

Summary
The principal signals that drive memory and cognitive impairment in Alzheimer’s disease (AD) remain elusive. Here, we revealed brain-wide cellular reactions to type I interferon (IFN-I), an innate immune cytokine aberrantly elicited by amyloid β plaques, and examined their role in cognition and neuropathology relevant to AD in a murine amyloidosis model. Using a fate-mapping reporter system to track cellular responses to IFN-I, we detected robust, Aβ pathology-dependent IFN-I activation in microglia and other cell types. Long-term blockade of IFN-I receptor (IFNAR) rescued both memory and synaptic deficits, and resulted in reduced microgliosis, inflammation, and neuritic pathology. Microglia-specific Ifnar1 deletion attenuated the loss of post-synaptic terminals by selective engulfment, whereas neural Ifnar1 deletion restored pre-synaptic terminals and decreased plaque accumulation. Overall, IFN-I signaling represents a critical module within the neuroinflammatory network of AD and prompts concerted cellular states that are detrimental to memory and cognition.
Keywords: interferon, memory impairment, neuroinflammation, synapse, microglia, Alzheimer’s disease
Graphical Abstract
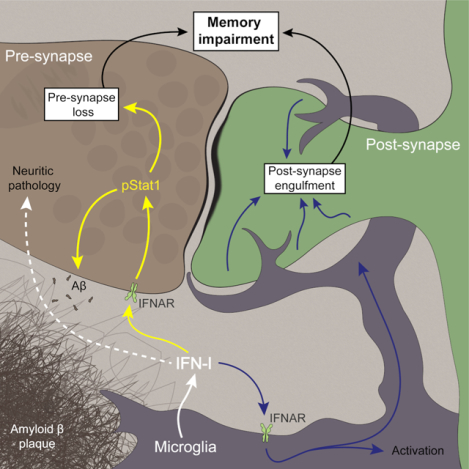
In brief
The signals that drive cognitive impairment in Alzheimer’s disease (AD) remain elusive. Type I interferon (IFN-I) activation is implicated in neurodegenerative conditions, including AD. Using cellular fate-mapping, receptor blockade, and conditional deletion models, Roy et al. demonstrate that IFN-I signaling in different brain cell types, including microglia and neural cells, critically affects synapses and causes cognitive impairment in an AD model.
Introduction
Alzheimer’s disease (AD) is the main cause of dementia, characterized clinically by memory impairment. Hallmarked by the deposition of amyloid β (Aβ) plaques and accumulation of neurofibrillary tangles, AD pathogenesis manifests with complex interactions between different brain cell types (De Strooper and Karran, 2016, Knopman et al., 2021). Collective histological, bioinformatic and molecular analyses have highlighted a perpetual activation of microglia, the brain’s resident immune cells, and connection of a number of AD risk polymorphisms and rare variants to microglia and innate immunity (Zhang et al., 2013, Huang et al., 2017). Despite an overwhelming consensus on the importance of neuroinflammatory responses, the core signal that disrupts cognition and memory in AD is not yet well understood.
Recently, we described a prominent antiviral immune response by microglia in multiple murine amyloid β models of AD, as well as human AD (Roy et al., 2020). At its center are type I interferons (IFN-I) and a large number of IFN-stimulated genes (ISGs), which usually confer an antiviral state in host cells. However, more light is being shed on the functions of these molecules in sterile central nervous system (CNS) inflammation. Recently, Hur et al. reported that IFITM3, an ISG, functions as an immune switch to increase γ-secretase activity, promoting APP cleavage and amyloid pathology (Hur et al., 2020). Meanwhile, microglial subsets with gene signatures of IFN response (“interferon-responsive microglia,” or IRMs) have been identified from single-cell RNA-seq (scRNA-seq) studies on murine amyloid β models and human AD brains (Sala Frigerio et al., 2019, Olah et al., 2020). Most recently, a subset of reactive astrocytes with a signature of IFN signaling was uncovered in the inflamed mouse brain (Hasel et al., 2021). Additionally, polymorphisms in several ISGs were collectively recognized as risk factors for AD (Salih et al., 2019), while an upregulated IFN response was reported in AD patients carrying the TREM2 R47H risk variant (Korvatska et al., 2020). In keeping with these reports, interferon-related functional genes were identified as trans-targets for other AD susceptibility alleles (Ramdhani et al., 2020). Given these significant findings, in-depth analysis of the functional contribution of IFN-I pathway to AD pathogenesis is warranted.
We previously focused on young 5XFAD mice, a widely studied Aβ model, in which microglia innately respond to amyloid fibrils harboring nucleic acids (NA), activate IFN-I response pathway, and promote acute, complement-dependent synapse elimination (Roy et al., 2020). Here, we examined the accrual of IFN-activated microglia over time and assessed their contribution to broader microglial heterogeneity in disease. To determine the effects of IFN-I on cognitive function, plaque pathology, and neuroinflammation, we performed a long-term blockade in older 5XFAD mice with abundant plaques. We further analyzed 5XFAD mice deficient of IFN-I receptor in different cell lineages to reveal cell type-specific roles of IFN-I signaling. Overall, we found that IFN-I signaling through multiple cell types is essential for memory impairment and synaptic damage during amyloidosis.
Results
Amyloidosis promotes progressive cellular activation by IFN-I in the brain
Previously, we observed an age-dependent increase of NA-containing plaques in 5XFAD brains (Roy et al., 2020). Nucleic acids, when complexed to amyloid, serve as an immunogenic stimulus that elicits an IFN-I response (Di Domizio et al., 2012). We thus detected microglia with active IFN-I signaling, marked by nuclear Stat1, exclusively near NA+ amyloid plaques in young 5XFAD brains. To examine the brain cells activated by IFN-I throughout the disease process, we first generated a fate-mapping reporter mouse line (MxG) by crossing Mx1-Cre mice with the ROSA26mT/mG strain, in which IFN-I exposure results in Mx1 (a well-known ISG) promoter-driven, permanent GFP expression in responsive cells (Propson et al., 2021). These mice were then bred into the 5XFAD background and examined at different ages to gauge IFN-I signaling in the brain.
At 3 months, only a small number of GFP+ cells were detected, most of which were identified as plaque-associated microglia, making up 21% of all microglia in plaque-bearing regions (Figures 1A and 1B). By 5 months, GFP+ microglia became more prevalent and, by 11 months of age, GFP+ cells represented a majority of the microglia in plaque-rich regions.
Figure 1: Progressive IFN-I signaling in 5XFAD brain.

A, GFP expression in the brains of 5XFAD:MxG mice at different ages. Representative brain sections of 5XFAD and non-transgenic (nTg) control mice at 3 months (n = 3 mice), 5 months (n = 3 mice), and 11 months (n = 2 mice) of age. Scale bar, 250 μm.
B, Confocal images of tissues from A co-labelled with Iba1, and quantification of % GFP+ microglia at 3 months (n = 75 cells from 3 mice), 5 months (n = 301 cells from 3 mice), and 11 months (n = 140 cells from 2 mice). Z-stack projections; scale bar, 20 μm. Single color images are shown in Figure S1G.
C, High-magnification image of Stat1 expression within Iba1+ microglia surrounding an amyloid plaque (location marked by an asterisk), including GFP+ cells (solid white box) and GFP− cells (dashed white box). Images at right: isolated Stat1 channels overlayed on DAPI+ nuclei (outlined) of GFP+ (top) and GFP− (bottom) subsets of plaque-associated microglia. Z-stack projection; scale bars, 3.75 μm. Single color images are shown in Figure S1H. Quantification of relative nuclear Stat1 signal in individual GFP+ and GFP− microglia in 5-month-old control (nTg;MxG) and 5XFAD;MxG mice (n = 138 total cells). Ordinary one-way ANOVA and Bonferroni’s multiple-comparisons tests were performed. ns, not significant; ***P < 0.001.
D, Image of a 5-month-old 5XFAD;MxG brain (representative of n = 3 mice) highlighting Axl expression in GFP+ (solid arrowheads) and GFP− (hollow arrowheads) of microglia. Scale bar, 20 μm.
E, Images of 5-month-old 5XFAD;MxG brains showing GFP+ and GFP− cells (left) and varying Axl expression (middle) among the plaque-associated Clec7a+ subset of microglia. Z-stack projections; scale bars, 15 μm. Quantification of microglial subtypes at 5 months using Iba1, Clec7a, and IFN-I-dependent GFP reporter expression (n = 408 cells from 3 mice).
F, High-magnification single-plane confocal images of plaques from 5XFAD:MxG mice (left, n = 3 mice at 5 months of age) as in A-E, or Thy1-eGFP reporter mice (right, n = 3 mice at 6 months of age). Plaques are labelled by methoxy-X04 (gray) and outlined (dashed lines). Insets show single channels overlapping the plaque areas. Scale bars, 10 μm.
All: Mice in all groups were analyzed in parallel as part of one experiment, and images are representative. See also Figure S1 for additional analysis relating to this figure.
