

Abstract
The present study describes our continued efforts in the discovery and characterization of a series of 2-sulfonamidebenzamides as allosteric modulators of MrgX1. MrgX1 has been shown to be an attractive target as a nonopioid receptor for the potential treatment of chronic pain. Working from our original compound, ML382, and utilizing iterative medicinal chemistry, we have identified key halogen substituents that improve MrgX1 potency by ∼8-fold. In addition, we have evaluated the compounds in Tier 1 drug metabolism and pharmacokinetics assays and have identified key compounds that impart improved potency and microsomal stability.
Keywords: Mas-related G-protein coupled receptor, MrgX1, Structure−activity relationship, Positive allosteric modulator, 2-Sulfonamidebenzamide
Chronic pain is a major health and economic problem worldwide with an estimated prevalence reaching epidemic levels of 20 to 25% of the population.1 In the United States, chronic pain affects over 116 million adults and costs up to $635 billion annually in treatment and lost productivity, more than those for cancer, heart disease, and diabetes combined.2 Chronic pain, especially nerve injury induced neuropathic pain and spontaneous pain, is challenging to treat and often refractory to current pharmacotherapies. Because the major analgesics (e.g., opioids) bind to receptors that are widely expressed throughout the central nervous system (CNS), dose-limiting adverse effects and risks of addiction and abuse present substantial barriers to their clinical use.3 Pain sensing neurons (a.k.a nociceptors) in dorsal root ganglion (DRG) play an essential role in pain transmission by detecting painful signals in the periphery such as skin and viscera. Therefore, targeting molecules specifically expressed in nociceptors may offer an opportunity for pain-selective pharmacologic interventions.
Mas-related G-protein coupled receptors (Mrg receptors) are a large family of orphan receptors expressed in small diameter sensory neurons and represent a set of potential targets for pain. Mrgs (also called Mrgprs) are a family of orphan G-protein coupled receptors (GPCRs) consisting of more than 50 members in the mouse genome.4 Our previous data have shown that Mrgs including mouse MrgC11 and human MrgX1 are specifically expressed in nociceptors in DRG and constitute an endogenous antipain pathway.5 The expression of many Mrgs, such as mouse MrgC11 and its human homologue MrgX1, is restricted to subsets of nociceptors in DRG, but not detected in the CNS (i.e., the spinal cord and brain) or in the rest of the body.4,6 Unfortunately, previous studies on MrgX1 were hampered as it became clear that human MrgX1 has binding and pharmacological profiles distinct from the binding and pharmacological profiles of rodent MrgC11, thus making it difficult to translate studies since it was not feasible to use traditional animal models to evaluate MrgX1 ligands in pain models. This roadblock was overcome with the generation of a transgenic mouse line where the MrgX1 gene is expressed in MrgC11-expressing DRG neurons.5 This new humanized mouse line makes it possible to examine MrgX1 modulators in animal models of pain. With this new mouse model, we were able to show that activating MrgX1 with a positive allosteric modulator (PAM) (ML382) lessened both evoked pain and spontaneous pain after injury, without any observable side effects.5 ML382 was the first reported MrgX1 PAM and herein, we report additional structure–activity relationship (SAR) studies culminating in the discovery of a next-generation MrgX1 PAM.7
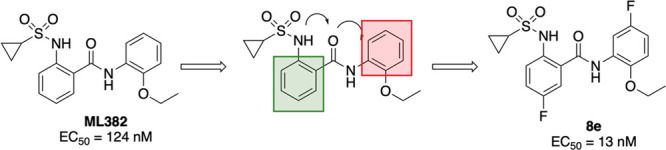
Our original medicinal chemistry efforts that led to the identification of ML382, 1 (Figure 1) started with a high-throughput screen of the NIH Molecular Library Small-Molecule Repository (MLSMR) compound collection that contained >300 000 compounds utilizing a triple addition protocol.7 The screen identified a 2-(sulfonamido)-N-phenylbenzamide scaffold that showed promise as a starting point for the initial SAR campaign. Our efforts centered around the cyclopropylsulfonamide (blue) and the 2-ethoxy (orange) moieties, and from that ML382 was identified (Figure 1).7 Although this molecule proved to be a beneficial tool compound, it did suffer from notable deficiencies. Namely, the compound did not possess suitable pharmacokinetic properties (high clearance due to oxidation of both phenyl rings) and, thus, was limited to intrathecal (i.t.) administration, and the overall MrgX1 potency was less than desired.5 Therefore, we have been working on the discovery and characterization of additional MrgX1 PAMs with improvement in these areas. The current work concentrated on the phenyl ring systems to minimize the oxidative metabolism, ring closing of the sulfonamide portion to five-membered ring systems as well as amide bioisosteres. The work culminated in the discovery of 8e, which enhanced the potency by 8-fold.
Figure 1.

Initial SAR around ML382, 1 and the current work.
