

Abstract
Drug-induced liver injury (DILI) is the leading cause of acute liver failure and a major concern in drug development. Altered bile acid homeostasis via inhibition of the bile salt export pump (BSEP) is one mechanism of DILI. Dasatinib, pazopanib, and sorafenib are tyrosine kinase inhibitors (TKIs) that competitively inhibit BSEP and increase serum biomarkers for hepatotoxicity in ∼25–50% of patients. However, the mechanism(s) of hepatotoxicity beyond competitive inhibition of BSEP are poorly understood. This study examined mechanisms of TKI-mediated hepatotoxicity associated with altered bile acid homeostasis. Dasatinib, pazopanib, and sorafenib showed bile acid-dependent toxicity at clinically relevant concentrations, based on the C-DILI assay using sandwich-cultured human hepatocytes (SCHH). Among several bile acid-relevant genes, cytochrome P450 (CYP) 7A1 mRNA was specifically upregulated by 6.2- to 7.8-fold (dasatinib) and 5.7- to 9.3-fold (pazopanib), compared with control, within 8 hours. This was consistent with increased total bile acid concentrations in culture medium up to 2.3-fold, and in SCHH up to 1.4-fold, compared with control, within 24 hours. Additionally, protein abundance of sodium taurocholate co-transporting polypeptide (NTCP) was increased up to 2.0-fold by these three TKIs. The increase in NTCP protein abundance correlated with increased function; dasatinib and pazopanib increased hepatocyte uptake clearance (CLuptake) of taurocholic acid, a probe bile acid substrate, up to 1.4-fold. In conclusion, upregulation of CYP7A1 and NTCP in SCHH constitute novel mechanisms of TKI-associated hepatotoxicity.
SIGNIFICANCE STATEMENT
Understanding the mechanisms of hepatotoxicity associated with tyrosine kinase inhibitors (TKIs) is fundamental to development of effective and safe intervention therapies for various cancers. Data generated in sandwich-cultured human hepatocytes, an in vitro model of drug-induced hepatotoxicity, revealed that TKIs upregulate bile acid synthesis and alter bile acid uptake and excretion. These findings provide novel insights into additional mechanisms of bile acid-mediated drug-induced liver injury, an adverse effect that limits the use and effectiveness of TKI treatment in some cancer patients.
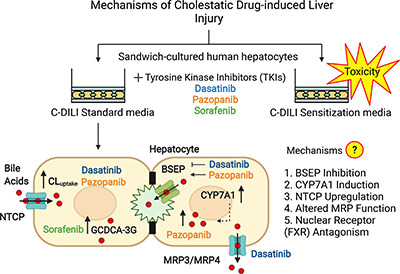
Introduction
Tyrosine kinase inhibitors (TKIs) are a breakthrough therapy in the treatment of several types of malignancies but have a high incidence of hepatotoxicity. As of April 2021, six TKIs (11%) approved for human use have boxed warnings for hepatotoxicity, and another 25 (46%) are labeled with warnings and precautions for hepatotoxicity. The majority of TKIs compete with ATP for binding to the conserved ATP-binding site within the kinase catalytic domain and display low selectivity among tyrosine kinases (Bhullar et al., 2018). This mechanism of action renders TKIs non-selective and prone to off-target effects. Dasatinib, pazopanib, and sorafenib are three TKIs commonly associated with hepatotoxicity as measured by liver function tests and elevated serum aminotransferase enzymes (Table 1).
TABLE 1.
Mechanism of Action and Disease Indications for Selected FDA-Approved Tyrosine Kinase Inhibitors.
| Tyrosine Kinase Inhibitors | Main Targets | Diseases | FDA Label; AST/ALT Elevationsa,b |
|---|---|---|---|
| Dasatinib | BCR-Abl, Src family, c-Kit, EphA2, Platelet-derived growth factor receptor (PDGFR) β | Chronic myeloid leukemia, acute lymphocytic leukemia | No warning; 50% of patients |
| Pazopanib | Vascular endothelial growth factor receptor (VEGFR) 1/2/3, PDGFRα/β, Fibroblast growth factor receptor (FGFR) 1/3, Kit, Lck, Fms, Itk | Renal cell carcinoma (RCC), soft-tissue sarcoma | Boxed Warning; 46–53% of patients |
| Sorafenib | VEGFR1/2/3, B-/C-Raf, mutant B-Raf, Kit, Flt3, RET, PDGFRβ | RCC, hepatocellular carcinoma, thyroid cancer | Warning and Precautions; 21–25% of patients |
aAST/ALT, aspartate/alanine aminotransferase; as reported by the US Food and Drug Administration (https://www.accessdata.fda.gov/scripts/cder/daf/).
Previous studies have attributed TKI-induced hepatotoxicity to reactive metabolite formation, time-dependent inhibition of cytochrome P450 (CYP) 3A4, increased reactive oxygen species, mitochondrial toxicity, and inhibition of the bile salt export pump (BSEP) (Jackson et al., 2018b). BSEP inhibition may lead to hepatic accumulation of toxic bile acids and cholestasis (Kenna et al., 2018). Dasatinib, pazopanib, and sorafenib competitively inhibit BSEP at IC50 values of 13.1, 10.3, and 8.0 µM, respectively, in membrane vesicle studies (Morgan et al., 2010; Morgan et al., 2013). Despite similar BSEP IC50 values, these three TKIs vary in the incidence and severity of hepatotoxicity (Table 1). Even though BSEP inhibition is an important mechanism of drug-induced liver injury (DILI) (Mosedale and Watkins, 2017), the correlation between BSEP inhibition and the incidence of DILI in humans is weak: 21% of compounds identified as “BSEP inhibitors” were associated with mild or no DILI, and 31% of compounds classified as “BSEP non-inhibitors” were linked with severe DILI (Kenna et al., 2018; Pedersen et al., 2013). This inadequate correlation is likely due to the fact that drug interactions causing altered bile acid homeostasis cannot be completely recapitulated in membrane vesicles, which lack key transporters, metabolic activity, and regulatory machinery, as well as other cellular constituents that are required for expression and membrane localization of transporters, and excretion of drugs and derived metabolites (Köck et al., 2014).
While BSEP is critical for hepatic bile acid excretion, bile acid homeostasis in hepatocytes is maintained in multiple ways. CYP7A1-mediated conversion of cholesterol to 7α-hydroxycholesterol is the first and rate-limiting step in bile acid synthesis in hepatocytes (Jelinek et al., 1990). The primary human bile acid species, cholic acid (CA) and chenodeoxycholic acid (CDCA), are synthesized in hepatocytes by CYP8B1 and CYP27A1 enzymes, respectively. (Chiang, 1998). Conjugation of CA and CDCA to taurine or glycine is catalyzed by the highly abundant hepatic enzymes, bile acid-CoA synthetase (BACS), and bile acid-CoA: amino acid N-acetyltransferase (BAAT) (O'Byrne et al., 2003). These unconjugated and conjugated bile acids are excreted via BSEP, the main canalicular bile acid efflux transporter (Gerloff et al., 1998; Noé et al., 2002). Secondary bile acid species are generated in the intestine, reabsorbed predominantly in the ileum, and returned to the liver via enterohepatic circulation (Chiang, 2013). Sodium taurocholate co-transporting polypeptide (NTCP) is the main hepatic basolateral uptake transporter for bile acids (Hagenbuch and Meier, 1994). Metabolism of bile acids to glucuronide and sulfate conjugates (Alnouti, 2009) and transport via multidrug resistance-associated proteins (MRPs), organic anion transporting polypeptides (OATPs), and organic solute transporter α/β also contribute to hepatic bile acid disposition and homeostasis (Beaudoin et al., 2020; Dawson et al., 2009). Intrahepatic bile acid concentrations are maintained by the farnesoid X receptor (FXR), which is activated by bile acids to repress CYP7A1 and NTCP transcription (Denson et al., 2001; Lu et al., 2000) and promote BSEP expression (Ananthanarayanan et al., 2001), thereby reducing synthesis and hepatic uptake of bile acids, and increasing bile acid efflux, respectively. Since several hepatic proteins regulate intracellular bile acid concentrations, TKI-mediated mechanisms of altered bile acid homeostasis other than competitive inhibition of BSEP warrant further investigation.
Hepatic membrane transport proteins are key determinants of the intracellular concentration of xenobiotics and endogenous substances, and influence the pharmacokinetics, efficacy, and toxicity of drugs (Chu et al., 2013; Giacomini et al., 2010). However, post-translational regulation of bile acid transporter localization and function, particularly by kinases, remains poorly understood (Crawford et al., 2018). Recently, TKIs such as nilotinib that target Lyn, a Src family kinase, were reported to inhibit OATP1B1 function and were associated with reduced phosphorylation of OATP1B1 (Hayden et al., 2021). The susceptibility of bile acid transporters to altered regulation and the clinical relevance of such perturbations to TKI-mediated hepatotoxicity has not been thoroughly investigated. Therefore, the objective of the current study was to investigate mechanisms of TKI-induced hepatotoxicity associated with altered bile acid homeostasis beyond the competitive inhibition of BSEP. Studies were conducted in sandwich-cultured human hepatocytes (SCHH), a commonly used in vitro model to investigate DILI mechanisms that maintain bile acid synthesis, secretion, and regulatory machinery for metabolic enzymes and transporters.
