

Summary
In this protocol, we describe the use of ChipCytometry to combine RNA in situ hybridization and antibody staining for multiplexed tissue imaging of human formalin-fixed and paraffin-embedded tissue samples. The advantages of ChipCytometry are long-term storage for re-interrogation and advanced image quality by high dynamic range imaging of staining and background. A titrated pretreatment of tissue samples bypasses challenges because of the retrieval of antigens on coverslips and achieves an optimal staining quality at the minimal expense of tissue integrity.
For complete details on the use and execution of this protocol, please refer to Jarosch et al. (2021).
Subject areas: Single Cell, Microscopy, Molecular Biology, Antibody, In Situ Hybridization
Graphical abstract
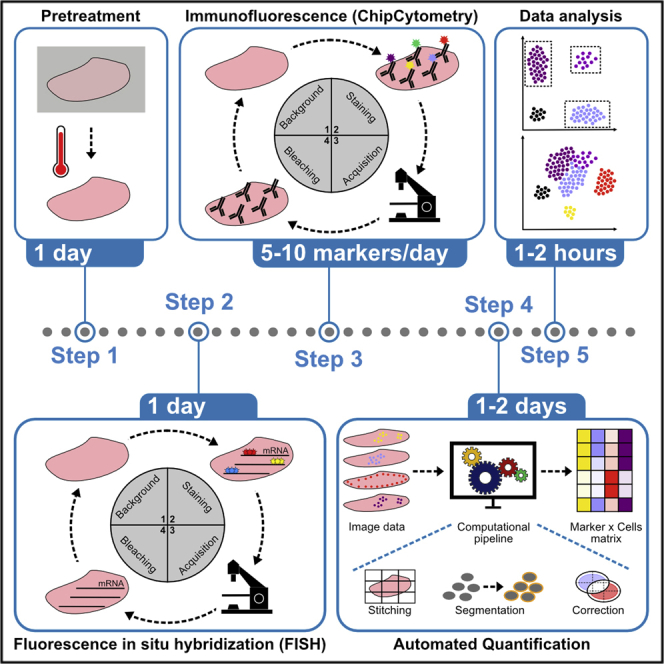
Highlights
-
•
This protocol describes use of ChipCytometry on human FFPE samples
-
•
This protocol combines mRNA staining via FISH with multiplexed immunofluorescence
-
•
This protocol provides an automatic image analysis pipeline for ChipCytometry data
-
•
The automated analysis pipeline can be applied on any multiplexed imaging data
In this protocol, we describe the use of ChipCytometry to combine RNA in situ hybridization and antibody staining for multiplexed tissue imaging of human formalin-fixed and paraffin-embedded tissue samples. The advantages of ChipCytometry are long-term storage for re-interrogation and advanced image quality by high dynamic range imaging of staining and background. A titrated pretreatment of tissue samples bypasses challenges because of the retrieval of antigens on coverslips and achieves an optimal staining quality at the minimal expense of tissue integrity.
Before you begin
The protocol describes the use of ChipCytometry to combine RNA in situ hybridization and antibody staining for deep tissue phenotyping of human FFPE samples. The protocol has been tested on several tissue types including colon, lung, tonsil, breast, kidney and pancreatic samples. FISH is described as an optional procedure. The overall protocol should also serve as a guideline for ChipCytometry including data export and automated analysis.
Institutional permissions
Inflamed colon tissue biopsies were kindly provided by Prof. Dr. Ernst Holler from patients who experienced GvHD after HSCT enrolled at the University hospital of Regensburg. All procedures were approved by local ethics committee (ethical committee of the University of Regensburg - 09/059 and 18-684482-101) and performed after informed, written consent of patients regarding use of the tissue samples.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-human pan-cytokeratin (AF488, 1:100) | BioLegend | RRID: AB_2616664 |
| Anti-human CD4 (AF488, 1:50) | R&D systems | RRID: AB_2728839 |
| Anti-human CD14 (AF488, 1:100) | Abcam | RRID: AB_2889158 |
| Anti-human CD68 (FITC, 1:100) | Santa Cruz | Cat#: sc-20060 FITC |
| Anti-human NF-kB (AF488, 1:50) | Abcam | Cat#: ab190205 |
| Anti-human Vimentin (AF488, 1:300) | BioLegend | RRID: AB_2650955 |
| Anti-human CD103 (AF488, 1:100) | Abcam | RRID: AB_2884944 |
| Anti-human Vinculin (AF488, 1:100) | Thermo Fisher Scientific | RRID: AB_2574473 |
| Anti-human CD45 (BUV395, 1:80) | BD Biosciences | RRID: AB_2744400 |
| Anti-human Foxp3 (PE, 1:30) | Thermo Fisher Scientific | RRID: AB_1944444 |
| Anti-human Ki-67 (PE, 1:50) | BD Biosciences | RRID: AB_2266296 |
| Anti-human CD45RA (PE, 1:600) | BioLegend | RRID: AB_314412 |
| Anti-human GATA-3 (PE, 1:50) | BD Biosciences | RRID: AB_1645330 |
| Anti-human CD8 (PE, 1:50) | Santa Cruz | RRID: AB_1120718 |
| Anti-human CD20 (PE, 1:200) | BD Biosciences | RRID: AB_10563904 |
| Anti-human CD45RO (PE, 1:150) | BioLegend | RRID: AB_314422 |
| Anti-human PD-L1 (PE, 1:200) | BioLegend | RRID: AB_940368 |
