

Abstract
Histone deacetylases (HDACs) 1–3 regulate chromatin structure and gene expression. These three enzymes are targets for cancer chemotherapy and have been studied for the treatment of immune disorders and neurodegeneration, but there is a lack of selective pharmacological tool compounds to unravel their individual roles. Potent inhibitors of HDACs 1–3 often display slow-binding kinetics, which causes a delay in inhibitor–enzyme equilibration and may affect assay readout. Here we compare the potencies and selectivities of slow-binding inhibitors measured by discontinuous and continuous assays. We find that entinostat, a clinical candidate, inhibits HDACs 1–3 by a two-step slow-binding mechanism with lower potencies than previously reported. In addition, we show that RGFP966, commercialized as an HDAC3-selective probe, is a slow-binding inhibitor with inhibitor constants of 57, 31, and 13 nM against HDACs 1–3, respectively. These data highlight the need for thorough kinetic investigation in the development of selective HDAC probes.
Keywords: Enzyme inhibitor, histone deacetylases, continuous assay, slow-binding kinetics, RGFP966
Inhibition of enzymes relies on the direct interaction of the inhibitor with its target. Extending the timespan of this interaction, also termed the residence time, is pursued in drug design to maximize a compound’s biological impact.1,2 Slow-binding inhibitors present low dissociation rates that translate into extended residence times and, often, higher potency.3−6 However, slow-binding kinetics also causes a delay in reaching equilibrium within assays, which is not always taken into account in medicinal chemistry campaigns.1,2 Disregarding slow-binding mechanisms may lead to an underestimation of the affinity not only against the desired target but also against off-targets that share structural similarity, such as members of the same enzyme family.1 Thus, understanding inhibitor kinetics is important to determine potency with accuracy and to assess subfamily and subclass selectivity.
Histone deacetylases (HDACs) are targeted by chemotherapy, and five HDAC inhibitors are in clinical use for the treatment of hematologic cancers.7 HDAC inhibitors are also undergoing clinical trials for treatment of dementia and muscular dystrophy7,8 and are being investigated in autoimmune diseases.9 Humans express 11 Zn2+-dependent HDACs, divided into class I (HDACs 1–3 and 8), class IIa (HDACs 4, 5, 7, and 9), class IIb (HDACs 6 and 10), and class IV (HDAC11).10 Class I HDACs, the targets of most therapies, remove Nε-acyllysine post-translational modifications from nuclear proteins, including histones and transcription factors,10−14 and thereby regulate gene expression.9 It is often unclear which isozymes are the relevant targets in each disease context because there is a lack of selective tool compounds to discriminate among class I HDACs. Most inhibitors used as probes inhibit multiple classes [e.g., SAHA (1); Figure 1], are class-I-selective [e.g., romidepsin (2)], or inhibit HDACs 1–3 [e.g., entinostat (3)];15 still compound 2 is the only recommended probe to study class I HDACs.16 Multiple HDAC inhibitors exhibit slow-binding kinetics. This was first reported for o-aminoanilides17 but was also found for trifluoromethyl ketones,18 hydroxamic acids,6,19 acylhydrazides,20,21 and compound 2.19,22 However, kinetic data are not available for all inhibitors used as probes, such as the claimed HDAC3-selective o-aminoanilide RGFP966 (4).23
Figure 1.

Structures of selected HDAC inhibitors.
Here we determine the potencies and selectivities of slow-binding inhibitors of HDACs 1–3 using standard end-point assays as well as discontinuous and continuous experiments that account for slow inhibitor–enzyme equilibration. We find that discontinuous assays often underestimate the potency and that the calculated selectivity is highly dependent on the assay format. Moreover, our data show that compound 4 is not an HDAC3-selective probe.
We selected four inhibitors for our investigations; compounds 1, 3, 4, and acylhydrazide 5.24 Compounds 1 and 3 inhibit HDACs 1–3 with similar potencies in end-point assays but display different kinetics of inhibition (fast- and slow-binding kinetics, respectively).15,17,25 Compounds 4 and 5, on the other hand, have been reported to exhibit selectivity for HDAC3,23,24 but their binding kinetics have not been investigated, although they are structurally similar to known slow-binding inhibitors.7,21
First, we evaluated the inhibition of HDACs 1–3 using a standard fluorescent assay based on a 7-amino-4-methylcoumarin (AMC)-coupled substrate.15,26 The substrate, Ac-Leu-Gly-Lys(Ac)-AMC, has similar Michaelis–Menten constants (KM) for the three enzymes in the low micromolar range, allowing investigation under comparable enzyme saturation conditions.15 The inhibitor and substrate were incubated with the enzyme for 30 min before development with a protease, providing end-point dose–response curves (Figure 2A). Compounds 1 and 3 showed similar potencies against the three enzymes, while 4 and 5 showed selectivity for HDAC3 as previously reported. However, the measured potency and selectivity of compound 4 differed significantly from those reported in the literature, with an IC50 of 514 ± 4 nM against HDAC3 (compared with the reported value of 80 nM) and a selectivity ratio of 3.5 versus HDAC2 (compared with <180) (Figure 2D).23 The data for compound 5 were in better agreement with those reported previously, with a 6.1 selectivity ratio versus HDAC1 (compared with 12.4).24
Figure 2.

