

Abstract
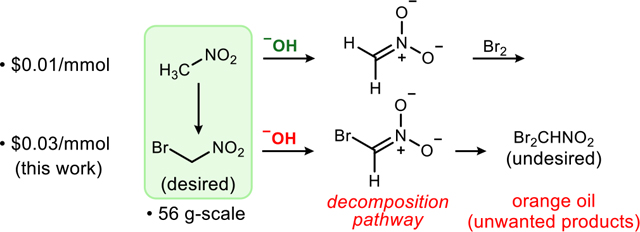
The accessibility of bromonitromethane has declined in recent years, limiting its viability as a reagent for chemical synthesis. Reinvestigation and optimization of a variety of preparations, and the development of safe operating principles, are described. The reproducible protocol described here leverages the effectiveness of hydroxide for nitromethane bromination, while respecting its incompatibility with the product it forms. This careful balance is achieved at scales up to 56 grams, resulting in a reproducible procedure that provides straightforward, sustainable, and affordable access to this critical reagent.
Graphical Abstract
INTRODUCTION:
Bromonitromethane is a versatile reagent used in the preparation of nitroheterocycles,1 nitrocyclopropanes,2 α-functionalized terminal nitroalkanes,3 and to a limited extent, 1,3-dipolar cycloaddition products.4 Advances in the synthesis of enantiopure amines, amides, and peptides have relied increasingly on the reagent,5,6 which was commercially available at reasonable cost for many years. More recently, our work leveraging bromonitromethane as a reagent7 was stymied by the need to contract with a supplier for amounts both smaller and more expensive (ranging USD $0.48 to >$6/mmol) than previously available. Nitromethane, however, remains readily available and stable in cost (less than USD $0.01/mmol). In this context, we turned to literature procedures describing the preparation of bromonitromethane from nitromethane. After some investment of time and effort, attempts to reproduce the most promising preparation were successful on a 13 to 56 -gram scale (Scheme 1). We detail here the key factors for reproducible production of bromonitromethane at laboratory scale. We also take the opportunity to highlight some safe operating procedures associated with the preparation and purification of reagents in this class. This robust, renewable, and inexpensive bromonitromethane preparation should alleviate any future supply obstacles, thereby stimulating further use of bromonitromethane in organic synthesis.
Scheme 1.

General outline of the optimized bromonitromethane synthesis from nitromethane.
A selection of bromonitromethane syntheses have been published, each varying significantly in the protocols described.8,9,10,11,12,13 Key differences include temperature (−15 °C to 35 °C), the addition rate of reagents, and the inclusion of sodium bromide in a substoichiometric amount. While all protocols report a yield between 60–90%, in our hands, we were unable to reproduce any single procedure consistently as described. A critical metric for success is the pre-distillation ratio of bromonitromethane to dibromonitromethane (and unconverted nitromethane); we sought high ratios that might eliminate the need for distillation for most applications. Procedures conducted in the −10 to −15 °C range led to freezing of the reaction mixture. This phase change prevented proper stirring, resulting in significant increases in dibrominated product. When operating at a higher temperature range, variable results were typical.
RESULTS AND DISCUSSION:
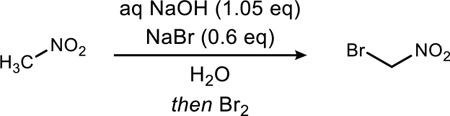
Most notable among the literature procedures replicated is a 1950’s patent12 in which extensive experimental detail is described. However, this procedure offers a wide acceptable temperature range (0 to 35 °C), as well as a bromine addition time of up to 2 minutes. Our attempts to replicate this procedure provided inconsistent results, particularly with varying ratios of mono-brominated to dibrominated product, despite careful control of addition rate and internal temperature (Table 1, entries 1–3). Inconsistency was further amplified when attempting to scale these bromination reactions (Table 1, entries 4–7). These collective experiences led us to refine the most promising method into a robust, operationally detailed procedure. This includes modifications and clarifications to the hydroxide/elemental bromine approach additions.
Table 1.