| Anti-human SMA (eFluor570, 1:500) | Thermo Fisher Scientific | RRID: AB_2573630 |
| Anti-human PD-1 (PE, 1:50) | BioLegend | RRID: AB_2566065 |
| Anti-human pSTAT3 (PE) | Cell Signaling Technology | RRID: AB_10859889 |
| Anti-human beta-Catenin (PE, 1:300) | Cell Signaling Technology | RRID: AB_10828097 |
| Anti-human CD133 (PE, 1:100) | BioLegend | RRID: AB_2632879 |
| Anti-human CD79a (PE, 1:100) | BioLegend | RRID: AB_1089076 |
| Anti-human Annexin A1 (PE, 1:100) | Abcam | Cat#: ab225512 |
| Anti-human CD57 (PE, 1:100) | BioLegend | RRID: AB_2562758 |
| Anti-human E-Cadherin (PE, 1:100) | Cell Signaling | RRID: AB_10950323 |
| Anti-human Muc2 (PE, 1:300) | Novus | Cat#: 34757PE |
| Anti-human CD123 (PE, 1:150) | BioLegend | RRID: AB_314580 |
| Anti-human CD45 (PerCP/Cy5.5, 1:50) | BioLegend | RRID: AB_893338 |
| Anti-human Ki-67 (PerCP/Cy5.5, 1:50) | BD Bioscience | RRID: AB_10611574 |
| Anti-human CD20 (PerCP/Cy5.5, 1:25) | BD Bioscience | RRID: AB_396990 |
| Anti-human CD45RA (PerCP/Cy5.5, 1:100) | BioLegend | RRID: AB_893357 |
| Anti-human CD56 (PerCP, 1:100) | Novus | Cat#: 33132PCP |
| Anti-human CD45RA (BV421, 1:100) | BioLegend | RRID: AB_10900421 |
| Anti-human CD3 (unconjugated, 1:150) | Thermo Fisher Scientific | RRID: AB_149924 |
| Anti-rabbit IgG (secondary, FITC, 1:200) | BioLegend | RRID: AB_893531 |
| Anti-rabbit IgG (secondary, PE, 1:300) | BioLegend | RRID: AB_2563484 |
| Biological samples | ||
| Biopsies from aHSCT patients | University hospital of Regensburg (Prof. Ernst Holler) | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Hoechst | Thermo Fisher Scientific | Cat#: H3570 |
| Ethanol absolute, 1% MEK | Carl Roth | Cat#: K928 |
| Ethanol 70%, 1% MEK | Carl Roth | Cat#: T913 |
| Roticlear ® | Carl Roth | Cat#: A538 |
| Tween20 | Carl Roth | Cat#: 9127 |
| Tris(hydroxymethyl)aminomethane (TRIS) | Carl Roth | Cat#: 9429 |
| Ethylenediaminetetraacetic acid (EDTA) | Carl Roth | Cat#: X986 |
| Sudan Back B | Sigma-Aldrich | Cat#: 199664 |
| True Black | Biotium | Cat#: 23007 |
| Dulbecco’s phosphate-buffered saline (DPBS) | PAN-Biotech | Cat#: P04-36050P |
| Sodium Borohydride | Sigma-Aldrich | Cat#: 71320 |
| Hydrogen peroxide (H2O2) | Sigma-Aldrich | Cat#: H1009 |
| Opal 520 | Akoya Bioscience | Cat#: FP1487001KT |
| Opal 570 | Akoya Bioscience | Cat#: FP1488001KT |
| Opal 650 | Akoya Bioscience | Cat#: FP1496001KT |
| Critical commercial assays | ||
| RNAscope Multiplex Fluorescent Reagent Kit v2 | ACDBio | Cat#: 323100 |
| RNAscope Probe Diluent | ACDBio | Cat#: 300041 |
| ZellSafe Chip kit FFPE | Canopy Bioscience | Cat#: 28050606/04 |
| Deposited data | ||
| Code and dataset for pipeline testing | Jarosch et al., (2021) | https://github.com/SebastianJarosch/ChipCytometry-Image-Processing |
| Software and algorithms | ||
| ImageJ 1.53c | Schindelin et al., (2012) | https://imagej.net/software/fiji/ |
| Affinity photo V1.8.3 | Serif Europe Ltd. 2020 | https://affinity.serif.com/ |
| FlowJo 10 | FlowJo LLC | https://www.flowjo.com |
| Prism 9 | GraphPad | https://www.graphpad.com |
| Automated fluorescence signal quantification | Jarosch et al., (2021) | Zenodo https://doi.org/10.5281/zenodo.5533411 |
| Other | ||
| ZellScannerONE | Canopy Bioscience | Cat#: 28050606/20-001 |
| Coverglass staining rack | Epredia | Cat#: 114 |
Materials and equipment
Antigen retrieval buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| TRIS | 10 mM | 1.21 g |
| EDTA | 1 mM | 0.37 g |
| ddH2O | n/a | 1 L |
| Total | n/a | 1 L |
Store up to two weeks at 4°C.
CRITICAL: Adjust pH to 8.5 using HCl. The exact pH is essential for an optimal retrieval of antigens.
Sudan Black B Solution
| Reagent | Final concentration | Amount |
|---|---|---|
| Sudan Black B | 0.1% (w/v) | 10 mg |
| 70% Ethanol (EtOH) | n/a | 10 mL |
| Total | 0.1% (w/v) | 10 mL |
Store long term at −20°C.