The synthesis of the compounds evaluated in this work is outlined in Scheme 1. The compounds that contain the main core 2-(sulfonamido)-N-benzamide were all synthesized via a common route. Namely, the anthranilic acid, 2, was coupled with an appropriate sulfonyl chloride under basic water conditions (NaHCO3, H2O) to yield the sulfonamide, 3. This compound was then coupled with an aniline (or amine) using T3P to yield the final compounds, 4–8.8,9 This synthetic scheme was modular in that the amide could be made first and then the sulfonamide as the final target. Next, the amide bioisostere compounds were synthesized individually as outlined. The benzimidazole was synthesized by the coupling of the sulfonamide benzoic acid, 3, with 1,2-diaminophenyl via a two-step protocol. First, the amide was formed using HATU followed by ring closure under heat and acidic conditions to yield 4k.10 The triazole was synthesized by reacting the hydrazine carbonyl, 10, with the cyano compound, 11, (K2CO3, BuOH, 150 °C) to yield the desired product, 4l.11 Finally, the oxadiazole was synthesized via a two-step protocol as outlined. The hydrazine carbonyl, 13, was coupled to the acid chloride, 12 to yield the penultimate intermediate, 14, which is then cyclized with POCl3 to yield the oxadiazole, 4m.
Scheme 1. Synthesis of MrgX1 PAMs.

Reagents and conditions: (a) NaHCO3, H2O, rt, 12 h; 25–80% (b) T3P, Et3N, CH2Cl2, rt, 6 h; 65% (c) HATU, iPr2NEt, THF, rt, 16 h; (d) AcOH, 80 °C, 16 h; 93% 2 steps (e) K2CO3, BuOH, 150 °C; (f) Et3N, CH2Cl2; 7% 2 steps (g) POCl3, 25%.
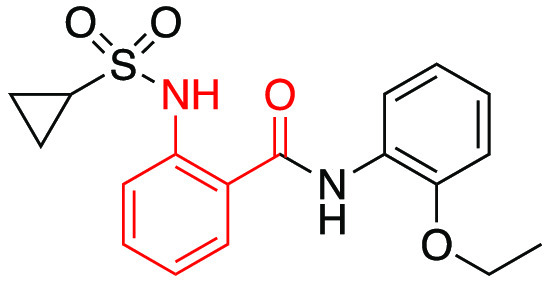
We envisioned closing the carbonyl of the amide with the sulfonamide to form a five-membered ring system as our first attempt to scaffold hop within this series. From our previous work, we knew the NH of the sulfonamide was not critical as alkylation of this nitrogen was tolerated. Thus, we started with commercially available indazole or aza-indazole derivatives (4a–c) (Table 1). We test the efficacy of the new compounds against MrgX1 stably expressing HEK293 cell line. The enhanced activation of MrgX1 by the compounds in the presence of agonist BAM8-22 is monitored by a Ca2+ imaging assay as described before.7 Unfortunately, these compounds did not show any activity against MrgX1. In fact, nearly all of our efforts to modify the benzamide phenyl ring led to inactive compounds. These included pyridine moieties (4d,e), expanding with a naphthyl moiety (4f,g) as well as changing the 1,2-relationship of the sulfonamide and the amide portion of the molecule (4h,i).
Table 1. Modification of the Benzamide Core Scaffold.
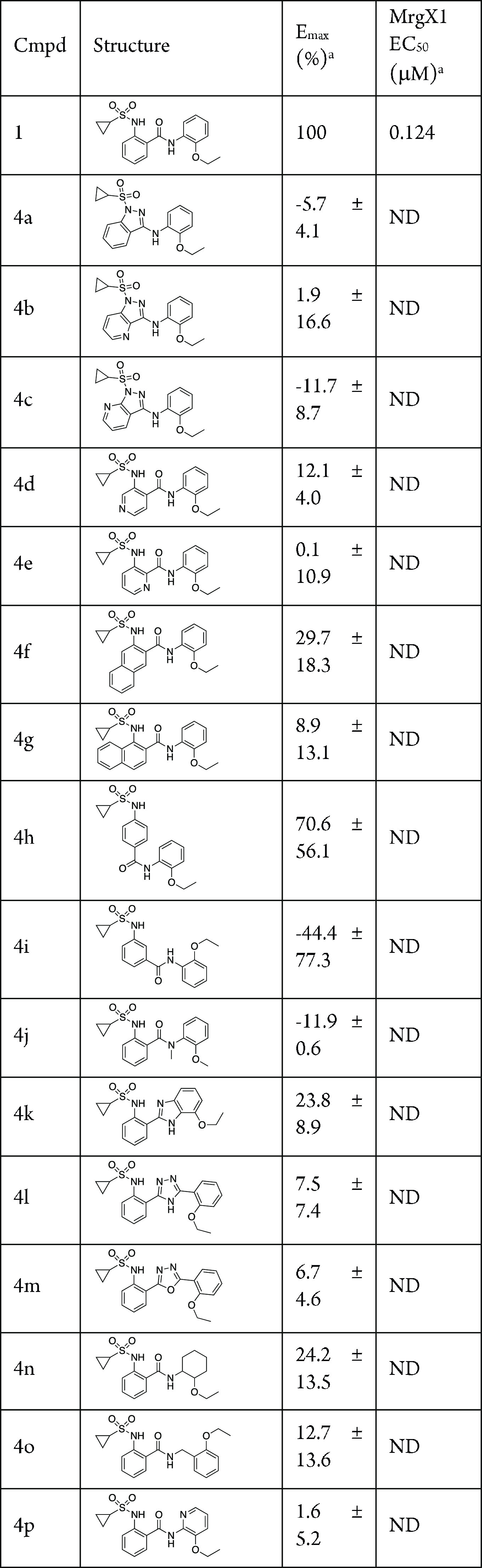
Assays were carried out in the presence of 10 nM BAM8-22; data are the mean ± SEM of n = 2 experiments; Emax values are normalized to the control compound ML382; ND, not determined, as Emax is too low.
