

Abstract
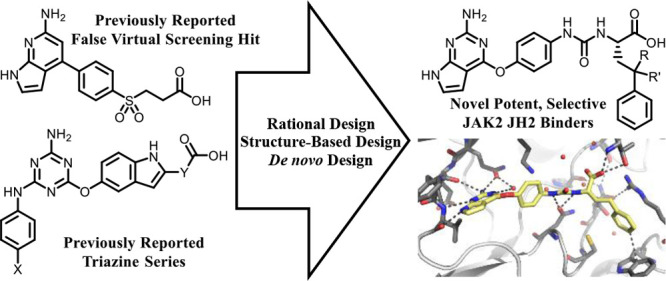
The Janus kinase 2 (JAK2) pseudokinase domain (JH2) is an ATP-binding domain that regulates the activity of the catalytic tyrosine kinase domain (JH1). Dysregulation of JAK2 JH1 signaling caused by the V617F mutation in JH2 is implicated in various myeloproliferative neoplasms. To explore if JAK2 activity can be modulated by a small molecule binding to the ATP site in JH2, we have developed several ligand series aimed at selectively targeting the JAK2 JH2 domain. We report here the evolution of a false virtual screen hit into a new JAK2 JH2 series. Optimization guided by computational modeling has yielded analogues with nanomolar affinity for the JAK2 JH2 domain and >100-fold selectivity for the JH2 domain over the JH1 domain. A crystal structure for one of the potent compounds bound to JAK2 JH2 clarifies the origins of the strong binding and selectivity. The compounds expand the platform for seeking molecules to regulate JAK2 signaling, including V617F JAK2 hyperactivation.
Keywords: JAK2 kinase, Pseudokinase domain, JH2/JH1 selectivity, Kinase inhibitors, Lead optimization
Janus Kinase 2 (JAK2) is a non-receptor tyrosine kinase necessary for the production of blood cells.1,2 Cytokine binding to an associated receptor initiates a signaling cascade through the kinase, resulting in phosphorylation of the signal transducer and activator of transcription (STAT) to induce hematopoiesis.1,3,4 JAKs acquire their namesake because they possess two conjugated ATP binding domains. The tyrosine kinase domain (JH1) is responsible for STAT phosphorylation,1 whereas the pseudokinase (JH2) domain regulates the JH1 catalytic activity.5−7 The V617F mutation in the JAK2 JH2 domain enables constitutive activation of the JAK-STAT pathway8 and is involved in various myeloproliferative disorders, including polycythemia vera, essential thrombocythemia, myelofibrosis,8−10 and some chronic myelomonocytic leukemias.11
Current drugs that target JAK2 tend to overcorrect for these disorders, leading to various forms of anemia.12−16 This is likely due to on-target toxicity through their inhibition of JAK2 JH1 activity, causing suppression of the JAK2 signaling pathway. One proposed strategy to circumvent this problem is to modulate the JAK-STAT signaling cascade by targeting the JAK2 JH2 domain; mutations around the JAK2 JH2 binding pocket that eliminate ATP binding have been shown to attenuate the effects of the V617F mutant without complete cessation of JAK2 signaling.17 The V617F mutant alters the conformation of residues Phe594 and Phe595,18,19 which could be modulated by a small molecule binding in the proximal ATP binding pocket of JAK2 JH2.18,20
The first step is to discover molecules that selectively and tenaciously bind to the ATP site in JAK2 JH2. We previously published several series that bind to JAK2 JH1 and JH2, including two series that show selectivity for JH2.21−25 Structural elements that promote binding to the JAK2 JH2 domain have been identified (Figure 1). The primary pharmacophore is a hydrogen-bonding donor–acceptor–donor motif (Figure 1, magenta), manifested as a diaminotriazole22 or a diaminotriazine25 scaffold that participates in hydrogen bonding to the hinge region. These heterocycles are connected to a phenyl or indole motif (Figure 1, green) and engage in cation−π interactions with Lys581.26 The aromatics are further linked to a carboxylic acid (Figure 1, blue) that forms hydrogen bonds with nearby residues and in many cases forms a salt-bridge with Arg715 which is not conserved in the JAK2 JH1 domain. Various structural elements were utilized as linkers of the aromatics to the carboxylic acid, which minimally affected binding to the JH2 domain (Figure 1, blue spheres). Finally, the triazines and triazoles possess an aryl sulfonamide or aryl nitrile moiety (Figure 1, red) that extends into solvent.
Figure 1.
