

Abstract

Lysine-specific demethylase 1 (LSD1/KDM1A) is a promising therapeutic target for the treatment of cancers. Several derivatives of tranylcypromine (trans-2-phenylcyclopropylamine) have been developed as LSD1 inhibitors. One such derivative is S2157; however, this compound has a high hERG channel inhibitory activity and a low microsomal stability, making it unsuitable as a drug candidate. Here, using an in silico hERG inhibition prediction model, we designed, synthesized, and evaluated a novel series of S2157 derivatives characterized by modifications of the benzyloxy and piperazine groups. Among the synthesized derivatives, a compound possessing 2-fluoropyridine and 2,8-diaza-spiro[4.5]decane groups (compound 10) showed the most desirable activities, and its eutomer, S1427, was isolated by the optical resolution of 10. In addition to potent LSD1 inhibitory activity, S1427 exhibited desirable hERG channel inhibition and microsomal stability profiles.
Keywords: Chromatin, Drug discovery, Epigenetics, Histone, Nucleosome
Lysine-specific demethylase 1 (LSD1 or KDM1A) is a flavin adenine dinucleotide (FAD) dependent demethylase that removes mono- and dimethylated groups at lysine 4 (H3K4) and lysine 9 (H3K9) of histone H3.1−3 Through its dual function of targeting both H3K4 and H3K9, LSD1 plays a pivotal role in maintaining cellular homeostasis by regulating gene transcription.4−7 The aberrant expression of LSD1 is present in various cancers, including T-cell acute lymphoblastic leukemia (T-ALL), acute myeloid leukemia, and glioblastoma, which is key in the self-renewal of cancer stem cells.8−11 The inhibition of LSD1 through gene knockdown or the use of small molecules has been shown to at least partially reduce the proliferation of cancer cells.12−16
Tranylcypromine is a nonselective small-molecule inhibitor of FAD-dependent amine oxidases, including LSD1.17−19 Tranylcypromine exerts its inhibitory activity by forming a covalent adduct with the flavin ring of FAD.17,18 To develop a potent LSD1-selective inhibitor, various tranylcypromine derivatives have been synthesized by various groups, including ours.12,20−24 Our approach has been to modify the phenyl ring and cyclopropylamine of tranylcypromine. For example, we reported one of the earliest LSD1-selective inhibitors, S2101, which we synthesized by introducing an ortho-benzyloxy group and two meta-fluorine atoms to the phenyl ring of tranylcypromine.25 Subsequently, we identified a more potent LSD1-selective inhibitor, S2157, which has (4-methylpiperazin-1-yl)ethanone as the cyclopropylamine substituent.26 We found that S2157 suppressed the growth of human T-ALL cells in a xenoplanted mouse cancer model and eradicated leukemia cells from the central nervous system as a result of the compound’s high brain penetration.27 Furthermore, we found that S2157 prevented the teratoma formation of induced pluripotent stem cells in vivo.28
Although we demonstrated that S2157 has potent pharmacological effects against T-ALL cells due to its LSD1-inhibiting activity, there is still room for improvement in its other properties. For instance, its liver microsomal stability and hERG channel inhibition profile are less than optimal; the percentage of S2157 that remains after 60 min of incubation with human, rat, or mouse microsomes is below 10%, and the half-maximum inhibitory concentration of the hERG channel (hERG IC50) is approximately 10 μM (Table 1). Here, with the help of an in silico hERG inhibition prediction model,30 we rationally designed, synthesized, and evaluated a series of S2157 derivatives with improved drug metabolism, pharmacokinetics, and off-target activity profiles.
Table 1. Optimization of the R1 Groupc.


Mean values of three independent experiments.
A probability of hERG inhibitory activity >50% at 10 μM was estimated using the AMED hERG SWC model.30
N.D., not done.