Moving to the amide portion of the molecule also did not provide any beneficial compounds. Attempts to alkylate the nitrogen (4j) or insert amide isosteres, such as benzimidazole (4k), triazole (4l), and oxadiazole (4m), were all inactive. Other efforts to remove the aryl group on the right-hand side (4n), addition of an additional methylene linker (4o), or a pyridine moiety (4p) were all unproductive changes to the core scaffold.
Having evaluated making core changes to the phenyl ring systems, we next moved to the ether moiety for modification (Table 2). First, the ethyl ether was investigated by introducing branching, elongating the chain, and addition of a trifluoromethyl group. Introduction of a branched group produced active compounds; however, these lost ∼25-fold activity compared to the ethyl group (5a, EC50 = 2.83 μM; 5b, EC50 = 3.14 μM). Adding an additional methylene group (propyl, 5c, EC50 = 0.506 μM) or the 2,2,2-trifluoroethyl group (5d, EC50 = 0.502 μM) led to active compounds, although not as potent as 1. Moving to the methoxyethyl (5e) lost all activity, but the cyclopropylmethyl group brought some activity back to the molecule (5f, EC50 = 1.36 μM). Interestingly, the 2,4-diethoxy analog, 5g, retained some potency compared with 1 (EC50 = 1.09 μM); however, the 4-methoxy analog, 5h, was inactive. Next, we moved to evaluate cyclized versions on the right-hand side of the molecule. The first analogs were cyclized via the amide nitrogen to impart rigidity into the molecule; however, these compounds, 5i–j, were not active, which is consistent with the methylated amide analog, 4j. Additional attempts to cyclize the ether analogs were also not productive changes, 5k–q, with the lone exception of the 2-methyl-7-benzofuran analog 5r (EC50 = 1.04 μM).
Table 2. Modifications of the Ether Moietya.
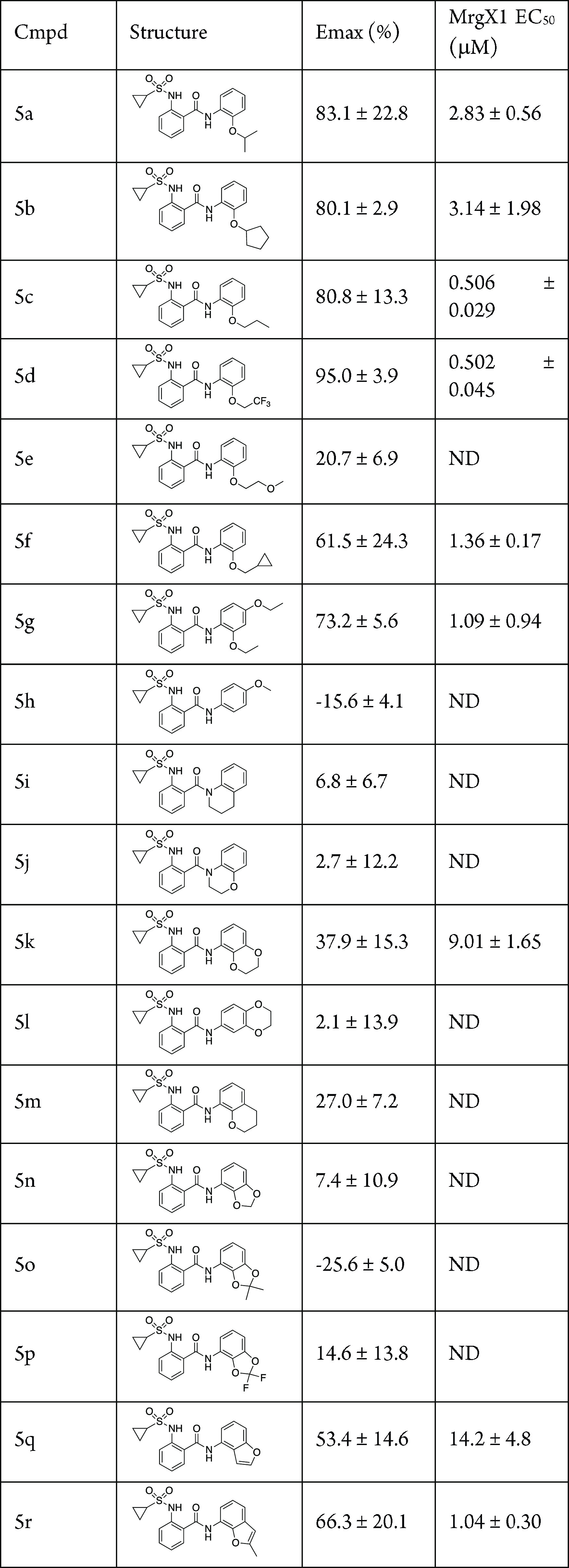
Assays were carried out in the presence of 10 nM BAM8-22; data are the mean ± SEM of n = 2 experiments; Emax values are normalized to the control compound ML382; ND, not determined, as Emax is too low.