Optimization of bromonitromethane preparation at decagram scale.a
| ||||
|---|---|---|---|---|
| entry | scale (g)b | NaBr | BrCH2NO2:Br2CHNO2 | yield (%)c (isolated) |
| 1 | 13 | – | 1.5:1 | – |
| 2 | 13 | – | 20:1 | – |
| 3 | 14 | – | 1.5:1 | – |
| 4 | 14 | – | 10.5 ± 0.7 : 1 (2) | 66d |
| 5 | 28 | – | 11.5 ± 4.3 : 1 (5) | 66d |
| 6e | 28 | – | 30:1 | 78 |
| 7 | 56 | – | 2.75 ± 0.5 : 1 (4) | – |
| 8 | 13 | 60 mol % | 21.5 ± 2.1 : 1 (2) | – |
| 9 | 28 | 60 mol % | 21:1 | – |
| 10e | 56 | 60 mol % | 27:1 | 79 |
| 11f | 28 | 60 mol % | 4.5:1 | – |
| 12f | 28 | 60 mol % | 7:1 | – |
| 13g | 28 | 60 mol % | 22:1 | – |
| 14g | 28 | 60 mol % | 20:1 | – |
Conditions: Entries 1–8: 0 °C using traditional stirring, with bro-mine added as a single addition; concentration was 740 mM; for any reaction leading to product ratios lower than 5:1, the material was discarded. Crude percent recoveries were >80% by mass for all reactions. Ratios in entries 4–5 and 7–8 are shown as mean ± stand-ard deviation. Parentheses indicate the number of experiments av-eraged for each entry.
Theoretical yield (in grams) of bromonitro-methane.
Reported yield is a distilled yield unless stated otherwise (Caution!16).
The crude reaction mixtures from entries 4 and 5 were combined and distilled together, leading to a 66% yield of bromonitromethane (15:1).
The reaction used mechanical stirring apparatus (see Supporting Information Figure S1).
Entries 11–12: dropwise addition of bromine.
Entries 13–14: single addition of bromine.
We focused on the use of aqueous sodium hydroxide-based formation of the nitromethane nitronate, and bromination using elemental bromine and sodium bromide. These studies focused not only on the specific formation of bromonitromethane, but also on the minimization of dibromonitromethane formation. The latter is formed presumably by competitive bromonitromethane deprotonation and bromination. The presumption is that this might occur if the exposure of bromonitromethane to the basic conditions is significant, and competitive with its own formation.
We found that the presence of sodium bromide increased the reproducibility of the results (Table 1, entry 8). Few examples have employed sodium bromide as a substoichiometric additive. However, when exploring other bromination protocols in the literature, we encountered several reports detailing the use of an alkali metal tribromide to provide instantaneous and selective bromination. Furthermore, it has been demonstrated that the use of liquid Br2 in the presence of aqueous KBr produces potassium tribromide in situ,14 and overall provides better brominating efficiency and selectivity.15 We hypothesized that this may translate well to our bromination protocol, and therefore carefully tested the use of sodium bromide. Indeed, the addition of sodium bromide provided more consistent reaction outcomes with increasing scale (Table 1, entries 8–10). Presumably, Br2 and NaBr in aqueous conditions are forming sodium tribromide in situ, perhaps attenuating the bromonium electrophilicity to better match the nucleophilicity of the nitronate.
Another key feature is the bromine addition rate. We found that a rapid single addition of bromine was essential to high selectivity for the mono-brominated product. When the bromine was added dropwise, selectivity suffered greatly (Table 1, c.f. entries 11–12 vs. 13–14). These results further support our hypothesis that the slow generation of bromonitromethane under basic conditions leads to the competitive dibrominated product. Therefore, a rapid addition favors selective bromonitromethane formation.
The internal reaction temperature was also evaluated (Table 2). Although the literature procedure states the reaction temperature can range from 0 °C to 35 °C,12 we found that lower temperatures consistently provide better selectivity for the mono-brominated product. When the reaction was conducted at 25 °C, the ratio of mono-brominated to dibrominated product decreased substantially (Table 2, entry 1). The temperature was systematically decreased to determine the ideal temperature range for high selectivity. As illustrated in Table 2, we found that temperatures starting below −5 °C are consistent, reproducible, and provide heightened selectivity for mono-brominated product in comparison to higher temperatures. These results hold true over multiple reaction scales (13-gram, 20-gram, 28-gram). Temperatures below −5 °C did not show any significant improvement, and instead were often detrimental to selectivity. It should be noted that on larger scales (above 28 grams) below −6 °C, the reaction mixture begins to freeze, resulting in poor stirring, and presumably slower incorporation of bromine throughout the mixture (Table 2, entries 11 and 12). This slower incorporation of bromine is likely similar to a dropwise addition approach, both resulting in significantly larger amounts of dibromonitromethane. This can be mitigated, however, with the use of a mechanical stirrer. If access to a mechanical stirrer is not feasible, several experiments can be conducted at 28 grams or less and pooled together for a larger scale distillation. Entries 12–13 in Table 2 demonstrate that high selectivity can be achieved at these lower temperatures so long as proper mixing is promoted. Ultimately, the temperature study demonstrated that as long as the reaction is started at −5 °C and does not spike higher than 20 °C, high selectivity for mono-brominated product is observed. This temperature spike (see max. temp, Table 2) occurs following the addition of bromine. It is important to note that all temperatures reported are measured internally to ensure consistency.
