

Abstract
Drug resistance impacts the effectiveness of many new therapeutics. Mutations in the therapeutic target confer resistance, however deciphering which mutations, often remote from the enzyme active site, drive resistance is challenging. In a series of Pneumocystis Jirovecii dihydrofolate reductase variants we elucidate which interactions are key bellwethers to confer resistance to trimethoprim using homology modeling, molecular dynamics and machine learning. Six molecular features involving mainly residues that did not vary, were the best indicators of resistance.
Graphical Abstract
Introduction
Mutations in the enzymatic drug target are a common cause of drug resistance.1 Such mutations are observed in response to treatment with antivirals, antibiotics, antifungals and chemotherapeutics.2–5 In enzymatic targets mutations can occur within or outside the active site. While mutations within the active site can confer resistance through steric and electrostatic alterations, how mutations outside the active site confer resistance is more difficult to ascertain.6–8 To determine how changes remote from the active site alter drug binding, we used molecular dynamics simulations (MD) combined with machine learning to identify interactions that distinguish resistant and susceptible variants. In previous studies a similar approach was used with HIV-1 protease variants in complex with a potent antiviral, darunavir.9, 10 This approach identified a sparse set of interactions that correlated strongly with resistance.9, 11 In this study we extend this strategy to an antifungal drug target, the dihydrofolate reductase (DHFR) enzyme in the fungus Pneumocystis Jirovecii.
P. Jirovecii is a fungus colonizing human lung epithelial cells. Although infections remains asymptomatic in healthy individuals, it is the cause of severe pneumocystis pneumonia in immunocompromised patients.12 Combination of the trimethoprim (TMP) and sulfamethoxazole are the standard for treatment for P. Jirovecii infections.13, 14 These broad-spectrum anti-infectives target two essential enzymes, dihydrofolate reductase (DHFR) and dihydropteroate synthase. DHFR is an essential enzyme in all living organisms, catalyzing a key step in the tetrahydrofolate pathway necessary for purine biosynthesis. This makes it an excellent antimicrobial drug target, as disruption of thymine synthesis leads to cell death in many microorganisms.15 TMP selectively targets DHFR from microbial pathogens over the mammalian DHFR. Mutations in DHFR have been associated with TMP resistance across multiple microbial pathogens.16 Although the overall sequence conservation of DHFR is low, the structure has a conserved fold, with most differences being located in loop regions.17 The core of DHFR is formed by an 8 stranded β-sheet, with α-helical regions on either side of the central β-sheet (Fig S1). To carry out its enzymatic function, DHFR needs to bind to both folate and the NADH co-factor. The folate binding site, which is also the primary drug binding site, is in a hydrophobic pocket formed by the two most N-Terminal helices and the β-sheet region. The catalytic aspartic acid is located on the most N-terminal helix as well. (Fig. S1B) Residues surrounding the catalytic aspartic acid are implicated in resistance not only against TMP but also against anti-malaria drugs such as pyrimethamine and cycloguanil.18, 19 20 In 2013 several clinical isolates of DHFR from P. Jirovecii (pjDHFR) with one or two mutations were characterized (Table S1), some of which exhibited significantly reduced binding affinity against TMP. The experimental inhibition constant (Ki) ranged from 38 nM to 15 μM;21 while the majority of variants retained a level of susceptibility to TMP (Ki < 1 μM) three variants were highly resistant with Kis greater than1 μM. The mutations F36C and S31F are located proximal to the catalytic aspartic acid 32 and lead to significant loss of TMP binding, but how distal changes confer resistance remained largely elusive.
Although hundreds of crystal structures of DHFR have been solved, no such structure has been solved for pjDHFR. In the present study a series of 20 variants of pjDHFR characterized by Queener et al.21 are modelled in complex with TMP, including wildtype and several variants with varying degrees of TMP resistance. Mutations were observed both within and outside the active site. Through MD simulations and machine learning we identified six structural features, three van der Waals (vdW) contacts and three hydrogen bonds, that are the bellwethers of resistance and can distinguish whether a particular variant exhibits significant levels of TMP resistance or not. These features could not have been predicted without this strategy of MD and machine learning and their location and varied interactions provide insights how remote changes can confer drug resistance.