Discontinuous inhibition of HDACs 1–3. (A) Dose–response curves measured after reaction of the enzyme and substrate for 30 min. (B) HDAC2 curves after preincubation with the enzyme for 0, 0.5, 1, or 2 h followed by addition of the substrate and reaction for 30 min. (C) HDAC1 and HDAC3 curves of compound 5 with and without preincubation. See Figure S1A,B for additional HDAC1 and HDAC3 curves. (D) IC50 data for compounds 4 and 5 and selectivity of HDAC3 inhibition. See Figure S1C for data for compounds 1 and 3. All data represent mean ± SD, n ≥ 2. *HDAC3 was incubated with the deacetylase activation domain (DAD) of NCoR2. †The selectivity was calculated vs the second-most-inhibited enzyme (HDAC2 for 4, HDAC1 for 5) by transformation of IC50 data into Ki values and calculation of the ratio (see data analysis in the Supporting Information). ‡Data were obtained in buffer without reducing agents or surfactants (see Figure S2 and preincubation methods in the Supporting Information for choice of buffer).
Compounds 3, 4, and 5 contain either o-aminoanilide or acylhydrazide Zn2+-binding groups, which are prevalent in inhibitors of class I HDACs that exhibit slow-binding kinetics.17,20,25 Since slow-binding inhibitors might equilibrate after periods of time longer than 30 min,27 the performed end-point assays do not necessarily occur at steady state.
To enable equilibration, assays were next performed with preincubation of the inhibitor and enzyme for 30 min, 1 h, or 2 h before substrate addition. Compound 1, an inhibitor that exhibits fast-on/fast-off binding kinetics, inhibited HDACs 1 and 2 with similar potencies within experimental error regardless of preincubation. The same trend was observed for compounds 3 and 4, with only slight changes in IC50 values in response to preincubation, which may indicate inhibitor–enzyme equilibration on the time scale of 0–30 min (Figures 2B,D and S1). Only compound 5 followed a slow-binding profile for inhibition of HDACs 1 and 2, with a substantial decrease in the IC50 values upon preincubation (Figure 2B–D). The selectivities of compounds 4 and 5 were calculated on the basis of each preincubation data set. Compound 4 maintained a similar selectivity for HDAC3 at all preincubation times, with just a slight loss in selectivity at 2 h, which is within the experimental error of the system (1.9 selectivity ratio vs HDAC2; Figure 2D). More strikingly, the selectivity of compound 5 dropped from 6.1 to 1.4 after 30 min and further decreased at the 1 and 2 h time points as a result of a decrease in IC50 for HDACs 1 and 2 while the potency against HDAC3 was maintained. Since HDAC3 experiments were carried out at an enzyme concentration of 1–5 nM, this difference in behavior might be explained by compound 5 already approaching stoichiometric inhibition of HDAC3 (tight binding) without preincubation, in which case a potential further increase in potency after preincubation would be detected for HDACs 1 and 2 only. Nonetheless, the data highlight the need for more thorough investigation of slow-binding inhibitor potency to assess the selectivity with better accuracy.
Preincubation data for slow-binding inhibitors can be fit to exponential decay functions in order to calculate slow binding kinetic and inhibition constants.27 Here this analysis could be performed for 3 against HDACs 2 and 3 and for 5 against HDACs 1 and 2, providing a rough estimate of the compound potencies and residence times (Figure S3). Through this analysis, compound 3 was calculated to have apparent inhibition constants of ∼73 nM (HDAC2) and ∼27 nM (HDAC3), while compound 5 exhibited even lower potency. Unfortunately, the selectivities could not be recalculated because of the lack of complete data sets for HDACs 1–3.
Next, we adapted the initial discontinuous assay conditions to a continuous format by adding the protease developer directly to the reaction mixture as previously reported.17,28 The protease concentration was optimized to obtain linear substrate conversion by the HDAC for 40–60 min while ensuring that deacetylation by the HDAC was the rate-limiting step (Figure S4).29 With these assay conditions in hand, we measured the enzyme kinetics to determine the Michaelis–Menten constant for each of our enzyme preparations (Figure S5) and recorded the enzyme activities at different concentrations of inhibitor. Fast-on/fast-off inhibitors such as compound 1 equilibrate rapidly to the steady state to afford linear progression curves at all inhibitor concentrations.6,17,18 Conversely, slow-binding inhibitors lower the reaction rate over time, resulting in bending assay progression curves. Fitting the apparent first-order rate constant for equilibration (kobs) to a linear or a hyperbolic function of inhibitor concentration reveals whether the inhibitor follows mechanism A or mechanism B of slow-binding kinetics, respectively (Figure 3A). In mechanism A, a single step of binding of the inhibitor to the enzyme is detected, with low overall on and off rates. In the more common mechanism B, rapid formation of an initial enzyme–inhibitor complex (EI) is found, followed by a slow transition to a more stable and long-lived enzyme–inhibitor complex (EI*).27,30
Figure 3.