Table 2.
Correlation of temperature control to reproducibility and high selectivity for bromonitromethane.a
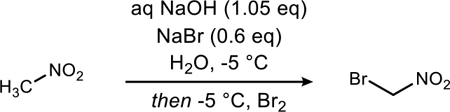
| ||||
|---|---|---|---|---|
| entry | scale (g)b | initial T (°C)c | maximum T (°C)d | BrCH2NO2:Br2CHNO2 |
| 1 | 13 | 25 | 43 | 3:1 |
| 2 | 28 | 0 | 14 | 20.5:1 |
| 3 | 28 | 0 | 12 | 15:1 |
| 4 | 13 | −5 | 19 | 20:1 |
| 5 | 28 | −5 | 5 | 16.5:1 |
| 6 | 28 | −6 | 10 | 20:1 |
| 7 | 28 | −7 | 13 | 22:1 |
| 8 | 28 | −8 | 3 | 20.5:1 |
| 9 | 13 | −10 | −5 | 41:1 |
| 10 | 13 | −10 | −7 | 41:1 |
| 11 | 13 | −10 | −7 | 23:1 |
| 12e | 20 | −15 | −10 | 20:1 |
| 13e | 20 | −15 | −10 | 24:1 |
Conditions: Traditional (magnetic) stirring unless noted otherwise, with bromine added as a single addition. Concentration was 740 mM. For any reaction lower than a 5:1 ratio, the material was discarded. Crude percent recoveries were >80% (mass) for all reactions.
Scale refers to the theoretical yield of bromonitromethane.
Initial temperature is the reaction temperature measured prior to bromine addition but after nitromethane addition (measured internally).
Maximum temperature is the maximum temperature measured either during or after bromine addition (measured internally).
Mechanical stirring used.
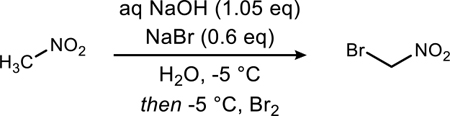
Following this sequence of optimization, we assessed the scalability of the final procedure (Table 3). For scales up to 28 grams, we found using the optimized conditions provides ratios greater than 20:1 (Table 3, entries 1–5). As the scale increased, it became substantially more important to ensure efficient mixing (Table 3, entries 4–5 versus 6–7). Below −5 °C, the solution can become slushy in consistency, but a mechanical stirrer ensures that fast addition of the elemental bromine is coupled with rapid mixing. In general, a mechanical stirrer is recommended at scales larger than 28 grams to achieve high selectivity (Table 3, entries 8–9). High yields, generally >70% (post distillation) were readily and reproducibly attained, (Table 3, entries 1, 3, 5, and 7–9). Percent recoveries of the crude mass were also consistently high (80–90%), allowing material to be pooled together for a larger scale distillation.16
Table 3.
Scaling of optimized procedure from 10 to 56 grams.a
| ||||
|---|---|---|---|---|
| entry | scale (g)b | BrCH2NO2:Br2CHNO2 | recovery (%)c | yield (%)c |
| 1 | 13 | 35.4 ± 9.8 : 1 (3) | 90.7 ± 1.5 | 77d |
| 2 | 13 | 21.5 ± 2.1 : 1 (2) | – | – |
| 3 | 26 | 40:1 | 83 | 77d |
| 4 | 28 | 20.8 ± 1.0 : 1 (3) | – | – |
| 5 | 28 | 32:1 | 80 | 73 |
| 6e | 28 | 15:1 | – | – |
| 7e | 28 | 16.5:1 | 84 | 81 |
| 8f | 56 | 18:1 | 88 | 68 |
| 9f | 56 | 27:1 | 81 | 79 |
Conditions: Temperature established at −5 °C with traditional (magnetic) stirring for entries 1–7, mechanical stirring for entries 8–9. Bromine was added as a single addition; concentration was 740 mM. Ratios in entries 1–2 and 4 are shown as mean ± standard deviation. Parentheses indicate the number of experiments averaged for each entry.