Results and Discussion
pjDHFR variants were sorted as either susceptible or resistant depending on whether the measured affinity, measured Queener et al.21, diverged significantly from wild type. Of the 20 pjDHFR variants, 11 showed a 3.5 to 400-fold loss in TMP potency and were classified as resistant. The remaining 8 variants and WT were labeled as susceptible (Table S1, Fig. S2). The term resistant in this case does not imply clinical resistance to TMP, but indicates mutations causing a statistically significant decrease in the TMP binding affinity (Table S1), whereas susceptible mutations were indistinguishable from wildtype.
Homology models of wildtype pjDHFR (Uniprot: Q9UUP5) and 19 variants bearing either single or double mutations were generated in complex with TMP. Without experimentally determined structures of pjDHFR, the DHFR structure from P. Carinii (PDB-ID: 1DYR)22 was used as a reference structure. P. Carinii DHFR had 62% sequence identity with the target sequence of P. Jirovecii. For each enzyme-inhibitor complex three 100 ns MD simulations were performed. Analysis of the carbon-alpha root mean squared deviation (RMSD) indicated that the simulated systems did not undergo major structural changes (Fig. S2). The distance between the center of mass of pjDHFR and TMP, even in the weakest binding variant (PJQ6: F36C/L65P; Ki=15.1 μM), remained stable in all simulations, indicating that TMP remained within the active site (Fig. S4). Thus, the simulations appear to be reasonable representations of the pjDHFR-TMP complexes.
Compared with wild type, all variants showed significantly more fluctuations (Fig. 1B, upper panel, Fig. S5). However, these changes did not correlate with binding affinity. Major differences were confined to loop regions outside of the active site. Strikingly, the F36C active site mutation (PJQ4) led to a significant increase in the fluctuations of the D100-V120 loop, a change that was not observed in any other variant. This increase in fluctuations was not observed in the more resistant double mutant variant F36C/L65P (ID: PJQ6). Thus, there is not a simple correlation between binding affinity and the RMSF of the backbone in the simulations.
Figure 1.
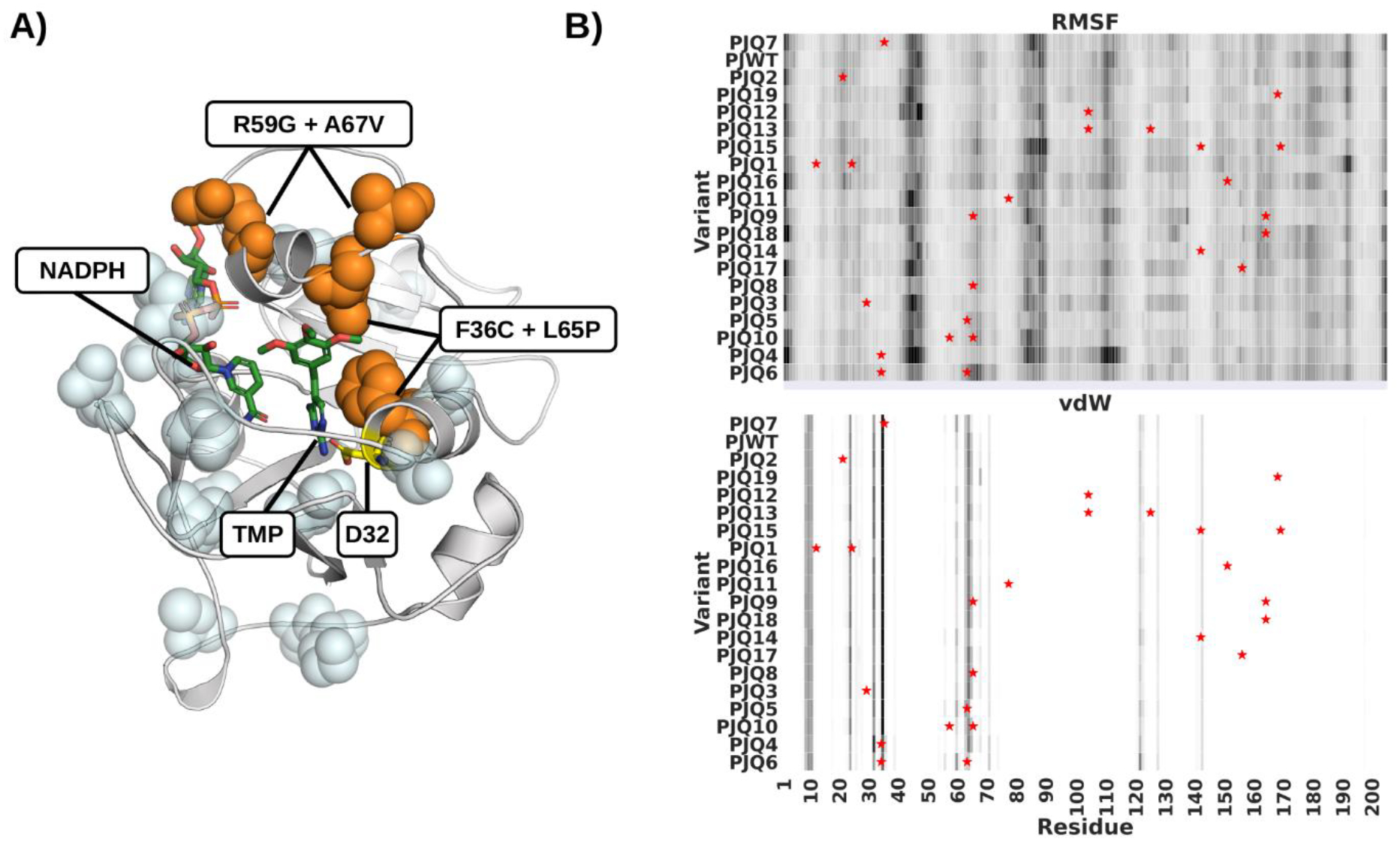