Models of slow-binding inhibition and continuous inhibition of HDACs 1–3 by compound 3. (A) Mechanisms A and B of competitive slow-binding inhibition and calculation of inhibitor constants (Ki) from kinetic data. (B) Continuous assay progression curves for the inhibition of HDACs 1–3 by different concentrations of 3. (C) Secondary plots of the apparent first-order rate constant for equilibration (kobs) vs inhibitor concentration and fits to mechanism B of slow-binding kinetics (see Table 1 for numerical data). Data represent mean ± SEM of individual experiments, with each experiment performed at least twice. *The HDAC3 preparation contained the DAD of NCoR2.
Compound 3 afforded the characteristic bending progression curves of a slow-binding inhibitor against all three HDACs (Figure 3B). The kobs data fitted well to mechanism B of HDAC inhibition (Figure 3C, hyperbolic relationship), which adds further insight into the previous kinetic analysis of this compound.25,31 The data revealed that compound 3 presents a fast first binding step with equilibrium constants (Ki,1) of ∼0.59 μM for HDACs 1 and 2 and ∼3.2 μM for HDAC3, which are similar to previous estimations of potency.25,31 When all of the kinetic constants were taken into account, the calculated potency (Ki) of 3 was <1 nM against HDAC1, ∼6 nM against HDAC2, and ∼39 nM against HDAC3 (Table 1). These values are much lower than those obtained from end-point experiments by us and others,32 including HDAC2 preincubation assays, and those calculated by previous fits to mechanism A of slow-binding kinetics.25,31 Our kinetic data may thus help explain the high potency of compound 3 in cellular assays as well as its long-lasting effect due to extended enzyme dissociation half-life (t1/2) (Table 1).25,32
Table 1. Calculated Rate Constants (kn), Inhibition Constants (Kn), and Dissociation Half-Lives (t1/2) for Compounds 3, 4, and 5a.
| HDAC1 | HDAC2 | HDAC3c | |
|---|---|---|---|
| Entinostat (3) | |||
| k2 (min–1) | 0.5 ± 0.1 | 0.28 ± 0.04 | 0.6 ± 0.5 |
| k–2 (min–1) | ∼7 × 10–9 | ∼3 × 10–3 | ∼7 × 10–3 |
| Ki,1 (nM) | 590 ± 400 | 590 ± 340 | ∼3200 |
| Ki (nM) | –b | ∼6 | ∼39 |
| t1/2 (min) | >105 | ≥240 | ≥95 |
| RGFP966 (4) | |||
| k2 (min–1) | 0.4 ± 0.1 | 0.38 ± 0.03 | 0.3 ± 0.1 |
| k–2 (min–1) | ∼2 × 10–2 | ∼7 × 10–3 | ∼5 × 10–3 |
| Ki,1 (nM) | 1300 ± 1000 | 1700 ± 400 | 700 ± 700 |
| Ki (nM) | ∼57 | ∼31 | ∼13 |
| t1/2 (min) | ≥36 | ≥95 | ≥131 |
| Compound 5 | |||
| k1 (nM–1·min–1) | (7.8 ± 0.4) × 10–5 | (5.1 ± 0.2) × 10–5 | |
| k–1 (min–1) | (3 ± 3) × 10–3 | (5 ± 2) × 10–3 | |
| Ki (nM) | ∼40 | ∼103 | |
| t1/2 (min) | ∼220 | ∼130 | |
| k2 (min–1) | 0.78 ± 0.08 | ||
| k–2 (min–1) | ∼4 × 10–11 | ||
| Ki,1 (nM) | 25 ± 6 | ||
| Ki (nM) | –b | ||
| t1/2 (min) | <105 | ||
The data correspond to mechanism B of slow-binding kinetics, except for the inhibition of HDACs 1 and 2 by compound 5, which follows mechanism A.
Ki was not determined because the value of k–2 obtained by data fitting approached zero.
The HDAC3 enzyme preparation contained the DAD of NCoR2.
Continuous assays with compound 4 revealed slow inhibition of HDACs 1–3, which was anticipated on the basis of its chemical structure but was not indicated in the discontinuous assays discussed above. Bending of assay progression curves was especially prominent for HDAC3 (Figure 4A), which emphasizes the higher sensitivity of continuous assays for detection of slow-binding profiles. Inhibition of HDACs 1–3 followed mechanism B of slow-binding kinetics and afforded inhibitor constants of ∼57 nM for HDAC1, ∼31 nM for HDAC2, and ∼13 nM for HDAC3 (Table 1). On the basis of these results, compound 4 exhibits a mere 2.4-fold selectivity toward HDAC3, which is similar to that obtained from preincubation (2 h time point in Figure 2D). On the other hand, the calculated potency against each enzyme is >10 times lower than the end-point estimate. Overall, our data indicate that compound 4 is a potent inhibitor of HDACs 1–3 with only a minor preference for HDAC3 inhibition.
Figure 4.