Scale refers to the theoretical yield of bromonitromethane.
Recovery is the % mass recovery of pre-distilled BrCH2NO2, and % yield is determined using distilled BrCH2NO2 (Caution!16).
The crude reaction mixtures from entries 1 and 3 were combined and distilled together, leading to a 77% yield of bromonitromethane (35:1).
Solution froze due to insufficient stirring (magnetic).
Mechanical stirring used.
Typical selectivity for the 28-gram scale reaction is greater than 20:1 favoring the mono-brominated product. Ultimately, we found that with the optimized procedures, the ratio of mono-brominated product was high enough that a purification via distillation was not necessary. However, if desired, the crude material can be further purified by vacuum distillation at 42 °C (5–18 Torr).16 Despite the large boiling point difference, enrichment of bromonitromethane was not easily achieved by fractional distillation in our experience.
The apparent contradiction that effective bromonitromethane formation uses a hydroxide base, and our discovery that bromonitromethane decomposes rapidly with hydroxide bases under phase transfer catalysis conditions,17 left us nonplussed. Consideration, if not resolution of this paradox was important to define safe operating procedures associated with bromonitromethane synthesis. Within the three dozen preparations executed for this study, at scales ranging from 66.8 mmol (10 g) to 374 mmol scale (56 g), only one showed evidence of a comparable decomposition. In that experiment (187 mmol scale), a viscous orange liquid was noted in the workup, after extraction. As we noted previously,17 when bromonitromethane is exposed to solid hydroxide base, an exothermic reaction occurs alongside deposition of an orange oil on the hydroxide solid. The success of the preparation detailed here may be due in part to the limited exposure of nascent bromonitromethane to hydroxide base. Furthermore, care during the workup/neutralization may be more important than usual.
We note an additional experience that may increase practice within the safe operating limits.18,19 In order to examine the behavior of α-bromonitronate in other reactions, the potassium salt of 1-bromo-1-nitropropane (2 mmol) was prepared in aqueous potassium hydroxide at ice-bath temperature, then cooled to −78 °C for lyophilization (Scheme 2, eq 1). Once dry, the solid was scraped and crushed into a free-flowing powder without incident in a glass flask. The solid was transferred to a screw-cap vial and moved to a drawer for temporary storage. Within minutes, and seemingly unprovoked, the vial shattered due to decomposition forces. This singular experience reaffirms that the isolation of desolvated nitronate salts should be avoided. This experience is reminiscent of reports that the dry sodium salt of nitromethane can react explosively with chlorine gas,20,21 and the 1,1,4,4-tetranitrobutane dinitronate (potassium) salt “occasionally exploded even while still wet”.22,23 A more rigorous analysis of a terminal nitroalkane’s process safety has been published,24 highlighting the generally wide safe operating parameters that align with our collective experience.25 Similarly, the nitration of the nitromethane sodium nitronate is reported to proceed without event in warm (50 °C) water.26 We have also repeated the formation of methazonic acid from the nitronate of nitromethane without event.27
Scheme 2.

Safety considerations for the preparation and use of bromonitroalkanes.
Overall, after careful exploration of temperature, addition rates, and additives, an optimal procedure for the synthesis of bromonitromethane has been developed that is consistently reproducible over a range of scales. Furthermore, this procedure yields material of high purity, such that further purification by distillation is optional for most applications.16 We detail a simple experimental setup that can be replicated in any lab setting using commonly available lab supplies. The resulting procedure provides straightforward and sustainable access to this important reagent at an affordable cost.
Supplementary Material
ACKNOWLEDGMENTS
Research reported in this publication was supported by the National Institute of General Medical Sciences and the National Heart, Lung, and Blood Institute of the National Institutes of Health (GM 063557, HL151223, HL151125 (F31 support for A.N.S.)).
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
SUPPORTING INFORMATION
The Supporting Information is available free of charge on the ACS Publications website.
full experimental details, pictures of the experimental set-up, and representative NMR spectra (PDF)
Contributor Information
Madelaine P. Thorpe, Department of Chemistry & Vanderbilt Institute of Chemical Biology, Vanderbilt University, Nashville, TN 37235-1822, USA.
Abigail N. Smith, Department of Chemistry & Vanderbilt Institute of Chemical Biology, Vanderbilt University, Nashville, TN 37235-1822, USA.