Materials and Methods
Chemicals, reagents, antibodies, and hepatocytes
Dasatinib (catalog #D-3307) and pazopanib (#S3012) were obtained from LC Laboratories (Woburn, MA) and Selleckchem (ThermoFisher Scientific, Pittsburgh, PA), respectively. Sorafenib tosylate was provided by Bayer HealthCare AG (Wuppertal, Germany). Bile acid species [CA (#1133503), CDCA (#C9377), glycocholic acid (GCA; #G2878), taurocholic acid (TCA; #T4009), glycochenodeoxycholic acid (GCDCA; #G7059), taurochenodeoxycholic acid (TCDCA; #T6260)], labetalol (#PHR1335), and DMSO (#41639) were purchased from Sigma-Aldrich (St. Louis, MO). Stable isotope-labeled internal standards CDCA-d4 (#C291902), TCA-d5 (#T008852), GCA-d5 (#G641357), GCDCA-d7 (#G641257), TCDCA-d5 (#T008133), and bile acid metabolites GCDCA-3-O-β-glucuronide (GCDCA-3G; #G641275) and GCDCA 3-sulfate (GCDCA-S; #G641270) were obtained from Toronto Research Chemicals. [3H]-TCA (#NET322250UC, > 97% radiochemical purity) was purchased from PerkinElmer, Inc. (Boston, MA). High-performance liquid chromatography grade acetonitrile, methanol, water, formic acid, and other reagents were purchased from Sigma-Aldrich (St. Louis, MO) or Fisher Scientific (Pittsburg, PA).
Antibodies for BSEP (F-6, #sc-74500), epidermal growth factor receptor (EGFR; D-20, #sc-31156), extracellular regulated kinase [ERK; C-16, #sc-93], vinculin (7F9, #sc-73614), and bovine anti-goat (#sc-2350) secondary antibody were obtained from Santa Cruz Biotechnology (Dallas, TX). Anti-NTCP (#ab 131084) and anti-Na+/K+ ATPase (#ab185065) antibodies were purchased from Abcam (Cambridge, MA). Anti-phospho-ERK (#MAB1018) antibody was purchased from R&D Systems (Minneapolis, MN), and HRP-conjugated goat anti-mouse (#115-035-003) and goat anti-rabbit (#111-035-144) secondary antibodies were from Jackson ImmunoResearch (West Grove, PA).
Cryopreserved Transporter Certified human hepatocytes (lots EGO, RVQ, and WID; Supplemental Table 1), QualGro seeding medium, thawing medium, overlay medium, culture medium, charcoal-stripped pooled human plasma, and the C-DILI assay kits were purchased from BioIVT (Baltimore, MD). Collagen I-coated BioCoat plates (24-well; #354408 and 96-well; #354407) and Matrigel Basement Membrane Matrix (#354234, lot 9091003) were obtained from Corning (Tewksbury, MA).
Sandwich-cultured human hepatocytes (SCHH)
Cryopreserved Transporter Certified human hepatocytes were cultured in a sandwich configuration in a 96-well plate format for cholestatic DILI assessment as described previously (Jackson and Brouwer, 2019; Jackson et al., 2018a), and in a 24-well format for mass spectrometry-based quantitation of bile acids, real-time quantitative PCR (RT-qPCR), membrane extraction, immunostaining, and evaluation of transporter function. Hepatocytes were thawed in QualGro thawing medium and diluted to a final density of 0.9 million cells/mL for 24-well plates or 0.8 million cells/mL for 96-well plates in QualGro seeding medium. Cells were seeded at a density of 0.45 million cells per well in a 24-well Collagen I-coated BioCoat plate or at 56,000 cells per well in a 96-well Collagen I-coated BioCoat plate (day 0). After 16-20 hours following seeding (day 1), hepatocytes were supplemented with ice-cold QualGro overlay medium containing 0.25 mg/mL of Matrigel to establish the sandwich-cultured configuration. On days 2–3, medium was replaced with warm QualGro culture medium.
Bile acid-mediated hepatotoxicity assessment
SCHH were established in a 96-well plate format and maintained for three days as described above. On day 4 of culture, SCHH were treated with controls and TKIs according to the C-DILI assay kit instructions (Jackson and Brouwer, 2019; Jackson et al., 2018a). SCHH were exposed to DMSO (0.1%; control), cyclosporine A (10 μM; negative control), imatinib (40 μM; direct toxicity control), troglitazone (75 μM; positive control for cholestatic toxicity) or TKIs at concentrations scaled from the reported maximum plasma concentration (Cmax, μM), as shown in Table 2. Compound stock solutions were diluted (1:1000) in either C-DILI culture medium or C-DILI sensitization medium. The C-DILI sensitization medium included free fatty acids and a pool of the most abundant human plasma bile acids at physiologically relevant concentrations as described by Jackson et al. (Jackson et al., 2018a). Each treatment was performed in triplicate using 125 μl of C-DILI culture medium (standard) or C-DILI sensitization medium per well. Following a 24-hour exposure period (day 5), cellular ATP and LDH release in cell culture medium were determined in parallel, using the CellTiter-Glo Luminescent Cell Viability Assay (#G7571, Promega) and the CytoTox-ONE Homogeneous Membrane Integrity Assay (#G7891, Promega), respectively.
TABLE 2.
Tyrosine Kinase Inhibitor (TKI) Concentrations Used to Assess Bile Acid-dependent Hepatotoxicity.
| TKI | Cmax a,b (μM) | Dose (mg) | Conc. 1 (μM, X-fold Cmax) |
Conc. 2 (μM, X-fold Cmax) |
Conc. 3 (μM, X-fold Cmax) |
|---|---|---|---|---|---|
| Dasatinib | 0.18 | 100 BID | 0.18 | 1.8 (10X) | 9.0 (50X) |
| Pazopanib | 132.80 | 800 QD | 0.66 (0.005X) | 6.6 (0.05X) | 66.4; 33.2 (0.5X; 0.25X) |
| Sorafenib | 4.30 | 400 BID | 0.43 (0.1X) | 4.3 | 43.0 (10X) |
a Cmax represents the highest mean or median steady-state total plasma concentration reported by the US FDA following chronic dosing.
b (Zhang et al., 2017).
Quantitation of TKI and bile acid concentrations
Sample preparation and bile acid quantification methodology was adapted from previously described methods (Jackson et al., 2016; Xie et al., 2015). SCHH (n = 3 or 4) were cultured for three days and exposed to 0.1% DMSO control, dasatinib (1.8 μM), pazopanib (6.6 μM), or sorafenib (4.3 μM) for 24 hours. The TKI concentrations for SCHH exposure were determined using the C-DILI assay and represent the lowest concentrations that caused bile acid-dependent toxicity. On day 5, cells were washed once with phosphate-buffered saline (PBS) and frozen at -80°C prior to analysis. Cells were lysed in 300 µl of acetonitrile containing six internal standards (50 nM each of GCA-d5, TCA-d5, TCDCA-d5, GCDCA-d7, and CDCA-d4 for bile acid quantitation, and 25 nM labetalol for TKI quantitation). Samples were transferred to a Whatman 96-well Unifilter 25 µm MBPP/0.45 µm PP filter plate (#7770-0062; Whatman) stacked on a 96-well deep well plate and shaken for 15 minutes. Lysate was filtered into the deep well plate by centrifugation (2390 × g, 2 minutes) and the filtrate was evaporated to dryness. Samples were reconstituted in 125 µl of 50% acetonitrile in water containing 0.1% formic acid and mixed for 10 minutes on a plate shaker. The reconstituted samples were transferred to a Millipore 0.45-µm filter plate (#MSHVN45; Millipore) and filtered into a 96-well plate (#5042-1386, Agilent Technologies) by centrifugation (2390 × g, 2 minutes). The plate was sealed with a silicone cap mat before liquid chromatography with tandem mass spectrometry (LC-MS/MS) analysis. Separate calibration curves were generated as a series of concentrations (0.1, 0.5, 1, 5, 10, 50, 100, 500, 1000, 5000 nM) for TKIs (dasatinib, pazopanib, and sorafenib) and bile acids (Supplemental Table 2) in charcoal-stripped pooled human plasma: PBS (1:1, v/v). Cell culture medium was also collected and frozen at -80°C prior to analysis. Analytes in 100 µl of standards, quality controls, and medium samples were extracted with 300 µl of acetonitrile containing internal standards using a similar process as described above.
Chromatographic separation was achieved using an ACQUITY BEH C18 column (1.7 μm, 100 mm × 2.1 mm internal dimensions; Waters, Milford, MA) and a ultra-high performance liquid chromatography tandem mass spectrometry system (Agilent 1290 Infinity II, Agilent Technologies, Santa Clara, CA). Mobile phase solvents used were 100% water (A) and 100% acetonitrile (B), both with 0.1% formic acid (v/v). The flow rate was 0.4 mL/min with the following mobile phase gradient: 0 − 1 minute (5% B), 1 − 5 minutes (5 − 25% B), 5 − 15.5 minutes (25 − 40% B), 15.5 − 17.5 minutes (40 − 95% B), 17.5 − 19 minutes (95% B), 19 − 19.5 minutes (95 − 5% B), and 19.5 − 21 minutes (5% B) for bile acid quantitation. For TKI measurement, the flow rate was 0.4 ml/min with the following gradient: 0 − 0.3 minutes (5% B), 0.3 − 3.0 minutes (5 − 95% B), 3.0 − 3.6 minutes (95% B), 3.6 − 3.7 minutes (95 − 5% B), 3.7 − 5.0 minutes (5% B). The column was maintained at 40°C and the injection volume of all samples was 10 μl. The mass spectrometer used was a SCIEX triple quadrupole 6500 instrument (AB Sciex, Framingham, MA) and was operated in negative ion mode for bile acid quantitation and positive ion mode for TKI quantitation. The data were acquired with multiple reaction monitoring (MRM), and the collision energy for each bile acid species and TKI is listed in Supplemental Table 2. Pazopanib and sorafenib samples that were above the upper limit of quantitation were diluted 20-fold with blank solvent containing labetalol internal standard. The lower limits of quantitation were: 0.1 nM for GCA, GCDCA, TCA, TCDCA and pazopanib; 0.5 nM for GCDCA-3G, dasatinib, and sorafenib; and 1 nM for GCDCA-S. The upper limit of quantitation was 5000 nM for GCA, 100 nM for pazopanib, and 1000 nM for all other bile acid species and TKIs.