Continuous inhibition of HDACs 1 and 3 by compounds 4 and 5. (A) (top) Continuous assay progression curves and (bottom) kobs secondary plots (mechanism B) for compound 4. (B) (top) Continuous assay progression curves and (bottom) kobs secondary plots (HDAC1 data fitted to mechanism A and HDAC3 data fitted to mechanism B) for compound 5. Data represent mean ± SEM of individual experiments, with each experiment performed at least twice. See Figure S6 for HDAC2 data and Table 1 for numerical data. *The HDAC3 preparation contained the DAD of NCoR2.
The slow-binding behavior of compound 5 against HDACs 1 and 2, which was identified by preincubation, was recapitulated by continuous assays. In addition, we also detected slow-binding inhibition of HDAC3 (Figure 4B).
Interestingly, secondary plots of kobs indicated that compound 5 follows mechanism A for inhibition of HDACs 1 and 2, whereas it inhibits HDAC3 through mechanism B (Figures 4B and S6B). These differences in kinetic mechanism are not uncommon and were reported previously for a trifluoromethyl ketone inhibitor18 as well as the natural product trapoxin A.6 As a result of data fitting, the calculated Ki values of compound 5 against HDACs 1 and 2 were ∼40 and ∼103 nM, respectively (Table 1), which are somewhat higher than indicated by the IC50 values obtained by end-point experiments (Figure 1D).24 Conversely, the data for inhibition of HDAC3 indicated pseudo-irreversible inhibition with very low off rates (k–2 ≈ 0) and a first step Ki,1 of 25 ± 6 nM. Thus, compound 5 is highly potent against HDACs 1–3, with enzyme residence times of >2 h (Table 1) and slow tight-binding behavior of HDAC3 inhibition. Future studies will reveal whether this kinetic profile could be exploited for selective inhibition of HDAC3 in a biological setting.
HDACs 1–3 form multiprotein complexes with diverse epigenetic functions,33 and reports have shown that the inhibitor potency and selectivity may differ between the free and complexed HDAC forms.34,35 Thus, inhibitor assays where the HDAC is accompanied by a complex partner likely produce results different from those for inhibition of free recombinant enzymes. Commercial HDAC3 includes the HDAC-interacting DAD of NCoR2, which is required for enzymatic activity, and this interaction can be further stabilized in vitro by inositol phosphates.36 Since standard experiments are performed with HDAC3 and the DAD in dynamic equilibrium, we also studied the effect of adding inositol hexaphosphate (InsP6) to HDAC3 experiments at a concentration shown to fully stabilize the complex.37 End-point inhibition data without preincubation remained similar to data recorded without InsP6 added. Conversely, preincubation of the inhibitor with enzyme and InsP6 provided stronger inhibition, with 10–20 times lower IC50 values for compounds 3, 4, and 5 after 2 h (Figures 5A and S7A,B). Addition of InsP6 enhanced the HDAC activity, allowing the experiments to be performed at 5 times lower enzyme concentration, but compound 5 still afforded stoichiometric (tight-binding) inhibition. These data suggest that the three compounds inhibit the activity of a stabilized HDAC3–NCoR2 complex with higher potency and/or slower kinetics compared with the more dynamic standard enzyme preparation. Such behavior appears to be different from the binding studies reported for the o-aminoanilide Cpd-60, which presents lower affinity for an HDAC3–NCoR2 complex prestabilized using InsP6.37
Figure 5.
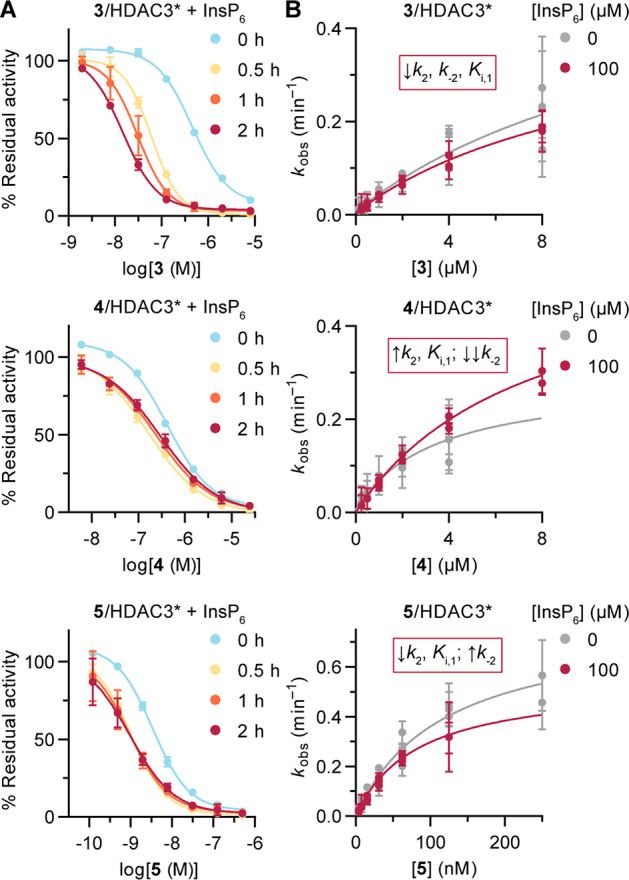
Inhibition of HDAC3 in the presence of inositol hexaphosphate (InsP6). (A) Curves for compounds 3, 4, and 5 with and without preincubation, measured in buffer without reducing agents or surfactants. See Figure S7A,B for curves for compound 1 and for IC50 and selectivity data. Data represent mean ± SD, n = 2. (B) kobs secondary plots for compounds 3, 4, and 5 with and without InsP6. Data represent mean ± SEM of individual experiments, with each experiment performed at least twice. See Figure S7C,D for progression curves and numerical data. A summary of data changes with InsP6 is provided for each compound. *The HDAC3 preparation contained the DAD of NCoR2.