Although the MxG reporter is primarily used as a fate-mapping tool to permanently mark IFN-I signaling events, we confirmed that GFP+ microglia tend to express more nuclear Stat1, the key marker of IFN-I activation and transcriptional driver of ISG expression (Cheon and Stark, 2009), compared to GFP− microglia in 5XFAD brains (Figure 1C). We previously showed that Axl protein, a receptor tyrosine kinase (RTK) and known ISG, is enriched in both Stat1+ microglia surrounding amyloid plaques in mouse, and neuritic plaques in human brains (Roy et al., 2020). In 5XFAD;MxG mice, we also detected significant overlap of GFP signal and expression of Axl in microglia (Figure 1D). Together, these observations indicate an ongoing IFN-I activation in an expanding subset of plaque-associated microglia during disease.
In β-amyloidosis, a subset of microglia adopt a disease-associated microglia (DAM) phenotype, marked by high Clec7a expression (Keren-Shaul et al., 2017, Krasemann et al., 2017). To estimate the heterogeneity of IFN responsiveness within the DAM population, we quantified proportions of microglia in plaque-loaded regions of 5-month-old 5XFAD;MxG brains using GFP and Clec7a expression, and found that roughly half of Clec7a+ cells were GFP+ (Figure 1E). This is consistent with elevated ISG transcripts detected in pooled Clec7a+ microglia transcriptome from APP-PS1 mice (Roy et al., 2020), and reveals a subpopulation within DAM marked by IFN-I responsiveness. Besides microglia, GFP+ astrocytes and blood vessels expanded in an age-dependent manner in plaque-rich regions of 5XFAD brains (Figure S1), suggesting IFN-I signaling goes beyond microglia amid accumulating parenchymal amyloidosis. Of note, we found that the MxG reporter yielded no GFP expression in neurons after brain administration of IFNβ, though numerous glial cells and blood vessels turned green (data not shown). Since neurons were not amenable to MxG labelling, we therefore relied on other methods to detect IFN-I signaling in neurons in this study.
While examining the spatial relationship between GFP-labelled cells and amyloid plaques, we noticed that, besides plaque-associated microglia, certain GFP signals were clearly visible within the inner zones of many amyloid plaques (Figures 1C and 1F; Figure S1A). Such a phenomenon was not observed in 5XFAD;Thy1-eGFP brains, in which a subset of cortical and hippocampal neurons are intensely labelled with GFP (Figure 1F; Figure S1B), hinting at differential participation by microglia and neurons in amassing plaque material.
Altogether, these findings demonstrated an age-dependent accrual of IFN-activated brain cells accompanying progressive β-amyloidosis. Particularly, microglia represented the foremost IFN-responsive cell type and displayed complex heterogeneity in their cellular response to amyloid plaques.
IFN-I blockade rescues memory and synaptic deficits
To examine the role of IFN-I in memory impairment, we implanted osmotic pumps with ventricular cannulae to administer an antibody that specifically blocks signaling of IFNα receptor (IFNAR) into 4-month-old 5XFAD mice for 30 days (Figure 2A). Mice were then subjected to Y maze and novel object recognition (NOR) assays to evaluate cognitive aspects related to memory loss, before brain tissues were harvested and subjected to detailed histological examination and gene expression analysis. As shown in Figure 2B, 5XFAD mice receiving isotype control IgG failed to show spatial novelty preference in the Y maze, as well as discriminate between novel and familiar objects in the NOR test, indicating severe deficits in both working memory and short-term reference memory retrieval, respectively. In contrast, 5XFAD mice receiving IFNAR blocking antibody behaved comparably to non-transgenic control mice, suggesting a full restoration of the memory deficits spurred by amyloid deposition.
Figure 2: Long-term IFN-I blockade rescues memory and synaptic deficits without altering plaque load.

A, Experimental scheme of long-term IFN-I blockade in 5XFAD mice. Mice were randomized, treated and assayed for memory impairment in three separate mixed-gender cohorts.
B, Results of behavioral assays. Ctrl + IgG, n = 13 mice; Ctrl + αIFNAR, n = 13 mice; 5XFAD + IgG, n = 12 mice; 5XFAD + αIFNAR, n = 12 mice. Mice were treated and analyzed in three separate cohorts.
C, High-magnification images of pre- and post-synaptic markers (synaptophysin and PSD95, respectively) in subicula of treated Ctrl and 5XFAD mice. Z-stack projection of 2 μm thickness; scale bar, 3 μm. Quantification of relative synaptic densities, and the density of functional synapse pairs (⩽200nm between puncta).
D, Images of cFos expression in dentate gyrus (DG) neurons of treated mice. Z-stack projections; scale bar, 150 μm. Quantification of relative density of cFos+ nuclei in the DG.
E, Histological determination of plaque burden in treated mice using 6E10 antibody to mark Aβ fibrils, and thioflavin S (ThioS) to mark dense core plaques. Epifluorescence images; scale bar, 500 μm. Quantifications of plaque load for both markers in relevant brain regions. Single color images are shown in Figure S2G.
All: Data represent means and s.e.m. For C, D, and E: Ctrl + IgG, n = 13 mice; Ctrl + αIFNAR, n = 11 mice; 5XFAD + IgG, n = 11 mice; 5XFAD + αIFNAR, n = 12 mice. Differences between groups were analyzed by ordinary one-way ANOVA and Bonferroni’s multiple-comparisons tests (B, C, and D) or by two-tailed t-tests (E). ns, not significant; **P <0.01; ***P < 0.001. For histology, mice from three separate treatment cohorts were combined and analyzed in parallel as part of one experiment, and images are representative. See also Figure S2 for additional analysis relating to this figure.
We then performed histological characterization of the brain tissues to explore the neurophysiological basis for the marked reversal of memory loss. Examination of synapse markers showed that 5XFAD mice administered control IgG had reduced puncta densities of synaptophysin and PSD95 proteins, which label pre- and post-synaptic terminals on excitatory neurons, respectively, as well as in functional co-localization (≤200 nm) of these markers (Figure 2C). Consistent with the outcome of the cognitive assays, these proteins were both significantly elevated in 5XFAD mice with IFN-I blockade. On a more functional level, we examined the expression of immediate-early gene cFos as a surrogate marker of basal neural activity in the hippocampal circuitry. 5XFAD mice showed a decrease in the number of cFos+ neurons in the dentate gyrus compared to control mice, a deficit reversed by IFN-I blockade (Figure 2D). These observations indicate a negative effect of IFN-I on synaptic function and overall neural activity in brains bearing amyloid plaques.
To understand the impact of IFN-I blockade on amyloid pathology, we stained plaques with thioflavin S (ThioS) and anti-Aβ antibody but failed to detect a significant difference in dense core plaque load, total Aβ deposition, or average plaque volume between 5XFAD mice with and without IFN-I blockade in any brain region (Figure 2E; Figures S2B and S2G). Microglia are the primary phagocytes of Aβ species, therefore modifying plaque burden. However, IFN-I blockade did not affect the amount of Aβ taken up by microglia, nor decrease the expression of microglial CD68, a lysosomal receptor involved in phagocytosis (Figures S2C and S2D).
Therefore, IFNAR signaling in the brain parenchyma damages memory and synapses during amyloidosis, and suppression of the pathway is sufficient to restore these deficits despite abundant plaque pathology.
IFN-I blockade reduces microgliosis, inflammation, and neuritic pathologies
Next, we examined glia and peri-plaque neuritic structures in the treated animals. IFN-I blockade effectively reduced Stat1 in the nuclei of both plaque-associated microglia and neurons, indicating broad suppression of excessive IFN-I signaling in these cells, thus validating the efficacy of the blockade strategy (Figure 3A). We saw a partial reduction of total Iba1+ area with IFN-I blockade in both cortex and subiculum, the latter an area with the earliest and densest plaque deposition (Figure 3B). In contrast, astrocyte reactivity markers were not significantly affected in either region (Figures S3A and S3B). Further scrutiny of microglia showed that, on a per-cell basis, IFN-I blockade partially but significantly reduced Axl and Clec7a expression, but not overall numbers of microglia as counted by PU.1+ nuclei (Figure 3C; Figure S3D), implying a selective attenuation of microglial activation.
Figure 3: IFN-I blockade ameliorates microgliosis, inflammation, dystrophic axons, and neuritic tau.