Cellular protein binding
Unbound cellular concentrations of TKIs in SCHH were estimated by measuring the fraction unbound in cells (fu,cell) using a human hepatoma cell line (HuH-7). Cellular protein binding was performed in HuH-7 cells based on equivalence established between fu,cell measured in human hepatocytes and HuH-7 cells (Riccardi et al., 2018). HuH-7 cells (JCRB0403) were purchased from Sekisui Xenotech (Kansas City, KS). Cells were cultured in 150 mm dishes at 37°C with 5% CO2 using maintenance medium composed of Dulbecco’s modified Eagle’s medium (DMEM, #11995-065, Thermo Fisher Scientific), 10% FBS (#F2442, Sigma-Aldrich), 100 U/ml of penicillin, and 100 μg/ml of streptomycin (#10378-016, Thermo Fisher Scientific). HuH-7 cells were diluted in PBS at 45 million cells per milliliter and homogenized using a TissueRuptor II (Qiagen, Germantown, MD) for 1-2 minutes at room temperature followed by a Kontes dounce tissue grinder (Kimble Chase Life Science, Vineland, NJ) with 5 strokes (tight pestle) until smooth. Equilibrium dialysis was performed using an HTD96 plate and dialysis membranes of molecular weight cut-off 12-14 kDa (HTDialysis, Gales Ferry, CT). Dialysis membranes were washed with distilled water, 20% ethanol in water (v/v), and stored in PBS overnight. TKIs (dasatinib, pazopanib, and sorafenib) or rosuvastatin (control) were added to the cell homogenate (600 µl) to achieve a final concentration of 2 µM and mixed thoroughly. A 100-µl aliquot of cell homogenate was added to the top “donor” chamber of the HTD96 plate and blank PBS was added to the bottom “receiver” chamber in triplicate. The HTD96 device was sealed with a gas permeable membrane and placed onto an orbital shaker at 200 rpm in a CO2 incubator (5% CO2/95% air, 75% relative humidity) for 5 hours at 37°C. Alongside the equilibrium dialysis, an abbreviated stability study was conducted at 37°C; a 25-µl aliquot of the remaining cell homogenate was precipitated in 200 µl of acetonitrile containing 25 nM labetalol internal standard at 0, 1, 2, and 5 hours. Each sample for stability was assessed in duplicate. After 5 hours of dialysis, 25 μl of cell homogenate and 25 μl of PBS were matrix-matched with blank PBS and blank cell homogenate, respectively, prior to precipitation in 200 μl of cold acetonitrile containing 25 nM of labetalol. Samples were mixed by vortex for 1 minute before centrifugation (3000 × g, 5 minutes). The supernatant was transferred into a 96-well plate (#5042-1386, Agilent Technologies) and peak area ratios were measured without a calibration curve using the LC-MS/MS conditions described above. The fraction unbound in diluted PBS (fu,d) and undiluted fu,cell, and stability as a percentage remaining were calculated using the following equations:
 |
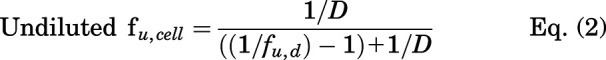 |
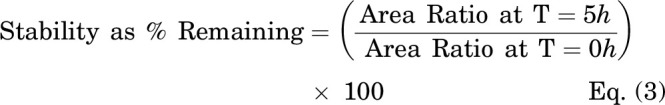 |
where area ratio is the peak area of analyte divided by the peak area of the internal standard (labetalol) and D is the dilution factor calculated based on the cell density and diameter (14.1 µm), i.e., 1012/[4/3 × 3.14 × (14.1/2)3 x 45 × 106] (Riccardi et al., 2018).
Gene expression studies using RT-qPCR
SCHH were established in a 24-well plate format, and on day 5 of culture, hepatocytes were treated with 0.1% DMSO control, dasatinib (1.8 μM), pazopanib (6.6 μM), or sorafenib (4.3 μM) for 8 hours. RNA was extracted from hepatocytes using the TRI Reagent according to the manufacturer’s protocol. The concentration and purity of isolated RNA were measured using a NanoDrop spectrophotometer. Reverse transcription of the RNA (2 µg) to cDNA was performed using the Applied Biosystems High-Capacity cDNA Reverse Transcription Kit. Gene expression of SLC10A1 (gene encoding NTCP), ABCB11 (gene encoding BSEP), CYP7A1, CYP8B1, CYP27A1, BAAT, BACS, and CYP3A4 was measured using RT-qPCR for each sample in triplicate with the QuntStudio 6 Flex System. The analyzed genes and the gene-specific TaqMan assays used for RT-qPCR are listed in Supplemental Table 3. Gene expression was calculated using the ΔΔCt method (Livak and Schmittgen, 2001), in which β-actin was used as the housekeeping gene.
Membrane protein extraction and immunostaining
On day 4, SCHH in a 24-well plate format were treated with 0.1% DMSO control, dasatinib (1.8 μM), pazopanib (6.6 μM), or sorafenib (4.3 μM) in warm QualGro culture medium for 24 hours. Membrane and cytosolic protein from each SCHH were extracted using the ProteoExtract Native Membrane Protein Extraction Kit (#444810, Millipore Sigma-Aldrich), following the manufacturer’s instructions. Briefly, cells were washed twice with cold wash buffer and treated with 200 μl of cold Extraction Buffer I containing protease and phosphatase inhibitors under gentle agitation (10 minutes, 4°C), followed by centrifugation (16,000 × g, 4°C, 15 minute). Subsequently, the supernatant (cytosolic fraction) was collected, and the pellet was re-suspended in 150 μl of cold Extraction Buffer II containing protease and phosphatase inhibitors. Following incubation under gentle agitation (30 minutes, 4°C), centrifugation was performed (16,000 × g, 4°C, 15 minutes) and the supernatant (membrane fraction) was collected. The total protein in the membrane and the cytosolic fraction was determined using the Pierce BCA Protein Assay Kit.
Proteins (30–35 μg) were mixed with NuPAGE LDS sample buffer and 50 mM of dithiothreitol and subjected to SDS-PAGE using NuPAGE 7% Tris-Acetate or 4–12% Bis-Tris gels. The proteins were transferred to a polyvinylidene difluoride membrane overnight at 15 V. Following the transfer, the membranes were blocked in tris-buffered saline/Tween 20 (TBS-T) buffer [(Tris-buffered saline) and 0.1% (v/v) Tween 20] containing 5% (w/v) nonfat dry milk (#1706404, Bio-Rad). The blocked membranes were incubated overnight at 4°C with primary antibodies [anti-BSEP (1:200), anti-NTCP (1:1000), anti-EGFR (1:200), anti-phospho-ERK (1:1000), anti-ERK (1:200), anti-vinculin (1:200), or anti-Na+/K+ ATPase (1:10,000)] diluted in TBS-T with 5% BSA (w/v). Subsequently, membranes were incubated with HRP-conjugated goat anti-mouse or goat anti-rabbit or bovine anti-goat secondary antibodies, diluted at 1:10,000 in blocking buffer, for 1 hour at room temperature. Chemiluminescent signal was detected using SuperSignal West Femto Maximum Sensitivity Substrate and captured with a Molecular Imager VersaDoc imaging system (BioRad, Hercules, CA).