Mutant Variants of pjDHFR. A) Homology model of pjDHFR in complex with NADPH and TMP. Amino acid substitutions that lead to a minor (<10 fold) decrease in TMP binding are highlighted in light blue. Substitutions that lead to a >10 fold decrease in TMP binding are labeled and highlighted in orange. B) Heatmap of pjDHFR C-alpha root-mean-squared fluctuations and vdW interactions with TMP calculated from the MD simulations. Rows presents variants, columns residues, darker colors indicate higher fluctuations and stronger vdW interactions. Variants are sorted by TMP Ki in descending order. Sites of mutations are marked by stars.
To investigate the changes in molecular interactions and dynamics that correlate with loss of binding affinity, physical features were calculated from the MD simulations. These included van der Waals interactions, hydrogen bonds and RMSF. To evaluate whether resistant and susceptible variants could be distinguished by concerted changes in dynamics and molecular interactions, principal component analysis was performed on these features. The first two principal components showed a significant separation between resistant and susceptible variants (Fig. 2A). To quantify this effect, a logistic regression model was trained on these principal components. The model was able to distinguish resistant variants with 85% accuracy. The coefficients of the regression model were βPC1= 0.036 and βPC2=0.093, corroborating that the separation of resistant and susceptible variants was driven by the distribution of variants along the second principal component. One outlier, PJQ7, was identified; this variant has a single amino acid substitution, S37T, close to the active site and the catalytic aspartic acid 32 (Fig. 1) and is more susceptible to TMP than the WT. The inclusion of additional principal components did not improve the segregation between resistant and susceptible variants, suggesting that the first and second principal components capture most of the changes that correlated with TMP resistance. PCA and regression performed better than comparable models trained on the interactions or a subset thereof directly (Table S2), suggesting that TMP resistance is not caused by changes in a specific set of interactions, but by concerted changes in multiple sites of the enzyme.