Alternatives: True black® solution has been tested and can be used as well for autofluorescence quenching.
PBST
| Reagent | Final concentration | Amount |
|---|---|---|
| Tween-20 | 0.1% (v/v) | 1 mL |
| PBS | n/a | 999 mL |
| Total | 0.1% (v/v) | 1 L |
Store up to two months at 4°C.
20× Sodium saline citrate (SSC) buffer (pH 7.00)
| Reagent | Final concentration | Amount |
|---|---|---|
| Sodium Chloride (NaCl) | 3 M | 175.3 g |
| Sodium citrate | 2.9 M | 88.2 g |
| ddH2O | n/a | 1 L |
| Total | n/a | 1 L |
Store up to two months at 21°C–24°C (RT).
Quenching buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Hydrogen Peroxide (35% v/v) | 4.5% (v/v) | 2.6 mL |
| 1 N NaOH | 24 mM | 480 μL |
| Phosphate buffered saline (PBS) | n/a | 16.9 mL |
| Total | n/a | 20 mL |
Prepare fresh for each usage and protect from light.
Step-by-step method details
Pretreatment of tissue samples
Timing: 1 day + drying and melting overnight
This step ensures the retrieval of antigens from FFPE tissue sections for later antibody staining, reduction of unspecific antibody binding and quenching of pre-existing tissue autofluorescence. The protocol provides two options to proceed with the tissue samples for optional RNA in-situ hybridization (Figure 1).
-
1.Deparaffinization and rehydration of tissue sections:
-
a.Cut 4–5 μm tissue sections of your tissues of interest and place them on 24 mm coated coverslips (included in the Zellkraftwerk Chip Kit).
-
b.Let the sections dry for at least 12 h at 21°C–24°C (RT) after cutting.
-
c.Transfer the coverslip into a coverslip staining rack for the subsequent procedures.
-
d.Incubate dry sections 12–16 h (overnight) at 60°C in a dried oven and increase the temperature to 70°C for 30 min the next day in order to completely melt the paraffin.
-
e.Incubate 3 × 10 min in Xylene (Roticlear®) at 21°C–24°C (RT).
-
f.Incubate 2 × 10 min in absolute Ethanol (EtOH) at 21°C–24°C (RT).
-
g.Incubate 5 min in 90% EtOH at 21°C–24°C (RT).
-
h.Incubate 5 min in 70% EtOH at 21°C–24°C (RT).
-
i.Incubate 5 min in 50% EtOH at 21°C–24°C (RT).
-
a.
Note: Keep the coverslips in the 50% EtOH until the antigen retrieval solution is heated up to 90°C (set the temperature for the water bath slightly higher in order to achieve this temperature within the retrieval solution). Depending on the heating speed of the water bath, it might be appropriate to switch on the water bath when the coverslips are transferred from the oven to Xylene.
Note: A station for deparaffinization and rehydration allows smooth processing of the samples. Especially the transfer from the oven to the first basin of Xylene needs to be as short as possible to avoid paraffin leftovers.
-
2.Antigen retrieval:
-
a.Rinse the coverslips in tap water.
-
b.Carefully transfer the coverslips from tap water into the pre-heated retrieval solution.
-
c.Incubate for 20 min at 90°C.
-
d.Remove the coverslips from the antigen retrieval solution and slowly transfer them to a jar filled with PBS at 21°C–24°C (RT).
-
a.
Note: From here on it is essential to prevent the tissue from drying out, since this would affect tissue integrity and stainability.
Figure 1.

Schematic overview of the two processing options provided in the protocol
After the general preprocessing (steps 1 and 2), the tissues can either be prepared for combined staining of mRNA and proteins (steps 3–5) or for proteins only (steps 6 and 7).
Proceed to step 6 if FISH is not performed.
Note: In general, RNA in-situ hybridization (Wang et al., 2012) is performed as described in the RNAScope protocol. Protease treatment was shown to destroy epitopes for subsequent antibody staining and has been skipped in order to ensure high antibody staining quality for ChipCytometry.
-
3.Probe hybridization for fluorescence in situ hybridization (FISH):
-
a.Remove the coverslips from the rack and place them horizontally on a piece of paper.
-
b.Create a hydrophobic barrier around the tissue using a hydrophobic barrier pen.Note: For all incubation steps performed on the coverslips, make sure that the whole tissue is covered with reagent. Usually 150–200 μl are an adequate volume depending on the tissue size. Washing steps are performed by rinsing the coverslips in 1 mL steps.
-
c.Incubate in H2O2 (included in the RNAScope Kit) for 10 min at 21°C–24°C (RT).
-
d.Wash (carefully rinse the coverslip) with 10 mL H2O.
-
e.Incubate with Sudan Black B for 10 min at 21°C–24°C (RT).
-
f.Carefully rinse coverslip with 1 mL 70% EtOH [10 times].CRITICAL: Washing of Sudan Black B is important to preserve high signal intensities. Excessive Sudan Black B leftover on the tissue may lead to reduced staining intensities.
-
g.Wash with 10 mL PBS.
-
h.Hybridize probes for 2 h at 40°C according to instructions of the RNAScope multiplex fluorescence kit.