Assessment of transporter function using B-CLEAR
On day 4, SCHH in a 24-well format were treated with 0.1% DMSO control, dasatinib (1.8 μM), pazopanib (6.6 μM,) or sorafenib (4.3 μM) in warm QualGro culture medium. Following a 24-hour exposure to TKIs (day 5), the biliary excretion index was measured using B-CLEAR technology (Brouwer et al., 2013; Liu et al., 1999; Swift et al., 2010). Cells were washed twice in standard Hanks' balanced salt solution (HBSS) or Ca2+-free HBSS containing 1 mM of egtazic acid (EGTA), prior to a 10-minute pre-incubation in standard or Ca2+-free HBSS buffer at 37°C. The standard HBSS buffer represents “Cells+Bile” or Plus (+) buffer containing Ca2+ and Mg2+, whereas the Ca2+-free HBSS represents “Cells” or Minus (−) buffer that disrupts tight junctions. At the end of the 10-minute incubation, cells were exposed to 2 μM of [3H]-TCA (200 nCi/ml) in standard HBSS for 10 minutes at 37°C. Following treatment, the buffer was removed, and cells were washed three times in ice-cold standard HBSS buffer. The plates were frozen at -20°C until processed further by lysis using 400 μl of 0.5% Triton X-100 and 0.005% Antifoam-A in PBS. Radioactivity of cell lysates was measured using Bio-Safe II counting cocktail (Research Products International Corp., Mt Prospect, IL) and a Tri-Carb 3100TR liquid scintillation analyzer (PerkinElmer Inc.). Total protein content was determined using Pierce BCA Protein Assay Kit following the manufacturer’s instructions. Accumulation of TCA in cells and bile canaliculi (Cells+Bile) and cells only (Cells) was measured in SCHH (n = 3) and normalized to total protein content per well. The biliary excretion index (BEI), which represents the percentage of total mass accumulated that is excreted into bile was calculated (Eq. 4). Additionally, the in vitro uptake clearance (CLuptake) was calculated at 10 minutes using Eq. 5.
 |
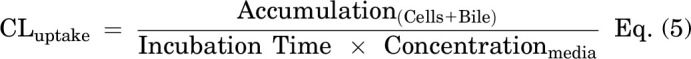 |
Data analysis
An ordinary one-way or repeated measures two-way ANOVA with Sidak’s, Dunnett’s, or Tukey’s multiple comparison correction was used for all statistical analyses (GraphPad Prism 7.03). Bile acid and TKI concentrations in SCHH and medium were analyzed with area ratio and calibration curves of analytes using MultiQuant software (AB Sciex, Framingham, MA). These concentrations were normalized to total protein content measured using the Pierce BCA Protein Assay Kit. In addition, cellular TKI concentrations were calculated by dividing the amount of analyte by the hepatocellular volume for human hepatocytes (7.69 μl/mg protein) (Qualyst Transporter Solutions Technical Application Bulletin; Lu et al., 2016). Western blot data were analyzed using ImageJ-based densitometry, normalized to Na+/K+ ATPase (loading control), and relative abundance of proteins in TKI-treated SCHH compared with 0.1% DMSO control was calculated.
Results
TKIs caused bile acid-dependent toxicity at clinically relevant concentrations in SCHH
Three TKIs were evaluated for bile acid-dependent toxicity at clinically relevant total concentrations (Table 2) in SCHH using the C-DILI assay. Results from control treatments (0.1% DMSO), cyclosporine A (CsA; negative control), imatinib (direct toxicity control), and troglitazone (TGZ; positive control for cholestatic drugs) were consistent with previous reports (Jackson and Brouwer, 2019). As shown in Fig. 1A, CsA (10 μM) did not cause toxicity in SCHH, as it did not change cellular ATP or LDH release in either medium relative to control, imatinib (40 μM) exhibited comparably high toxicity in both standard and sensitization media, and troglitazone (TGZ; 75 μM) was toxic only in the sensitization medium.
Fig. 1.

Bile acid-dependent toxicity from tyrosine kinase inhibitors in sandwich-cultured human hepatocytes (SCHH). Bile acid-dependent toxicity was measured in SCHH (lots EGO and WID) in Sensitization Medium (black bars, closed circles) and Standard Medium (white bars, open circles). SCHH were treated with (A) controls including 0.1% DMSO control, cyclosporine A (CsA; 10 μM), imatinib (40 μM) or troglitazone (TGZ; 75 μM) or (B) dasatinib, pazopanib, or sorafenib for 24 hours. ATP content and LDH release were normalized to 0.1% DMSO control. Data are shown as mean and standard deviation (n = 3). Statistically significant differences were determined by a repeated measures two-way ANOVA, with multiple comparisons corrected using the Sidak test (* P value < 0.05, **<0.0001, standard versus sensitization medium). #Precipitation was observed at 66.4 μM pazopanib, and therefore, the highest concentration was decreased to 33.2 μM for lot EGO. Imatinib and TGZ ATP values <2% of control are not visible in Fig. 1A.
Dasatinib, pazopanib, and sorafenib decreased SCHH viability and reduced membrane integrity, as observed by lower ATP content and higher LDH release, when treated with C-DILI sensitization medium (Fig. 1B). ATP levels expressed as a percentage of DMSO control were significantly decreased in SCHH lot EGO exposed to the sensitization medium compared with the standard medium by dasatinib (1.8 and 9.0 μM), pazopanib (6.6 μM), and sorafenib (4.3 μM). These concentrations corresponded to 10- and 50-fold Cmax, respectively, for dasatinib, 0.05-fold Cmax for pazopanib, and Cmax for sorafenib. The differential decline in ATP content in SCHH exposed to sensitization medium was confirmed with an increase in LDH release (relative to DMSO control) for dasatinib (1.8 and 9.0 μM), pazopanib (6.6 μM), and sorafenib (4.3 μM). Similarly, in SCHH lot WID, dasatinib (1.8 and 9.0 μM), pazopanib (6.6 μM), and sorafenib (4.3 μM) significantly decreased cellular ATP levels and increased LDH release in the sensitization medium compared with the standard medium. Higher exposure to sorafenib (43 μM) showed high toxicity in the standard and sensitization media in both SCHH lots (Fig. 1B). Taken together, SCHH treated for 24 hours with dasatinib 1.8 μM (and 9.0 μM), pazopanib 6.6 μM, and sorafenib 4.3 μM exhibited bile acid-dependent toxicity based on the C-DILI assay; these TKI concentrations, which were only toxic in the sensitization media, were used in subsequent studies.
Unbound cellular concentrations of TKIs were below the reported IC50 for BSEP inhibition
Cellular TKI concentrations were measured in SCHH following a 24-hour treatment with dasatinib (1.8 μM), pazopanib (6.6 μM), and sorafenib (4.3 μM). LC-MS/MS analysis of TKI-treated SCHH revealed that the total cellular concentrations of dasatinib, pazopanib and sorafenib were 5–51, 213–288, and 296–587 µM, respectively, in three lots of SCHH (Fig. 2). Cellular TKI concentrations in SCHH were calculated assuming that the BEI of unchanged parent was minimal based on pilot studies and published data. For example, the BEI of sorafenib in SCHH after a 10-minute incubation was reported as ≤ 11% (Swift et al., 2013). The cellular protein binding of dasatinib, pazopanib, and sorafenib was calculated as 99.08%, 99.36%, and 99.90% bound, respectively, using Eq. 2, in which D was 15.15. Based on the calculated fu,cell, the unbound cellular concentrations of dasatinib (0.05-0.47 µM), pazopanib (1.36-1.84 µM), and sorafenib (0.30-0.59 µM) were below the reported BSEP IC50 values (Table 3). The cellular protein binding of rosuvastatin was 85.8%, consistent with previous data (Riccardi et al., 2018). Stability was acceptable with > 70% remaining for the TKIs and rosuvastatin (control), as measured by Eq. 3.
Fig. 2.

Cellular concentration of tyrosine kinase inhibitors (TKIs) in sandwich-cultured human hepatocytes (SCHH) treated for 24 hours. TKI concentrations were measured in SCHH (lots EGO, RVQ, and WID) treated with dasatinib (1.8 µM), pazopanib (6.6 µM), or sorafenib (4.3 µM) using liquid chromatography with tandem mass spectrometry. Concentrations of each TKI were calculated, normalized to total protein and hepatocellular volume, and plotted as mean ± standard deviation (n = 3 or 4).
TABLE 3.
Total cellular tyrosine kinase inhibitor (TKI) concentrations, fraction bound, and unbound TKI concentrations in three lots of SCHH compared with published BSEP IC50 values.
| Cellular TKI Concentrations (µM, Mean ± standard deviation) | |||
|---|---|---|---|
| Lot/TKI | Dasatinib | Pazopanib | Sorafenib |
| EGO | 27 ± 10 | 288 ± 30 | 517 ± 41 |
| RVQ | 51 ± 5 | 257 ± 11 | 587 ± 36 |
| WID | 5 ± 1 | 213 ± 19 | 296 ± 17 |
| Cellular Fraction Unbound (fu,cell) | 0.0092 | 0.0064 | 0.0010 |
| EGO (unbound) | 0.25 ± 0.09 | 1.84 ± 0.19 | 0.52 ± 0.04 |
| RVQ (unbound) | 0.47 ± 0.05 | 1.64 ± 0.07 | 0.59 ± 0.04 |
| WID (unbound) | 0.05 ± 0.01 | 1.36 ± 0.12 | 0.30 ± 0.02 |
| Reported BSEP IC50a | 13.1 | 10.3 | 8.0 |
BID, twice daily dosing; QD, daily dosing.
a (Morgan et al., 2010; Morgan et al., 2013)
TKIs increased expression of CYP7A1 mRNA
To further understand the mechanism driving bile acid-dependent toxicity, mRNAs were quantified for CYP7A1, CYP8B1, CYP27A1, BAAT, and BACS, the primary bile acid-synthesizing and conjugating enzymes, and the main bile acid transporters SLC10A1 and ABCB11. Since TKIs are metabolized by CYP3A4, CYP3A4 mRNA also was measured. In both SCHH lots (EGO and WID), SLC10A1, CYP8B1, CYP27A1, BAAT, BACS, and CYP3A4 mRNA did not significantly differ from the DMSO control after an 8-hour treatment with dasatinib, pazopanib or sorafenib (Fig. 3A). In contrast, CYP7A1 mRNA was significantly induced by dasatinib (6.2 to 7.8-fold), pazopanib (5.7 to 9.3-fold) and sorafenib (1.9 to 3.9-fold). ABCB11 mRNA showed inconsistent trends in both SCHH lots treated with TKIs (Fig. 3A).