We then performed continuous inhibition assays with compounds 3, 4, and 5, where the HDAC3/DAD preparation was premixed with InsP6 before addition to the assay plate. The impact of complex stabilization was less pronounced in these experiments. All three inhibitors maintained mechanism B of slow-binding kinetics, although changes in the kinetic constants were observed in each case (Figures 5 and S7C,D). Compound 3 gained affinity in the first binding step and showed slower kinetics for the transition to the more stable EI* complex, affording an estimated Ki of 24 nM. Compound 4 exhibited a decrease in affinity for the first step but gained substantial overall potency because of the very low off rates (k–2 ≈ 0). Finally, compound 5 gained affinity in the first step and remained a slow, tight-binding inhibitor of the stabilized HDAC3–DAD complex.
Our data underpin the need for detailed kinetic characterization of potent class I HDAC inhibitors, which is of particular importance for the determination of subclass selectivity. Compound 4 was reported and commercialized as an HDAC3-selective probe and has been employed in numerous studies currently associated with specific HDAC3 biology.23,38−40 Here we have shown that this compound is not appropriate as an HDAC3-selective probe, which calls into question the biological functions assigned to HDAC3 based on the use of compound 4.
A recent report by Liu and co-workers identified compound 6 (Figure 1) as a highly selective inhibitor of HDAC3.41 Compound 6 contains an o-methylthiobenzamide Zn2+-binding group and exhibits slow-binding mechanism A for HDAC3 inhibition with Ki ≈ 24 nM (calculated from kinetic data).41 HDACs 1 and 2 are inhibited in end-point experiments with IC50 values of 20 and 31 μM, respectively, but no kinetic analyses are provided for those enzymes. Even though there is a large difference in IC50 values based on discontinuous experiments, interrogation of the inhibition of HDACs 1 and 2 in a continuous fashion might be appropriate.
HDACs 1–3 form multiprotein complexes in the cell, in which they have different sensitivities to inhibitors.34 Here stabilization of the HDAC3–NCoR2 interaction with the “molecular glue” InsP6 led to changes in kinetic data that were compound-specific and resulted in a substantial increase in the measured potency after preincubation. These effects might also appear when HDACs 1 and 2 are studied in the presence of corepressors, which would add a new layer of complexity to inhibitor selectivity assessments. Therefore, future HDAC probe characterization may benefit from kinetic analysis not only against the free enzymes but also against reconstituted complexes. One potential solution may be to measure binding to a library of HDAC complexes by time-resolved Förster resonance energy transfer (TR-FRET), as proposed recently.37 These analyses will ensure the development of robust and reproducible tool compounds for HDAC research, which are of importance for the development of epigenetic therapies against neurodegeneration and immune disorders.
In conclusion, we have shown that entinostat (3) is an inhibitor of HDACs 1–3 with nanomolar potency that follows mechanism B of slow-binding kinetics and that acylhydrazide 5 exhibits subclass differences in kinetic behavior with a preference for inhibition of HDAC3. Importantly, we report that RGFP966 (4) is a potent slow-binding inhibitor of HDACs 1–3 and not an HDAC3-selective probe. It is our hope that these findings will assist in future experimental design toward the elucidation of class I HDAC function and subsequent drug development.
Acknowledgments
This project received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation Programme (Grant Agreement CoG-725172-SIRFUNCT to C.A.O.).
Glossary
Abbreviations
- DAD
deacetylase activation domain
- HDAC
histone deacetylase
- kobs
apparent first-order rate constant for equilibration
- NCoR2
nuclear receptor corepressor 2
Biographies
Carlos Moreno-Yruela obtained his M.Sc. degree in drug discovery from the University of Surrey (UK) in 2015. After a short scholarship at the Spanish Research Council in Zaragoza (Spain), he started his Ph.D. studies in 2016 in the laboratory of Prof. Christian A. Olsen, where he worked on peptide microarrays to develop histone deacetylase (HDAC) probes. He defended his thesis in 2019, and his work was selected to represent Denmark at the 2021 EFMC Young Medicinal Chemists’ Symposium. His current research as a postdoctoral fellow focuses on the development of tools to characterize HDAC activity and cellular function.
Christian A. Olsen received his M.Sc. from the Technical University of Denmark (DTU) and his Ph.D. from the Danish University of Pharmaceutical Sciences. After independently working on peptidomimetics, he did his postdoctoral fellowship with Prof. Ghadiri at Scripps Research. In 2010 he returned to a faculty position at DTU, and in 2014 he accepted his current position as professor at the University of Copenhagen. He has received the EFMC award for young medicinal chemists in academia (2014) and an ERC Consolidator grant (2016). His research interests include foldamers, HDAC inhibitors, quorum sensing modulators, and investigation of lysine acylation.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00702.