Michael S. Crocker, Department of Chemistry & Vanderbilt Institute of Chemical Biology, Vanderbilt University, Nashville, TN 37235-1822, USA.
Jeffrey N. Johnston, Department of Chemistry & Vanderbilt Institute of Chemical Biology, Vanderbilt University, Nashville, TN 37235-1822, USA.
REFERENCES
- 1.John J; Thomas J; Parekh N; Dehaen W Tandem Organocatalyzed Knoevenagel Condensation/1,3-Dipolar Cycloaddition towards Highly Functionalized Fused 1,2,3-Triazoles, Eur. J. Org. Chem. 2015, 2015, 4922. [Google Scholar]
- 2.a) Hayashi Y; Yamazaki T; Nakanishi Y; Ono T; Taniguchi T; Monde K; Uchimaru T Asymmetric Nitrocyclopropanation of α-Substituted α,β-Enals Catalyzed by Diphenylprolinol Silyl Ether for the Construction of All-Carbon Quaternary Stereogenic Centers, Eur. J. Org. Chem. 2015, 2015, 5747. [Google Scholar]; b) Zhao B-L; Du D-M Chiral Squaramide-Catalyzed Michael/Alkylation Cascade Reaction for the Asymmetric Synthesis of Nitro-Spirocyclopropanes, Eur. J. Org. Chem. 2015, 2015, 5350. [Google Scholar]
- 3.a) Concellón JM; Rodríguez-Solla H; Concellón C; García-Granda S; Díaz MR Efficient Addition Reaction of Bromonitromethane to Aldehydes Catalyzed by NaI: A New Route to 1-Bromo-1-nitroalkan-2-ols under Very Mild Conditions, Org. Lett. 2006, 8, 5979. [DOI] [PubMed] [Google Scholar]; b) Schwieter KE; Johnston JN Enantioselective Addition of Bromonitromethane to Aliphatic N-Boc Aldimines Using a Homogeneous Bifunctional Chiral Organocatalyst, ACS Catal. 2015, 5, 6559. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Victoria T Lim SVT, Stephens Amanda B., and Jeffrey N. Johnston Enantioselective Synthesis of α-Bromonitroalkanes for Umpolung Amide Synthesis: Preparation of tert-Butyl ((1R)-1-(4-(benzyloxy)phenyl)-2-bromo-2-nitroethyl)carbamate, Org. Synth. 2016, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hwu JR; Sambaiah T; Chakraborty SK New transformations of 2-nitro-2,3-dihydrofurans to multi-functionalized dihydrofurans, Tetrahedron Lett. 2003, 44, 3167. [Google Scholar]
- 5.a) Zhang J.-m. Bromonitromethane: A Versatile Reagent in Organic Synthesis, Synlett 2009, 2009, 1692. [Google Scholar]; b) Inokuma T; Takemoto Y; Tsukanov SV; Johnston JN; Vishe M; Johnston JN In Encyclopedia of Reagents for Organic Synthesis 2015, p 1. [Google Scholar]
- 6.a) Fishwick BR; Rowles DK; Stirling CJM Bromonitromethane - a Versatile Electrophile, J. Chem. Soc., Perkin Trans. 1 1986, 1171. [Google Scholar]; b) Fishwick BR; Rowles DK; Stirling CJM Bromonitromethane, a Versatile Electrophile - Reactions with Thiolates, J. Chem. Soc., Chem. Commun. 1983, 834. [Google Scholar]; c) Fishwick BR; Rowles DK; Stirling CJM Bromonitromethane, a Versatile Electrophile - Reactions with Feebly Basic Nucleophiles, J. Chem. Soc., Chem. Commun. 1983, 835. [Google Scholar]
- 7.Nugent BM; Yoder RA; Johnston JN Chiral proton catalysis: A catalytic enantioselective direct aza-Henry reaction, J. Am. Chem. Soc. 2004, 126, 3418. [DOI] [PubMed] [Google Scholar]
- 8.Dotsenko VV; Krivokolysko SG Reaction of 3-Aryl-2-Cyanoprop-2-ene- Thioamides With Bromonitromethane: A new Method for the Synthesis of Functionalized 1,2,4-Thiadiazoles, Chem. Heterocycl. Compd. (Hoboken, NJ, U. S.) 2014, 50, 557. [Google Scholar]
- 9.Fishwick BR; Rowles DK; Stirling CJM Bromonitromethane - a Versatile Electrophile, J. Chem. Soc., Perkin Trans. 1 1986, 1171. [Google Scholar]
- 10.Ling J; Laugeois M; Michelet V; Ratovelomanana-Vidal V; Vitale MR Palladium(0)-Catalyzed Dearomatization of 2-Nitrobenzofurans through Formal (3+2) Cycloadditions with Vinylcyclopropanes: A Straightforward Access to Cyclopenta[b]benzofurans, Synlett 2018, 29, 928. [Google Scholar]