Fig. 3.

Gene expression analysis by RT-qPCR and quantitative analysis of bile acid concentrations in sandwich-cultured human hepatocytes (SCHH) treated with tyrosine kinase inhibitors (TKIs). (A) mRNA was measured in SCHH (lots EGO and WID) after an 8-hour incubation with 0.1% DMSO control (DMSO Ctrl), dasatinib (1.8 μM), pazopanib (6.6 μM), or sorafenib (4.3 μM). Threshold cycle (CT) values of each gene of interest (SLC10A1, ABCB11, CYP7A1, CYP8B1, CYP27A1, BAAT, BACS, and CYP3A4) were normalized to the housekeeping gene β-actin (ACTB) and compared with 0.1% DMSO control. Data are plotted as mean ± standard deviation (n = 3). Statistically significant differences were determined by a repeated measures two-way ANOVA with Dunnett’s multiple comparison test (*P value < 0.05, **<0.0001, TKI versus control). (B) Bile acid concentrations were quantified in SCHH (lots EGO, RVQ and WID) treated with 0.1% DMSO control, dasatinib (1.8 µM), pazopanib (6.6 µM) or sorafenib (4.3 µM) for 24 hours and in medium from each well using liquid chromatography with tandem mass spectrometry (LC-MS/MS). Bile acid concentrations in “Cells+Bile” were normalized to total protein. Average fold change (n = 3 or 4) of each bile acid species [glycocholic acid (GCA), glycochenodeoxycholic acid (GCDCA), GCDCA-3-O-β-glucuronide (GCDCA-3G), GCDCA 3-sulfate (GCDCA-S), taurochenodeoxycholic acid (TCDCA), and taurocholic acid (TCA)] was calculated, compared with 0.1% DMSO control in SCHH and medium, and plotted as a heat map (red = increase, blue = decrease). (C) Average total bile acid species in “Cells+Bile” and medium were plotted based on SCHH lot and treatment. BA, bile acids.
TKIs caused modest changes in bile acids in SCHH and medium after a 24-hour exposure in standard medium (Fig. 3B and 3C), consistent with induction of CYP7A1 mRNA. Dasatinib increased the concentrations of six bile acid species in the medium up to 2.7-fold (GCA; lot RVQ), and pazopanib increased GCA, TCDCA, and metabolites of GCDCA (GCDCA-3G and GCDCA-S) in SCHH up to 1.8-fold (lot RVQ). GCDCA-3G was increased up to 2.7-fold with sorafenib treatment in SCHH and medium (Fig. 3B). GCA was the most abundant bile acid present in SCHH and medium (Fig. 3C). An increase of up to 2.3-fold in total bile acid concentrations in the medium was observed with dasatinib treatment compared with control. Pazopanib increased total bile acids in SCHH up to 1.4-fold; total bile acids were higher relative to control in all three SCHH lots (Fig. 3C). Total bile acids in SCHH and medium appeared to be unchanged or slightly decreased by sorafenib. Changes in each individual bile acid species in SCHH and medium are reported in Supplemental Fig. 1. CA and CDCA concentrations in SCHH and medium were below the lower limit of quantitation.
Since phosphorylation and activation of ERK repress CYP7A1 expression (Byun et al., 2018; Song et al., 2009), the phosphorylation status of ERK also was examined. Dasatinib, pazopanib, and sorafenib treatment for 24 hours inhibited ERK phosphorylation in lot RVQ (Supplemental Fig. 2).
Hepatic NTCP membrane protein abundance was increased by TKIs
To investigate the role of bile acid transporters in TKI-mediated cholestatic hepatotoxicity, the effect of TKIs on bile acid transport protein abundance was evaluated using western blot analysis. NTCP membrane protein abundance was increased up to 2.0-fold by all TKIs tested except for sorafenib in lot RVQ (Fig. 4). BSEP membrane protein abundance showed an increasing trend in all SCHH lots treated with dasatinib and pazopanib: however, this trend was not significant. Additionally, abundance of the EGFR membrane protein, which is known to co-traffic to the basolateral membrane with NTCP (Wang et al., 2016), was unchanged by TKI treatment.
Fig. 4.

Effect of tyrosine kinase inhibitors (TKIs) on the membrane protein abundance of hepatic bile acid transporters in sandwich-cultured human hepatocytes (SCHH). SCHH were treated with 0.1% DMSO control (Ctrl), dasatinib (1.8 μM), pazopanib (6.6 μM) or sorafenib (4.3 μM) for 24 hours. Abundance of bile salt export pump (BSEP), sodium taurocholate co-transporting polypeptide (NTCP), epidermal growth factor receptor (EGFR), and Na+/K+ ATPase (loading control) was evaluated using immunostaining of membrane fractions harvested from SCHH lots EGO, RVQ, and WID. Densitometry was performed using ImageJ and BSEP, NTCP, and EGFR signals were normalized to Na+/K+ ATPase. All treatments were performed in triplicate except in lot WID (n = 2). Statistically significant differences in lots EGO and RVQ were assessed using repeated measures two-way ANOVA with Dunnett’s multiple comparison test (*, P < 0.05, **, P < 0.0001, TKI versus control).
Dasatinib and pazopanib increased bile acid uptake in SCHH
TKI-mediated changes in bile acid transporter function were assessed using the B-CLEAR assay in SCHH pretreated with TKIs for 24 hours. Dasatinib increased TCA accumulation in “Cells+Bile” (Standard HBSS Buffer) and in “Cells” (Ca2+-free HBSS Buffer) compared with DMSO control (Fig. 5). Consistent with these findings, the TCA uptake clearance was increased up to 1.4-fold by dasatinib in three SCHH lots (Table 4). Interestingly, the BEI trended up to one-third lower in dasatinib-treated SCHH compared with DMSO control even though TCA uptake was increased. Pazopanib also increased TCA accumulation in “Cells+Bile” in SCHH lots EGO and RVQ consistent with an increase in CLuptake up to 1.3-fold. In contrast to dasatinib, pazopanib treatment increased the BEI in three SCHH lots relative to control by up to 1.4-fold. Sorafenib treatment resulted in a minor decrease in TCA accumulation in “Cells+Bile” and CLuptake (Supplemental Fig. 3).
Fig. 5.

Effect of tyrosine kinase inhibitors (TKIs) on the function of hepatic bile acid transporters in sandwich-cultured human hepatocytes (SCHH). The function of bile acid transporters was measured using B-CLEAR technology in SCHH (lots EGO, RVQ, and WID); [3H]-taurocholate (TCA) biliary excretion index (BEI; %) and uptake clearance (CLuptake; μl/min/mg protein) values (Table 4) were calculated using Eqs. 4 and 5, respectively. SCHH were pretreated with 0.1% DMSO control (DMSO Ctrl), dasatinib (1.8 μM), pazopanib (6.6 μM), or sorafenib (4.3 μM) for 24 hours. Accumulation of 2 μM [3H]-TCA (200 nCi/ml) was measured in “Cells+Bile” and “Cells” using standard (+ Ca2+; black bars) and Ca2+-free (− Ca2+; white bars) Hank’s balanced salt solution (HBSS) buffer. Data are plotted as mean ± standard deviation (n = 3). Statistically significant differences were assessed using an ordinary two-way ANOVA with Dunnett’s multiple comparison test (*, P < 0.05, **, P < 0.0001, TKI versus DMSO control).
TABLE 4.
Effect of tyrosine kinase inhibitors on uptake clearance (CLuptake) and biliary excretion index (BEI) of [3H]-taurocholate (TCA).
| DMSO Ctrl | Dasatinib | Pazopanib | ||||
|---|---|---|---|---|---|---|
| TCA BEI (%) | TCA CLuptake (µL/min/mg protein) |
TCA BEI (%) | TCA CLuptake (µL/min/mg protein) |
TCA BEI (%) | TCA CLuptake (µL/min/mg protein) |
|
| Lot EGO | 41.4 | 16.1 ± 1.6 | 26.1 | 23.2 ± 1.6 | 56.2 | 21.5 ± 1.5 |
| Lot RVQ | 59.6 | 13.4 ± 0.3 | 44.3 | 15.8 ± 0.9 | 71.0 | 15.9 ± 0.9 |
| Lot WID | 60.9 | 15.1 ± 1.3 | 49.4 | 17.8 ± 0.3 | 72.6 | 14.2 ± 0.5 |
Discussion
DILI remains the leading cause of acute liver failure and a major adverse event in drug development. Mechanisms of DILI include oxidative stress, mitochondrial dysfunction, apoptosis, immune-mediated reactions, reactive metabolite formation, time-dependent inhibition of CYP3A4, and altered bile acid homeostasis (Mosedale and Watkins, 2017). Sensitization of SCHH with bile acid mixtures is one approach that has been reported to assess DILI (Chatterjee et al., 2014; Jackson et al., 2018a; Ogimura et al., 2011; Oorts et al., 2016). Recently, the C-DILI assay was introduced as a mechanism-based in vitro method using SCHH to evaluate the bile acid-dependent toxicity of compounds, and to assess their potential for cholestatic DILI (Jackson et al., 2018a). This assay integrates several mechanisms of cholestatic toxicity, including increased bile acid synthesis, metabolism, FXR antagonism, and altered transport (i.e., increased uptake, decreased biliary excretion and/or decreased basolateral efflux). In the present study, dasatinib and sorafenib concentrations tested were scaled from the reported Cmax values, assuming higher hepatic exposure, and established the lowest concentration at which bile acid-dependent toxicity was observed. Since the reported Cmax for pazopanib is high, in vitro test concentrations were scaled down due to solubility limitations. Based on the results of the C-DILI assay, clinically relevant concentrations (dasatinib: 1.8 μM equivalent to 10-fold Cmax; pazopanib: 6.6 μM equivalent to 0.05-fold Cmax; sorafenib: 4.3 μM equivalent to Cmax) were selected for further investigation. Unbound cellular TKI concentrations were below the IC50 reported for BSEP inhibition, suggesting that mechanisms other than direct BSEP inhibition may contribute to TKI-mediated hepatotoxicity and warrant further investigation.