To help identify compounds with low hERG inhibitory activities and high LSD1 inhibitory activities, we used a recent in silico hERG inhibition prediction model (the AMED hERG SVM model)29 (see the Supporting Information for details). The two parent compounds used for virtual compound generation are shown in Figure S1. The predicted hERG channel inhibition probabilities of the virtual compounds generated from compounds A and B in Figure S1 are respectively shown in Tables S1 and S2. After considering the results obtained with the model, as well as the drug-likeness and the synthetic accessibility, we synthesized a series of S2157 derivatives with modifications at one of two positions (R1 and R2; see Tables 1 and 2, respectively, and Supporting Information Scheme 1). The LSD1 and hERG inhibitory activities of the derivatives were measured in vitro using the peroxidase-coupled reaction method30 and the automated patch-clamp assay in hERG-expressing HEK293 cells,31 respectively (see the Supporting Information for details).
Table 2. Optimization of the R2 Group.

Mean values of three independent experiments.
The introduction of various substituents to the phenyl group of S2157 was accomplished by subjecting 2,4-difluorophenol to an addition–elimination reaction,25 followed by cyclopropanation between ethyldiazoacetate and the allyl group of the phenyl group, hydrolysis of the ethylester, and Curtius rearrangement; the derivatives obtained were mainly trans-2-aryl-cyclopropyl-1-amine derivatives. All the substitutions at the R1 and R2 positions were accomplished following a literature procedure,25 and the conditions are summarized in Supporting Information Scheme 1.
Early in the study (see Table S3 and the Supporting Information for details), compound 1 was found to exhibit a slightly improved microsomal stability (liver microsome stability at 60 min; 5.1%, 13%, and 4.7% remaining in human, rat, and mouse microsomes, respectively) compared to that of S2157 (1.3%, 0.7%, and 0.7%), albeit with an approximately twofold reduction of its LSD1-inhibiting potency (Ki = 1.3 ± 0.43 μM versus 0.75 ± 0.26 μM, respectively; kinact/Ki = 2800 M–1 s–1 versus 6000 M–1 s–1, respectively; see Table 1). We therefore decided to start our structural modifications using 1. Table 1 summarizes our optimization of the R1 group, where the R2 group was fixed as N-methoxyethyl piperazine.
As compared with 1, extending the methylene linkage between the phenyl and aryl groups in compounds 2 and 3 did not reduce the hERG inhibitory activity. In contrast, substituting 2-fluoropyridine at the R1 position (4) resulted in a marked reduction in the hERG inhibitory activity without a reduction in the LSD1 inhibitory activity. Compound 6 with a pyrazole at R1 showed an overall profile that was comparable to that of 4, but mild degradation was observed during high-performance liquid chromatography analysis. Overall, a correlation (R2 = 0.54) was observed between the predicted and measured hERG channel inhibitory activities (Figure S2). Thus, this prediction may be useful for compound screening, but the actual measurement of the hERG inhibitory activities of the selected compounds is still necessary.
Considering that the introduction of 2-fluoropyridine at the R1 position resulted in a reduction of the hERG channel inhibitory activity, we analyzed the binding modes of compounds 4 and 5 (both of which possessed a 2-fluoropyridine group) toward LSD1 by X-ray crystallography. To understand the binding modes of 4 and 5 toward LSD1, we determined their cocrystal structures at 2.94 and 2.91 Å, respectively (Table S4). The electron density maps for 4 (Figure 1A) and 5 (Figure S3-A) revealed that the N-methoxyethyl piperazine portions (R2) of 4 and 5 were lacking in the crystal structures, which was consistent with observations for other N-alkylated derivatives, including S2157 (PDB ID 6KGP).26
Figure 1.