Figure 2.
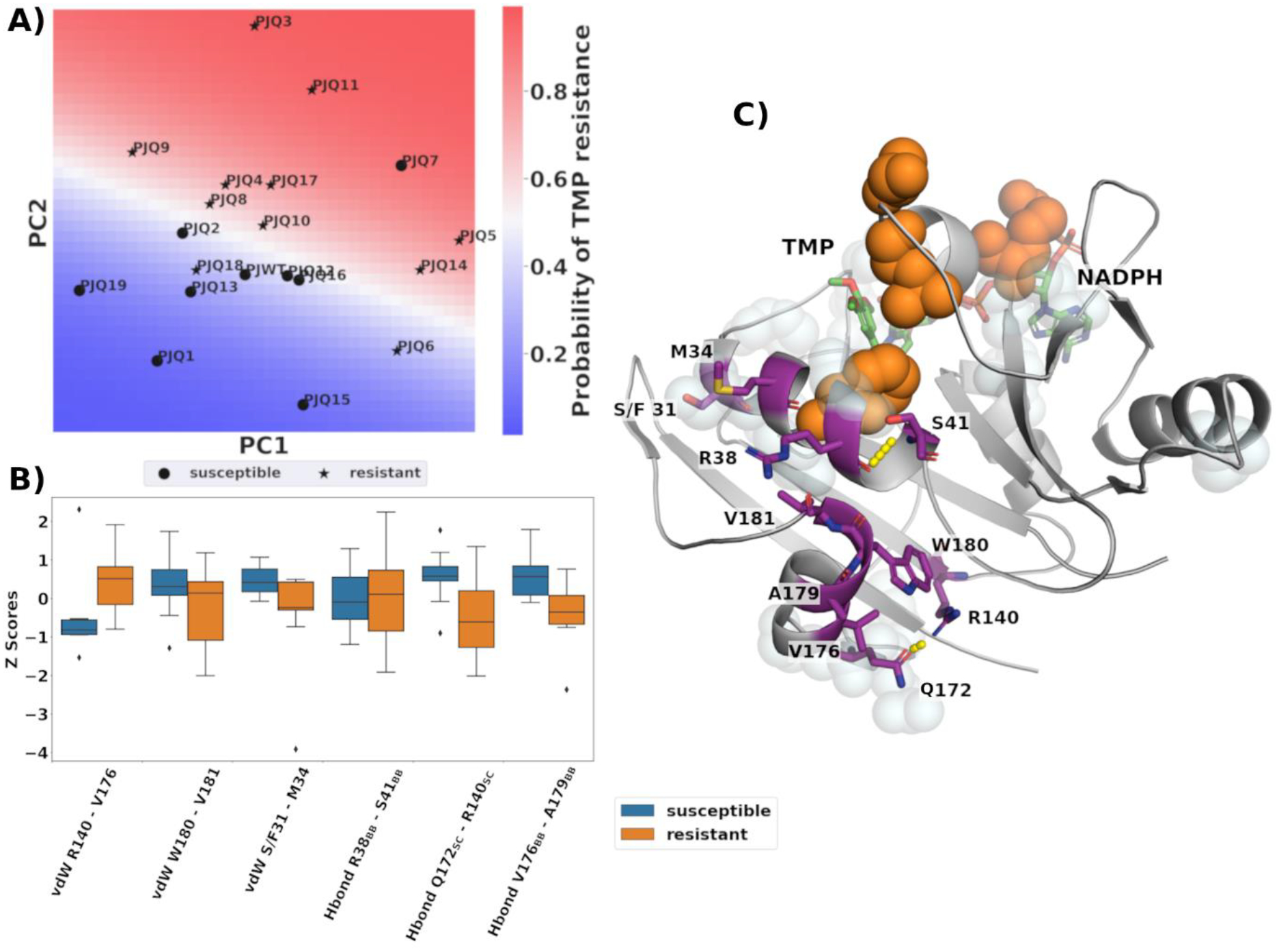
PCA of the molecular interactions distinguishes susceptible versus resistant pjDHFR A) Distribution of susceptible and resistant DHFR variants on the first and second principal components. Classification probabilities are indicated by blue-white-red color gradient. B) Boxplot comparing the distribution of the 6 interactions with the largest loading of PC2 for susceptible and resistant variants. Interactions are sorted based on their absolute loading, from highest (left) to lowest (right). C) Spatial distribution of the 6 interactions with the largest loading in PC2. Amino acids contributing to the interactions are shown in purple. Sites of mutations are shown as light blue spheres, the major mutations F36C, R59G, L65P, A67V are emphasized in orange. TMP and the NADPH cofactor are shown in green.