A novel finding of this work was the marked upregulation of CYP7A1 mRNA by dasatinib, pazopanib and sorafenib. This upregulation was specific for CYP7A1 and was consistent with the observed increase in total bile acid concentrations with dasatinib and pazopanib. The more modest ∼2 to 4-fold induction of CYP7A1 mRNA with an 8-hour sorafenib treatment was insufficient to increase bile acid concentrations after 24 hours. Previous studies showed that overexpression of CYP7A1 in HepG2 cells markedly activated the classic pathway of bile acid biosynthesis, increased CYP7A1 activity, and resulted in an approximately 2-fold increase in total bile acids synthesized after CYP7A1 overexpression (Pandak et al., 2001). CYP7A1 is repressed by ERK phosphorylation via growth factor-activated receptor tyrosine kinases in a mitogen-activated protein kinase (MAPK)- and Src-dependent manner (Byun et al., 2018; Song et al., 2009). Dasatinib may inactivate ERK indirectly via EGFR and PDGFR inhibition or directly by Src inhibition. Pazopanib inhibits fibroblast growth factor receptor (FGFR) 4 and Src, and sorafenib inhibits platelet-derived growth factor receptor (PDGFR), vascular endothelial growth factor receptor (VEGFR), and B-/C-Raf (Karaman et al., 2008). Inhibition of the receptor tyrosine kinases, Raf and Src would lead to ERK inactivation and increased CYP7A1 mRNA. The mechanism of the pazopanib-mediated increase in CYP7A1 mRNA agrees with previous reports in cynomolgus monkeys; FGFR4 inactivation leads to CYP7A1 upregulation (Pai et al., 2012). Other TKIs that are FGFR4 inhibitors, such as FGF401, also increase CYP7A1 mRNA in cancer cell lines and hepatotoxicity was reported in clinical trials (Liu et al., 2020; Weiss et al., 2019). Taken together, increased bile acid synthesis and CYP7A1 de-repression may contribute to increased bile acids in SCHH and define a novel mechanism of dasatinib- and pazopanib-induced hepatotoxicity.
Gene expression studies revealed that dasatinib, pazopanib, and sorafenib did not affect CYP3A4 mRNA. This suggested that these TKIs had no major acute or direct effect on several nuclear receptors, such as pregnane X (PXR) or constitutively active receptor (CAR) and hepatocyte nuclear factor 4 (HNF4) (Honkakoski and Negishi, 2000; Tirona et al., 2003; Zollner and Trauner, 2009). Although concurrent induction of BSEP and suppression of NTCP are thought to protect hepatocytes from hydrophobic bile acid-mediated toxicity (Anwer, 2004), NTCP protein, not mRNA, was invariably increased with dasatinib, pazopanib and sorafenib treatment. Since FXR agonism represses SLC10A1 mRNA (Denson et al., 2001) and FXR antagonism de-represses CYP7A1 mRNA (Lu et al., 2000) to maintain bile acid homeostasis via negative feedback regulation, direct modulation of FXR was examined using a luciferase reporter assay in HepG2 cells. No significant direct FXR agonism or antagonism by TKIs was detected (Supplemental Methods and Supplemental Fig. 4). This novel finding of increased NTCP protein was not due to an increase in SLC10A1 mRNA or in EGFR protein, which co-traffics with NTCP to the membrane (Wang et al., 2016). Thus, the increase in NTCP membrane protein does not appear to be transcriptionally driven or mediated by trafficking but may be due to decreased internalization and degradation. It is also noteworthy that the increased abundance of NTCP may explain case reports of hepatitis B viral reactivation in patients administered dasatinib (Ando et al., 2015). The precise mechanism of increased NTCP abundance may be phosphorylation-dependent and is the subject of ongoing investigations.
The disposition of bile acids in hepatocytes relies on multiple transporters; NTCP is primarily responsible for basolateral uptake, but OATPs may also contribute. Dasatinib inhibits OATP1B1 and OATP1B3 without affecting membrane protein abundance (Pahwa et al., 2017); pazopanib and sorafenib are also potent OATP1B1 inhibitors in vitro (Hu et al., 2014). Therefore, increased NTCP protein abundance in hepatocytes exposed to dasatinib would be expected to increase bile acid uptake, consistent with the observed increase in TCA CLuptake measured using the B-CLEAR assay. Pazopanib increased TCA CLuptake in two out of three SCHH lots. Pazopanib also increased “Cell+Bile” accumulation of bile acids, consistent with increased hepatocyte bile acid uptake and enhanced bile acid synthesis; GCA, GCDCA-3G, GCDCA-S and TCDCA were increased to the greatest extent.
Biliary excretion of bile acids in humans is dominated by BSEP and, to a lesser extent, by MRP2. The biliary excretion index (BEI) of TCA was decreased by dasatinib treatment despite increased bile acid uptake and enhanced bile acid synthesis. BSEP inhibition would not be expected in SCHH because the unbound cellular dasatinib concentrations in this study were more than an order of magnitude lower than the reported IC50 value of 13.1 µM (Morgan et al., 2013). There are several possible explanations for this apparent discrepancy. Membrane vesicles prepared from insect Sf9 cells overexpressing BSEP may overestimate the relevant BSEP IC50 in human hepatocytes. Alternatively, a metabolite of dasatinib formed in SCHH may be a more potent inhibitor of BSEP than dasatinib. The increased concentrations of bile acids in the medium with a modest decrease or little change in bile acids in SCHH exposed to dasatinib could be attributed to higher basolateral efflux of bile acids, possibly via MRP3 and/or MRP4 (Keppler, 2011). In contrast, pazopanib increased the BEI of TCA, which is expected with increased bile acid uptake and increased cellular bile acids. It is noteworthy that pazopanib is a competitive inhibitor of MRP4 (Morgan et al., 2013). The sorafenib-mediated increase in the bile acid metabolite GCDCA-3G in SCHH and medium may be due to increased bile acid glucuronidation via uridine 5′-diphospho-glucuronosyltransferase (UGTs) and/or altered transport via MRPs. Inhibition of MRP2 and MRP4 by sorafenib is consistent with previous in vitro studies (Hu et al., 2009). In general, sorafenib treatment of SCHH resulted in only modest changes in bile acid disposition based on the B-CLEAR assay, which is consistent with less notable alterations in NTCP and BSEP mRNA and protein, and CYP7A1 mRNA, and total bile acid content in SCHH and medium. BSEP inhibition as reported in membrane vesicles was not reflected in SCHH pretreated with pazopanib and sorafenib due to much lower unbound concentrations in SCHH compared with the IC50 values determined in membrane vesicles. One limitation of this study is the absence of albumin in the culture medium, which might lead to higher total and unbound intracellular concentrations.
In conclusion, these results implicate novel mechanisms of bile acid-mediated dasatinib and pazopanib hepatotoxicity (Fig. 6) that are associated with CYP7A1 mRNA induction and enhanced NTCP-mediated bile acid uptake in human hepatocytes. These novel findings demonstrate the relationship between altered bile acid homeostasis, hepatic transporter regulation, and tyrosine kinases targeted by these TKIs in SCHH. Furthermore, these data suggest that competitive inhibition of BSEP, as reported in membrane vesicles, is not the only mechanism of altered bile acid homeostasis associated with these TKIs in human hepatocytes. This work emphasizes that multiple factors may contribute to DILI and highlights the importance of using in vitro systems such as SCHH with intact cellular regulatory machinery to investigate DILI mechanisms.
Fig. 6.

Novel mechanisms of tyrosine kinase inhibitor (TKI)-induced hepatic injury. Data presented herein demonstrate that dasatinib and pazopanib increase sodium taurocholate co-transporting polypeptide (NTCP) membrane protein abundance and the uptake clearance (CLuptake) of bile acids (red circles) in addition to increasing bile acid synthesis via induction of CYP7A1 mRNA in human hepatocytes. While dasatinib promoted basolateral efflux of bile acids into medium, presumably via multidrug resistance-associated protein (MRP) 3 and/or MRP4, pazopanib increased bile acids in human hepatocytes. Dasatinib reduced the biliary excretion index and pazopanib promoted bile salt export pump (BSEP)-mediated biliary excretion. Sorafenib increased cellular levels of GCDCA-3-O-β-glucuronide (GCDCA-3G). These novel findings contribute to TKI-induced hepatic injury. BC, bile canaliculus; TJ, tight junctions.