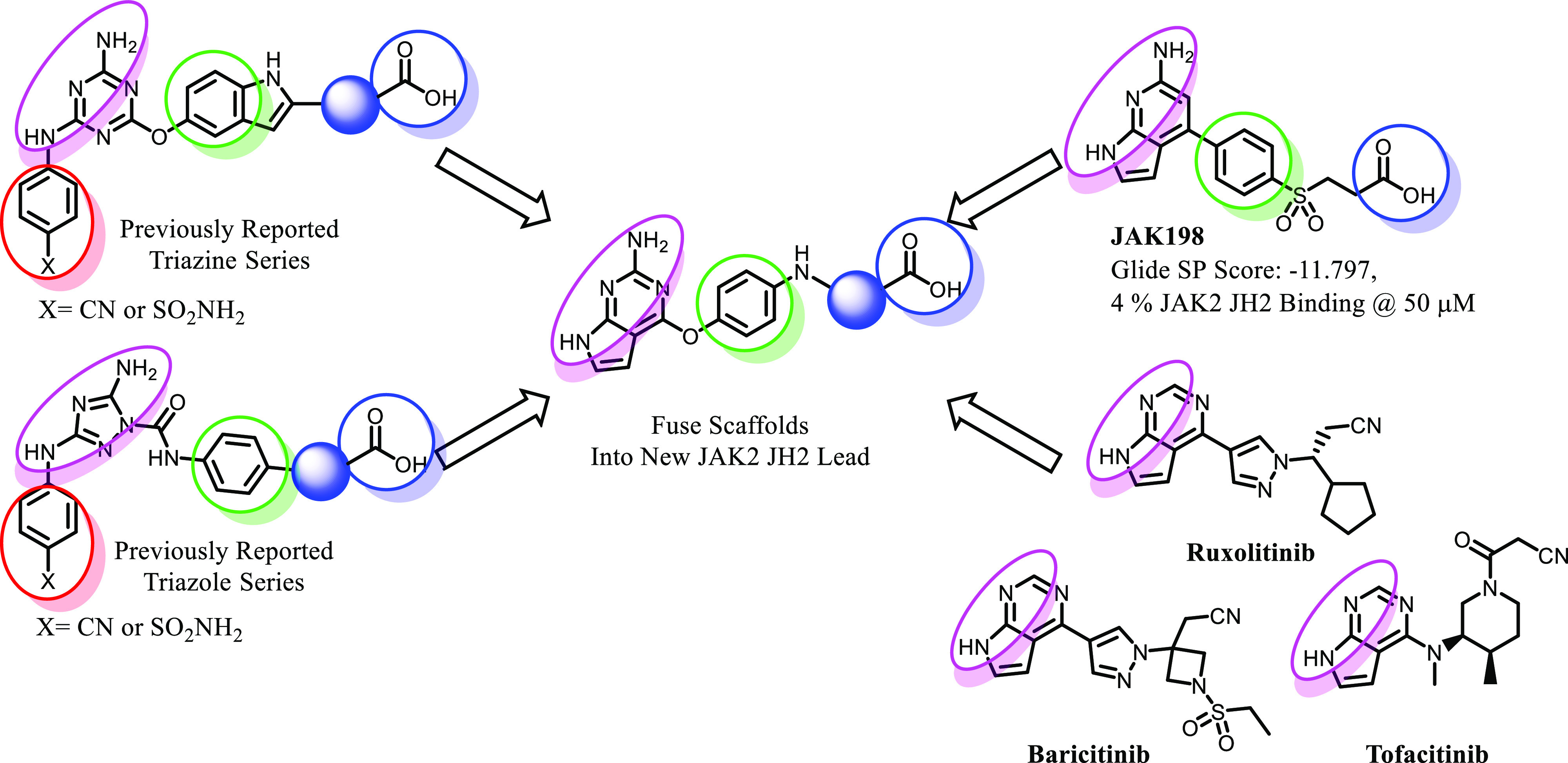
Structural elements that promote binding to the JAK2 JH2 domain. Shown are a donor–acceptor–donor motif (magenta), an aromatic pharmacophore (green) attached to a terminal carboxylate (blue) through a variable linker (blue sphere), and a solvent-exposed aryl nitrile or sulfonamide moiety (red). These structural elements were adopted from our previously reported triazine and triazole series and used to reconfigure the false virtual screen hit JAK198 with further inspiration from several JAK2 drugs.
With this guidance, we have reconsidered previously reported compounds from a virtual screen, in particular, the highest scoring analogue JAK198 (Figure 1).23 Though it and analogues were predicted to bind strongly to JAK2 JH2 by docking with Glide,27−29 they exhibited little or no binding to JAK2 JH2 when tested using a fluorescence polarization (FP) assay.23JAK198 possesses three of the four pharmacophore elements described above: the hydrogen-bond donor–acceptor–donor motif (Figure 1, magenta), the phenyl ring (Figure 1, green), and the terminal carboxylic acid (Figure 1, blue). The aryl nitrile or sulfonamide moiety is absent in JAK198 and could explain, in part, the poor binding. However, the JAK2 inhibitors ruxolitinib, baricitinib, and tofacitinib each possess a related donor–acceptor motif within their pyrrolopyrimidine scaffolds (Figure 1, magenta). While these inhibitors act on the JH1 domain of JAK2, the pyrrolopyrimidine core should be viable for binding to the hinge region of both JH1 and JH2.30,31 Therefore, it was expected that a pyrrolopyrimidine could be incorporated into new JAK2 JH2 ligands.