To evaluate which interactions drive the separation of variants, the loading of the second principal component was examined. 3 protein-protein van der Waals (vdW) interactions and 3 hydrogen bonds interactions contribute most to the distribution on the second principal component. They include vdW interactions between R140 and V176, W180 and V181 and the variable residue S/F31 and M34. The hydrogen bonds are formed between the backbone of R38 and S41, the sidechain of Q172 and R140 and the backbone of V176 and A179. (Fig. 2) PCA is agnostic to the labels of the input features, nevertheless we found that four out of six interactions showed significant differences between wildtype and susceptible variants. Only one of these interactions involves a variable residue, S31F, the remaining interactions did not include variable residues. These interactions form a contiguous region on the enzyme structure (Fig. 2C). This region consists of two alpha helices. The first helix. Formed by residues K30-S41 is part of the active site. This helix includes the catalytic D32 (Fig. 2C). The second alpha helix is formed by residues Q172-V181, remote from the active site, but forms contacts with the first alpha helix. Except for the backbone hydrogen bond between residues R38 and S41, five of the interactions showed significant differences between TMP susceptible and resistant variants. One of these interactions is in the first alpha helix and the rest are within the remote alpha helix, indicating that changes in features remote from the active site can be indicative bellwethers of levels of resistance.
To evaluate the predictive performance of the regression model, a stratified cross validation procedure was used. The dataset was split 50:50 into training and test sets, conserving the ratio of resistant and susceptible variants. The descriptors of the training set were normalized by subtracting the mean and dividing by their standard deviation. Principal component analysis was performed, and the logistic regression model was fit to the two components with the highest explained variance. This split was performed 50 times on randomized train/test splits. Resistant and susceptible variants were labeled correctly in 64% and 66% of the cases, respectively (Fig. S6). Overall, the predictions performed significantly better than random chance with an area under the curve value of 0.7 for the receiver operating characteristic curve (Fig. S7).
Conclusion
DHFR is a key target for the antimicrobial drug TMP, however mutations in DHFR often lead to resistance. In this study homology modeling, molecular dynamics and machine learning was used on a series of P. Jirovecii DHFR variants to investigate the molecular mechanisms of TMP resistance. Analysis of the eigenvector used in PCA indicated, that changes in intra-molecular interactions surrounding the catalytic residue contribute significantly to the separation of resistant and susceptible variants. This analysis specifically revealed six molecular features (3 hydrogen bonds and 3 van der Waal interactions) that were bellwethers of resistance. These features localized on two proximal helices (residues K30-S41 and Q172-V181). Helix K30-S41 is part of the active site and contains that catalytic aspartic acid. Its amino acid composition is relatively conserved across different species (Fig S1A), whereas helix Q172-V181 showed little sequence conservation, only present in P. Jirovecii and closely related organisms. Our findings are supported by mass spectroscopy studies of TMP resistant DHFR from pathogenic Escherichia Coli where drug resistance mutations modulate the structural integrity of helix K30-S41.23 We demonstrate, that most drug resistance mutations alter interactions with TMP by impacting these two alpha helices, demonstrating the tremendous utility of molecular dynamics for establishing relationships between an enzyme’s structure and its interaction with a drug.