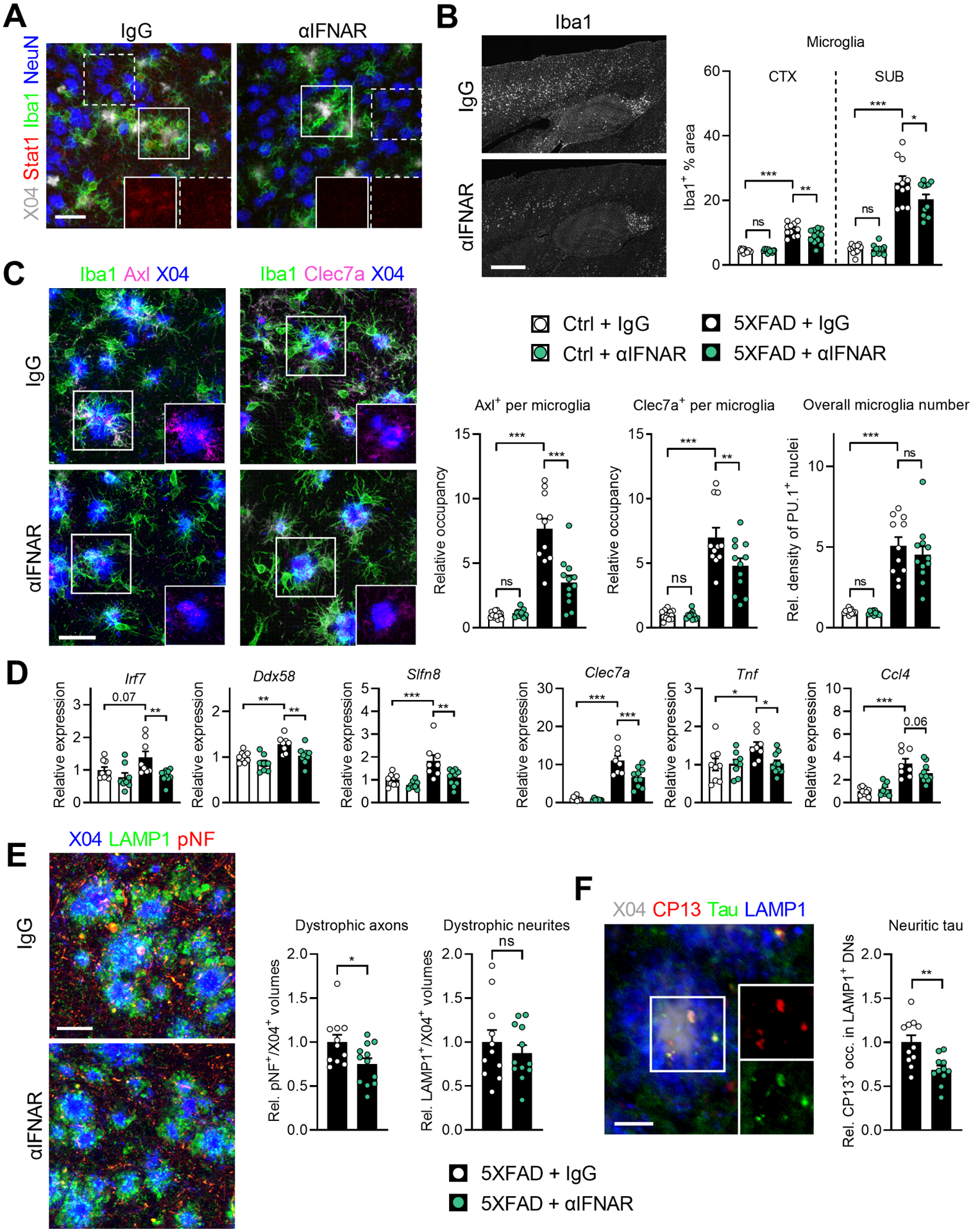
A, Representative images of Stat1 signals in different cell types of 5XFAD mice treated with IgG (n = 11 mice) or αIFNAR (n = 12 mice). Insets: isolated Stat1 channels from boxed areas showing microglia (solid boxes) and neuronal nuclei (dashed boxes) in each condition. Z-stack projections; scale bar, 30 μm.
B, Iba1 staining in the cortex and hippocampus of treated 5XFAD mice. Epifluorescence images; scale bar, 500 μm. Quantifications of % Iba1 area.
C, Images of plaque-associated microglia in treated 5XFAD mice expressing Axl and Clec7a (isolated in insets). Z-stack projections; scale bar, 30 μm. Quantification of microglial occupancy of both markers, and quantification of overall subicular microglia numbers by PU.1+ nuclei density.
D, Gene expression alterations with αIFNAR treatment as measured by Nanostring analysis on bulk hippocampal tissues. Ctrl + IgG, n = 9 mice; Ctrl + αIFNAR, n = 8 mice; 5XFAD + IgG, n = 8 mice; 5XFAD + αIFNAR, n = 10 mice.
E, Images and quantification of LAMP1+ dystrophic neurites (DNs) and phospho-neurofilament+ (pNF+) dystrophic axons (DAs) surrounding X04+ amyloid plaques in subicula of treated mice. Z-stack projections; scale bar, 20 μm. Single color images are shown in Figure S3F.
F, Image of endogenous tau foci inside LAMP1+ DNs in the subiculum of a 5XFAD animal (insets: isolated channels for CP13+ p-tau and total tau) and quantification of CP13+ occupancy in DNs after treatment with IgG or αIFNAR. Z-stack projection; scale bar, 30 μm. 5XFAD + IgG, n = 11 mice; 5XFAD + αIFNAR, n = 11 mice.
All: Data represent means and s.e.m. For B, C, and E: Ctrl + IgG, n = 13 mice; Ctrl + αIFNAR, n = 11 mice; 5XFAD + IgG, n = 11 mice; 5XFAD + αIFNAR, n = 12 mice. Differences between groups were analyzed by ordinary one-way ANOVA and Bonferroni’s multiple-comparisons tests (B, C, and D) or by two-tailed t-tests (E and F). ns, not significant; *P < 0.05; **P <0.01; ***P < 0.001. Mice from three separate treatment cohorts were combined and analyzed in parallel as part of one experiment, and images are representative. See also Figure S3 for additional analysis relating to this figure.
We next analyzed gene expression in bulk hippocampal tissues from the experimental cohorts and found that IFN-I blockade not only significantly lowered the expression of ISGs, such as Irf7, Ddx58 and Slfn8, as expected, but also decreased the expression of Clec7a, Tnf and Ccl4 (Figure 3D), suggesting a broad dampening effect on neuroinflammation in general.
Dystrophic neurites (DNs) surrounding amyloid plaques represent another hallmark of AD. We found that IFN-I blockade did not affect LAMP1+ DNs nor microglial barrier formation, the latter known to be crucial in DN biogenesis (Figure 3E; Figure S3E). In contrast, both phospho-neurofilament+ dystrophic axons and phosphorylated endogenous tau within DNs were partially but significantly diminished by IFN-I blockade in 5XFAD brain (Figures 3E and 3F), implying an IFN-mediated mechanism in promoting these pathologies.
Selective microglial Ifnar1 deletion alters microglial activation and prevents post-synaptic loss
All nucleated mammalian cells express IFNAR and thus can respond to the cytokine. Although our analysis identified microglia as the earliest and primary responders to IFN-I, how instrumental microglia are in mediating IFN-I’s overall effects in the brain is not known. We bred Ifnar1fl/fl with Cx3cr1-CreERT2 mice, then crossed with the 5XFAD strain to generate mice lacking IFN-I receptor selectively in microglia (here termed 5XFAD;MKO). When FACS-sorted CD11b+ and Cd11b− cells from the brains of MKO mice were analyzed, a significant reduction of Ifnar1 was detected, in conjunction with decreased ISGs, only in the Cd11b+ population, confirming the selective deletion in microglia (Figure S4A). Accordingly, Stat1 protein was noticeably absent in plaque-associated microglia in 5XFAD;MKO brains (Figure 4A).
Figure 4: Selective microglial Ifnar1 ablation decreases microglial reactivity and rescues post-synaptic loss.