Acknowledgments
The authors would like to thank Drs. Kenneth R. Brouwer, Raju Khatri, and James J. Beaudoin for their input with the C-DILI assay, and Dr. Jenni Küblbeck for advice on the FXR reporter assay. The authors also thank Drs. Lee M. Graves, Paul B. Watkins, Klarissa D. Jackson, Dong Fu, and Jacqueline B. Tiley for their valuable feedback and input. The visual abstract and Figure 6 were created with Biorender.
This work was presented, in part, as follows:
Saran C, Sundqvist L, Ho H, Niskanen J, Honkakoski P, and Brouwer KLR (2021) Novel Bile Acid-Dependent Mechanisms of Hepatotoxicity Associated with Tyrosine Kinase Inhibitors. ASCPT Annual Meeting (Virtual); Mar 10-13, 2021
Saran C, Sundqvist L, Ho H, Niskanen J, Honkakoski P, and Brouwer KLR (2021) Novel Bile Acid-Dependent Mechanisms of Hepatotoxicity Associated with Tyrosine Kinase Inhibitors. International Transporter Consortium 4 Workshop (Virtual); Apr 19-21, 2021
Saran C, Sundqvist L, Ho H, Niskanen J, Honkakoski P, and Brouwer KLR (2021) Novel Bile Acid-Dependent Mechanisms of Hepatotoxicity Associated with Tyrosine Kinase Inhibitors. ISSX Annual Meeting (Virtual); Sep 13-17, 2021
Abbreviations
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- ATP
adenosine triphosphate
- BAAT
bile acid-CoA: amino acid N-acetyltransferase
- BACS
bile acid-CoA synthetase
- BEI
biliary excretion index
- BSEP
bile salt export pump
- CA
cholic acid
- CDCA
chenodeoxycholic acid
- CYP
cytochrome P450
- DILI
drug-induced liver injury
- EGFR
epidermal growth factor receptor
- ERK
extracellular regulated kinase
- FGFR
fibroblast growth factor receptor
- FXR
farnesoid X receptor
- GCA
glycocholic acid
- GCDCA
glycochenodeoxycholic acid
- HBSS
Hanks’ balanced salt solution
- MAPK
mitogen-activated protein kinase
- MRP
multidrug resistance-associated protein
- NTCP
sodium taurocholate co-transporting polypeptide
- OATP
organic anion transporting polypeptide
- PDGFR
platelet-derived growth factor receptor
- RCC
renal cell carcinoma
- SCHH
sandwich-cultured human hepatocytes
- TCDCA
taurochenodeoxycholic acid
- TKI
tyrosine kinase inhibitor
- UGT
uridine 5'-diphospho-glucuronosyltransferase
- VEGFR
vascular endothelial growth factor receptor
Authorship Contributions
Participated in research design: Saran, Honkakoski, Brouwer.
Conducted experiments: Saran, Sundqvist, Ho, Niskanen.
Performed data analysis: Saran, Sundqvist, Ho, Niskanen.
Wrote or contributed to the writing of the manuscript: Saran, Sundqvist, Ho, Niskanen, Honkakoski, Brouwer.
Footnotes
This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R35 GM122576. The Biomarker Mass Spectrometry Core Facility at the University of North Carolina at Chapel Hill, which is supported by the National Institute of Environmental Health Sciences of the National Institutes of Health under award number P30ES010126, provided instrumentation used in the analysis of some samples reported in this publication. Funding for the FXR reporter assay was provided by an Academy of Finland grant (332660) to Prof. Paavo Honkakoski.
Financial Disclosure: Prof. Kim L.R. Brouwer is a coinventor of the sandwich-cultured hepatocyte technology for quantification of biliary excretion (B-CLEAR) and related technologies, which have been licensed exclusively to BioIVT.
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Alnouti Y (2009) Bile acid sulfation: a pathway of bile acid elimination and detoxification. Toxicol Sci 108:225–246. [DOI] [PubMed] [Google Scholar]
- Ananthanarayanan M, Balasubramanian N, Makishima M, Mangelsdorf DJ, Suchy FJ (2001) Human bile salt export pump promoter is transactivated by the farnesoid X receptor/bile acid receptor. J Biol Chem 276:28857–28865. [DOI] [PubMed] [Google Scholar]
- Ando T, Kojima K, Isoda H, Eguchi Y, Honda T, Ishigami M, Kimura S (2015) Reactivation of resolved infection with the hepatitis B virus immune escape mutant G145R during dasatinib treatment for chronic myeloid leukemia. Int J Hematol 102:379–382. [DOI] [PubMed] [Google Scholar]
- Anwer MS (2004) Cellular regulation of hepatic bile acid transport in health and cholestasis. Hepatology 39:581–590. [DOI] [PubMed] [Google Scholar]
- Beaudoin JJ, Brouwer KLR, Malinen MM (2020) Novel insights into the organic solute transporter alpha/beta, OSTα/β: From the bench to the bedside. Pharmacol Ther 211:107542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhullar KS, Lagarón NO, McGowan EM, Parmar I, Jha A, Hubbard BP, Rupasinghe HPV (2018) Kinase-targeted cancer therapies: progress, challenges and future directions. Mol Cancer 17:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwer KLR, Keppler D, Hoffmaster KA, Bow DAJ, Cheng Y, Lai Y, Palm JE, Stieger B, Evers R; International Transporter Consortium (2013) In vitro methods to support transporter evaluation in drug discovery and development. Clin Pharmacol Ther 94:95–112. [DOI] [PubMed] [Google Scholar]
- Bunchorntavakul C, Reddy KR (2017) Drug hepatotoxicity: newer agents. Clin Liver Dis 21:115–134. [DOI] [PubMed] [Google Scholar]
- Byun SKim DHRyerson DKim YCSun HKong BYau PGuo GXu HEKemper B, et al. (2018) Postprandial FGF19-induced phosphorylation by Src is critical for FXR function in bile acid homeostasis. Nat Commun 9:2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S, Richert L, Augustijns P, Annaert P (2014) Hepatocyte-based in vitro model for assessment of drug-induced cholestasis. Toxicol Appl Pharmacol 274:124–136. [DOI] [PubMed] [Google Scholar]
- Chiang JY (1998) Regulation of bile acid synthesis. Front Biosci 3:d176–d193. [DOI] [PubMed] [Google Scholar]
- Chiang JY (2013) Bile acid metabolism and signaling. Compr Physiol 3:1191–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu XKorzekwa KElsby RFenner KGaletin ALai YMatsson PMoss ANagar SRosania GR, et al. ; International Transporter Consortium (2013) Intracellular drug concentrations and transporters: measurement, modeling, and implications for the liver. Clin Pharmacol Ther 94:126–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford RR, Potukuchi PK, Schuetz EG, Schuetz JD (2018) Beyond competitive inhibition: Regulation of ABC transporters by kinases and protein–protein interactions as potential mechanisms of drug–drug interactions. Drug Metab Dispos 46:567–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson PA, Lan T, Rao A (2009) Bile acid transporters. J Lipid Res 50:2340–2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denson LA, Sturm E, Echevarria W, Zimmerman TL, Makishima M, Mangelsdorf DJ, Karpen SJ (2001) The orphan nuclear receptor, shp, mediates bile acid-induced inhibition of the rat bile acid transporter, ntcp. Gastroenterology 121:140–147. [DOI] [PubMed] [Google Scholar]
- Gerloff T, Stieger B, Hagenbuch B, Madon J, Landmann L, Roth J, Hofmann AF, Meier PJ (1998) The sister of P-glycoprotein represents the canalicular bile salt export pump of mammalian liver. J Biol Chem 273:10046–10050. [DOI] [PubMed] [Google Scholar]
- Giacomini KMHuang SMTweedie DJBenet LZBrouwer KLRChu XDahlin AEvers RFischer VHillgren KM, et al. ; International Transporter Consortium (2010) Membrane transporters in drug development. Nat Rev Drug Discov 9:215–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagenbuch B, Meier PJ (1994) Molecular cloning, chromosomal localization, and functional characterization of a human liver Na+/bile acid cotransporter. J Clin Invest 93:1326–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden ERChen MPasquariello KZGibson AAPetti JJShen SQu JOng SSChen TJin Y, et al. (2021) Regulation of OATP1B1 function by tyrosine kinase-mediated phosphorylation. Clin Cancer Res 27:4301–4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honkakoski P, Negishi M (2000) Regulation of cytochrome P450 (CYP) genes by nuclear receptors. Biochem J 347:321–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S, Chen Z, Franke R, Orwick S, Zhao M, Rudek MA, Sparreboom A, Baker SD (2009) Interaction of the multikinase inhibitors sorafenib and sunitinib with solute carriers and ATP-binding cassette transporters. Clin Cancer Res 15:6062–6069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S, Mathijssen RH, de Bruijn P, Baker SD, Sparreboom A (2014) Inhibition of OATP1B1 by tyrosine kinase inhibitors: in vitro–in vivo correlations. Br J Cancer 110:894–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson JP, Brouwer KR (2019) The C-DILI™ assay: An integrated in vitro approach to predict cholestatic hepatotoxicity. Methods Mol Biol 1981:75–85. [DOI] [PubMed] [Google Scholar]