- 11.Mahasneh AS Tin(II) Chloride Mediated Addition Reaction of Bromonitromethane to Aldehydes, Z. Naturforsch., B: Chem. Sci. 2005, 60, 416. [Google Scholar]
- 12.Slagh HR Production of monobromonitromethane. US2632776A, March 3, 1953. [Google Scholar]
- 13.Nocito V; Bedell LJ; Levinson MI Method of preparing halogenated nitroalcohols. US4922030A, May 1, 1990. [Google Scholar]
- 14.Bellucci G; Bianchini R; Vecchiani S Comparison of molecular bromine and tribromide ion as brominating reagents. 2. Kinetic and product investigation of the bromination of 3-substituted cyclohexenes, J. Org. Chem. 1986, 51, 4224. [Google Scholar]
- 15.Kumar L; Sharma V; Mahajan T; Agarwal DD Instantaneous, Facile and Selective Synthesis of Tetrabromobisphenol A using Potassium Tribromide: An Efficient and Renewable Brominating Agent, Org. Process Res. Dev. 2010, 14, 174. [Google Scholar]
- 16. Caution! The distillation of flammable liquids, including nitroalkanes, requires additional precautions: Coetzee JF; Chang T-H Recommended methods for the purification of solvents and tests for impurities: nitromethane, Pure Appl. Chem. 1986, 58, 1541. Consult the latest available information in addition to descriptions provided here (see Supporting Information). Undistilled bromonitromethane prepared by this procedure is typically used in our applications, with results indistinguishable from the use of distilled material.
- 17.Schwieter KE; Johnston JN Enantioselective synthesis of D-α-amino amides from aliphatic aldehydes, Chem. Sci. 2015, 6, 2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.a) Product Safety Assessment Nitromethane. [Online Early Access]. Published Online: 2014. https://businessdocbox.com/Green_Solutions/76290845-Product-safety-assessment-nitromethane.html (accessed November 11, 2021).; b) Material Safety Data Sheet Bromonitromethane. [Online Early Access]. https://fscimage.fishersci.com/msds/98039.htm (accessed 11/11/2021).
- 19.Markofsky SB In Ullmann’s Encyclopedia of Industrial Chemistry 2011. [Google Scholar]
- 20.Stirling CJM An Explosive Mixture, Chemi. Brit. 1986, 22, 524. [Google Scholar]
- 21.Pigou PE; Stirling CJM Pathways in the Reactions of Nitronate Ions with Sulfonyl Halides, J. Chem. Soc., Perkin Trans. 2 1988, 725. [Google Scholar]
- 22.Feuer H; Colwell CE; Leston G; Nielsen AT Synthesis of α,α,ι,ι-Tetranitroalkanes1, J. Org. Chem. 1962, 27, 3598. [Google Scholar]
- 23.For general comments about polynitro compounds, see: Noble P; Borgardt FG; Reed WL Chemistry of the Aliphatic Polynitro Compounds and Their Derivatives, Chem. Rev. 1964, 64, 19. [Google Scholar]
- 24.Tsukanov SV; Johnson MD; May SA; Rosemeyer M; Watkins MA; Kolis SP; Yates MH; Johnston JN Development of an Intermittent-Flow Enantioselective Aza-Henry Reaction Using an Arylnitromethane and Homogeneous Brønsted Acid–Base Catalyst with Recycle, Org. Process Res. Dev. 2016, 20, 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.See also: Nguyen NV; Baum K Preparation of 1,2-Dibromodinitroethylene and 1,1-Dibromodinitroethylene, Tetrahedron Lett. 1992, 33, 2949. [Google Scholar]
- 26.Matthews V; Kubler D Notes- Improved Synthesis of Salts and Esters of Nitroacetic Acid, J. Org. Chem. 1960, 25, 266. [Google Scholar]
- 27.Lecco MT Ueber die Methazonsäure, Berichte der deutschen chemischen Gesellschaft 1876, 9, 705. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.