-
i.Wash with RNAScope wash buffer.Note: To prevent the tissues from drying out during the incubation, place a wet tissue paper in the incubator underneath the sample slides and cover it with a lid to create a humidified chamber.
-
a.
-
4.Chip loading for FISH:
-
a.Wipe off any liquid from the coverslip that is not covering the tissue itself.
-
b.Remove the adhesive film from the ZellSafeTM tissue chip.
-
c.Transfer the coverslip to the chip and make sure that the whole tissue is located within the visible window of the chip.
-
d.Gently press together the chip and the coverslip to assure that the system is leakproof.
-
e.Rinse the chip with 5 mL 5× Sodium saline citrate (SSC) buffer and remove any air bubbles.
-
a.
Note: Air bubbles should be strictly avoided and removed immediately in case of appearance.
-
5.FISH signal amplification, acquisition and quenching:
-
a.Scan background of the tissue with the ChipCytometry microscope while it is in 5× SSC buffer (chip can be stored in 5× SSC up to 16 h at 4°C).
-
b.Rinse the chip with 10 mL wash buffer.
-
c.Follow the instructions of the RNAScope in situ hybridization kit for signal development.Note: Opal dyes compatible with the fluorescent channels of the Zellscanner instrument are listed in Table 1.
-
d.Rinse the chip with 5 mL PBS.
-
e.Acquire signals in the corresponding channels with the ChipCytometry microscope.
-
f.Rinse the chip with 5 mL quenching buffer.
-
g.Incubate 30 min under white light at 21°C–24°C (RT), exchange buffer every 5 min to avoid formation of air bubbles in the chip.
-
h.Rinse with 20 mL PBST.
-
a.
Table 1.
Filter set specifications
| Filter set | Excitation [nm] | Emission [nm] | Exposure time [ms] | Tested dyes |
|---|---|---|---|---|
| FS395 | 364–366 | 381–403 | 1,000 | Hoechst 33342, DAPI, BUV395 |
| FS421 | 370–410 | 440–485 | 50 | BV421, eF450, PB, Hoechst, DAPI |
| FS488 | 450–490 | 500–550 | 500 | FITC, AF488, GFP, Opal520 |
| FS560 | 525–575 | 570–640 | 300 | PE, eF570, Opal570 |
| FSPerCP | 456–484 | 672–748 | 300 | PerCP, PerCP-Cy5.5, Opal650 |
| ALL (Bleach) | 390–644 | – | 20,000 | – |
BV = Brilliant Violet, BUV = Brilliant Ultraviolet, eF = eFluor, PB = Pacific Blue, DAPI = 4′,6-Diamidino-2-phenylindole, FITC = Fluorescein isothiocyanate, AF = Alexa Fluor, GFP = Green Fluorescent Protein, PE = Phycoerythrin, PerCP = Peridinin-Chlorophyll-Protein.
Proceed with cyclic immunofluorescence via ChipCytometry (step 8). Steps 6 and 7 represent the preprocessing for the option without FISH staining.
-
6.Chip loading for antibody staining only:
-
a.Wipe off any liquid from the coverslip that is not covering the tissue itself.
-
b.Transfer the coverslip on the ChipCytometry chip according to manufactureŕs instructions making sure that the whole tissue is located within the visible window of the chip.
-
c.Rinse the chip with 5 mL PBS and remove any air bubbles.
-
a.
Note: Air bubbles should be strictly avoided and removed immediately in case of appearance.
-
7.Pre-treatments enhancing specificity and quality of antibody staining.
-
a.Blocking:
-
i.Add 500 μL of 5% goat serum in PBST into the chip.
-
ii.Incubate for 1–2 h at 21°C–24°C (RT).
-
iii.Rinse with 1 mL PBS [5 times].
-
i.
-
b.Autofluorescence quenching:
-
i.Add 500 μL 0.1% Sudan Black B solution into the chip, incubate 10 min at 21°C–24°C (RT).
-
ii.Rinse with 1 mL of 70% ethanol, incubate 1 min at 21°C–24°C (RT).
-
iii.Wash (gently push) with 1 mL of 70% ethanol, wait 1 min [2 times].
-
iv.Wash (push faster) with 1 mL of 70% ethanol, wait 1 min [2 times].Note: The washing of Sudan Black B within the chip is more difficult than on the coverslip (compare steps 3e and f). Repeat the washing procedure if there are visible components of the Sudan Black B solution still inside the chip.
-
v.Rinse with 1 mL PBST [3 times]. Force liquid flow by pushing PBST through the channel.
-
vi.Rinse with 1 mL PBST, wait 1 min [3 times].CRITICAL: Washing of Sudan Black B is important to keep signal intensities high. If too much Sudan Black B is still on the tissue, this may lead to reduced staining intensities.
-
i.
-
a.
Cyclic immunofluorescence via ChipCytometry
This step describes the general procedure of ChipCytometry as cyclic immunofluorescence method. The specifications for individual channels can be found in Table 1.
Optional: Photobleaching before the first background acquisition can further reduce autofluorescence of the sample and might be considered if the tissue is prone to high autofluorescence.
-
8.
Acquire a background image in all required channels using the ChipCytometry microscope.
-
9.
Rinse the chip with 5 mL PBST in order to establish a liquid flow through the chip.
-
10.Staining:
-
a.Prepare 300 μL antibody master mix per chip containing all antibodies in titrated concentration, mix by pipetting [10 times].