Supporting figures, schemes, and tables; supporting methods, experimental procedures, HPLC traces, and copies of NMR spectra (PDF)
The authors declare no competing financial interest.
Special Issue
Published as part of the ACS Medicinal Chemistry Letters special issue “Epigenetics 2022”.
Supplementary Material
References
- Copeland R. A.; Pompliano D. L.; Meek T. D. Drug-target residence time and its implications for lead optimization. Nat. Rev. Drug Discovery 2006, 5 (9), 730–739. 10.1038/nrd2082. [DOI] [PubMed] [Google Scholar]
- Holdgate G. A.; Meek T. D.; Grimley R. L. Mechanistic enzymology in drug discovery: a fresh perspective. Nat. Rev. Drug Discovery 2018, 17 (2), 115–132. 10.1038/nrd.2017.219. [DOI] [PubMed] [Google Scholar]
- Basavapathruni A.; Jin L.; Daigle S. R.; Majer C. R.; Therkelsen C. A.; Wigle T. J.; Kuntz K. W.; Chesworth R.; Pollock R. M.; Scott M. P.; Moyer M. P.; Richon V. M.; Copeland R. A.; Olhava E. J. Conformational adaptation drives potent, selective and durable inhibition of the human protein methyltransferase DOT1L. Chem. Biol. Drug Des. 2012, 80 (6), 971–980. 10.1111/cbdd.12050. [DOI] [PubMed] [Google Scholar]
- Chang A.; Schiebel J.; Yu W.; Bommineni G. R.; Pan P.; Baxter M. V.; Khanna A.; Sotriffer C. A.; Kisker C.; Tonge P. J. Rational optimization of drug-target residence time: insights from inhibitor binding to the Staphylococcus aureus FabI enzyme-product complex. Biochemistry 2013, 52 (24), 4217–4228. 10.1021/bi400413c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland R. A. The drug-target residence time model: a 10-year retrospective. Nat. Rev. Drug Discovery 2016, 15 (2), 87–95. 10.1038/nrd.2015.18. [DOI] [PubMed] [Google Scholar]
- Moreno-Yruela C.; Fass D. M.; Cheng C.; Herz J.; Olsen C. A.; Haggarty S. J. Kinetic Tuning of HDAC Inhibitors Affords Potent Inducers of Progranulin Expression. ACS Chem. Neurosci. 2019, 10 (8), 3769–3777. 10.1021/acschemneuro.9b00281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho T. C. S.; Chan A. H. Y.; Ganesan A. Thirty Years of HDAC Inhibitors: 2020 Insight and Hindsight. J. Med. Chem. 2020, 63 (21), 12460–12484. 10.1021/acs.jmedchem.0c00830. [DOI] [PubMed] [Google Scholar]
- Kristensen H. M. E.; Madsen A. S.; Olsen C. A.. Inhibitors of the Zinc Dependent Histone Deacetylases. In Epigenetic Drug Discovery; Sippl W., Jung M., Eds.; Methods and Principles in Medicinal Chemistry, Vol. 74; Wiley-VCH, 2019; pp 155–184. [Google Scholar]
- Falkenberg K. J.; Johnstone R. W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discovery 2014, 13 (9), 673–691. 10.1038/nrd4360. [DOI] [PubMed] [Google Scholar]
- Yang X. J.; Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 2008, 9 (3), 206–218. 10.1038/nrm2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen A. S.; Olsen C. A. Profiling of Substrates for Zinc-dependent Lysine Deacylase Enzymes: HDAC3 Exhibits Decrotonylase Activity In Vitro. Angew. Chem. 2012, 124 (36), 9217–9221. 10.1002/ange.201203754. [DOI] [PubMed] [Google Scholar]
- Wei W.; Liu X.; Chen J.; Gao S.; Lu L.; Zhang H.; Ding G.; Wang Z.; Chen Z.; Shi T.; Li J.; Yu J.; Wong J. Class I histone deacetylases are major histone decrotonylases: evidence for critical and broad function of histone crotonylation in transcription. Cell Res. 2017, 27 (7), 898–915. 10.1038/cr.2017.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H.; Zhang D.; Weng Y.; Delaney K.; Tang Z.; Yan C.; Qi S.; Peng C.; Cole P. A.; Roeder R. G.; Zhao Y. The regulatory enzymes and protein substrates for the lysine beta-hydroxybutyrylation pathway. Sci. Adv. 2021, 7 (9), eabe2771 10.1126/sciadv.abe2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Yruela C.; Zhang D.; Wei W.; Baek M.; Liu W.; Gao J.; Dankova D.; Nielsen A. L.; Bolding J. E.; Yang L.; Jameson S. T.; Wong J.; Olsen C. A.; Zhao Y. Class I histone deacetylases (HDAC1–3) are histone lysine delactylases. Sci. Adv. 2022, 8 (3), eabi6696 10.1126/sciadv.abi6696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradner J. E.; West N.; Grachan M. L.; Greenberg E. F.; Haggarty S. J.; Warnow T.; Mazitschek R. Chemical phylogenetics of histone deacetylases. Nat. Chem. Biol. 2010, 6 (3), 238–243. 10.1038/nchembio.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The inhibitors apicidin, entinostat, SAHA, trichostatin A, and panobinostat are rated “historical compounds” and are no longer recommended at chemicalprobes.org (accessed November 2021).