- Jackson JP, Freeman KM, Friley WW, St. ClaireIii RL, Black C, Brouwer KR (2016) Basolateral efflux transporters: A potentially important pathway for the prevention of cholestatic hepatotoxicity. Appl In Vitro Toxicol 2:207–216. [Google Scholar]
- Jackson JP, Freeman KM, St Claire RL, Black CB, Brouwer KR (2018a) Cholestatic drug induced liver injury: A function of bile salt export pump inhibition and farnesoid X receptor antagonism. Appl In Vitro Toxicol 4:265–279. [Google Scholar]
- Jackson KD, Durandis R, Vergne MJ (2018b) Role of cytochrome P450 enzymes in the metabolic activation of tyrosine kinase inhibitors. Int J Mol Sci 19:2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelinek DF, Andersson S, Slaughter CA, Russell DW (1990) Cloning and regulation of cholesterol 7 alpha-hydroxylase, the rate-limiting enzyme in bile acid biosynthesis. J Biol Chem 265:8190–8197. [PMC free article] [PubMed] [Google Scholar]
- Karaman MWHerrgard STreiber DKGallant PAtteridge CECampbell BTChan KWCiceri PDavis MIEdeen PT, et al. (2008) A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol 26:127–132. [DOI] [PubMed] [Google Scholar]
- Kenna JGTaskar KSBattista CBourdet DLBrouwer KLRBrouwer KRDai DFunk CHafey MJLai Y, et al. ; International Transporter Consortium (2018) Can bile salt export pump inhibition testing in drug discovery and development reduce liver injury risk? An international transporter consortium perspective. Clin Pharmacol Ther 104:916–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keppler D (2011) Multidrug resistance proteins (MRPs, ABCCs): importance for pathophysiology and drug therapy. Handb Exp Pharmacol 201:299–323. [DOI] [PubMed] [Google Scholar]
- Köck K, Ferslew BC, Netterberg I, Yang K, Urban TJ, Swaan PW, Stewart PW, Brouwer KLR (2014) Risk factors for development of cholestatic drug-induced liver injury: inhibition of hepatic basolateral bile acid transporters multidrug resistance-associated proteins 3 and 4. Drug Metab Dispos 42:665–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, LeCluyse EL, Brouwer KR, Gan LS, Lemasters JJ, Stieger B, Meier PJ, Brouwer KLR (1999) Biliary excretion in primary rat hepatocytes cultured in a collagen-sandwich configuration. Am J Physiol 277:G12–G21. [DOI] [PubMed] [Google Scholar]
- Liu Y, Cao M, Cai Y, Li X, Zhao C, Cui R (2020) Dissecting the role of the FGF19-FGFR4 signaling pathway in cancer development and progression. Front Cell Dev Biol 8:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402–408. [DOI] [PubMed] [Google Scholar]
- Lu TT, Makishima M, Repa JJ, Schoonjans K, Kerr TA, Auwerx J, Mangelsdorf DJ (2000) Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol Cell 6:507–515. [DOI] [PubMed] [Google Scholar]
- Lu Y, Slizgi JR, Brouwer KR, Claire RL, Freeman KM, Pan M, Brock WJ, Brouwer KLR (2016) Hepatocellular disposition and transporter interactions with tolvaptan and metabolites in sandwich-cultured human hepatocytes. Drug Metab Dispos 44:867–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan RE, Trauner M, van Staden CJ, Lee PH, Ramachandran B, Eschenberg M, Afshari CA, Qualls CW Jr, Lightfoot-Dunn R, Hamadeh HK (2010) Interference with bile salt export pump function is a susceptibility factor for human liver injury in drug development. Toxicol Sci 118:485–500. [DOI] [PubMed] [Google Scholar]
- Morgan RE, van Staden CJ, Chen Y, Kalyanaraman N, Kalanzi J, Dunn RT 2nd, Afshari CA, Hamadeh HK (2013) A multifactorial approach to hepatobiliary transporter assessment enables improved therapeutic compound development. Toxicol Sci 136:216–241. [DOI] [PubMed] [Google Scholar]
- Mosedale M, Watkins PB (2017) Drug-induced liver injury: Advances in mechanistic understanding that will inform risk management. Clin Pharmacol Ther 101:469–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noé J, Stieger B, Meier PJ (2002) Functional expression of the canalicular bile salt export pump of human liver. Gastroenterology 123:1659–1666. [DOI] [PubMed] [Google Scholar]
- O’Byrne J, Hunt MC, Rai DK, Saeki M, Alexson SE (2003) The human bile acid-CoA:amino acid N-acyltransferase functions in the conjugation of fatty acids to glycine. J Biol Chem 278:34237–34244. [DOI] [PubMed] [Google Scholar]
- Ogimura E, Sekine S, Horie T (2011) Bile salt export pump inhibitors are associated with bile acid-dependent drug-induced toxicity in sandwich-cultured hepatocytes. Biochem Biophys Res Commun 416:313–317. [DOI] [PubMed] [Google Scholar]
- Oorts M, Baze A, Bachellier P, Heyd B, Zacharias T, Annaert P, Richert L (2016) Drug-induced cholestasis risk assessment in sandwich-cultured human hepatocytes. Toxicol In Vitro 34:179–186. [DOI] [PubMed] [Google Scholar]
- Pahwa S, Alam K, Crowe A, Farasyn T, Neuhoff S, Hatley O, Ding K, Yue W (2017) Pretreatment with rifampicin and tyrosine kinase inhibitor dasatinib potentiates the inhibitory effects toward OATP1B1- and OATP1B3-mediated transport. J Pharm Sci 106:2123–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai RFrench DMa NHotzel KPlise ESalphati LSetchell KDWare JLauriault VSchutt L, et al. (2012) Antibody-mediated inhibition of fibroblast growth factor 19 results in increased bile acids synthesis and ileal malabsorption of bile acids in cynomolgus monkeys. Toxicol Sci 126:446–456. [DOI] [PubMed] [Google Scholar]
- Pandak WM, Schwarz C, Hylemon PB, Mallonee D, Valerie K, Heuman DM, Fisher RA, Redford K, Vlahcevic ZR (2001) Effects of CYP7A1 overexpression on cholesterol and bile acid homeostasis. Am J Physiol Gastrointest Liver Physiol 281:G878–G889. [DOI] [PubMed] [Google Scholar]
- Pedersen JM, Matsson P, Bergström CA, Hoogstraate J, Norén A, LeCluyse EL, Artursson P (2013) Early identification of clinically relevant drug interactions with the human bile salt export pump (BSEP/ABCB11). Toxicol Sci 136:328–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qualyst Transporter Solutions Technical Application Bulletin (2011) TAB Biol 005v2. [Google Scholar]
- Riccardi KRyu SLin JYates PTess DLi RSingh DHolder BRKapinos BChang G, et al. (2018) Comparison of species and cell-type differences in fraction unbound of liver tissues, hepatocytes, and cell lines. Drug Metab Dispos 46:415–421. [DOI] [PubMed] [Google Scholar]
- Shah RR, Morganroth J, Shah DR (2013) Hepatotoxicity of tyrosine kinase inhibitors: clinical and regulatory perspectives. Drug Saf 36:491–503. [DOI] [PubMed] [Google Scholar]
- Song KH, Li T, Owsley E, Strom S, Chiang JY (2009) Bile acids activate fibroblast growth factor 19 signaling in human hepatocytes to inhibit cholesterol 7alpha-hydroxylase gene expression. Hepatology 49:297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swift B, Nebot N, Lee JK, Han T, Proctor WR, Thakker DR, Lang D, Radtke M, Gnoth MJ, Brouwer KLR (2013) Sorafenib hepatobiliary disposition: mechanisms of hepatic uptake and disposition of generated metabolites. Drug Metab Dispos 41:1179–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swift B, Pfeifer ND, Brouwer KLR (2010) Sandwich-cultured hepatocytes: an in vitro model to evaluate hepatobiliary transporter-based drug interactions and hepatotoxicity. Drug Metab Rev 42:446–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirona RGLee WLeake BFLan LBCline CBLamba VParviz FDuncan SAInoue YGonzalez FJ, et al. (2003) The orphan nuclear receptor HNF4alpha determines PXR- and CAR-mediated xenobiotic induction of CYP3A4. Nat Med 9:220–224. [DOI] [PubMed] [Google Scholar]
- Wang X, Wang P, Wang W, Murray JW, Wolkoff AW (2016) The Na(+)-taurocholate cotransporting polypeptide traffics with the epidermal growth factor receptor. Traffic 17:230–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss AAdler FBuhles AStamm CFairhurst RAKiffe MSterker DCenteleghe MWartmann MKinyamu-Akunda J. et al. (2019) FGF401, a first-in-class highly selective and potent FGFR4 inhibitor for the treatment of FGF19-driven hepatocellular cancer. Mol Cancer Ther 18:2194–2206. [DOI] [PubMed] [Google Scholar]
- Xie GWang YWang XZhao AChen TNi YWong LZhang HZhang JLiu C, et al. (2015) Profiling of serum bile acids in a healthy Chinese population using UPLC-MS/MS. J Proteome Res 14:850–859. [DOI] [PubMed] [Google Scholar]
- Zhang JSalminen AYang XLuo YWu QWhite MGreenhaw JRen LBryant MSalminen W, et al. (2017) Effects of 31 FDA approved small-molecule kinase inhibitors on isolated rat liver mitochondria. Arch Toxicol 91:2921–2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zollner G, Trauner M (2009) Nuclear receptors as therapeutic targets in cholestatic liver diseases. Br J Pharmacol 156:7–27. [DOI] [PMC free article] [PubMed] [Google Scholar]