-
b.Centrifuge master mix for 10 min 16.000 × g (or max speed) at 4°C to remove dye or antibody aggregates.
-
c.Transfer 290 μL of the mix into the chip without touching the bottom of the tube.
-
d.Incubate 8–14 h at 4°C (shorter incubation times are possible depending on the epitope).
-
a.
-
11.
Rinse the chip with 20 mL PBST.
-
12.
Acquire the signal in the dedicated channels using the ChipCytometry microscope.
-
13.
Photo bleach each position for 20 s using the ChipCytometry microscope.
Note: All photobleaching steps are performed with the built-in HBO lamb (Zeiss HBO 100 Microscope Illuminator with a 364 nm long pass filter to protect epitope damage from UV light).
-
14.
Wash the chip with 5 mL PBST.
Note: Repeat steps 8–14 until all markers (see Table 2 for a list of validated markers) have been stained on a chip.
Pause point: After bleaching and washing, the chip can be kept at 4°C for several months. However, for some markers the staining intensity may decrease over time. If the chip is kept for more than seven days, rinse with 2 mL Zellkraftwerk Storage buffer (included in the ZellSafe Chip Kit).
Table 2.
Validated antibodies ChipCytometry with human FFPE samples
| Epitope | Conjugate | Filter set | Clone | Company | Cat number | Dilution | Incubation | Localization |
|---|---|---|---|---|---|---|---|---|
| CD14 | AF488 | FS488 | EPR3653 | Abcam | ab133335 | 1:100 | o.n. 4°C | Surface |
| CD103 | AF488 | FS488 | EPR4166(2) | abcam | ab225152 | 1:100 | o.n. 4°C | Surface |
| CD4 | AF488 | FS488 | polyclonal | R&D systems | FAB8165G | 1:50 | o.n. 4°C | Surface |
| CD68 | FITC | FS488 | KP1 | Santa-Cruz | sc-20060 FITC | 1:100 | o.n. 4°C | Surface |
| Collagen IV | AF488 | FS488 | Thermo Fisher | 53-9871-80 | 1:100 | o.n. 4°C | Surface | |
| Cytokeratin (Pan) | AF488 | FS488 | C11 | BioLegend | 628608 | 1:100 | o.n. 4°C | intracellular |
| NF-kb | AF488 | FS488 | E379 | abcam | ab190205 | 1:50 | o.n. 4°C | intranuclear |
| Vimentin | AF488 | FS488 | O91D3 | BioLegend | 677809 | 1:300 | o.n. 4°C | Surface |
| Vinculin | AF488 | FS488 | 7F9 | Invitrogen | 53-9777-82 | 1:100 | o.n. 4°C | Surface |
| CD45 | BUV395 | FS395 | HI30 | BD Bioscience | 563791 | 1:80 | o.n. 4°C | Surface |
| Annexin A1 | PE | FS560 | EPR19342 | abcam | ab225512 | 1:100 | o.n. 4°C | Surface |
| beta-Catenin | PE | FS560 | L54E2 | Cell Signaling | 6898S | 1:300 | o.n. 4°C | intranuclear |
| CD117 | PE | FS560 | 104D2 | Thermo Fisher | 12-1178-41 | 1:100 | o.n. 4°C | Surface |
| CD123 | PE | FS560 | 6H6 | BioLegend | 306006 | 1:150 | o.n. 4°C | Surface |
| CD133 | PE | FS560 | clone 7 | BioLegend | 372803 | 1:100 | o.n. 4°C | Surface |
| CD20 | PE | FS560 | H1 | BD Bioscience | 561174 | 1:200 | o.n. 4°C | Surface |
| CD45RA | PE | FS560 | HI100 | BioLegend | 304108 | 1:600 | o.n. 4°C | Surface |
| CD45RO | PE | FS560 | UCHL1 | BioLegend | 304206 | 1:150 | o.n. 4°C | Surface |
| CD57 | PE | FS560 | HNK-1 | BioLegend | 359611 | 1:100 | o.n. 4°C | Surface |
| CD79a | PE | FS560 | HM47 | BioLegend | 333503 | 1:100 | o.n. 4°C | Surface |
| CD8 | PE | FS560 | C8/144B | SantaCruz | sc53212 PE | 1:50 | o.n. 4°C | Surface |
| E-Cadherin | PE | FS560 | 24E10 | Cell Signaling | 7559S | 1:100 | o.n. 4°C | Surface |
| Foxp3 | PE | FS560 | 236A/E7 | eBioscience | 12-4777-42 | 1:30 | o.n. 4°C | intranuclear |
| GATA-3 | PE | FS560 | L50-823 | BD Pharmingen | 560074 | 1:50 | o.n. 4°C | intranuclear |
| Ki-67 | PE | FS560 | B56 | BD Bioscience | 556027 | 1:50 | o.n. 4°C | intranuclear |
| Mast Cell Chymase | PE | FS560 | CC1 | SantaCruz | sc-59586 | 1:100 | o.n. 4°C | Surface |
| Mast Cell Tryptase | PE | FS560 | G3 | SantaCruz | sc-33676 | 1:200 | o.n. 4°C | Surface |
| Muc2 | PE | FS560 | SPM296 | Novus | 34757PE | 1:300 | o.n. 4°C | Intracellular |
| PD-1 | PE | FS560 | NAT105 | BioLegend | 367404 | 1:50 | o.n. 4°C | Surface |
| PD-L1 | PE | FS560 | 29E.2A3 | BioLegend | 329706 | 1:200 | o.n. 4°C | Surface |
| pSTAT3 | PE | FS560 | D3A7 | Cell Signaling | 8119 | 1:150 | o.n. 4°C | intranuclear |
| SMA | eF570 | FS560 | 1A4 | eBioscience | 41-9760-80 | 1:500 | o.n. 4°C | Surface |
| CD20 | PerCP/Cy5.5 | FSPerCP | H1 | BD Bioscience | 558021 | 1:25 | o.n. 4°C | Surface |
| CD45 | PerCP/Cy5.5 | FSPerCP | HI30 | BioLegend | 304028 | 1:50 | o.n. 4°C | Surface |
| CD45RA | PerCP/Cy5.5 | FSPerCP | HI100 | BioLegend | 304122 | 1:100 | o.n. 4°C | Surface |
| CD56 | PerCP | FSPerCP | 123C3.D5 | Novus | 33132PCP | 1:100 | o.n. 4°C | Surface |