Having established the SAR around the benzamide and the ether moieties of the molecule, we next turned our attention to substitutions on the phenyl rings. From the previous SAR, the substitutions chosen were single methyl or halogen groups, as it was evident that larger modifications were not tolerated. For these studies we used both the cyclopropylsulfonamide and the ethylsulfonamide as it was shown in our previous work that these two were well tolerated. In addition, we used the ethyl and the 2,2,2-trifluoroethyl as these were also active moieties. The right-hand selections were also based on those that are commercially available as the corresponding anilines. The compounds tested for this portion of the SAR are shown in Table 3. The 4-fluoro derivatives, 6a,b, were not as active as the parent compounds; however, the 5-fluoro derivatives, 6c,d, were very potent (6c, EC50 = 0.014 μM; 6d, EC50 = 0.285 μM).
Table 3. Right-Hand Halogen SARa.

Assays were carried out in the presence of 10 nM BAM8-22; data are the mean ± SEM of n = 2 experiments; Emax values are normalized to the control compound ML382; ND, not determined, as Emax is too low.
The left-hand portion of the molecule afforded more analogs as there were more commercially available starting materials to utilize (Table 4). In general, the 5-substitued analogs produced potent MrgX1 PAMs (7a–l), many that were more potent than 1, and the ethoxy analogs were more potent than the 2,2,2-trifluoromethyl analogs. Some of the highlighted compounds were 5-methyl, 7a (EC50 = 0.103 μM), 7g (EC50 = 0.098 μM), and 7i (EC50 = 0.054 μM), although nearly all of the compounds showed potency <1 μM. The 4-substituted analogs (Me, F, or Cl) were significantly less potent and in most cases were inactive altogether.
Table 4. Analysis of the Left-Hand Phenyl Ring Systema.
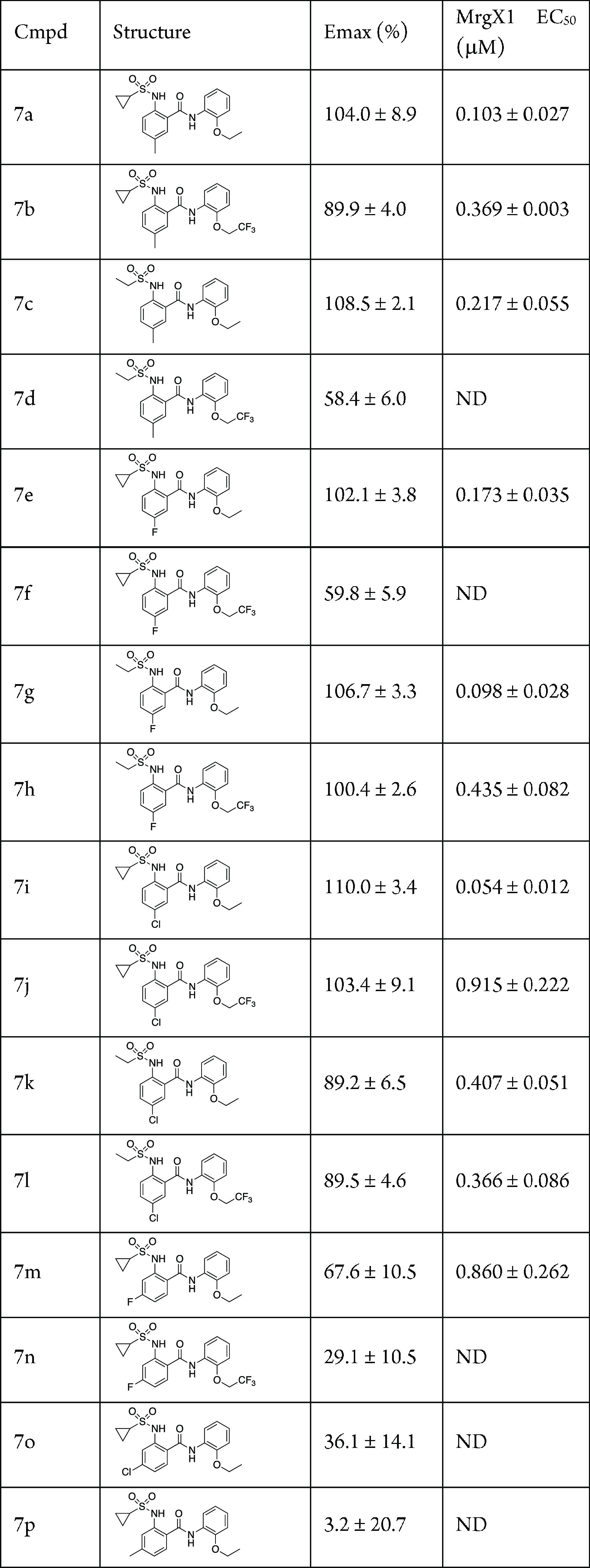
Assays were carried out in the presence of 10 nM BAM8-22; data are the mean ± SEM of n = 2 experiments; Emax values are normalized to the control compound ML382; ND, not determined, as Emax is too low.
Having established the optimal substitution patterns on the individual phenyl ring systems, we next investigated the matching of the best pairs from the above study. As we only had one active right-hand substitution pattern, this was kept constant, and we varied the left-hand portion. All of the synthesized analogs were active, and the matched pairs were equipotent with the other analogs. For example, the 5-chloro, 8a (EC50 = 0.055 μM), 5-methyl, 8c (EC50 = 0.069 μM), and 5-fluoro, 8e (EC50 = 0.013 μM) were all of similar activity (as were the corresponding ethylsulfonamide analogs). These analogs emphasize the steric environment for the benzamide portion of the molecule as other, larger groups (naphthyl and indazole, Table 1) were all inactive, whereas the smaller substituents were tolerated. In