Supplementary Material
Acknowledgments
This research was supported by NIGMS R01 GM135919.
Footnotes
Supporting Information Available:
Description of the Methods used in this Study. Full list of References. Tabulated description of the DHFR variants (Table S1, Fig. S2). Comparison of regression methods to predict TMP resistance (Table S2) Sequence conservation of DHFR and overview of secondary structure and features (Fig. S1). Cα RMSD of all DHFR simulations (Fig. S3). Distribution of DHFR-TMP center of mass distances calculated over all simulations (Fig. S4). DHFR Cα RMSF for all simulations (Fig. S5). Confusion matrix showing of the logistic regression model (Fig. S6). Metrics evaluating the performance of the regression model (Fig. S7).
Data and Software Availability
The code to calculate molecular features, the full set of features and the regression analysis are made available on Github (https://github.com/SchifferLab/ROBUST).
References
- 1.Blair JM; Webber MA; Baylay AJ; Ogbolu DO; Piddock LJ, Molecular Mechanisms of Antibiotic Resistance. Nat Rev Microbiol 2015, 13, 42–51. [DOI] [PubMed] [Google Scholar]
- 2.Kurt Yilmaz N; Swanstrom R; Schiffer CA, Improving Viral Protease Inhibitors to Counter Drug Resistance. Trends Microbiol 2016, 24, 547–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Loeffler J; Stevens DA, Antifungal Drug Resistance. Clin Infect Dis 2003, 36, S31–41. [DOI] [PubMed] [Google Scholar]
- 4.Hussain M; Galvin HD; Haw TY; Nutsford AN; Husain M, Drug Resistance in Influenza A Virus: the Epidemiology and Management. Infect Drug Resist 2017, 10, 121–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vasan N; Baselga J; Hyman DM, A View on Drug Resistance in Cancer. Nature 2019, 575, 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prabu-Jeyabalan M; Nalivaika E; Schiffer CA, Substrate Shape Determines Specificity of Recognition for HIV-1 Protease: Analysis of Crystal Structures of Six Substrate Complexes. Structure 2002, 10, 369–81. [DOI] [PubMed] [Google Scholar]
- 7.Romano KP; Ali A; Royer WE; Schiffer CA, Drug Resistance against HCV NS3/4A Inhibitors is Defined by the Balance of Substrate Recognition Versus Inhibitor Binding. Proc Natl Acad Sci U S A 2010, 107, 20986–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matthew AN; Leidner F; Lockbaum GJ; Henes M; Zephyr J; Hou S; Rao DN; Timm J; Rusere LN; Ragland DA; Paulsen JL; Prachanronarong K; Soumana DI; Nalivaika EA; Kurt Yilmaz N; Ali A; Schiffer CA, Drug Design Strategies to Avoid Resistance in Direct-Acting Antivirals and Beyond. Chem Rev 2021, 121, 3238–3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leidner F; Kurt Yilmaz N; Schiffer CA, Deciphering Complex Mechanisms of Resistance and Loss of Potency through Coupled Molecular Dynamics and Machine Learning. J Chem Theory Comput 2021, 17, 2054–2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Whitfield TW; Ragland DA; Zeldovich KB; Schiffer CA, Characterizing Protein-Ligand Binding Using Atomistic Simulation and Machine Learning: Application to Drug Resistance in HIV-1 Protease. J Chem Theory Comput 2020, 16, 1284–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lockbaum GJ; Leidner F; Rusere LN; Henes M; Kosovrasti K; Nachum GS; Nalivaika EA; Bolon DNA; Ali A; Kurt Yilmaz N; Schiffer CA, Correction to Structural Adaptation of Darunavir Analogues against Primary Mutations in HIV-1 Protease. ACS Infect Dis 2019, 5, 1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gottlieb MSSHM; Fan PT; Saxon A; Weisman JD; Pozalski I, Pneumocystis Pneumonia --- Los Angeles. https://www.cdc.gov/mmwr/preview/mmwrhtml/june_5.htm (Accessed Jun/08/2021)