We initially sought to modify JAK198 to better mimic the triazines described above. An aminopyrrolopyrimidine core was selected because it possessed the desired donor–acceptor–donor motif (Figure 1, magenta). This core could then be attached to a phenyl group via an ether linkage instead to better mimic the triazine series. Since the indole hydrogen is hydrogen-bonded to Asn678 in this series,25 we also sought to attach an amino group to the phenyl group (Figure 1, green). This resultant aniline could then provide a synthetic handle to connect the terminal carboxylic acid through various linkers, completing the redesign of JAK198 into a new series.
For the synthesis of the new compounds, we started from the pyrrolopyrimidine and built to the right (Scheme 1). Commercially available 4-chloro-7H-pyrrolo[2,3-d]pyrimidin-2-amine (1) was subjected to an SNAr reaction with 4-nitrophenol. Harsh conditions were required to facilitate this substitution reaction, including microwave irradiation at 100 °C for 1.5 h and the use of DABCO as a catalyst.32 Even under these conditions, the nitro group was required to activate the phenol in conjunction with potassium carbonate to deprotonate the phenol to enable conversion to product. Less acidic phenols required stronger bases, which likely also resulted in deprotonation of the pyrrolopyrimidine. This would produce a negative charge on the pyrrolopyrimidine and thus deactivate it to nucleophilic attack, explaining the poor conversion to product. Upon completion of the SNAr, intermediate 2 was subject to a Bechamp reduction to reduce the nitro group into aniline 3 using ammonium chloride as the proton source.
Scheme 1. Synthesis of Intermediate 3 and Subsequent Attachment of Carboxylic Acids to 3.

Aniline 3 was then used to synthesize analogues terminating in carboxylic acids through various linkers (Scheme 1). This includes sulfonamide 4, aimed at mimicking the sulfone motif of JAK198, made by treating 3 with the corresponding sulfonyl chloride followed by saponification using NaOH. A pair of amide analogues (5, 6) was also made by treating aniline 3 with cyclic anhydrides. Finally, two urea analogues (7, 8) were prepared by coupling 3 to unprotected glycine or β-alanine using CDI.
The binding affinities of compounds 4–8 to the wild-type (WT) JAK2 JH2 domain were then evaluated using a previously described fluorescence polarization assay21 (Table 1), and the non-selective JAK2 JH2 ligand JNJ7706621(25) was used as a positive control. Upon comparison to the previously reported analogue JAK198,23 the sulfonamide analogue 4 showed a binding of 18% at 50 μM. Replacement of the sulfonamide by an amide yielded significant improvement; analogue 5 has a Kd of 18.8 ± 0.2 μM, comparable to those of previously reported triazines.25 Curiously, the extension of the alkyl chain of 5 by a single methylene unit to give 6 significantly reduced the binding to JAK2 JH2. Furthermore, conversion of the amides 5 and 6 to the corresponding ureas 7 and 8 gave equivalent binding. Overall, the progress made by 5 and 7 illustrates that a few modest changes can transform a false hit from virtual screening into a novel series of JAK2 JH2 binders.
Table 1. Binding of Initial Pyrrolopyrimidines to WT JAK2 JH2a.

Binding affinities (Kd) using a fluorescence polarization (FP) assay21 were measured for analogues that exhibited greater than 50% binding at 50 μΜ, whereas weaker binders are shown only as% binding at 50 μΜ. Kd values are represented as averages of two assays in quadruplicate ± SEM.
From ref (25).
From ref (23).
Given these promising results, analogues 5 and 7 were used for further development. Since the urea-containing 7 incorporates a glycine residue, a simple diversification strategy was pursued by replacement of glycine with other amino acids. This strategy not only allowed access to a wide variety of commercially available substituents but also enabled direction of the substituents toward different regions of the JAK2 JH2 domain. These include a pocket adjacent to Phe595 and Phe594, which are implicated in V617F hyperactivation,18,19,33 or a groove defined by Asn673, Cys675, Arg715, and Trp718. Accessing either of these regions brings advantages. The former feature does not exist in known crystal structures of the JAK2 JH1 domain, as it is blocked by a salt-bridge between Lys882 and Glu898. In the latter case, residues Asn673, Cys675, and Arg715 in the JAK2 JH2 domain are replaced with Asp976, Ala978, and Pro1017 in JAK2 JH1. As such, accessing either of these features could improve binding affinity and selectivity for the JAK2 JH2 domain over the JH1 domain.