| Ki-67 | PerCP/Cy5.5 | FSPerCP | B56 | BD Bioscience | 561284 | 1:50 | o.n. 4°C | intranuclear |
| CD45RA | BV421 | FS421 | HI100 | BioLegend | 304129 | 1:100 | o.n. 4°C | Surface |
| CD3 | unconjugated | – | SP7 | Thermo Scientific | RM-9107-S1 | 1:150 | o.n. 4°C | Surface |
| anti-Rabbit | 2nd FITC | FS488 | polyclonal | BioLegend | 406403 | 1:200 | 2 h RT | secondary |
| anti-Rabbit | 2nd PE | FS560 | polyclonal | BioLegend | 406421 | 1:300 | 2 h RT | secondary |
eF = eFluor, FITC = Fluorescein isothiocyanate, AF = Alexa Fluor, PE = Phycoerythrin, PerCP = Peridinin-Chlorophyll-Protein, o.n. = overnight (8–14 h).
Expected outcomes
The integrity of the tissue should stay stable over the course of pre-treatments. Any signs of tissue detachment or disruption hint to an error in sample handling (see troubleshooting section for more details). The Sudan Black B incubation may lead to some small black particles on the tissue which do not interfere with the staining. If such an interference is observed, the number of washing steps with 70% ethanol can be increased. The staining should result in high-contrast images with clear shape of the surface membrane or intranuclear location depending on the marker. Signals from FISH staining should have a scattered shape (Figure 2).
Figure 2.

Expected outcome images from a ChipCytometry experiment
Upper row shows the correlation of CD3 staining via FISH and antibody staining. Lower row shows exemplary markers from a ChipCytometry experiment including mutually exclusive surface markers (CD3/CD20 and CD4/CD8) as well as intranuclear staining (FoxP3/nuclei). Scale bars indicate 10 μm.
Quantification and statistical analysis
This section should provide a step-by-step guidance for the automated analysis of ChipCytometry imaging data using an ImageJ (Schindelin et al., 2012) pipeline for signal quantification.
-
1.Adjust images and export them for quantification:
-
a.Find the ideal combination between contrast and background adjustment in the ZellExplorer App of the ChipCytometry instrument (Figure 3).
-
b.Select ‘grayscale’, ‘16 bit’ and ‘also generate tiff’ options.
-
c.Apply the settings to all positions.
-
d.Copy the scan-folder to another location and rename it according to the corresponding marker.
-
a.
-
2.Installation instructions for the image analysis pipeline.
-
a.ImageJ Macro:
-
i.Download Fiji (https://imagej.net/software/fiji/).
-
ii.Download the scripts from the GitHub repository (https://github.com/SebastianJarosch/ChipCytometry-Image-Processing).
-
iii.In ImageJ run Plugins/Install Plugin… and select the automatic_image_processing.ijm file from the repository.
-
i.
-
b.Optional ImageJ plugins:
-
i.BaSIC: In Fiji run Help/Update Fiji/Manage Update Sites and add “BaSiC” to the update sites. Apply changes and restart ImageJ. For further instructions check the original repository: https://github.com/marrlab/BaSiC.
-
ii.StarDist: In Fiji run Help/Update Fiji/Manage Update Sites and add “CSBDeep” and “StarDist” to the update sites. Apply changes and restart ImageJ. For further instructions check the original repository: https://github.com/stardist/stardist-imagej/
-
i.
-
a.
Note: Changes on the installation instructions depending on package/code updates in the future will be provided in the documentation of the pipeline on our repository (https://github.com/SebastianJarosch/ChipCytometry-Image-Processing).
-
3.Automatic quantification of signal intensities:
-
a.Run the script “automatic_image_processing.ijm” in ImageJ and select the folder that contains all the exported images as input directory.
-
b.Go through the selection process of parameters and analyses (Figure 4).
-
c.Select “ChipCytometry” as input data type.
-
d.Specify properties of the tissue sample.
-
i.The organism is just meant to be specified for documentation.
-
ii.The tissue type defines some parameters in the later processing (Table 3).
-
iii.Tile properties for the size of the area can be extracted from the overview panel in the Cell Explorer App.
-
iv.Shading correction corrects individual tiles derived from scanning microscopy. For this option, the BaSIC plugin (Peng et al., 2017) needs to be installed. The correction is dependent on the “LDRFL.png” images which are part of the ChipCytometry output structure.Note: Shading correction has not been extensively tested yet, but preliminary assessments show an improvement in reducing artifacts deriving from double-bleaching of the edges at each position (Figure 5).
-
v.“Clean folder” removes all unnecessary files to save disk space. Only the TIFF and PNG files are kept for each position and marker.
-
i.
-
e.Select the markers to quantify. Also specify if some of the markers contain intranuclear staining or RNA in situ hybridization (FISH).Note: For intranuclear markers, minimum filter and spatial spillover correction are not necessary and will not be performed once a marker has been defined as intranuclear. Furthermore, this marker will not be considered for aggregate removal.
-
f.Select the pre-processing and analysis steps you would like to perform.
-
i.“Extract erythrocytes” uses an early background in the PerCP marker to estimate erythrocyte numbers, taking advantage of their autofluorescence.
-
ii.“Aggregate removal” (not recommended yet) tries to detect large blobs in the images as aggregates.
-
iii.“Create merge image” provides an overview about the tissue architecture, and up to seven markers can be merged in this step. Markers can be selected after the image stitching is finished. By default, vimentin, SMA, nuclei and pan-cytokeratin are selected. The intensity of a marker within the merged image can be weighted (1 = merge of the full-intensity image).
-
iv.“Segmentation” allows to define individual cells either by thresholding or neuronal network segmentation with StarDist (Weigert et al., 2020). Furthermore, the markers for nuclei and epithelial cell staining can be selected here.Note: There might be some degree of under-segmentation via manual thresholding. The segmentation via the pre-trained neuronal network showed higher performance in terms of absolute number of quantified cells but might tend to over-segmentation dependent on the tissue type (Figure 6). It is generally recommended to carefully check the segmentation for each individual sample analyzed. Models trained on ChipCytometry data directly could further improve segmentation performance with the StarDist plugin for special tissue types.
-
v.“FL value calculation” selects and specifies preprocessing steps.
-
vi.“Marker consistency check” controls if images are available for all positions in all markers. An error will be thrown in case of any inconsistency.
-
vii.“Measure tissue size” measures the size of the tissue according to the segmentation marker. This is very helpful for later quantification and allows quantifying counts per mm2.
-
viii.“Spatial spillover correction” performs spatial spillover correction and lets you specify parameters for the threshold (the signal gets excluded when > threshold % of the signal are present in one of the quadrants) and the minimum intensity (signals below this value will not be taken into account).Note: For preliminary testing of the pipeline, it is recommended to reduce the dataset to a small area of the tissue and to reduce the number of analysis steps (e.g., stitching only or quantification without spillover correction) in order to save computing time. Furthermore, export settings can be validated on the stitched images of the whole sample and adjusted if necessary before running the signal quantification.
-
i.
-
a.
-
4.
Conversion of intensity data to the FCS file format for quantitative analysis for example using writeFCS in MATLAB (https://de.mathworks.com/matlabcentral/fileexchange/42603-writefcs-fname-data-text-other) or fcswrite in python (https://github.com/ZELLMECHANIK-DRESDEN/fcswrite).
Note: For more information on the conversion, please refer to our GitHub repository where you will find the instructions and code to convert your generated signal intensities into FCS files.
-
5.
Gating cell populations for quantification using flow cytometry software (e.g., FlowJo).
Figure 3.

Exemplary images showing the process of finding the ideal values for the background (BG) factor and the contrast adjusted in the ZellExplorer App
The numbers above each image indicate (BG factor, contrast) values. The red frame indicates the ideal BG/contrast values for this CD3 staining. The scale bar indicates 10 μm.
Figure 4.

Specifying parameters for automatic quantification
The graphical interface allows the user to specify parameters from data format via the markers to be analyzed until the specific analysis steps are selected.
Table 3.
Tissue specific analysis parameters
| Analysis step | Cell suspension | Spleen/LN | Colon/stomach/pancreas/breast |
|---|---|---|---|
| Separate epithelial cell quantification | no | optional | optional |
| ROI enlargement after segmentation | 1 μm | 1 μm | 1.5 μm |
| Min size / max size / min circularity | 50 / 2000 / 0.75 | 70 / 400 / 0.55 | 70 / 400 / 0.55 |
| Calculation of the tissue size | no | optional | optional |
| Spatial spillover correction | no | optional | optional |
ROI = Region of interest.
Figure 5.

Exemplary image shows the effect of shading correction with BaSIC plugin
The rectangles mark the corrected areas. The scale bar indicates 100 μm.
Figure 6.

Nuclei segmentation options
Nuclear segmentation based on manual thresholding (left) or using the pre-trained neuronal network from StarDist (right). The scale bar indicates 20 μm.
Limitations
The combination of antibody staining and RNA in situ hybridizations allows scientists to add an additional layer of information to ChipCytometry experiments. However, the treatment conditions for the in-situ hybridization might affect the staining quality of the following antibody staining, which needs to be considered in the experimental planning. Furthermore, the throughput of the method is still limited and it needs to be improved in order to allow an even broader applicability of the described method.
Troubleshooting
Problem 1
Low tissue integrity is preserved after antigen-retrieval (step 2) or the whole tissue detaches from the coverslip during this step.
Potential solution
The antigen-retrieval has been tightly titrated and it is essential to stick to the temperatures and incubation times. Check whether the actual temperature in the antigen retrieval is at 90°C. Always calibrate the pH instrument before measuring the retrieval solution, as a wrong pH can lead to tissue detachment.