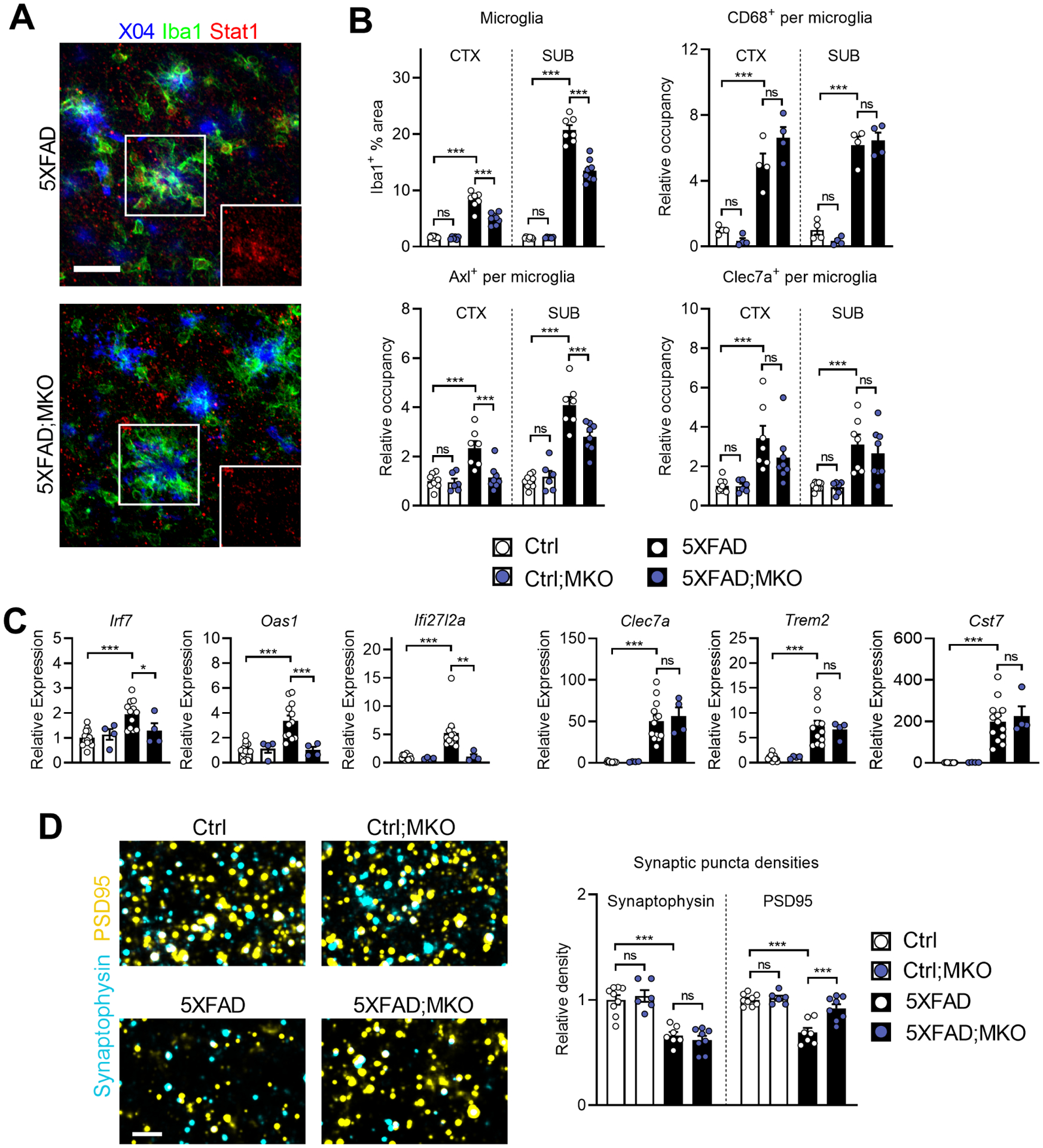
A, Representative images of Stat1 expression in subicula of 5XFAD;MKO mice (n = 8 mice) compared to Ifnar1-sufficient 5XFAD mice (n = 7 mice). Insets show isolated Stat1 channel of boxed areas. Z-stack projections; scale bar, 30 μm.
B, Quantifications of total % Iba1+ area and activation marker occupancy by region. For Iba1, Axl, and Clec7a: Ctrl, n = 9 mice; Ctrl;MKO, n = 6 mice; 5XFAD, n = 7 mice; 5XFAD;MKO, n = 8 mice. For Cd68: n = 4 mice per genotype.
C, Relative expression of ISGs and microglial activation markers analyzed by qPCR on bulk hippocampal tissues. Ctrl, n = 13 mice; Ctrl;MKO, n = 4 mice; 5XFAD, n = 12 mice; 5XFAD;MKO, n = 4 mice.
D, High-magnification confocal images of pre- and post-synaptic puncta (synaptophysin and PSD95, respectively) in subiculum. Z-stack projections of 2 μm thickness; scale bar, 3 μm. Quantification of relative synaptic puncta densities. Ctrl, n = 9 mice; Ctrl;MKO, n = 6 mice; 5XFAD, n = 7 mice; 5XFAD;MKO, n = 8 mice.
All: Data represent means and s.e.m. Differences between groups were analyzed by ordinary one-way ANOVA and Bonferroni’s multiple-comparisons tests. ns, not significant; *P < 0.05; **P <0.01; ***P < 0.001. Brain tissues were collected over time, combined and analyzed in parallel as part of one experiment, and images are representative. See also Figure S4 for additional analysis relating to this figure.
Similar to IFN-I blockade, microglia in 5-month-old 5XFAD;MKO mice showed a reduction of Iba1+ area and significantly less Axl expression on a per-cell basis (Figure 4B). However, Clec7a and CD68 expression were not affected, and neither were overall microglial numbers (Figure 4B; Figure S4B). 5XFAD;MKO mice expressed significantly less ISG transcripts, such as Irf7, Oas1 and Ifi2712a, while maintaining the expression of classical DAM markers, such as Clec7a, Trem2 and Cst7 (Figure 4C). These findings suggest that microglial IFN-I signaling selectively regulates a subset of molecular changes observed in plaque-associated microglia.
Examination of synaptic markers in subiculum yielded an unexpected effect – while both pre- and post-synapses were reduced in normal 5XFAD brains, PSD95+ puncta density, but not synaptophysin+, was restored in 5XFAD;MKO (Figure 4D). In addition to the subiculum, CA1 and CA3 regions of the hippocampus showed similar trends for these synaptic markers (Figure S4C). Independent analysis of Vglut1 and GluR, another pair of excitatory pre- and post-synaptic markers, respectively, showed reestablishment of GluR, but not Vglut1, in 5XFAD;MKO mice (Figure S4C), corroborating a selective effect of microglial IFN-I signaling on post-terminals of excitatory synapses. We also examined dystrophic neuronal structures and found that neuritic endogenous p-tau signals were significantly reduced by ablating microglial IFN-I signaling, despite comparable axonal dystrophy (Figure S5E). Altogether, these results reveal selective functions of IFN-I signaling in microglial activation and synapse modification.
Selective neural Ifnar1 deletion reduces amyloid plaques and restores pre-synaptic terminals
To understand the importance of IFN-I signaling in non-microglial cells in the brain, we bred Ifnar1fl/fl and Nestin-Cre mice to generate 5XFAD mice with the type I IFN receptor ablated in neuroectodermal-derived cells, including neurons and glial cells other than microglia (here termed 5XFAD;NKO). Consistent with the conditional deletion, Stat1 signal in 5XFAD;NKO neuronal nuclei was selectively reduced (Figure 5A). Of note, we did not detect overt accumulation of LC3B-II or protein hyper-ubiquitination in the brains of adult Ifnar1fl/fl;Nestin-Cre mice as reported (Ejlerskov et al., 2015), nor decreased expression of endogenous or transgenic full-length amyloid precursor protein (APP) with conditional Ifnar1 ablation, as reported to be affected by germline Ifnar1 deletion (Minter et al., 2016) (Figure S5A).
Figure 5: Selective neural Ifnar1 ablation decreases plaque load and rescues pre-synaptic loss.
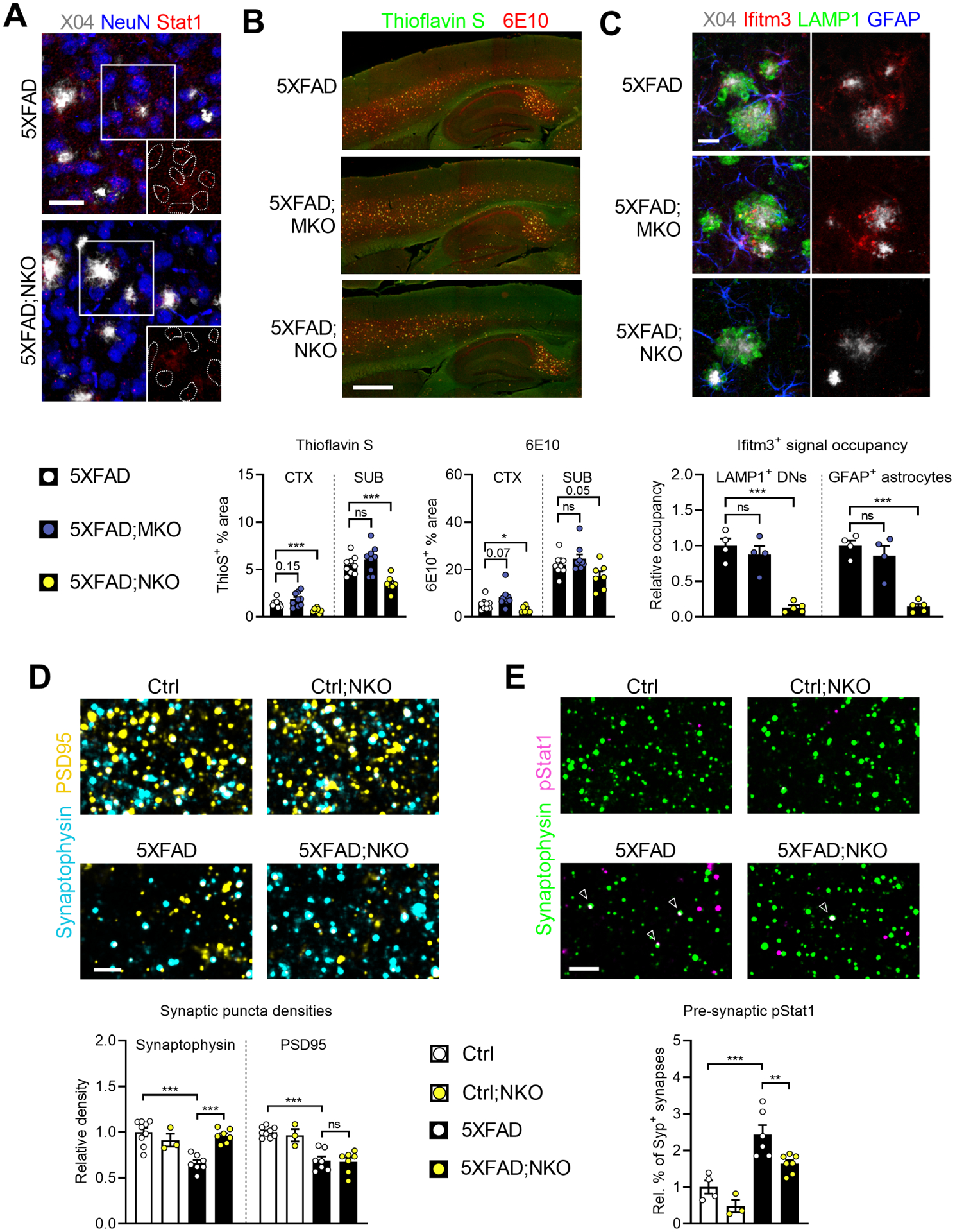
A, Representative images of neuronal Stat1 expression in subicula of 5XFAD;NKO mice (n = 7 mice) compared to Ifnar1-sufficient 5XFAD mice (n = 7 mice). Insets show isolated Stat1 channels in outlined NeuN+ neuronal nuclei. Z-stack projections; scale bar, 30 μm.