To gauge whether the added substituents could access these features, de novo design was employed using BOMB,34 generating d- and l-phenylalanyl and d- and l-homophenylalanyl analogues of compound 7. The predicted orientations of the substituents in the JAK2 JH2 domain are shown in Figure 2. Generally, the d-isomers placed the phenyl group toward Phe594, whereas the l-isomers did the opposite, placing the phenyl group toward Trp718. As such, there was motivation to pursue both enantiomers.
Figure 2.

Pyrrolopyrimidine (a) d- and (b) l-phenylalanine or (c) d- and (d) l-homophenylalanine analogues modeled via BOMB in the WT JAK2 JH2 domain starting from the JAK2 JH2/JAK67 crystal structure (PDB ID: 6XJK(26)). The d-isomers tended toward Phe594, whereas the l-isomers tended toward Trp718.
Following the modeling with BOMB, the four analogues were synthesized (Scheme 2). The chiral integrity of these amino acids was verified by attachment of the chiral resolving agents (R)- or (S)-1-phenylethan-1-amine to analogue 11 using HBTU. Differences in the NMR of the two synthesized diastereomers were compared, and no racemization was observed (see Supporting Information for further details). These analogues were thus tested for their binding to the JAK2 JH2 domain (Table 2). The results showed that analogues expressing the l-amino acid gave stronger binding compared to their d-analogues. The l-phenylalanine 9 had an improved binding affinity by a log unit, while the l-homophenylalanine 11 exhibited almost a two log unit improvement. Conversely, the d-phenylalanine 10 yielded only around a 2-fold improvement in binding affinity, whereas the d-homophenylalanine 12 had weaker affinity than the original glycyl analogue (7). These data indicate that the phenethyl group is an important addition for improving binding affinity and that there is a strong dependence on the stereochemistry of the attachment.
Scheme 2. Synthesis of Amino Acid Derivatives of Analogue 7.

Table 2. Binding of d- and l-Pyrrolopyrimidine Analogues to WT JAK2 JH2a.

To fully elucidate the binding mode of analogue 11, we obtained a crystal structure for its complex with JAK2 JH2 (Figure 3). It shows an extensive network of hydrogen-bonding, cation−π, and aryl–aryl interactions. As expected, the pyrrolopyrimidine subunit is hydrogen-bonded with the hinge region of the JAK2 JH2 domain (Figure 3A) in similar fashion to our previously reported triazine and triazole analogues.22,25 There is a tight cation−π interaction between the phenoxy ring and Lys581. Interestingly, the urea moiety, while initially installed to mimic the hydrogen bonding found with the planar indole, possesses a dihedral angle of 55.7° in the crystal, out-of-plane with the attached phenolic ether (Figure 3B); however, it is also hydrogen-bonded to Asn678 as for the indole.25 Importantly, the crystal structure confirmed that the amino-acid side chain of 11 is directed toward Trp718 (Figure 3B), as predicted with BOMB. The terminal phenyl group participates in a T-shaped aryl–aryl interaction with Trp718, and it is also parallel to Arg715, forming a second cation−π interaction. The distance between the ipso carbon of the phenyl group and Nε of Arg715 is 3.6 Å. These striking interactions explain the origin of the much-enhanced binding for 11 (Table 2). Addition of the phenethyl group also rearranges the side chain of Asn673 to form new hydrogen bonds with Pro700, Ile702, and Arg715. Two orientations of Asn673 appear in the crystal, reflecting some flexibility in this region.
Figure 3.

Crystal structure of 11 bound to the WT JAK2 JH2 domain (PDB ID: 7T1T, 2.08 Å resolution). Shown are (a) the entire ligand bound to the JAK2 JH2 domain, including interactions with key residues, and (b) the l-homophenylalanine urea moiety, with its orientation and interactions with binding-site residues.
To seek further gains in affinity, additional side chains were screened (Table 3). Extension of the phenyl group of 11 with a methylene (13) or an ether oxygen (14) yielded modest losses in affinity. Tying the phenyl group into biaryl motifs (analogues 15–17) yielded further losses in affinity, similar to that for phenylalanine analogue 9. This indicated that some flexibility is desired to properly position the phenyl group near Trp718. To further enrich the SAR, l-amino acids containing heteroatoms were also explored, namely the histidine (18), glutamine (19), and asparagine (20) alternatives. Glutamine derivative 19 showed activity similar to that of glycine (7), whereas deletion of a single methylene unit to produce the asparagine 20 yielded a 5-fold improvement in binding affinity. A possible explanation is that the side chain of glutamine 19 is extending out to solvent and is not interacting with the binding site, whereas interactions are enabled with the asparagine variant (20), possibly a hydrogen bond between Asn673 and the carbonyl oxygen of 20. To build on this observation, we created a hybrid analogue by adding the asparagine carbonyl group of 20 to homophenylalanine analogue 11. This yielded the best JAK2 JH2 binder, 21, which exhibited 96 nM affinity. The best previously reported result was 346 nM for a triazole analogue.22
Table 3. Binding Affinities (Kd) of Derivatives of Analogue 11 to WT JAK2 JH2a.