Problem 2
Dye aggregates affect the acquisition of high-quality images during cyclic immunofluorescence (step 12) by interference with the actual staining as white blobs (Figure 7A).
Figure 7.

Troubleshooting
(A) Extreme example of accumulated dye aggregates in a surface marker staining. The actual staining on the cell surface is barely visible but still present as visualized in the zoom-in on the right.
(B) Background intensity increases with time during the acquisition. Shown are 5 consecutive positions of a CD8 staining.
(C) Repositioning problem of the background image for the position on the right. The intranuclear staining can still be visually detected, but the background is not subtracted properly.
(D) Dust particle (marked with a dashed line) moved in the chip between the background and staining acquisition. By autofluorescence in the background channel, it generates a dark area in the subtracted image as well as an autofluorescence artifact at the new position. Scale bars indicate 50 μm.
Potential solution
Dye aggregates should be removed by centrifugation of the staining master mix. However, for some aggregation-prone antibodies it might be necessary to centrifuge the stock solution and transfer the supernatant into a new storage tube. Filtering of the antibody might also be an option to avoid the transfer of dye aggregates into the chip.
Problem 3
Low staining quality due to background intensity shading in parts of the chip. Here the background is getting brighter during the cyclic immunofluorescence acquisition (step 12) and is not matching the background before the staining (Figure 7B).
Potential solution
In case of shading, additional washing of the chip might help. This shade usually arises from unbound antibodies which are still on the chip and are fading back from the outlet position of the chip. Therefore, titrating down the antibody dilution might help as well, since less unbound antibody would be present inside the chip.
Problem 4
Positions of the chip have been shifted in either background or staining cycles leading to a mismatch between staining and background (Quantification step 1). Ultimately, staining artifacts can be generated because the background is not subtracted correctly for individual positions (Figure 7C).
Potential solution
Manually readjust the x and y shift of the corresponding positions in ZellExplorer App. If many positions are affected, all positions can be shifted automatically (this might take some time).
Another reason for a mismatch between background and staining cycles or even between staining cycles might be that the chip was not correctly positioned within the microscope. The scan app will throw an error only for the first position in this case, so make sure to check the chip position if an error appears.
Problem 5
Moving dust particles accumulate inside the chip, generating artifact either in the background (resulting in false negative signals) or in the staining cycle (resulting in false positive signals). In most of the cases, they cannot be removed once they entered the chip (Figure 7D).
Potential solution
Sterile filtering of buffers (PBS, PBST, water) and storage in smaller aliquots (100–250 mL) might help to minimize the chance for dust particles to get into the chip via the buffer. In addition, the inlet should be covered whenever possible, for example with the lid of a 15 mL tube.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dirk Busch (dirk.busch@tum.de).
Materials availability
This study did not generate new unique reagents.
Acknowledgments
We thank members of the Busch laboratory for experimental help as well as critical discussion. This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) SFB1371/1 -395357507 (project P04), SFB1321/1 -329628492 (project P17), and SFB- TRR 338/1 2021 - 452881907 (project A01). We thank the team of Prof. Ernst Holler from the University of Regensburg for kindly providing human tissue samples for method establishment.
Author contributions
J.K. and S.J. established FFPE tissue processing for the ChipCytometry platform. S.J. developed the automated workflow for quantification of staining intensities. S.J. and S.W. established the mRNA in-situ hybridization in combination with ChipCytometry. E.D. contributed to data interpretation. S.J., E.D., and D.H.B. wrote the protocol. All authors read and reviewed the protocol.
Declaration of interests
D.H.B. is co-founder of STAGE Cell Therapeutics GmbH (now Juno Therapeutics/Bristol-Myers Squibb) and T Cell Factory B.V. (now Kite/Gilead). D.H.B. has a consulting contract with and receives sponsored research support from Juno Therapeutics/BMS.
Contributor Information
Sebastian Jarosch, Email: sebastian.jarosch@tum.de.
Dirk H. Busch, Email: dirk.busch@tum.de.
Data and code availability
The datasets and code generated during this study are available on GitHub https://github.com/SebastianJarosch/ChipCytometry-Image-Processing.
References
- Jarosch S., Köhlen J., Sarker R.S., Steiger K., Janssen K.-P., Christians A., Hennig C., Holler E., D'Ippolito E., Busch D.H. Multiplexed imaging and automated signal quantification in formalin-fixed paraffin-embedded tissues by ChipCytometry. Cell Rep. Methods. 2021;1:100104. doi: 10.1016/j.crmeth.2021.100104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng T., Thorn K., Schroeder T., Wang L., Theis F.J., Marr C., Navab N. A BaSiC tool for background and shading correction of optical microscopy images. Nat. Commun. 2017;8:14836. doi: 10.1038/ncomms14836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F., Flanagan J., Su N., Wang L.C., Bui S., Nielson A., Wu X., Vo H.T., Ma X.J., Luo Y. RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J. Mol. Diagn. 2012;14:22–29. doi: 10.1016/j.jmoldx.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigert M., Schmidt U., Haase R., Sugawara K., Myers G. EEE Winter Conference on Applications of Computer Vision (WACV) 2020. Star-convex polyhedra for 3D object detection and segmentation in microscopy; pp. 3655–3662. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets and code generated during this study are available on GitHub https://github.com/SebastianJarosch/ChipCytometry-Image-Processing.