B, Histological determination of plaque burden. Epifluorescence images; scale bar, 500 μm. Quantification (below) of % area of plaque markers by brain region. 5XFAD, n = 9 mice; 5XFAD;MKO, n= 9 mice; 5XFAD;NKO, n= 7 mice.
C, Images (top) and quantification (bottom) of Ifitm3 signals localized inside LAMP1+ DNs and GFAP+ astrocytes in subiculum. Z-stack projections; scale bar, 20 μm. 5XFAD, n = 4 mice; 5XFAD;MKO, n = 4 mice; 5XFAD;NKO, n = 5 mice.
D, High-magnification confocal images of pre- and post-synaptic puncta (synaptophysin and PSD95, respectively) in subiculum. Z-stack projections of 2 μm thickness; scale bar, 3 μm. Ctrl, n= 9 mice; Ctrl;NKO, n= 3 mice; 5XFAD, n= 7 mice; 5XFAD;NKO, n= 7 mice.
E, High-magnification confocal images and quantification (below) of pStat1+ signals co-localized with Syp+ pre-synaptic boutons in subiculum. Z-stack projections of 2 μm thickness; scale bar, 3 μm. Ctrl, n= 4 mice; Ctrl;NKO, n= 3 mice; 5XFAD, n= 6 mice; 5XFAD;NKO, n= 7 mice.
All: Data represent means and s.e.m. Differences between groups were analyzed by ordinary one-way ANOVA and Bonferroni’s multiple-comparisons tests (D and E), or by two-tailed t-tests (B and C). ns, not significant; *P < 0.05; **P <0.01; ***P < 0.001. Brain tissues were collected over time, combined and analyzed in parallel as part of one experiment, and images are representative. See also Figure S5 for additional analysis relating to this figure.
In contrast to IFN-I blockade and microglia-specific Ifnar1 ablation, 5XFAD;NKO mice at 5 months accumulated fewer ThioS+ and 6E10+ plaques in different brain regions (Figure 5B). However, measurement of Aβ inside microglial CD68+ vesicles indicated unaltered plaque phagocytosis by microglia (Figure S5B). In line with immunoblotting results on whole brain lysates, APP expression in dystrophic neurites was unchanged (Figure S5I). Hence, plaque density alteration is unlikely to have resulted from changes in APP expression or microglial clearance, but may be mediated by some intermediate process.
Since IFITM3 is known to function as an inflammation-triggered switch to enhance Aβ production (Hur et al., 2020), we investigated the possible involvement of this ISG. First, we confirmed a sensitive and dose-dependent induction of Ifitm3 protein in primary neurons by IFNβ (Figure S5C). Further, we detected the upregulation of Ifitm3 protein in dystrophic neurites, a known site of heightened Aβ production and release, as well as in astrocytes in 5XFAD brains (Figure S5D). On the other hand, a selective diminution of Ifitm3 was detected in 5XFAD;NKO, but not 5XFAD;MKO, mice (Figure 5C). Neural IFN-I signaling did not have a major impact on overall dystrophic neuronal structures (Figure S5E).
On synaptic regulation, 5XFAD;NKO displayed an opposing phenotype to 5XFAD;MKO, such that synaptophysin+ puncta density, but not PSD95+, was restored, implying a neural-intrinsic and IFN-I-dependent regulation of excitatory pre-synaptic bouton density during disease (Figure 5D). This was further confirmed by analyzing other hippocampal areas and examining independent excitatory synaptic markers (Figures S4C and S4D). By contrast, quantification of inhibitory pre-synaptic markers (VGAT+ and GAD65+ puncta) showed no comparable rescue from the deficits observed in the disease model (Figure S4E). Activity-dependent events shape neuronal networks in part by elimination of inactive synapses, a mechanism critical for proper configuration of circuits. In post-natal brain, Stat1 signaling at inactive excitatory pre-synaptic terminals is instrumental for synapse refinement (Yasuda et al., 2021). To gauge the relevance of this axis in pathogenic pre-synaptic loss during β-amyloidosis, we employed an antibody against Stat1 phosphorylated at Tyr701 (pStat1) and detected enhanced frequency of pStat1+ pre-synaptic boutons in 5XFAD brain (Figures S5F and S5G), suggesting a potential functional involvement. In 5XFAD;NKO subicula, a significantly lower percentage of Syp+ pre-synapses were pStat1 positive compared to 5XFAD (Figure 5E). This observation was specific to pStat1, as pStat3 (Tyr705), a key signaling mediator for multiple proinflammatory cytokines, displayed no enrichment in diseased synapses compared to control (Figures S5F and S5G). In accordance, fewer pStat1+ puncta were present in the nuclei of CA1 neurons, which project their axons to the subiculum (Figure S5H).
Overall, these findings reveal pathogenic effects of IFN-I signaling in non-microglial brain cells on plaque formation and synaptic pathology.
IFN-I signaling promotes microglia-mediated synaptic engulfment
Previously we showed that post-synaptic loss in 3-month-old 5XFAD brain is coupled with IFN-stimulated uptake by microglia (Roy et al., 2020). We did not in that study detect any change in overall pre-synaptic density nor microglial pre-synaptic engulfment, implying that synapse loss is restricted to the post-synaptic compartment early in disease, and is IFN- and microglia-dependent. Given the concurrent deficits in pre- and post-synaptic elements (Figure 2C) and seemingly differential cellular requirements for IFN-I signaling (Figures 4 and 5) in mid-stage 5XFAD mice, we sought to further investigate microglial involvement in synapse modification.
First, synaptic engulfment assays showed that microglia in 5-month-old 5XFAD brain selectively engulfed enhanced amounts of PSD95+ puncta, an activity dependent on extracellular IFN-I (Figure 6A), and necessarily and exclusively mediated by IFN-I signaling in microglia (Figure 6B). Consistent with this, more PSD95 was detected inside GFP+ microglia over GFP− counterparts in plaque-rich regions of 5XFAD;MxG mice (see Figure 1) at 5 months (Figure 6C). In contrast, no enhanced synaptophysin signal was detected inside microglia from 5XFAD brains, with or without IFN-I signaling, compared to control mice (Figures 6A and 6B), suggesting a selective post-synaptic elimination by microglia, persisting at different stages of disease.
Figure 6: Post-synapses are preferentially engulfed by IFN-I-stimulated Axl+ microglia.