- 13.Maschmeyer G; Helweg-Larsen J; Pagano L; Robin C; Cordonnier C; Schellongowski P, ECIL Guidelines for Treatment of Pneumocystis Jirovecii Pneumonia in non-HIV-Infected Haematology Patients. J Antimicrob Chemother 2016, 71, 2405–13. [DOI] [PubMed] [Google Scholar]
- 14.Huang YS; Yang JJ; Lee NY; Chen GJ; Ko WC; Sun HY; Hung CC, Treatment of Pneumocystis Jirovecii Pneumonia in HIV-Infected Patients: a Review. Expert Rev Anti Infect Ther 2017, 15, 873–892. [DOI] [PubMed] [Google Scholar]
- 15.Ahmad SI; Kirk SH; Eisenstark A, Thymine Metabolism and Thymineless Death in Prokaryotes and Eukaryotes. Annu Rev Microbiol 1998, 52, 591–625. [DOI] [PubMed] [Google Scholar]
- 16.Wrobel A; Arciszewska K; Maliszewski D; Drozdowska D, Trimethoprim and other Nonclassical Antifolates an Excellent Template for Searching Modifications of Dihydrofolate Reductase Enzyme Inhibitors. J Antibiot (Tokyo) 2020, 73, 5–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cody V; Schwalbe CH, Structural Characteristics of Antifolate Dihydrofolate Reductase Enzyme Interactions. Crystallogr Rev 2006, 12, 301–333. [Google Scholar]
- 18.Yuthavong Y; Tarnchompoo B; Vilaivan T; Chitnumsub P; Kamchonwongpaisan S; Charman SA; McLennan DN; White KL; Vivas L; Bongard E; Thongphanchang C; Taweechai S; Vanichtanankul J; Rattanajak R; Arwon U; Fantauzzi P; Yuvaniyama J; Charman WN; Matthews D, Malarial Dihydrofolate Reductase as a Paradigm for Drug Development against a Resistance-Compromised Target. Proc Natl Acad Sci U S A 2012, 109, 16823–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lombardo MN; G-Dayanandan N; Wright DL; Anderson AC, Crystal Structures of Trimethoprim-Resistant DfrA1 Rationalize Potent Inhibition by Propargyl-Linked Antifolates. ACS Infect Dis 2016, 2, 149–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tamer YT; Gaszek IK; Abdizadeh H; Batur TA; Reynolds KA; Atilgan AR; Atilgan C; Toprak E, High-Order Epistasis in Catalytic Power of Dihydrofolate Reductase Gives Rise to a Rugged Fitness Landscape in the Presence of Trimethoprim Selection. Mol Biol Evol 2019, 36, 1533–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Queener SF; Cody V; Pace J; Torkelson P; Gangjee A, Trimethoprim Resistance of Dihydrofolate Reductase Variants from Clinical Isolates of Pneumocystis Jirovecii. Antimicrob Agents Chemother 2013, 57, 4990–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Champness JN; Achari A; Ballantine SP; Bryant PK; Delves CJ; Stammers DK, The Structure of Pneumocystis Carinii Dihydrofolate Reductase to 1.9 Å Resolution. Structure 1994, 2, 915–24. [DOI] [PubMed] [Google Scholar]
- 23.Cammarata M; Thyer R; Lombardo M; Anderson A; Wright D; Ellington A; Brodbelt JS, Characterization of Trimethoprim Resistant E. Coli Dihydrofolate Reductase Mutants by Mass Spectrometry and Inhibition by Propargyl-Linked Antifolates. Chemical science 2017, 8, 4062–4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The code to calculate molecular features, the full set of features and the regression analysis are made available on Github (https://github.com/SchifferLab/ROBUST).