- Chou C. J.; Herman D.; Gottesfeld J. M. Pimelic Diphenylamide 106 Is a Slow, Tight-binding Inhibitor of Class I Histone Deacetylases. J. Biol. Chem. 2008, 283 (51), 35402–35409. 10.1074/jbc.M807045200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen A. S.; Olsen C. A. A potent trifluoromethyl ketone histone deacetylase inhibitor exhibits class-dependent mechanism of action. MedChemComm 2016, 7 (3), 464–470. 10.1039/C5MD00451A. [DOI] [Google Scholar]
- Kitir B. l.; Maolanon A. R.; Ohm R. G.; Colaço A. R.; Fristrup P.; Madsen A. S.; Olsen C. A. Chemical editing of macrocyclic natural products and kinetic profiling reveal slow, tight-binding histone deacetylase inhibitors with picomolar affinities. Biochemistry 2017, 56 (38), 5134–5146. 10.1021/acs.biochem.7b00725. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Stowe R. L.; Pinello C. E.; Tian G.; Madoux F.; Li D.; Zhao L. Y.; Li J. L.; Wang Y.; Wang Y.; Ma H.; Hodder P.; Roush W. R.; Liao D. Identification of histone deacetylase inhibitors with benzoylhydrazide scaffold that selectively inhibit class I histone deacetylases. Chem. Biol. 2015, 22 (2), 273–284. 10.1016/j.chembiol.2014.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Peterson Y. K.; Inks E. S.; Himes R. A.; Li J.; Zhang Y.; Kong X.; Chou C. J. Class I HDAC Inhibitors Display Different Antitumor Mechanism in Leukemia and Prostatic Cancer Cells Depending on Their p53 Status. J. Med. Chem. 2018, 61 (6), 2589–2603. 10.1021/acs.jmedchem.8b00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robers M. B.; Dart M. L.; Woodroofe C. C.; Zimprich C. A.; Kirkland T. A.; Machleidt T.; Kupcho K. R.; Levin S.; Hartnett J. R.; Zimmerman K.; Niles A. L.; Ohana R. F.; Daniels D. L.; Slater M.; Wood M. G.; Cong M.; Cheng Y. Q.; Wood K. V. Target engagement and drug residence time can be observed in living cells with BRET. Nat. Commun. 2015, 6, 10091. 10.1038/ncomms10091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malvaez M.; McQuown S. C.; Rogge G. A.; Astarabadi M.; Jacques V.; Carreiro S.; Rusche J. R.; Wood M. A. HDAC3-selective inhibitor enhances extinction of cocaine-seeking behavior in a persistent manner. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (7), 2647–2652. 10.1073/pnas.1213364110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClure J. J.; Zhang C.; Inks E. S.; Peterson Y. K.; Li J.; Chou C. J. Development of Allosteric Hydrazide-Containing Class I Histone Deacetylase Inhibitors for Use in Acute Myeloid Leukemia. J. Med. Chem. 2016, 59 (21), 9942–9959. 10.1021/acs.jmedchem.6b01385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauffer B. E.; Mintzer R.; Fong R.; Mukund S.; Tam C.; Zilberleyb I.; Flicke B.; Ritscher A.; Fedorowicz G.; Vallero R.; Ortwine D. F.; Gunzner J.; Modrusan Z.; Neumann L.; Koth C. M.; Lupardus P. J.; Kaminker J. S.; Heise C. E.; Steiner P. Histone deacetylase (HDAC) inhibitor kinetic rate constants correlate with cellular histone acetylation but not transcription and cell viability. J. Biol. Chem. 2013, 288, 26926–26943. 10.1074/jbc.M113.490706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegener D.; Hildmann C.; Riester D.; Schwienhorst A. Improved fluorogenic histone deacetylase assay for high-throughput-screening applications. Anal. Biochem. 2003, 321 (2), 202–208. 10.1016/S0003-2697(03)00426-3. [DOI] [PubMed] [Google Scholar]
- Copeland R. A.Slow binding inhibitors. In Evaluation of Enzyme Inhibitors in Drug Discovery, 2nd ed.; Wiley-Interscience, 2013; pp 203–244. [Google Scholar]
- Moreno-Yruela C.; Stahl Madsen A.; Olsen C. A. Kinetic characterization of inhibitors of histone deacetylases (HDACs) and sirtuins. Protoc. Exch. 2019, 10.21203/rs.2.13042/v1. [DOI] [Google Scholar]
- Moreno-Yruela C.; Olsen C. A. High-throughput screening of histone deacetylases and determination of kinetic parameters using fluorogenic assays. STAR Protoc. 2021, 2 (1), 100313. 10.1016/j.xpro.2021.100313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison J. F.; Walsh C. T. The behavior and significance of slow-binding enzyme inhibitors. Adv. Enzymol. Relat. Areas Mol. Biol. 2006, 61, 201–301. 10.1002/9780470123072.ch5. [DOI] [PubMed] [Google Scholar]