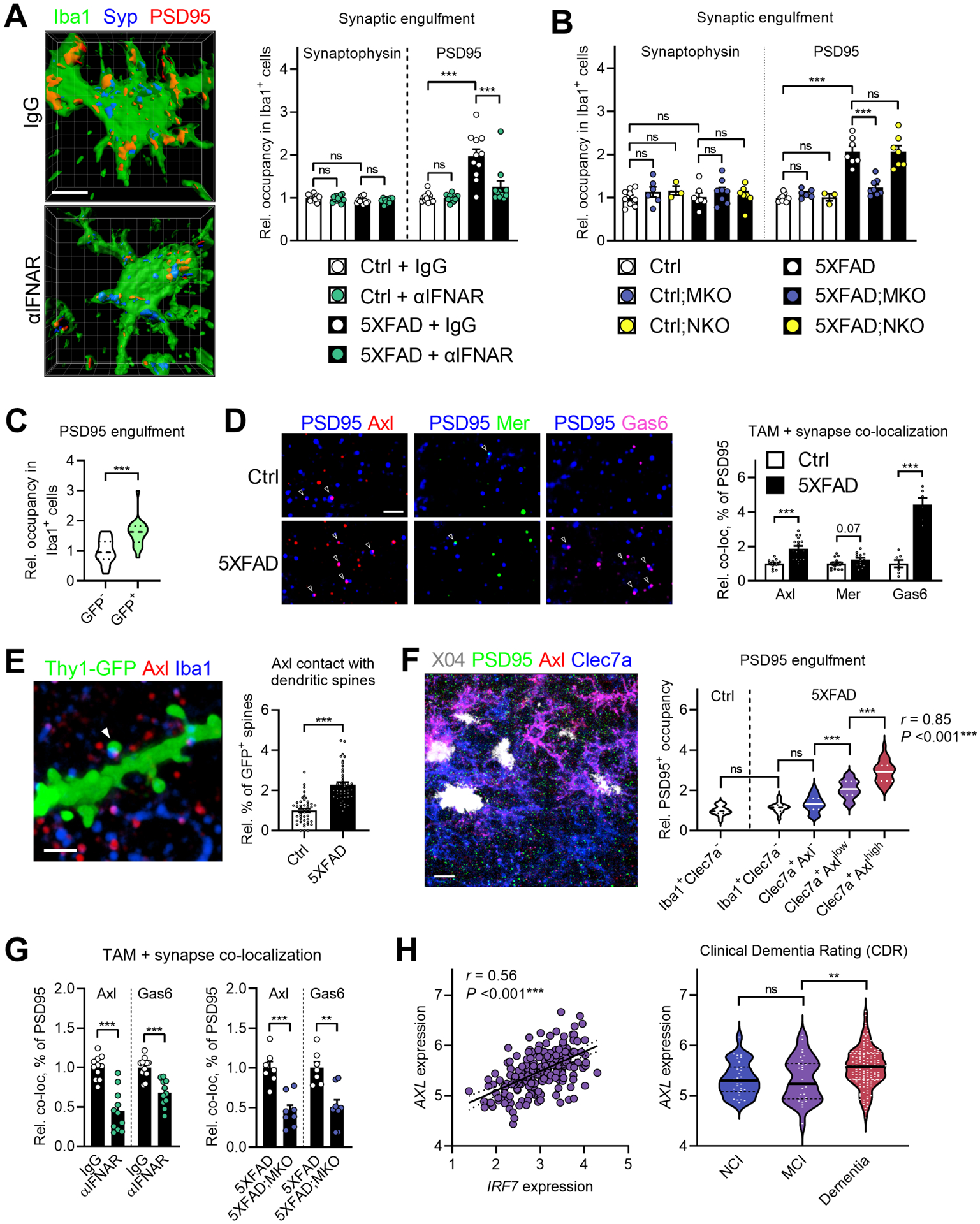
A, 3D renderings of microglia engulfing synaptic markers in subicula of 5XFAD mice subjected to IFN-I blockade from Figure 2. Scale bar, 5 μm. Quantification of microglial engulfment of these markers. Ctrl + IgG, n = 13 mice; Ctrl + αIFNAR, n = 11 mice; 5XFAD + IgG, n = 11 mice; 5XFAD + αIFNAR, n = 11 mice.
B, Quantification of microglial engulfment of synaptic markers in subicula of mice with conditional Ifnar1 deletion. Ctrl, n = 9 mice; Ctrl;MKO, n = 6 mice; Ctrl;NKO, n = 3 mice; 5XFAD, n = 7 mice; 5XFAD;MKO, n = 8 mice; and 5XFAD;NKO, n = 7 mice.
C, Quantification of relative uptake of PSD95+ synaptic puncta by GFP− (n = 19 cells) and GFP+ microglia (n = 21 cells) from 5-month-old 5XFAD;MxG mice (n = 3 mice).
D, High-magnification images of PSD95+ synaptic puncta in proximity (⩽200 nm; arrows) to Axl, Mer, and Gas6 in Ctrl and 5XFAD brains. Scale bar, 2 μm. Quantification of co-localization, as relative percent of PSD95+ puncta, of TAM molecules with PSD95 (Axl + PSD95: n = 13 Ctrl images, n = 17 5XFAD images; Mer + PSD95: n = 14 Ctrl images, n = 15 5XFAD images; Gas6 + PSD95: n = 7 Ctrl images, n = 6 5XFAD images). Data is representative of multiple animals in two independent experiments.
E, High-magnification confocal image of Axl+ microglial processes contacting dendritic spines (arrow) in a 5XFAD;Thy1-eGFP mouse at 5 months. Scale bar, 2 μm. Quantification of relative frequency of observed contacts between control (n = 42 dendrites >10 μm long from n = 8 mice) and 5XFAD mice (n = 44 dendrites >10 μm long from n = 8 mice).
F, (left) Confocal image of Clec7a+ microglia with varying degrees of Axl expression in relation to PSD95 in a 5-month-old 5XFAD animal. Scale bar, 20 μm. (right) Histological analysis of single microglia in both control and 5XFAD brains stratified by Clec7a and Axl expression. Ctrl Iba1+Clec7a−, n = 17 cells; 5XFAD Iba1+Clec7a−, n = 19 cells; 5XFAD Clec7a+Axl−, n = 37 cells; 5XFAD Clec7a+Axllow, n = 35 cells; 5XFAD Clec7a+Axlhigh, n = 32 cells; all cells combined from n = 2 Ctrl mice and n = 3 5XFAD mice at 5 months. Pearson r was calculated by correlation analysis of Axl expression and PSD95+ uptake in all Clec7a+ cells.
G, Quantified co-localization of Axl and Gas6 with PSD95, as relative percent of PSD95, in 5XFAD mice treated with IgG (n = 11 mice) or αIFNAR (n = 11 mice), and in microglial Ifnar1 conditional deletion lines (5XFAD, n = 7 mice; 5XFAD;MKO, n = 8 mice).
H, AXL expression in human AD correlated with IRF7 expression (left), and plotted by Clinical Dementia Rating (CDR) classification (right). Both plots: n = 195 total subjects. For IRF7: Pearson correlation (r) was calculated, and simple linear regression line (solid) with 95% CI intervals (dashed) were plotted. For CDR: ‘NCI’=no cognitive impairment, CDR=0, n = 30 subjects; ‘MCI’=mild cognitive impairment, CDR=0.5, n = 30 subjects; ‘Dementia’, CDR>0.5, n = 135 subjects.
All: Data represent means and s.e.m. (A, B, D, E, and G) or presented as violin plots with medians (dashed lines) and quartiles (dotted lines) (C, F, and H). Differences between groups were analyzed by ordinary one-way ANOVA and Bonferroni’s multiple-comparisons tests (A, B, F, and H), or by two-tailed t-tests (C-E, and G). ns, not significant; **P <0.01; ***P < 0.001. For mouse data, brain tissues were collected over time, combined and analyzed in parallel as part of one experiment, and images are representative. See also Figure S6 for additional analysis relating to this figure.
We previously showed that IFN-I-activated microglia rapidly remove dendritic spines in a complement C3-dependent manner (Roy et al., 2020). Although IFNβ is sufficient in inducing many members of the complement cascade in wild-type mice, blockade of extracellular IFN-I or genetic IFN-I receptor ablation in 5XFAD mice did not reduce complement transcription (Figures S6A and S6B), consistent with unchanged C3 protein in astrocytes (Figure S3B), indicating that signals other than IFN-I may play a role in chronic complement activation in older 5XFAD mice.
Perineuronal nets (PNN) are extracellular matrix structures that enwrap and stabilize synapses on inhibitory interneurons, loss of which in 5XFAD is known to be mediated by microglia (Crapser et al., 2020). Although Wisteria floribunda agglutinin (WFA) staining confirmed a significant reduction of PNN structures in disease, IFN-I blockade did not appear to affect this deficit (Figure S6C). This finding is in line with the persistent deficit of inhibitory synapses in the mice (Figure S2A; Figure S4E) and argues against a direct link between IFN-I and PNN modification.
Axl is a member of the TAM (Tyro3, Axl, and Mer) family of RTKs that plays important roles in phagocytosis of apoptotic cells (Lemke, 2019). Recently, a plaque-centric expression pattern of TAM receptors and their ligand Gas6 was reported to engage microglia with amyloid plaques in a largely Mer-dependent manner (Huang et al., 2021). Given the high sensitivity of microglial Axl to IFN-I signaling, we investigated its relation to synapses, together with Mer and Gas6. Employing high-magnification confocal imaging, we detected specific, punctate signals for Gas6 as well as both Axl and Mer in wild-type brain (Figure S6D), which displayed non-random co-localization with synaptic puncta (Figure S6E), indicating a physiological interaction of TAM molecules with synapses. In diseased brain, we observed notable Gas6 deposition on amyloid plaques and enhanced Mer expression in plaque-associated microglia (Figures S6F and S6G), in agreement with Huang et al (Huang et al., 2021). However, unlike Axl, Mer expression in microglia, as well as extent of Gas6 deposition on plaques, were not IFN-I-dependent. At the synaptic level, we found substantial Gas6 deposition on PSD95+ synaptic terminals in 5XFAD brains, which was accompanied by significantly increased Axl, but not Mer, co-localization with PSD95 (Figure 6D). To visualize the physical relationship between Axl and synapses, we analyzed dendritic spines of 5XFAD mice containing the Thy1-eGFP reporter. High-magnification confocal imaging revealed the formation of contact points between GFP+ dendritic spines and Axl+ microglial processes, which were significantly more frequent along dendrites in 5XFAD mice (Figure 6E), indicating enhanced engagement of Axl with synapses in disease.