The potent analogues 11 and 21 were then evaluated for selectivity by also measuring their binding to the V617F mutant of JAK2 JH2 and the WT JH1 domain (Table 4). As expected, the binding affinities are similar for WT and V617F JAK2 JH2 domains, and these analogues show much weaker binding to JAK2 JH1, with Kd values of ∼35 μM. As such, the selectivities of these compounds for the JAK2 JH2 domain over the JH1 domain were greater than 100-fold. Indeed, analogue 21 is the most selective compound found to-date, with a 360-fold preference for binding the JAK2 JH2 domain over the JH1 domain.
Table 4. Binding Affinities (Kd) of Analogues 11 and 21 to WT JAK2 JH2, V617F JH2, and JH1a.

The selectivity of these analogues was also examined through docking with Glide SP.27 For the JAK2 JH2 WT and JH2 V617F domains, analogues docked and scored similarly for both, consistent with their similar binding affinities. The carboxylate of each compound formed a salt-bridge with Arg715 and/or a hydrogen bond with Thr555. Additionally, the terminal phenyl group was often found to form an aryl–aryl interaction with Trp718, as seen in Figure 3 for analogue 11. In contrast, analogues were found to score considerably worse in the JAK2 JH1 domain by >2.5 kcal/mol (Table S2). This difference in score can be attributed to the difference in binding poses between JAK2 JH2 and JH1 (Figure S8). If they had similar poses, the carboxylate on the ligands would be located near Asp976 and Asp994, which would be highly repulsive. Instead, the carboxylate of the ligand was predicted to either interact with Lys882 or Arg980, orienting the terminal phenyl away from Trp1020 (JAK2 JH2 equivalent Trp718).
Since analogues 11 and 21 were found to be potent and highly selective for the JAK2 JH2 domain, we proceeded with preliminary evaluation of the effects of these compounds on kinase activity in human erythroleukemia (HEL) cells. These cells express only the V617F mutant of JAK2 and, as such, constitutively phosphorylate STAT5. Since the JAK2 JH2 domain is intracellular, we also decided to synthesize and test methyl ester prodrugs of 11 and 21 (see SI) in case the carboxylic acids impeded cell permeability. The JAK2 JH2 affinity for both prodrugs was found to be 18% at 50 μM, indicating the importance of the carboxylic acid for binding. Upon treatment of HEL cells with analogues 11, 21, and their respective methyl esters, no inhibition of phosphorylation of STAT5 was observed via Western blot at concentrations up to 75–80 μM after 3-h incubation (Figure S9). Ruxolitinib was used as a positive control and did yield complete inhibition of STAT5 phosphorylation at 2 μM. Thus, the four compounds are not penetrating the cells, or their binding is insufficient to affect the constitutive activation of the V617F JAK2 in HEL cells.
Analogues 11, 21, and their corresponding methyl esters were thus assayed in preliminary PAMPA experiments to explore further if permeability is a problem with these compounds. The two carboxylic acids were found to be impermeable in these assays, and the methyl ester of analogue 21 was also impermeable, likely due to its poor solubility. The methyl ester of analogue 11 was found to have a permeability of 3.57 × 10–6 cm/s, a value between the medium- (3.08 × 10–6 cm/s) and high-permeability controls (4.53 × 10–6 cm/s) diclofenac and chloramphenicol, respectively. Preliminary LCMS of the cell extracts indicates that the methyl ester of 11 is hydrolyzed to 11, but that 11 is present in relatively minute quantities in the cell lysates as compared to the medium, indicating poor permeability. PAMPA does not appear to be a good predictor of cell permeability for this series, a phenomenon more clearly illustrated in our more developed triazole series.35 Permeability does appear to be a major contributing factor for poor cellular activity of the triazole series. Nevertheless, the present work sets the stage for additional advances by providing molecules that bind strongly and selectively to JAK2 JH2 and a structural basis for their activity.
Selective targeting of the JAK2 JH2 domain is of therapeutic interest due to its reported potential to circumvent negative side-effects associated with conventional targeting of the JAK2 JH1 domain.17 Given the structural similarity between these two domains, it is challenging to discover JAK2 JH2-selective ligands.22−25 Herein, a combination of structure-based and de novo design strategies was employed to transform a false hit from virtual screening into a new series of selective JAK2 JH2 binders. The convergent design approach was successful in yielding compounds with both greater potency and selectivity than previously reported.21−25,36
These improvements were facilitated without significantly affecting the molecular weight, and these compounds had reduced tPSA compared to our previously reported ligands (150–167 Å2 in this series vs 165–203 Å2 in our previous series). This can be attributed largely to the discovered phenethyl motif, which is more efficient at improving binding to the JAK2 JH2 domain over the JAK2 JH1 domain than the aryl nitrile or aryl sulfonamide of previous series. Indeed, the ligand efficiency (LE) improved as the side chain evolved. Analogue 7 has an LE of 0.252 kcal/mol-HAC, which was reduced to 0.237 kcal/mol-HAC with the phenylalanine analogue 9. Incorporation of one additional methylene unit to analogue 11 improved the LE to 0.263 kcal/mol-HAC, and the further addition of the oxygen in analogue 21 increased it further to 0.277 kcal/mol-HAC, representing a net improvement in LE over the course of this series. Though permeability does appear to remain an issue for these ligands, they represent a step in the right direction toward improving their physiochemical properties as our ligands develop. Further exploration of analogues is needed in search of compounds that may reduce the hyperactivation of the V617F JAK2 in cell culture.
Acknowledgments
This work was supported by the U.S. National Institutes of Health (GM32136). This research made use of the Chemical and Biophysical Instrumentation Center at Yale University (RRID: SCR_021738). The authors thank Dr. Brandon Mercado at the Chemical and Biophysical Instrumentation Center at Yale University for assistance with X-ray data collection.
Glossary
Abbreviations
- JAK
Janus kinase
- JH2
pseudokinase domain
- ATP
adenosine triphosphate
- JH1
kinase domain
- SAR
structure–activity relationship
- STAT
signal transducer and activator of transcription
- SNAr
nucleophilic aromatic substitution
- DABCO
1,4-diazabicyclo[2.2.2]octane
- DMF
N,N-dimethylformamide
- CDI
N,N′-carbonyldiimidazole
- NMM
N-methylmorpholine
- THF
tetrahydrofuran
- DMSO
dimethylsulfoxide
- TEA
triethylamine
- FP
fluorescence polarization
- Kd
dissociation constant
- WT
wild type
- BOMB
Biochemical and Organic Model Builder
- PDB
Protein Databank
- vdW
van der Waals
- HEL
human erythroleukemia
- tPSA
topological polar surface area
- LE
ligand efficiency
- HAC
heavy atom count
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00051.
The authors declare the following competing financial interest(s): Yale University has submitted a preliminary patent application on the compounds.
Notes
All data are available in the main text or Supporting Information. Coordinates for the crystal structure (Figure 3) have been deposited in the Protein Data Bank with ID 7T1T.
Supplementary Material
References
- Kisseleva T.; Bhattacharya S.; Braunstein J.; Schindler C. W. Signaling through the JAK/STAT Pathway, Recent Advances and Future Challenges. Gene 2002, 285, 1–24. 10.1016/S0378-1119(02)00398-0. [DOI] [PubMed] [Google Scholar]
- Neubauer H.; Cumano A.; Muller M.; Wu H.; Huffstadt U.; Pfeffer K. Jak2 Deficiency Defines an Essential Developmental Checkpoint in Definitive Hematopoiesis. Cell 1998, 93, 397–409. 10.1016/S0092-8674(00)81168-X. [DOI] [PubMed] [Google Scholar]