addition to steric considerations, these analogs also show that electronic factors are not as important (electron donating vs electron withdrawing). Overall, the SAR effort around this scaffold showed little tolerance for major scaffold changes, or even smaller changes around the ether substituent (Table 5). This work highlights many of the known challenges in the allosteric modulator field, namely the steep and flat SAR associated with allosteric modulator drug discovery.12−14
Table 5. SAR of the Matched Pairs of Phenyl Substituentsa.
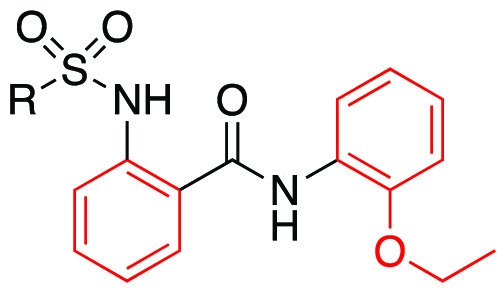

Assays were carried out in the presence of 10 nM BAM8-22; data are the mean ± SEM of n = 2 experiments; Emax values are normalized to the control compound ML382; ND, not determined, as Emax is too low.
Having identified several compounds that showed EC50 values of <1 μM, we next profiled select compounds in in vitro drug metabolism and pharmacokinetics (DMPK) assays to assess their human and mouse liver microsomal intrinsic clearance and plasma protein binding (Table 6).15,16 All the compounds evaluated displayed high intrinsic clearance and were predicted to have high hepatic clearance in vivo (CLHEP > 75% QH), with a few notable exceptions (5d, 7e, 7g). From a previous metabolite identification study (unpublished results), we anticipated that the major metabolite on 1, was oxidation of the left-hand phenyl ring. Thus, it was disappointing that the compounds that incorporated a halogen substituent on that ring did not translate to improved clearance. Although, the metabolism could shift from oxidation of the phenyl ring being the major metabolite to potentially O-dealkylation in the new compounds. Using equilibrium dialysis, the plasma protein binding of the selected compounds was determined in human and mouse plasma. The results revealed that all the tested compounds were highly protein bound in human plasma (%fu < 0.5). However, the compounds showed a better plasma protein binding profile in mouse plasma with several compounds displaying moderate free fraction (>2%). We profiled select compounds for selectivity against the closely related MrgX2 receptor. All the compounds tested were selective against MrgX2 (Supplemental Table 1), except for 7i. In addition, we tested select compounds (6c, 8c, and 8e) against the Psychoactive Drug Screening Panel at the University of North Carolina, Chapel Hill.17 This panel consists of 45 receptors and transporters that are of importance in the CNS. All three compounds were active against PBR (peripheral benzodiazepine receptor) (Ki, 6c = 271 nM, 8c = 28 nM, 8e = 71 nM), although this receptor does not cause concern for the use of these compounds as tools. In addition, 6c was active against D5 and H1 (>4000 nM), and 8c was active against H1 (1089 nM), showing these compounds are generally selective for MrgX1 (Supplemental Table 2).
Table 6. MrgX2 Selectivity and In Vitro DMPK Properties of Selected Compounds.
| Intrinsic
Clearance (mL/min/kg)c |
Plasma
Protein Binding (%fu)b |
|||||
|---|---|---|---|---|---|---|
| Cmpd | hCLINT | hCLHEPa | mCLINT | mCLHEPa | Human | Mouse |
| 1 | 61.9 | 15.2 | 1065 | 83.1 | 0.4 | 1.7 |
| 5c | 115.6 | 17.1 | 4888.6 | 88.5 | <0.3 | 1.5 |
| 5d | 40.2 | 13.4 | 979.1 | 82.5 | 0.4 | 2.2 |
| 5r | 74.9 | 15.8 | 1457.3 | 84.9 | <0.3 | 2.6 |
| 6b | 80.0 | 16.1 | 1210.7 | 83.9 | 0.5 | 2.1 |
| 6c | 79.1 | 16.0 | 1659 | 85.5 | 0.4 | 2.2 |
| 7a | 75.1 | 15.9 | 410.2 | 73.9 | 0.3 | 4.3 |
| 7e | 45.4 | 13.9 | 383.2 | 72.9 | 0.4 | 4.1 |
| 7g | 36.0 | 12.9 | 269.0 | 67.5 | 0.2 | 4.1 |
| 7i | 129.2 | 17.4 | 1470.4 | 85.0 | 0.1 | 0.1 |
| 8a | 138.4 | 17.6 | >2400 | >86.8 | 0.1 | 0.3 |
| 8c | 114.3 | 17.1 | 1101.6 | 83.3 | 0.3 | 1.2 |
| 8d | 69.3 | 15.6 | 1004.3 | 82.7 | 0.3 | 1.4 |
| 8e | 59.4 | 15.0 | 979.5 | 82.5 | 0.2 | 1.1 |
| 8f | 69.1 | 15.6 | 1235.7 | 84.0 | 0.2 | 0.1 |
Predicted hepatic clearance based on intrinsic clearance in human and mouse liver microsomes using the well-stirred organ CL model (binding terms excluded).
%fu = percent fraction unbound.
In vitro DMPK studies were performed at Q2 Solutions, Indianapolis, IN.
We evaluated a smaller set of the most potent compounds (6c, 8c, 8e) in a rat IV cassette study to assess their ability to cross the blood–brain barrier and evaluate their in vivo clearance (Table 7).18,19 The cassette study was done in a 5-in-1 format where five compounds are dosed in a single cassette (IV, 0.25 mg/kg) and then evaluated for their plasma and brain concentrations. These compounds had varying levels of brain penetrance with 6c being the best (Kp = 0.57). However, 8e, had the better overall plasma profile with low clearance (CL = 14.1 mL/min/kg) and better overall plasma exposure (AUC = 304 h·ng/mL). Finally, we further profiled 6c and 8e in a discrete mouse PK study as this would be the species for the in vivo pain assay. The compounds were dosed IP (2 mg/kg) and the results are shown in Table 7. As can be seen, the discrete mouse results are similar to the rat cassette in terms of Kp, and 8e has better overall exposure.
Table 7. In Vivo Rat Cassette and Discrete Mouse PK.
| IV Bolus
(0.25 mg/kg)a | ||||
|---|---|---|---|---|
| Cmpd | CL (mL/min/kg) | T1/2 (h) | AUC (h·ng/mL) | Vss (L/kg) |
| 6c | 36.7 | 0.48 | 119 | 0.52 |
| 8c | 63.1 | 0.42 | 66.2 | 1.2 |
| 8e | 14.1 | 0.41 | 304 | 0.34 |
| Brain:Plasma
partitioning (0.25 mg/kg, t = 15 min)a |
||||
|---|---|---|---|---|
| Plasma (ng/mL) | Brain (ng/g) | Kpb | ||
| 6c | 79.6 | 23.6 | 0.29 | |
| 8c | 61.5 | 35.1 | 0.57 | |
| 8e | 297 | 55.9 | 0.19 | |
| Discrete
Mouse PK (IP, 2 mg/kg)c | ||||
|---|---|---|---|---|
| T1/2 (h) | Tmax (h) | Cmax (ng/mL) | AUC (h·ng/mL) | |
| 6c | 0.18 | 0.17 | 259 | 69 |
| 8e | 0.12 | 0.17 | 1282 | 426 |
| Brain:Plasma
ratio |
||||
|---|---|---|---|---|
| B:P | ||||
| 6c | 0.32 | |||
| 8e | 0.20 | |||
In vivo DMPK studies were performed at Pharmaron Laboratories, Louisville, KY. Cassette dosing (5-in-1) dose of 0.25 mg/kg (IV) of each test article to male Sprague–Dawley rats. Formulation: DMSO:PEG400:EtOH:Saline (5:48:10:37). Plasma blood sampling at 0.0833, 0.25, 0.5, 1, 3, 4, 8, and 24 h and Day 2 0.25 h post dose. Brain sample on Day 2 at 0.25 h.
Kp = total brain/plasma ratio.
Formulation: 10% DMSO/10% cremphor EL/30% PEG400/50% water.
In summary, we have identified additional 2-sulfonamidebenzamides as selective positive allosteric modulators of MrgX1. This work is an extension of our previous report, and the compounds identified represent an ∼8-fold increase in potency. The SAR highlighted the steep and flat SAR around this scaffold, a feature that is well-documented in the allosteric modulator discovery field. Compounds were also profiled in our Tier 1 DMPK assays and possess favorable plasma protein binding in mice; however, the compounds displayed high intrinsic clearance in both species tested. Selected compounds were also profiled in in vivo PK studies, and they displayed moderate brain penetration; however, 8e displayed improved in vivo clearance and plasma concentrations. Because of the limitations in the PK properties, these newly identified compounds would be limited to nonoral dosage regimens (e.g., intraperitoneal, subcutaneous, or intrathecal); however, they do represent a major improvement in potency compared to the previously known MrgX1 PAMs.
Acknowledgments
The authors thank Q2 Solutions (Indianapolis, IN, USA) for the in vitro DMPK experiments and Pharmaron (Louisville, KY) for the in vivo DMPK experiments.
Glossary
ABBREVIATIONS
- MrgX1
Mas-related G-protein coupled receptor member X1
- HATU
1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate, N-[(Dimethylamino)-1H-1,2,3-triazolo-[4,5-b]pyridin-1-ylmethylene]-N-methylmethanaminium hexafluorophosphate N-oxide
- T3P
propylphosphonic anhydride
- HEK293
human embryonic kidney 293 cells
- EC50
half-maximal effective concentration
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00100.
Experimental procedures and characterization for all final compounds, description of in vitro studies, and selectivity tables (PDF)
Author Contributions
C.R.H. and X.D. drafted and corrected the paper with input from all authors. S.S., A.K.V., C.D.A., A.A.J., and A.I.W. performed the chemical synthesis, and C.R.H. oversaw the medicinal chemistry and target selection and interpreted the biological data. Q.P. performed the cellular assays, and X.D. oversaw the experiments. C.R.H. oversaw the PK experiments.
This work was generously supported by a grant from the US National Institutes of Health (NIDA: R33DA045303) to C.R.H.
The authors declare no competing financial interest.
Supplementary Material
References
- Gold M. S.; Gebhart G. F. Nociceptor sensitization in pain pathogenesis. Nat. Med. 2010, 16, 1248–1257. 10.1038/nm.2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meghani S. H.; Polomano R. C.; Tait R. C.; Vallerand A. H.; Anderson K. O.; Gallagher R. M. Advancing a national agenda to eliminate disparities in pain care: directions for health policy, education, practice, and research. Pain Med. 2012, 13, 5–28. 10.1111/j.1526-4637.2011.01289.x. [DOI] [PubMed] [Google Scholar]
- Raja S. N.; Haythornthwaite J. A. Combination therapy for neuropathic pain - which drugs, which combination, which patients?. N. Eng. J. Med. 2005, 352, 1373–1375. 10.1056/NEJMe058039. [DOI] [PubMed] [Google Scholar]
- Dong X.; Han S.-k.; Zylka M. J.; Simon M. I.; Anderson D. J. A diverse family of GPCRs expressed in specific subsets of nociceptive sensory neurons. Cell 2001, 106, 619–632. 10.1016/S0092-8674(01)00483-4. [DOI] [PubMed] [Google Scholar]
- Li Z.; Tseng P.-Y.; Tiwari V.; Xu Q.; He S.-Q.; Wang Y.; Zheng Q.; Han L.; Wu Z.; Blobaum A. L.; Cui Y.; Tiwari V.; Sun S.; Cheng Y.; Huang-Lionnet J. H. Y.; Geng Y.; Xiao B.; Peng J.; Hopkins C.; Raja S. N.; Guan Y.; Dong X. Targeting human Mas-related G protein-coupled receptor X1 to inhibit persistent pain. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E1996–E2005. 10.1073/pnas.1615255114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S.-Q.; Han L.; Li Z.; Xu Q.; Tiwari V.; Yang F.; Guan X.; Wang Y.; Raja S. N.; Dong X.; Guan Y. Temporal changes in Mrgc expression after spinal nerve injury. Neurosci. 2014, 261, 43–51. 10.1016/j.neuroscience.2013.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen W.; Wang Y.; Li Z.; Tseng P.-Y.; McManus O. B.; Wu M.; Li M.; Lindsley C. W.; Dong X.; Hopkins C. R. Discovery and characterization of 2-(cyclopropanesulfonamido)-N-(2-ethoxyphenyl)benzamide, ML382: a potent and selective positive allosteric modulator of MrgX1. ChemMedChem. 2015, 10, 57–61. 10.1002/cmdc.201402277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunetz J. R.; Xiang Y.; Baldwin A.; Ringling J. General and scalable amide bond formation with epimerization-prone substrates using T3P and pyridine. Org. Lett. 2011, 13, 5048–5051. 10.1021/ol201875q. [DOI] [PubMed] [Google Scholar]
- Llanes Garcia A. T3P: a convenient and useful reagent in organic synthesis. Synlett 2007, 2007, 1328–1329. 10.1055/s-2007-980339. [DOI] [Google Scholar]
- Zhu G.-D.; Gong J.; Gandhi V. B.; Liu X.; Shi Y.; Johnson E. F.; Donawho C. K.; Ellis P. A.; Bouska J. J.; Ostrling D. J.; Olson A. M.; Park C.; Luo Y.; Shoemaker A.; Giranda V. L.; Penning T. D. Discovery and SAR of orally efficacious tetrahydropyridopyridazinone PARP inhibitors for the treatment of cancer. Bioorg. Med. Chem. 2012, 20, 4635–4645. 10.1016/j.bmc.2012.06.021. [DOI] [PubMed] [Google Scholar]
- Potts K. T. The chemistry of 1,2,4-triazoles. Chem. Rev. 1961, 61, 87–127. 10.1021/cr60210a001. [DOI] [Google Scholar]
- Bertron J. L.; Cho H. P.; Garcia-Barrantes P. M.; Panarese J. D.; Salovich J. M.; Nance K. D.; Engers D. W.; Rook J. M.; Blobaum A. L.; Niswender C. M.; Stauffer S. R.; Conn J. P.; Lindsley C. W. The discovery of VU0486846: steep SAR from a series of M1 PAMs based on a novel benzomorpholine series. Bioorg. Med. Chem. Lett. 2018, 28, 2175–2179. 10.1016/j.bmcl.2018.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utley T.; Haddenham D.; Salovich J. M.; Zamorano R.; Vinson P. N.; Lindsley C. W.; Hopkins C. R.; Niswender C. M. Synthesis and SAR of a novel metabotropic glutamate receptor 4 (mGlu4) antagonist: Unexpected ‘molecular switch’ from a closely related mGlu4 positive allosteric modulator. Bioorg. Med. Chem. Lett. 2011, 21, 6955–6969. 10.1016/j.bmcl.2011.09.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melancon B. J.; Hopkins C. R.; Wood M. R.; Emmitte K. A.; Niswender C. M.; Christopoulos A.; Conn P. J.; Lindsley C. W. Allosteric Modulation of Seven Transmembrane Spanning Receptors: Theory, Practice, and Opportunities for Central Nervous System Drug Discovery. J. Med. Chem. 2012, 55, 1445–1464. 10.1021/jm201139r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obach R. S. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: an examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab. Dispos. 1999, 27, 1350–1359. [PubMed] [Google Scholar]
- Chang G.; Steyn S. J.; Umland J. P.; Scott D. O. Strategic Use of Plasma and Microsome Binding To Exploit in Vitro Clearance in Early Drug Discovery. ACS Med. Chem. Lett. 2010, 1, 50–63. 10.1021/ml900012h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besnard J.; Ruda G. F.; Setola V.; Abecassis K.; Rodriguiz R. M.; Huang X.-P.; Norval S.; Sassano M. F.; Shin A. I.; Webster L. A.; Simeons F. R. C.; Stojanovski L.; Prat A.; Seidah N. G.; Constam D. B.; Bickerton G. R.; Read K. D.; Wetsel W. C.; Gilbert I. H.; Roth B. L.; Hopkins A. L. Automated design of ligands to polypharmacological profiles. Nature 2012, 492, 215–220. 10.1038/nature11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges T. M.; Morrison R. D.; Byers F. W.; Luo S.; Daniels J. S. Use of a novel rapid and resource-efficient cassette dosing approach to determine the pharmacokinetics and CNS distribution of small molecule 7-transmembrane receptor allosteric modulators in rat. Pharmacol. Res. Perspect. 2014, 2, e00077 10.1002/prp2.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith N. F.; Raynaud F. I.; Workman P. The application of cassette dosing for pharmacokinetic screening in small-molecule cancer drug discovery. Mol. Cancer Ther. 2007, 6, 428–440. 10.1158/1535-7163.MCT-06-0324. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.