To explore the role of Axl in synapse uptake, we examined PSD95 engulfment by microglia from control and 5XFAD brains, particularly Clec7a+ plaque-associated microglia with varying expression of Axl, and detected robust per-cell correlation of Axl and PSD95 occupancy in microglia (Figure 6F). To test whether Axl and Gas6 localization to synapses is dependent on IFN-I signaling, we measured the frequency of Axl/PSD95 and Gas6/PSD95 co-localization in 5XFAD mice treated with IgG or αIFNAR and found that co-localization of both Axl receptor and Gas6 ligand to synapses was reduced with IFN-I blockade, a finding mirrored by microglial conditional Ifnar1 deletion (Figure 6G). Furthermore, hippocampal Axl mRNA expression was positively correlated with post-synapse engulfment in 5XFAD (Figure S6J). Altogether, these data obtained from AD animal model analysis identified an IFN-instructed synapse elimination program in microglia that compromises memory.
To gain insight into the effects of IFN-I signaling on cognitive deficits associated with human AD, we queried the clinical and gene expression data from Mt. Sinai Brain Bank (MSBB), which shows profound upregulation of ISGs in human AD brain tissues (Roy et al., 2020). Subjects rated with Clinical Dementia Rating (CDR) as ‘Dementia’ (CDR > 1) displayed significant upregulation of 458 genes (with fold change >1.15, P < 0.01) compared to cognitively healthy individuals (CDR = 0). Pathway analysis of these genes revealed that clinical dementia was strongly associated with multiple modules of gene expression changes (Figure S6H). Highly represented among AD-enriched modules were pathways directly related to IFN-I signaling and innate immune cascades leading to IFNα/β production (Figure S6H), indicating the centrality of IFN-I pathway within the broader neuroinflammatory network. IRF7 is a master transcriptional regulator of IFN-I response, expression of which is significantly increased in AD (Roy et al., 2020). In MSBB, while immune-related processes were enriched in genes positively associated with IRF7 (r > 0.6) across all human subjects, numerous pathways governing neuronal function and synaptic transmission were enriched in genes inversely correlated with IRF7 expression (r < −0.6; Figure S6I). Furthermore, expression of human AXL showed high correlation with IRF7 expression and was significantly elevated in AD patients (Figure 6H), a finding consistent with our results from AD mice. In total, these data pinpoint IFN-I pathway as central to the neuroinflammatory program and highlight its antagonistic effect on cognition in human AD.
Discussion
Beyond antiviral function, IFN-I is linked to cognitive and neuropsychiatric dysfunction in various clinical contexts (Fritz-French and Tyor, 2012, Valentine et al., 1998, Asnis and De La Garza, 2005, Lieb et al., 2006). Previous studies describe that IFN-I modifies the brain through microglia activation, neural stem cell dysfunction, and disruption of whole-brain functional network connectivity (Zheng et al., 2014, Hayley et al., 2013, Wachholz et al., 2016, Dipasquale et al., 2016). Of the neuropathological features of AD, synapse loss appears early and correlates most strongly with dementia thus represents a key step of the disease process (Koffie et al., 2011). This current study reveals discrete and coordinated actions of IFN-I-stimulated brain cells in compromising synapses and causing memory impairment under the sterile inflammatory condition initiated by AD pathology.
Multiple microglia populations identified by scRNA-seq analyses correspond to different activation states under various physiological or pathological conditions (Prinz et al., 2021). Parallel to DAM, IRMs represent a subset of microglia arising in models of AD and brain aging (Sala Frigerio et al., 2019, Olah et al., 2020, Hammond et al., 2019). In line with our findings that microglial IFN-I response universally accompanies brain amyloidosis in vivo (Roy et al., 2020), a proteome analysis revealed that IFN-I pathway is activated early and persists in microglia of murine Aβ models (Sebastian Monasor et al., 2020). Using an IFN-sensitive reporter system, we documented here a plaque-dependent, brain-wide, and profound accrual of brain cells responding to IFN-I signaling activation in the 5XFAD model. In young mice, a sparse population of microglia were the principal IFN-I-responsive cell type, consistent with the results obtained with Stat1 staining (Roy et al., 2020). By 5 months, about half of plaque-associated Clec7a+ microglia expressed GFP, revealing a further layer of microglial heterogeneity. Given the higher percentage of cells turning green at older ages, microglia seemed to be activated by IFN-I continuously as amyloidosis progresses. Further, our data suggest that IFN-I signaling in microglia is neither required for transition to DAM, nor does it in turn significantly affect DAM markers after activation, as over 90% of GFP+ microglia retained Clec7a expression.
As IFN-I-responsive microglia accumulated, other brain cells became GFP+, revealing a more complex and wider IFN-I response than previously appreciated. Specifically, we detected IFN-I-responsive astrocytes that expanded in an Aβ pathology-dependent manner. Recently, a specialized subset of astrocytes expressing ISGs were observed near blood vessels, ventricles, and meningeal layers upon acute systemic inflammation (Hasel et al., 2021). They were also detected in multiple disease models, including 5XFAD. Using the MxG reporter system, we found that GFP+ astrocytes arose within regions bearing the most plaque pathology, namely subiculum, entorhinal cortex, cortical layer V, hippocampus, and thalamus. How IFN-I-responsive astrocytes engage with inflammatory signals from plaque-associated microglia is unknown. Future studies using conditional astrocytic IFNAR ablation may help elucidate the function of this subtype among the heterogeneous astrocyte activation states.
Many brain cell types participate in plaque formation and neuritic pathology, a process marked by a multicellular co-expression network of plaque-induced genes (PIGs) (Chen et al., 2020). We found that 22 of the 57 PIG module are CNS ISGs (Roy et al., 2020, Li et al., 2018, Cho et al., 2013), many of which overlap with markers of DAM and neurotoxic reactive astrocytes (Liddelow et al., 2017), further highlighting a profound influence of IFN-I in dysregulating the cellular network. The size of such effect is consistent with the magnitude of cellular responsivity revealed by the MxG reporter but significantly higher than the percentage of IRMs and IFN-responsive astrocytes detected by scRNA-seq or snRNA-seq, respectively (Sala Frigerio et al., 2019, Olah et al., 2020, Hasel et al., 2021). The discrepancy may stem from the differential reliance on protein vs. mRNA detection and additional technical limitations.
Our scrutiny of amyloid plaques formed in the presence of GFP-labeled microglia and neurons revealed a preferential incorporation of microglial protein content inside plaque cores. This unexpected finding is pertinent to the role of microglia in promoting dense-core plaque development (Huang et al., 2021, Spangenberg et al., 2019, Baik et al., 2016). Two processes may contribute to such a phenomenon: first, microglia may ingest and release Aβ aggregates in a more compact form, or as protein complexes that further seed plaques (Venegas et al., 2017, d’Errico et al., 2022); or second, after taking up Aβ, plaque-associated microglia may undergo cell death, as documented by serial intravital two-photon microscopy (Baik et al., 2016), and subsequently affix to the plaque. Although our findings align with the second process, whether and how microglial cell death directly contributes to plaque growth necessitates further investigation.
We obtained apparently conflicting results on whether IFN-I signaling affects plaque pathology: blocking IFN-I receptor did not, while neural Ifnar1 deletion partially reduced plaque load. The latter observation was correlated with significantly tempered Ifitm3 expression, in keeping with the activity of Ifitm3 in promoting APP cleavage and amyloid pathology (Hur et al., 2020). Paradoxically, Ifitm3 was unaltered with IFN-I blockade, implying a difference between extracellular IFN-I blocking and genetic ablation. One possible explanation comes from an observation with therapeutic αIFNAR antibodies that IFN-I has more persistent effects in cells devoid of negative IFN-I regulators ISG15 and USP18 (Altman et al., 2020). Cortical neurons do not express Isg15 or Usp18 (both are ISGs) even after IFN-I exposure, in contrast to microglia (Li et al., 2018, Cho et al., 2013). Given the sensitive neuronal response of Ifitm3 induction by IFN-I, it is plausible that, under IFNAR blockade, residual IFN-I receptor signaling was sufficient to maintain the expression of neuronal Ifitm3.