- O’Shea J. J.; Holland S. M.; Staudt L. M. JAKs and STATs in Immunity, Immunodeficiency, and Cancer. N. Engl. J. Med. 2013, 368 (2), 161–170. 10.1056/NEJMra1202117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staerk J.; Constantinescu S. N. The JAK-STAT Pathway and Hematopoietic Stem Cells from the JAK2 V617F Perspective. JAK-STAT 2012, 1 (3), 184–190. 10.4161/jkst.22071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M. I. N.; Cheng A.; Candotti F.; Zhou Y.; Hymel A.; Fasth A.; Notarangelo L. D.; O’Shea J. J. Complex Effects of Naturally Occurring Mutations in the JAK3 Pseudokinase Domain: Evidence for Interactions between the Kinase and Pseudokinase Domains. Mol. Cell. Biol. 2000, 20 (3), 947–956. 10.1128/MCB.20.3.947-956.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saharinen P.; Silvennoinen O. The Pseudokinase Domain Is Required for Suppression of Basal Activity of Jak2 and Jak3 Tyrosine Kinases and for Cytokine-Inducible Activation of Signal Transduction. J. Biol. Chem. 2002, 277 (49), 47954–47963. 10.1074/jbc.M205156200. [DOI] [PubMed] [Google Scholar]
- Kung J. E.; Jura N. Prospects for Pharmacological Targeting of Pseudokinases. Nature Rev. Drug Disc. 2019, 18, 501–526. 10.1038/s41573-019-0018-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine R. L.; Wadleigh M.; Cools J.; Ebert B. L.; Wernig G.; Huntly B. J.P.; Boggon T. J.; Wlodarska I.; Clark J. J.; Moore S.; Adelsperger J.; Koo S.; Lee J. C.; Gabriel S.; Mercher T.; D’Andrea A.; Frohling S.; Dohner K.; Marynen P.; Vandenberghe P.; Mesa R. A.; Tefferi A.; Griffin J. D.; Eck M. J.; Sellers W. R.; Meyerson M.; Golub T. R.; Lee S. J.; Gilliland D. G. Activating Mutation in the Tyrosine Kinase JAK2 in Polycythemia Vera, Essential Thrombocythemia, and Myeloid Metaplasia with Myelofibrosis. Cancer Cell 2005, 7, 387–397. 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- Campbell P. J.; Green A. R. The Myeloproliferative Disorders. N. Engl. J. Med. 2006, 355, 2452–2466. 10.1056/NEJMra063728. [DOI] [PubMed] [Google Scholar]
- Vainchenker W.; Dusa A.; Constantinescu S. N. JAKs in Pathology: Role of Janus Kinases in Hematopoietic Malignancies and Immunodeficiencies. Semin. Cell Dev. Biol. 2008, 19, 385–393. 10.1016/j.semcdb.2008.07.002. [DOI] [PubMed] [Google Scholar]
- Chen E.; Staudt L. M.; Green A. R. Janus Kinase Deregulation in Leukemia and Lymphoma. Immunity 2012, 36 (4), 529–541. 10.1016/j.immuni.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadina M.; Le M. T.; Schwartz D. M.; Silvennoinen O.; Nakayamada S.; Yamaoka K.; O’Shea J. J. Janus Kinases to Jakinibs: From Basic Insights to Clinical Practice. Rheumatology 2019, 58, i4–i16. 10.1093/rheumatology/key432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assadiasl S.; Fatahi Y.; Mosharmovahed B.; Mohebbi B.; Nicknam M. H. Baricitinib: From Rheumatoid Arthritis to COVID-19. J. Clin. Pharmacol. 2021, 61 (142), 1274–1285. 10.1002/jcph.1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison C.; Kiladjian J.-J.; Al-Ali H. K.; Gisslinger H.; Waltzman R.; Stalbovskaya V.; McQuitty M.; Hunter D. S.; Levy R.; Knoops L.; Cervantes F.; Vannucchi A. M.; Barbui T.; Barosi G. JAK Inhibition with Ruxolitinib versus Best Available Therapy for Myelofibrosis. N. Engl. J. Med. 2012, 366 (9), 787–798. 10.1056/NEJMoa1110556. [DOI] [PubMed] [Google Scholar]
- Blair H. A. Fedratinib: First Approval. Drugs 2019, 79 (15), 1719–1725. 10.1007/s40265-019-01205-x. [DOI] [PubMed] [Google Scholar]
- Dhillon S. Tofacitinib: A Review in Rheumatoid Arthritis. Drugs 2017, 77 (18), 1987–2001. 10.1007/s40265-017-0835-9. [DOI] [PubMed] [Google Scholar]
- Hammarén H. M.; Ungureanu D.; Grisouard J.; Skoda R. C.; Hubbard S. R.; Silvennoinen O. ATP Binding to the Pseudokinase Domain of JAK2 Is Critical for Pathogenic Activation. Proc. Natl. Acad. Sci. U. S. A. 2015, 112 (15), 4642–4647. 10.1073/pnas.1423201112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dusa A.; Mouton C.; Pecquet C.; Herman M.; Constantinescu S. N. JAK2 V617F Constitutive Activation Requires JH2 Residue F595: A Pseudokinase Domain Target for Specific Inhibitors. PLoS One 2010, 5 (6), e11157 10.1371/journal.pone.0011157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnanasambandan K.; Magis A.; Sayeski P. P. The Constitutive Activation of Jak2-V617F Is Mediated by a π Stacking Mechanism Involving Phenylalanines 595 and 617. Biochemistry 2010, 49, 9972–9984. 10.1021/bi1014858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy E.; Dusa A.; Colau D.; Motamedi A.; Cahu X.; Mouton C.; Huang L. J.; Shiau A. K.; Constantinescu S. N. Uncoupling JAK2 V617F Activation from Cytokine-Induced Signalling by Modulation of JH2 AC Helix. Biochem. J. 2016, 473, 1579–1591. 10.1042/BCJ20160085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton A. S.; Deiana L.; Puleo D. E.; Cisneros J. A.; Cutrona K. J.; Schlessinger J.; Jorgensen W. L. JAK2 JH2 Fluorescence Polarization Assay and Crystal Structures for Complexes with Three Small Molecules. ACS Med. Chem. Lett. 2017, 8, 614–617. 10.1021/acsmedchemlett.7b00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liosi M.-E.; Krimmer S. G.; Newton A. S.; Dawson T. K.; Puleo D. E.; Cutrona K. J.; Suzuki Y.; Schlessinger J.; Jorgensen W. L. Selective Janus Kinase 2 (JAK2) Pseudokinase Ligands with a Diaminotriazole Core. J. Med. Chem. 2020, 63, 5324–5340. 10.1021/acs.jmedchem.0c00192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutrona K. J.; Newton A. S.; Krimmer S. G.; Tirado-Rives J.; Jorgensen W. L. Metadynamics as a Postprocessing Method for Virtual Screening with Application to the Pseudokinase Domain of JAK2. J. Chem. Inf. Model. 2020, 60, 4403–4415. 10.1021/acs.jcim.0c00276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puleo D. E.; Kucera K.; Hammaren H. M.; Ungureanu D.; Newton A. S.; Silvennoinen O.; Jorgensen W. L.; Schlessinger J. Identification and Characterization of JAK2 Pseudokinase Domain Small Molecule Binders. ACS Med. Chem. Lett. 2017, 8, 618–621. 10.1021/acsmedchemlett.7b00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton A. S.; Liosi M.; Henry S. P.; Deiana L.; Faver J. C.; Krimmer S. G.; Puleo D. E.; Schlessinger J.; Jorgensen W. L. Indoloxytriazines as Binding Molecules for the JAK2 JH2 Pseudokinase Domain and Its V617F Variant. Tetrahedron Lett. 2021, 77, 153248. 10.1016/j.tetlet.2021.153248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turupcu A.; Tirado-Rives J.; Jorgensen W. L. Explicit Representation of Cation-π Interactions in Force Fields with 1/R4 Nonbonded Terms. J. Chem. Theory Comput. 2020, 16, 7184–7194. 10.1021/acs.jctc.0c00847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K.; Shaw D. E.; Francis P.; Shenkin P. S. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- Halgren T. A.; Murphy R. B.; Friesner R. A.; Beard H. S.; Frye L. L.; Pollard W. T.; Banks J. L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- Friesner R. A.; Murphy R. B.; Repasky M. P.; Frye L. L.; Greenwood J. R.; Halgren T. A.; Sanschagrin P. C.; Mainz D. T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- Williams N. K.; Bamert R. S.; Patel O.; Wang C.; Walden P. M.; Wilks A. F.; Fantino E.; Rossjohn J.; Lucet I. S. Dissecting Specificity in the Janus Kinases: The Structures of JAK-Specific Inhibitors Complexed to the JAK1 and JAK2 Protein Tyrosine Kinase Domains. J. Mol. Biol. 2009, 387 (1), 219–232. 10.1016/j.jmb.2009.01.041. [DOI] [PubMed] [Google Scholar]
- Davis R. R.; Li B.; Yun S. Y.; Chan A.; Nareddy P.; Gunawan S.; Ayaz M.; Lawrence H. R.; Reuther G. W.; Lawrence N. J.; Schönbrunn E. Structural Insights into JAK2 Inhibition by Ruxolitinib, Fedratinib, and Derivatives Thereof. J. Med. Chem. 2021, 64, 2228–2241. 10.1021/acs.jmedchem.0c01952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lembicz N. K.; Grant S.; Clegg W.; Griffin R. J.; Heath S. L.; Golding B. T. Facilitation of Displacements at the 6-Position of Purines by the Use of 1,4-Diazabicyclo [2.2.2] Octane as Leaving Group. J. Chem. Soc. Perkin Trans. 1 1997, 1, 185–186. 10.1039/a608207f. [DOI] [Google Scholar]
- Bandaranayake R. M.; Ungureanu D.; Shan Y.; Shaw D. E.; Silvennoinen O.; Hubbard S. R. Crystal Structures of the JAK2 Pseudokinase Domain and the Pathogenic Mutant V617F. Nat. Struct. Mol. Biol. 2012, 19 (8), 754–760. 10.1038/nsmb.2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L. Efficient Drug Lead Discovery and Optimization. Acc. Chem. Res. 2009, 42 (6), 724–733. 10.1021/ar800236t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liosi M.-E.; Ippolito J. E.; Henry S. P.; Krimmer S. G.; Newton A. S.; Cutrona K. J.; Olivarez R. A.; Mohanty J.; Schlessinger J.; Jorgensen W. L.. Insights on JAK2Modulation by Potent, Selective, and Cell-Permeable Pseudokinase-Domain Ligands. J. Med. Chem. 2022, submitted for publication [DOI] [PMC free article] [PubMed]
- McNally R.; Li Q.; Li K.; Dekker C.; Vangrevelinghe E.; Jones M.; Chène P.; Machauer R.; Radimerski T.; Eck M. J. Discovery and Structural Characterization of ATP-site Ligands for the Wild-Type and V617F mutant JAK2 Pseudokinase Domain. ACS Chem. Biol. 2019, 14, 587–593. 10.1021/acschembio.8b00722. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.