- Becher I.; Dittmann A.; Savitski M. M.; Hopf C.; Drewes G.; Bantscheff M. Chemoproteomics reveals time-dependent binding of histone deacetylase inhibitors to endogenous repressor complexes. ACS Chem. Biol. 2014, 9 (8), 1736–1746. 10.1021/cb500235n. [DOI] [PubMed] [Google Scholar]
- Hu E.; Dul E.; Sung C. M.; Chen Z.; Kirkpatrick R.; Zhang G. F.; Johanson K.; Liu R.; Lago A.; Hofmann G.; Macarron R.; de los Frailes M.; Perez P.; Krawiec J.; Winkler J.; Jaye M. Identification of novel isoform-selective inhibitors within class I histone deacetylases. J. Pharmacol. Exp. Ther, 2003, 307 (2), 720–728. 10.1124/jpet.103.055541. [DOI] [PubMed] [Google Scholar]
- Millard C. J.; Watson P. J.; Fairall L.; Schwabe J. W. R. Targeting Class I Histone Deacetylases in a “Complex” Environment. Trends Pharmacol. Sci. 2017, 38 (4), 363–377. 10.1016/j.tips.2016.12.006. [DOI] [PubMed] [Google Scholar]
- Bantscheff M.; Hopf C.; Savitski M. M.; Dittmann A.; Grandi P.; Michon A. M.; Schlegl J.; Abraham Y.; Becher I.; Bergamini G.; Boesche M.; Delling M.; Dumpelfeld B.; Eberhard D.; Huthmacher C.; Mathieson T.; Poeckel D.; Reader V.; Strunk K.; Sweetman G.; Kruse U.; Neubauer G.; Ramsden N. G.; Drewes G. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat. Biotechnol. 2011, 29 (3), 255–265. 10.1038/nbt.1759. [DOI] [PubMed] [Google Scholar]
- Fuller N. O.; Pirone A.; Lynch B. A.; Hewitt M. C.; Quinton M. S.; McKee T. D.; Ivarsson M. CoREST Complex-Selective Histone Deacetylase Inhibitors Show Prosynaptic Effects and an Improved Safety Profile To Enable Treatment of Synaptopathies. ACS Chem. Neurosci. 2019, 10 (3), 1729–1743. 10.1021/acschemneuro.8b00620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millard C. J.; Watson P. J.; Celardo I.; Gordiyenko Y.; Cowley S. M.; Robinson C. V.; Fairall L.; Schwabe J. W. Class I HDACs share a common mechanism of regulation by inositol phosphates. Mol. Cell 2013, 51 (1), 57–67. 10.1016/j.molcel.2013.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne N. C.; Mazitschek R. Resolving the Deceptive Isoform and Complex Selectivity of HDAC1/2 Inhibitors. SSRN 2021, 10.2139/ssrn.3960267. [DOI] [PubMed] [Google Scholar]
- Wells C. E.; Bhaskara S.; Stengel K. R.; Zhao Y.; Sirbu B.; Chagot B.; Cortez D.; Khabele D.; Chazin W. J.; Cooper A.; Jacques V.; Rusche J.; Eischen C. M.; McGirt L. Y.; Hiebert S. W. Inhibition of histone deacetylase 3 causes replication stress in cutaneous T cell lymphoma. PLoS One 2013, 8 (7), e68915 10.1371/journal.pone.0068915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leus N. G.; van der Wouden P. E.; van den Bosch T.; Hooghiemstra W. T. R.; Ourailidou M. E.; Kistemaker L. E.; Bischoff R.; Gosens R.; Haisma H. J.; Dekker F. J. HDAC 3-selective inhibitor RGFP966 demonstrates anti-inflammatory properties in RAW 264.7 macrophages and mouse precision-cut lung slices by attenuating NF-kappaB p65 transcriptional activity. Biochem. Pharmacol. 2016, 108, 58–74. 10.1016/j.bcp.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janczura K. J.; Volmar C. H.; Sartor G. C.; Rao S. J.; Ricciardi N. R.; Lambert G.; Brothers S. P.; Wahlestedt C. Inhibition of HDAC3 reverses Alzheimer’s disease-related pathologies in vitro and in the 3xTg-AD mouse model. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (47), E11148–E11157. 10.1073/pnas.1805436115. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Liu J.; Yu Y.; Kelly J.; Sha D.; Alhassan A. B.; Yu W.; Maletic M. M.; Duffy J. L.; Klein D. J.; Holloway M. K.; Carroll S.; Howell B. J.; Barnard R. J. O.; Wolkenberg S.; Kozlowski J. A. Discovery of Highly Selective and Potent HDAC3 Inhibitors Based on a 2-Substituted Benzamide Zinc Binding Group. ACS Med. Chem. Lett. 2020, 11 (12), 2476–2483. 10.1021/acsmedchemlett.0c00462. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.