A key AD pathology is dystrophic pre-synaptic neurites surrounding amyloid plaques, which accumulate APP as well as β- and γ-secretases, and serve as localized sites of Aβ generation and release (Sadleir et al., 2016, Sanchez-Varo et al., 2012). As we have shown, neural IFN-I signaling was required for expression of Ifitm3, which is known to enhance γ-secretase activity. β-secretase expression is also reportedly regulated by interferon and Stat1 (Cho et al., 2007, Cho et al., 2009), implying a sweeping effect of IFN-I on APP processing. Overall, our results support a feed-forward Aβ-plaque-IFN-Aβ loop whereby inflammation stimulates factors that further exacerbate AD pathology.
We discovered that synaptophysin+ boutons were selectively diminished by IFN-I signaling in neural cells, in sharp contrast to the regulation of post-synaptic densities. A Jak2-Stat1 axis serves as a major neuron-autonomous determinant to eliminate inactive synapses in vivo (Yasuda et al., 2021). Stat1 functions not only as a negative regulator of spatial memory formation in wild-type mice but is also a key mediator of Aβ-induced learning and memory deficits (Hsu et al., 2014, Tai et al., 2011). In 5XFAD brain, we detected increased pStat1 localized to pre-synapses as well as the nuclei of their corresponding neurons, indicating a likely two-way communication between synapses and nuclei of the affected neurons. We found both signals reduced upon IFN-I blockade or genetic ablation. Hence, the evidence strongly supports a neuronal IFN-Stat1 axis that pathogenically modulates the pre-synapse in AD, analogous to the process of synapse refinement during neurodevelopment. It should be noted that, while pre- and post-synaptic compartments were differentially rescued in the NKO and MKO lines, cFos+ neurons in the hippocampus were not restored in either, as they were after αIFNAR blockade. Therefore, IFN-I signaling in multiple CNS cell types contributes to whole-network disruption.
While microglia prune synapses during normal CNS development, excessive removal can result in pathological synapse loss in diverse neurological and neuropsychiatric diseases (Wilton et al., 2019). In β-amyloidosis models, germline C3 deficiency protects from loss of synapses and neurons (Shi et al., 2017, Wu et al., 2019), and microglia engulf C1q-tagged post-synaptic components early in the disease (Hong et al., 2016). Despite the strong relationship between IFN-I and complement in young 5XFAD mice, we found unexpectedly that IFN-I signaling became dispensable in complement expression in 5-month-old mice, likely reflecting the influences of other prevailing proinflammatory signals. In contrast, IFN-I blockade was effective to blunt Tnf and Clec7a expression, similar to the treatment effects in 10- to 12-month-old APPNL-G-F mice (Roy et al., 2020).
Besides complement, microglia use myriad other surface receptors to engulf or otherwise limit synapses (Filipello et al., 2018, Cheadle et al., 2020, Li et al., 2020, Scott-Hewitt et al., 2020). Several synapse-eliminating receptors recognize a common neuronal cue: phosphatidylserine (PS), the “eat-me” signal for phagocytosis (Peet et al., 2020). As principal myeloid phagocytic receptors, Axl and Mer detect PS exposed on apoptotic cells via their ligand Gas6, which has high affinity towards PS (Lemke, 2019). In CNS, Mer facilitates astrocytic phagocytosis of synapses in developing and adult brain (Chung et al., 2013), and engages plaques in AD (Huang et al., 2021). Yet, the function of Axl in the brain, despite its prominent upregulation in AD, was unknown. Here, we demonstrated a highly IFN-I-dependent Axl expression in microglia surrounding amyloid plaques. As reported (Huang et al., 2021), we found Mertk mRNA positively correlated with dense-core plaques. However, IFN-I blockade and microglial Ifnar1 deletion effectively reduced Axl expression but failed to modify the plaques, in line with Axl deficiency in APP/PS1 model (Huang et al., 2021). On the other hand, we showed direct contact between Axl and synapses along with enrichment of synaptic material inside Axl+ microglia, all of which were IFN-I-dependent in β-amyloidosis. Further, Axl mRNA, but not Mertk mRNA, was positively correlated with post-synapse engulfment. These results imply a distinct role of Axl in impairing synapses. Consistent with this notion, Gas6 overexpression worsens behavior of APP/PS1 mice despite reduced plaque burden (Owlett et al., 2022). In line with the elevated AXL expression in human AD (Roy et al., 2020), both soluble AXL and GAS6 are increased in cerebrospinal fluid of AD patients (Sainaghi et al., 2017, Mattsson et al., 2013). Altogether, these findings warrant further characterization of a pathogenic involvement of Axl in AD via examining the impact of microglial-specific Axl deficiency and dissecting TAM receptor-ligand interactions in diseased brain.
Limitations of the study
Our investigation into microglial heterogeneity is limited by the irreversible nature of the fate-mapping reporter, which does not assess real-time IFN-I signaling. The cellular IFN-I response in AD should be further investigated in detail using temporal reporters for real-time tracking and by improved spatial phenotypic profiling in the future. Despite the evidence of Axl co-localization with synapse and preferential synaptic engulfment by Axl+ microglia, our findings on the function of Axl are largely correlational at this time. Another limitation of this study is that evaluation of cognition, synapses and AD pathology was primarily performed on 5-month-old 5XFAD mice. The functional impact of IFN-I at later stages of β-amyloidosis remains to be elucidated.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Wei Cao (Wei.Cao@uth.tmc.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All data including behavior data and microscopy data generated in this study are available from the lead contact on request. Nanostring data are provided in Table S2. Human transcriptomic data (MSBB) is publicly available from AMP-AD (see Key Resources Table).
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-Aβ (clone 126E10 | Novus Biologicals | Cat# NBP2-62566 |
| Chicken anti-GFP | Abcam | Cat# ab13970 (RRID: AB_300798) |
| Goat anti-Iba1 | Novus Biologicals | Cat# NB100-1028 (RRID: AB_521594) |
| Rabbit anti-Iba1 | Wako | Cat# 019-19741 (RRID: AB_839504) |
| Rat anti-PU.1 | Novus Biologicals | Cat# MAB7124 |
| Rat anti-Clec7a (clone R1-8g7) | Invivogen | Cat# mabg-mdect (RRID: AB_2753143) |
| Goat anti-Axl | R&D Systems | Cat# AF854 (RRID: AB_355663) |
| Rat anti-CD68 (clone FA-11) | BioRad | Cat# MCA1957GA (RRID: AB_324217) |
| Rabbit anti-Olig2 | Millipore | Cat# AB9610 (RRID: AB_570666) |
| Rat anti-LAMP1 (clone 1D4B) | BD Biosciences | Cat# 553792 (RRID: AB_2134499) |
| Mouse anti-GFAP (clone GA5) | Millipore | Cat# MAB360 (RRID: AB_11212597) |
| Mouse anti-PSD95 (clone 6G6-1C9) | Millipore | Cat# MAB1596 (RRID: AB_2092365) |
| Rabbit anti-PSD95 | Invitrogen | Cat# 51-6900 (RRID: AB_2533914) |
| Rabbit anti-synaptophysin | Abcam | Cat# MA5-14532 (RRID: AB_10983675) |
| Goat anti-synaptophysin | R&D Systems | Cat# AF5555 (RRID: AB_2198864) |
| anti-Vglut1 | Synaptic Systems | Cat# 135302 (RRID: AB_887877) |
| Rabbit anti-GluR2/3/4 | Thermo | Cat# PA5-17314 (RRID: AB_10985891) |
| Anti-VGAT | Synaptic Systems | Cat# 131011 (RRID: AB_887872) |
| Mouse anti-GAD65 | BD Pharmingen | Cat# 559931 (RRID: AB_397380) |
| Rabbit anti-cFos | Millipore | Cat# ABE457 (RRID: AB_2631318) |
| Rabbit anti-Fragilis (anti-Ifitm3) | Abcam | Cat# ab15592 (RRID: AB_2122095) |
| Rabbit anti-Stat1 (clone D1K9Y) | Cell Signaling | Cat# 14994 (RRID: AB_2799965) |
| Rabbit anti-Stat1, pTyr701 (clone 58D6) | Cell Signaling | Cat# 9167 (RRID: AB_561284) |
| Rabbit anti-Stat3, pTyr705 (clone D3A7) | Cell Signaling | Cat# 9145 (RRID: AB_2491009) |
| Mouse anti-NeuN (clone A60) | Millipore | Cat# MAB377 (RRID: AB_2298772) |
| Mouse anti-pNF (clone SMI31) | Biolegend | Cat# 801601 (RRID: AB_2564641) |
| Mouse anti-p-Tau (CP13) | Peter Davies lab (Polydoro et al., 2009) | N/A |
| Rabbit anti-human Tau (K9JA) | Agilent (Dako) | Cat# A0024 (RRID: AB_10013724) |
| Rat anti-C3 (clone 11H9) | Hycult | Cat# HM1045 (RRID: AB_10129042) |
| Goat anti-Mer | R&D Systems | Cat# AF591 (RRID: AB_2098565) |
| Goat anti-Gas6 | R&D Systems | Cat# AF986 (RRID: AB_2263130) |
| