto-PGE2 could be a way for the host to mitigate the effects of Cryptococcus-derived (or exogenously added) PGE2. This was a eureka moment for our study because we realized that conversion of PGE2 into 15-keto-PGE2 was actually required for the growth of Cryptococcus and that if host eicosanoid signaling was being affected it was through a 15-keto-PGE2 receptor rather than a PGE2 receptor.
Figure 1.
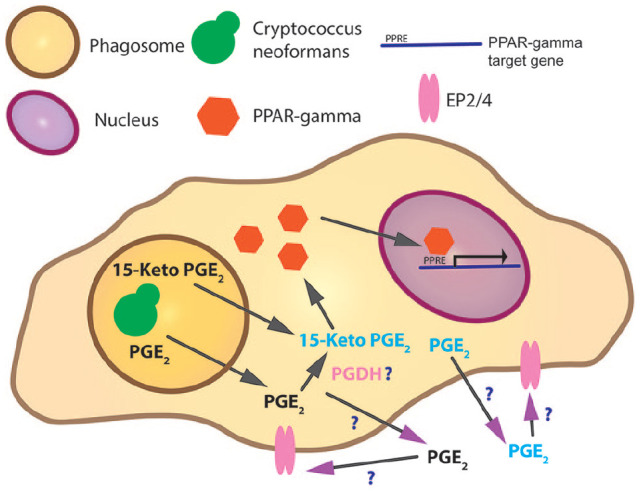
Evidenced and potential pathways of eicosanoid and PPAR-gamma interactions during Cryptococcus infection of macrophages.
Note. During infection, Cryptococcus resides within the phagosome. Prostaglandin E2 or 15-keto-PGE2 is produced by the fungus. Generation of 15-keto-PGE2 is either by Cryptococcus or the host—or perhaps both. 15-Keto PGE2 is a partial agonist to PPAR-gamma. PPAR-gamma is a cytosolic eicosanoid receptor; on ligand binding and activation, PPAR-gamma translocates to the nucleus where it binds to and activates genes with a PPRE target motif. In addition, prostaglandin E2 may bind its extracellular receptors EP2/4 on macrophages or other cells. PPAR = peroxisome proliferator–activated receptor; PPRE = peroxisome proliferator hormone response elements; PGDH = 15-hydroxy prostaglandin dehydrogenase.
Our experiments had shown that Cryptococcus-derived 15-keto-PGE2 promoted cryptococcal growth and that any host receptors involved were likely to be intracellular (Figure 1). While searching for putative receptors, we found that 15-keto-PGE2 had been reported to be an agonist for peroxisome proliferator–activated receptor gamma (PPARγ)—an intracellular receptor that is known to control inflammatory responses. PPARγ is a cytosolic receptor that has a variety of ligands including many lipid eicosanoids. Ligand binding leads to the formation of a heterodimer between PPARγ and retinoid X receptor (RXR). Following heterodimer formation, the PPARγ/RXR complex translocates to the nucleus and acts as a transcription factor controlling the expression of genes which possess a peroxisome proliferator hormone response element. 17
Through in vivo experiments with a transgenic zebrafish PPARγ reporter, we found that 15-keto-PGE2 was unable to activate the PPARγ reporter itself; however, when 15-keto-PGE2 was added in combination with a full PPARγ agonist troglitazone, the level of PPARγ activation was reduced compared with a troglitazone-only control. This indicated that 15-keto-PGE2 could interact with PPARγ in some capacity either as a partial agonist (a partial agonist is an agonist that binds to a receptor with a weak affinity and as a result does not fully activate the receptor) or an antagonist. To resolve this question, we proved that the effects of 15-keto-PGE2 were reversed by a known PPARγ antagonist. From these data, we concluded that 15-keto-PGE2 is a partial agonist to PPARγ, a finding that is supported by a previous study 18 (Figure 1). Interestingly, we settled on this conclusion through interpretation of our data and it was only afterward that we became aware of other partial agonists against PPARγ.19,20 The protein structure of PPARγ has evolved to provide different binding sites for full and partial agonists within the PPARγ ligand-binding domain (LBD)—full agonists bind to and stabilize the H12 alpha-helix of the LBD which produces a binding site for PPARγ transactivators. In contrast, partial agonists do not interact with the H12 alpha-helix and as a result do not provide stabilization of this region but binding still produces PPARγ activation to varying magnitudes. 21 Partial agonism is a mechanism that allows great flexibility in transcription factor function, rather than modulating the full gamut of PPARγ-controlled genes, a partial agonist will only activate a subset of these genes. This means a receptor like PPARγ can produce a variety of different transcriptional responses depending on the partial agonists present.
Future Perspectives
Where is PGE2 metabolized into 15-keto-PGE2 during infection? PGE2 is quickly metabolized into 15-keto-PGE2 in living cells (Figure 1). In higher organisms, this reaction is performed by PGDH. It is therefore possible that PGE2 produced by Cryptococcus is metabolized into 15-keto-PGE2 by the host. 15-keto-PGE2 has been detected in the supernatant of Cryptococcus cultures so it is also likely that Cryptococcus possesses an enzyme similar in function to PGDH.
What is the effect of PPARγ activation by 15-keto-PGE2 on host cells—specifically host macrophages? We have found that 15-keto-PGE2 is a partial agonist to PPARγ; this means that agonist binding only modulates a subset of PPARγ-controlled genes (Figure 1). Identifying this subset in host cells will be essential to understand how 15-keto-PGE2 enables Cryptococcus to cause infection.