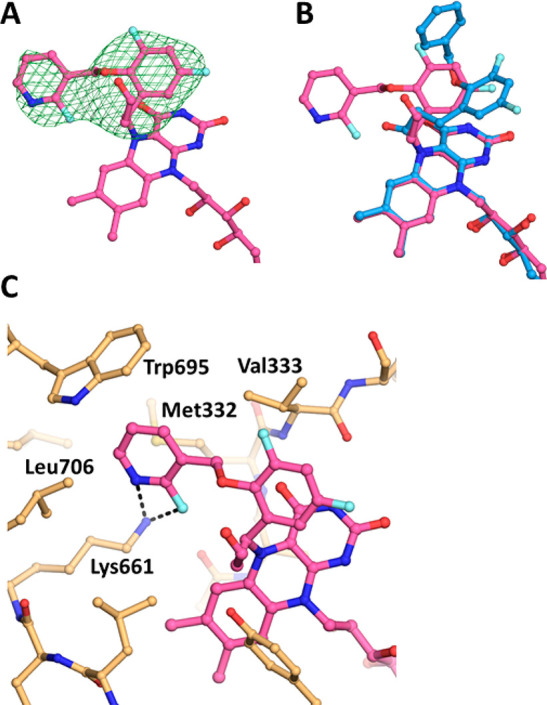
Crystal structure analysis of LSD1 complexed with 4. (A) Electron density map of the FAD–4 adduct. The mFo–DFc map at +2.5 σ, which was calculated without the compound portion of the adduct, is shown as a green mesh. (B) Superimposition of the structures of the FAD–4 and FAD–S2157 adducts. The FAD–4 and FAD–S2157 adducts are shown as magenta and cyan sticks, respectively. The nitrogen, oxygen, and fluorine atoms are shown in dark blue, red, and light blue, respectively. (C) Interactions of the FAD–4 adduct. The adduct is shown as in panel B, and the protein residues are shown as orange sticks.
Interestingly, the adducts formed between FAD and these derivatives were different from that formed between FAD and S2157.26 First, the covalent bond-forming carbon atom in the cyclopropane of the derivatives was different. In the FAD–S2157 adduct, a carbonyl carbon of S2157 was bonded to N5 of FAD, whereas in the adducts with 4 or 5 a carbon attached to the phenyl ring that was bonded to FAD. Second, in the FAD–S2157 adduct, the benzyloxy group of S2157 extended toward the opening of the catalytic cavity. In contrast, in the FAD adducts with 4 and 5, the fluoropyridine groups of 4 and 5 extended toward a side pocket in the catalytic cavity formed by Met332, Trp695, Leu706, and Lys661 (Figure 1B and C, respectively). In addition, atoms in the benzyloxy group of S2157 did not interact with the side-chain nitrogen atom of Lys661, whereas the fluorine and nitrogen atoms of the fluoropyridine ring of 4 and the fluorine atom of 5 did interact with that atom (Figures 1C and S3B). Furthermore, due to the steric hindrance caused by the fluoropyridine ring, the conformations of the Met332 side-chain in the structures of LSD1–4 (Figure 1C) and LSD1–5 (Figure S3B) were different from that in LSD1–S2157. We attribute these differences to the interactions of the different R1 moieties with the surrounding residues, including Lys661, and the effects of the different R2 moieties on the position of the derivative with respect to the FAD structure before the formation of the covalent bond.
Table 2 summarizes our optimization of the R2 group, where R1 was fixed as the moiety used in compound 4. The substitution of spiro-fused pyrrolo-piperidines at the R2 position (9 and 10) resulted in improvements in the LSD1-inhibitory activity (kinact/Ki = 7500 and 6300 M–1 s–1, respectively) and the liver microsomal stability as compared with those of 4 and S2157, although the hERG-inhibitory activity of the derivatives was sensitive to the position of the pyrrole nitrogen. The substitution of bicyclic piperazine and piperidine at the R2 position (11 and 12, respectively) also resulted in a low hERG-inhibitory activity, but the liver microsomal stability was poor. With these findings in mind, compound 10 was selected for enantiomeric characterization. As a class, S2157 derivatives contain a chiral structural motif at the 1,2-trans-substituted cyclopropyl group. Therefore, the optical resolution of 10 was achieved by chiral column separation to afford 10a (S1427) and 10b (S1428) with >95% enantiomeric excess. Despite several attempts, we were unable to successfully determine the absolute stereochemistry.
Table 3 summarizes the in vitro activities of enantiomers 10a (S1427) and 10b (S1428). S1427 showed a value for kinact/Ki against LSD1 more than 100× higher than that of S1428, but both enantiomers exhibited similar liver microsomal stabilities and hERG inhibition profiles. In addition to S2157,26 both S1427 and S1428 exhibited selectivity for LSD1 over monoamine oxidase (MAO)-A and MAO-B, and their Ki values for MAO-A and MAO-B were both above 250 μM (Table S5). X-ray crystallography confirmed that S1427 formed an adduct with FAD (Figure 2 and Table S4) in a manner almost identical to that of 4 (Figure 1C), suggesting that differences at the R2 position did not affect the adduct structure.
Table 3. Summary of the Activities of the Enantiomers of 10.

| compound | chiral form | LSD1 IC50 (μM)a | kinact (s–1)a | Ki (μM)a | kinact/Ki (M–1 s–1) | liver microsomal stability (% remaining after 60 min, human/rat/mouse) | hERG inhibition IC50 (μM)a |
|---|---|---|---|---|---|---|---|
| 10a (S1427) | (−) | 0.39 ± 0.0040 | 0.0014 ± 3.0 × 10–4 | 0.080 ± 0.046 | 18 000 | 38/62/62 | >30 |
| 10b (S1428) | (+) | 28 ± 0.0019 | 0.00076 ± 1.1 × 10–4 | 4.5 ± 1.8 | 170 | 45/55/73 | >30 |
Mean values of three independent experiments.
Figure 2.
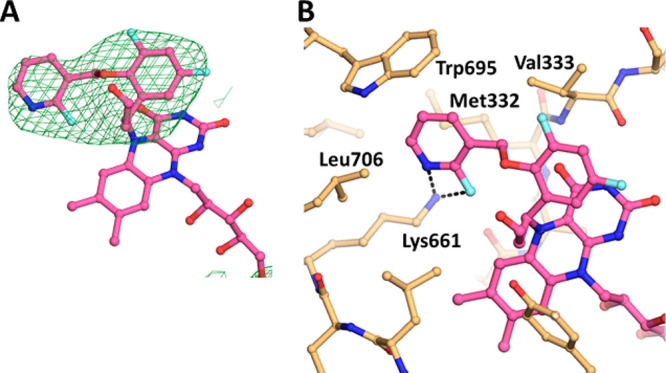
Crystal structure analysis of LSD1 complexed with S1427. (A) Electron density map of the FAD–S1427 adduct. (B) Interactions of the FAD–S1427 adduct. Figures are depicted as in Figure 1A and C.
Subsequently, we conducted a mouse pharmacokinetics study using S1427 and S2157 (Table 4). Significantly more exposure (area under the concentration–time curve from 0 to the last measurement; AUC0–∞), a higher maximum serum concentration, and a longer serum half-life were observed for S1427 compared with S2157 (Table 4). Finally, we treated human T-ALL Jurkat cells with S1427 or S1428 and examined their effects on LSD1-dependent transcriptional regulation. As expected, the expression of NOTCH3, which is one of the genes downregulated in Jurkat cells by the chemical inhibition of LSD1,27 was significantly reduced by treatment with S1427, as was observed with S2157 (Figure 3). However, the effect of the less-active enantiomer, S1428, was comparable with that of the control (DMSO-treated cells).
Table 4. Comparison of the Pharmacokinetic Profiles of S2157 and S1427a.
| parameter | S2157 | S1427 (10a) |
|---|---|---|
| AUC0–∞ (μM·h) | 0.98 | 5.09 |
| Cmax (μM) | 0.96 | 3.03 |
| Tmax (h) | 0.25 | 0.50 |
| T1/2 (h) | 0.95 | 3.90 |
Compounds were administered via a single intraperitoneal injection at a dosage of 10 mg/5 mL per kilogram (n = 3) to an ICR-strain mouse (eight weeks old, male).
Figure 3.

Downregulation of NOTCH3 expression by S1427. Jurkat cells were cultured with 10 μM test compound for 12 h. The expression level of NOTCH3 normalized to GAPDH was quantified by real-time PCR and statistically analyzed using the independent-samples one-sided t test (*p < 0.01). Data are presented as the mean ± SD (n = 3).
In conclusion, a systematic SAR study that used an in silico hERG channel inhibition prediction model and started from the tranylcypromine derivative S2157 led to the identification of the novel LSD1 inhibitor S1427. Compared with the parent compound, S1427 had triple the kinact/Ki value against LSD1 (kinact/Ki = 18,000 vs 6000 M–1 s–1), a lower hERG-inhibitory activity (IC50 > 30 μM vs 10 μM), and a significantly better liver microsomal stability in vitro. Together with the newly obtained cocrystal X-ray structures, the accumulated knowledge of the SARs and off-target profiles of the tranylcypromine derivatives is expected to offer valuable insights for the future development of LSD1 inhibitors as therapeutic agents.
Acknowledgments
We thank the staff of beamline BL26B2 at SPring-8 (Hyogo, Japan) and beamline X06DA at Swiss Light Source (Villigen, Switzerland) for their help with the collection of the X-ray diffraction data and Dr. Keiji Ogura (RIKEN) for his technical support and useful discussion about the in silico hERG inhibition prediction model. This study was supported by the RIKEN Program for Drug Discovery and Medical Technology Platforms, the Japan Society for the Promotion of Science (20H03388 and 20K21406), and the Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research) from the Japan Agency for Medical Research and Development (AMED) under Grant JP21am0101113. This study was also partially supported by the AMED Drug Discovery Informatics project.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00120.
In silico docking, chemistry, and biological evaluations (PDF)
Author Contributions
# These authors contributed equally to this work.
The authors declare no competing financial interest.
Special Issue
Published as part of the ACS Medicinal Chemistry Letters special issue “Epigenetics 2022”.
Supplementary Material
References
- Shi Y.; Lan F.; Matson C.; Mulligan P.; Whetstine J.; Cole P.; Casero R.; Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Metzger E.; Wissmann M.; Yin N.; Muller J.; Schneider R.; Peters A.; Gunther T.; Buettner R.; Schule R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 2005, 437, 436–439. 10.1038/nature04020. [DOI] [PubMed] [Google Scholar]
- Mosammaparast N.; Shi Y. Biochemical and molecular mechanisms of histone demethylases. Annu. Rev. Biochem. 2010, 79, 155–179. 10.1146/annurev.biochem.78.070907.103946. [DOI] [PubMed] [Google Scholar]
- Varier R.; Timmers H. Histone lysine methylation and demethylation pathways in cancer. Biochim. Biophys. Act. Rev. Cancer 2011, 1815, 75–89. 10.1016/j.bbcan.2010.10.002. [DOI] [PubMed] [Google Scholar]
- Lan F.; Nottke A.; Shi Y. Mechanisms involved in the regulation of histone lysine demethylases. Curr. Opin. Cell Biol. 2008, 20, 316–325. 10.1016/j.ceb.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amente S.; Lania L.; Majello B. The histone LSD1 demethylase in stemness and cancer transcription programs. Biochim. Biophys. Act. Gene Regul Mech 2013, 1829, 981–986. 10.1016/j.bbagrm.2013.05.002. [DOI] [PubMed] [Google Scholar]
- Forneris F.; Binda C.; Battaglioli E.; Mattevi A. LSD1: Oxidative chemistry for multifaceted functions in chromatin regulation. Trends Biochem. Sci. 2008, 33, 181–189. 10.1016/j.tibs.2008.01.003. [DOI] [PubMed] [Google Scholar]
- Wada T.; Koyama D.; Kikuchi J.; Honda H.; Furukawa Y. Overexpression of the shortest isoform of histone demethylase LSD1 primes hematopoietic stem cells for malignant transformation. Blood 2015, 125, 3731–3746. 10.1182/blood-2014-11-610907. [DOI] [PubMed] [Google Scholar]
- Schenk T.; Chen W.; Gollner S.; Howell L.; Jin L.; Hebestreit K.; Klein H.; Popescu A.; Burnett A.; Mills K.; Casero R.; Marton L.; Woster P.; Minden M.; Dugas M.; Wang J.; Dick J.; Muller-Tidow C.; Petrie K.; Zelent A. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat. Med. 2012, 18, 605–611. 10.1038/nm.2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris W.; Huang X.; Lynch J.; Spencer G.; Hitchin J.; Li Y.; Ciceri F.; Blaser J.; Greystoke B.; Jordan A.; Miller C.; Ogilvie D.; Somervaille T. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell 2012, 21, 473–487. 10.1016/j.ccr.2012.03.014. [DOI] [PubMed] [Google Scholar]
- Suva M.; Rheinbay E.; Gillespie S.; Patel A.; Wakimoto H.; Rabkin S.; Riggi N.; Chi A.; Cahill D.; Nahed B.; Curry W.; Martuza R.; Rivera M.; Rossetti N.; Kasif S.; Beik S.; Kadri S.; Tirosh I.; Wortman I.; Shalek A.; Rozenblatt-Rosen O.; Regev A.; Louis D.; Bernstein B. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell 2014, 157, 580–594. 10.1016/j.cell.2014.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa H.; Umehara T. Structural insight into inhibitors of flavin adenine dinucleotide-dependent lysine demethylases. Epigenetics 2017, 12, 340–352. 10.1080/15592294.2017.1290032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morera L.; Lübbert M.; Jung M. Targeting histone methyltransferases and demethylases in clinical trials for cancer therapy. Clinical Epigenetics 2016, 8, 57. 10.1186/s13148-016-0223-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole P. Chemical probes for histone-modifying enzymes. Nat. Chem. Biol. 2008, 4, 590–597. 10.1038/nchembio.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister T.; England K.; Hopkinson R.; Brennan P.; Kawamura A.; Schofield C. Recent progress in histone demethylase inhibitors. J. Med. Chem. 2016, 59, 1308–1329. 10.1021/acs.jmedchem.5b01758. [DOI] [PubMed] [Google Scholar]
- Zheng Y.; Ma J.; Wang Z.; Li J.; Jiang B.; Zhou W.; Shi X.; Wang X.; Zhao W.; Liu H. A systematic review of histone lysine-specific demethylase 1 and its inhibitors. Med. Res. Rev. 2015, 35, 1032–1071. 10.1002/med.21350. [DOI] [PubMed] [Google Scholar]
- Lee M.; Wynder C.; Schmidt D.; McCafferty D.; Shiekhattar R. Histone H3 lysine 4 demethylation is a target of nonselective antidepressive medications. Chem. Biol. 2006, 13, 563–567. 10.1016/j.chembiol.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Schmidt D.; McCafferty D. trans-2-Phenylcyclopropylamine is a mechanism-based inactivator of the histone demethylase LSD1. Biochemistry 2007, 46, 4408–4416. 10.1021/bi0618621. [DOI] [PubMed] [Google Scholar]
- Fioravanti R.; Romanelli A.; Mautone N.; Di Bello E.; Rovere A.; Corinti D.; Zwergel C.; Valente S.; Rotili D.; Botrugno O.; Dessanti P.; Vultaggio S.; Vianello P.; Cappa A.; Binda C.; Mattevi A.; Minucci S.; Mercurio C.; Varasi M.; Mai A. Tranylcypromine-Based LSD1 Inhibitors: Structure-Activity Relationships, Antiproliferative Effects in Leukemia, and Gene Target Modulation. ChemMedChem. 2020, 15, 643–658. 10.1002/cmdc.201900730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelamegam R.; Ricq E.; Malvaez M.; Patnaik D.; Norton S.; Carlin S.; Hill I.; Wood M.; Haggarty S.; Hooker J. Brain-Penetrant LSD1 Inhibitors Can. Block Memory Consolidation. Acs Chem. Neurosci. 2012, 3, 120–128. 10.1021/cn200104y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M. N. A.; Tsumoto H.; Itoh Y.; Ota Y.; Suzuki M.; Ogasawara D.; Nakagawa H.; Mizukami T.; Miyata N.; Suzuki T. Design, synthesis, and biological activity of N-alkylated analogue of NCL1, a selective inhibitor of lysine-specific demethylase 1. MedChemComm 2015, 6, 407–412. 10.1039/C4MD00330F. [DOI] [Google Scholar]
- Zhou C.; Wu F.; Lu L.; Wei L.; Pai E.; Yao Y.; Song Y. Structure activity relationship and modeling studies of inhibitors of lysine specific demethylase 1. PLoS One 2017, 12, e0170301. 10.1371/journal.pone.0170301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz-Fincke J.; Hau M.; Barth J.; Robaa D.; Willmann D.; Kurner A.; Haas C.; Greve G.; Haydn T.; Fulda S.; Lubbert M.; Ludeke S.; Berg T.; Sippl W.; Schule R.; Jung M. Structure-activity studies on N-Substituted tranylcypromine derivatives lead to selective inhibitors of lysine specific demethylase 1 (LSD1) and potent inducers of leukemic cell differentiation. Eur. J. Med. Chem. 2018, 144, 52–67. 10.1016/j.ejmech.2017.12.001. [DOI] [PubMed] [Google Scholar]
- Maes T.; Mascaro C.; Tirapu I.; Estiarte A.; Ciceri F.; Lunardi S.; Guibourt N.; Perdones A.; Lufino M.; Somervaille T.; Wiseman D.; Duy C.; Melnick A.; Willekens C.; Ortega A.; Martinell M.; Valls N.; Kurz G.; Fyfe M.; Castro-Palomino J.; Buesa C. ORY-1001, a potent and selective covalent KDM1A inhibitor, for the treatment of acute leukemia. Cancer Cell 2018, 33, 495–511. 10.1016/j.ccell.2018.02.002. [DOI] [PubMed] [Google Scholar]
- Mimasu S.; Umezawa N.; Sato S.; Higuchi T.; Umehara T.; Yokoyama S. Structurally designed trans-2-phenylcyclopropylamine derivatives potently inhibit histone demethylase LSDI/KDM1. Biochemistry 2010, 49, 6494–6503. 10.1021/bi100299r. [DOI] [PubMed] [Google Scholar]
- Niwa H.; Sato S.; Handa N.; Sengoku T.; Umehara T.; Yokoyama S. Development and structural evaluation of N-alkylated trans-2-phenylcyclopropylamine-based LSD1 inhibitors. ChemMedChem. 2020, 15, 787–793. 10.1002/cmdc.202000014. [DOI] [PubMed] [Google Scholar]
- Saito S.; Kikuchi J.; Koyama D.; Sato S.; Koyama H.; Osada N.; Kuroda Y.; Akahane K.; Inukai T.; Umehara T.; Furukawa Y. Eradication of central nervous system leukemia of T-cell origin with a brain-permeable LSD1 Inhibitor. Clin. Cancer Res. 2019, 25, 1601–1611. 10.1158/1078-0432.CCR-18-0919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osada N.; Kikuchi J.; Umehara T.; Sato S.; Urabe M.; Abe T.; Hayashi N.; Sugitani M.; Hanazono Y.; Furukawa Y. Lysine-specific demethylase 1 inhibitors prevent teratoma development from human induced pluripotent stem cells. Oncotarget 2018, 9, 6450–6462. 10.18632/oncotarget.24030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura K.; Sato T.; Yuki H.; Honma T. Support Vector Machine model for hERG inhibitory activities based on the integrated hERG database using descriptor selection by NSGA-II. Sci. Rep. 2019, 9, 12220. 10.1038/s41598-019-47536-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa H.; Kikuchi M.; Sato S.; Watanabe H.; Umezawa N.; Kato M.; Hisamatsu Y.; Umehara T.; Higuchi T. Structure-based identification of potent lysine-specific demethylase 1 inhibitor peptides and temporary cyclization to enhance proteolytic stability and cell growth-inhibitory activity. J. Med. Chem. 2021, 64, 3707–3719. 10.1021/acs.jmedchem.0c01371. [DOI] [PubMed] [Google Scholar]
- Golden A.; Li N.; Chen Q.; Lee T.; Nevill T.; Cao X.; Johnson J.; Erdemli G.; Ionescu-Zanetti C.; Urban L.; Holmqvist M. IonFlux: A microfluidic patch clamp system evaluated with human Ether-a-go-go related gene channel physiology and pharmacology. Assay Drug Dev. Technol. 2011, 9, 608–619. 10.1089/adt.2010.0362. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.