Do other Cryptococcus-derived eicosanoids promote virulence? Our study has focused on PGE2/15-keto-PGE2 production by Cryptococcus. We also tested PGD2 but found this had no effect on infection. Cryptococcus produces many more eicosanoids which could have synergistic effects to PGE2/15-keto-PGE2 or completely different effects. In view of our findings, future studies in this area should also consider metabolites which could be produced from Cryptococcus eicosanoids within the host.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: R.J.E. was supported by a British Infection Association postdoctoral fellowship. S.A.J. was supported by Medical Research Council and Department for International Development Career Development Award Fellowship MR/J009156/1. S.A.J. was additionally supported by a Krebs Institute Fellowship, Medical Research Foundation grant R/140419 and Medical Research Council Center grant (G0700091).
ORCID iD: Robert J. Evans  https://orcid.org/0000-0003-0678-6510
https://orcid.org/0000-0003-0678-6510
References
- 1. Gibson JF, Johnston SA. Immunity to Cryptococcus neoformans and C. gattii during cryptococcosis. Fungal Genet Biol. 2015;78:76-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rudman J, Evans RJ, Johnston SA. Are macrophages the heroes or villains during cryptococcosis. Fungal Genet Biol. 2019;132:103261. [DOI] [PubMed] [Google Scholar]
- 3. Voelz K, Lammas DA, May RC. Cytokine signaling regulates the outcome of intracellular macrophage parasitism by Cryptococcus neoformans. Infect Immun. 2009;77:3450-3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wiesner DL, Specht CA, Lee CK, et al. Chitin recognition via chitotriosidase promotes pathologic type-2 helper T cell responses to cryptococcal infection. PLoS Pathog. 2015;11(3):e1004701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Evans RJ, Pline K, Loynes CA, et al. 15-keto-prostaglandin E2 activates host peroxisome proliferator-activated receptor gamma (PPAR-gamma) to promote Cryptococcus neoformans growth during infection. PLoS Pathog. 2019;15(3):e1007597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Noverr MC, Erb-Downward JR, Huffnagle GB. Production of eicosanoids and other oxylipins by pathogenic eukaryotic microbes. Clin Microbiol Rev. 2003;16(3):517-533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Erb-Downward JR, Noggle RM, Williamson PR, Huffnagle GB. The role of laccase in prostaglandin production by Crypto-coccus neoformans. Mol Microbiol. 2008;68(6):1428-1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cox GM, McDade HC, Chen SC, et al. Extracellular phospholipase activity is a virulence factor for Cryptococcus neoformans. Mol Microbiol. 2001;39(1):166-175. [DOI] [PubMed] [Google Scholar]
- 9. Zhu X, Williamson PR. Role of laccase in the biology and virulence of Cryptococcus neoformans. FEMS Yeast Res. 2004;5(1):1-10. [DOI] [PubMed] [Google Scholar]
- 10. Evans RJ, Li Z, Hughes WS, Djordjevic JT, Nielsen K, May RC. Cryptococcal phospholipase B1 is required for intracellular proliferation and control of titan cell morphology during macrophage infection. Infect Immun. 2015;83(4):1296-1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sabiiti W, Robertson E, Beale MA, et al. Efficient phagocytosis and laccase activity affect the outcome of HIV-associated cryptococcosis. J Clin Invest. 2014;124:2000-2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bojarczuk A, Miller KA, Hotham R, et al. Cryptococcus neoformans intracellular proliferation and capsule size determines early macrophage control of infection. Sci Rep. 2016;6:21489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shen L, Liu Y. Prostaglandin E2 blockade enhances the pulmonary anti-Cryptococcus neoformans immune reaction via the induction of TLR-4. Int Immunopharmacol. 2015;28(1):376-381. [DOI] [PubMed] [Google Scholar]
- 14. Voelz K, Johnston SA, Smith LM, Hall RA, Idnurm A, May RC. “Division of labour” in response to host oxidative burst drives a fatal Cryptococcus gattii outbreak. Nat Commun. 2014;5:5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ohno H, Morikawa Y, Hirata F. Studies on 15-hydroxyprostaglandin dehydrogenase with various prostaglandin analogues. J Biochem. 1978;84(6):1485-1494. [DOI] [PubMed] [Google Scholar]
- 16. Tai HH, Ensor CM, Tong M, Zhou H, Yan F. Prostaglandin catabolizing enzymes. Prostaglandins Other Lipid Mediat. 2002;68-69:483-493. [DOI] [PubMed] [Google Scholar]
- 17. Lemberger T, Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: a nuclear receptor signaling pathway in lipid physiology. Annu Rev Cell Dev Biol. 1996;12:335-363. [DOI] [PubMed] [Google Scholar]
- 18. Chou WL, Chuang LM, Chou CC, et al. Identification of a novel prostaglandin reductase reveals the involvement of prostaglandin E2 catabolism in regulation of peroxisome proliferator-activated receptor gamma activation. J Biol Chem. 2007;282(25):18162-18172. [DOI] [PubMed] [Google Scholar]
- 19. Atanasov AG, Wang JN, Gu SP, et al. Honokiol: a non-adipogenic PPARgamma agonist from nature. Biochim Biophys Acta. 2013;1830(10):4813-4819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bhalla K, Hwang BJ, Choi JH, et al. N-Acetylfarnesylcysteine is a novel class of peroxisome proliferator-activated receptor gamma ligand with partial and full agonist activity in vitro and in vivo. J Biol Chem. 2011;286(48):41626-41635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kroker AJ, Bruning JB. Review of the structural and dynamic mechanisms of PPARgamma partial agonism. PPAR Res. 2015;2015:816856. [DOI] [PMC free article] [PubMed] [Google Scholar]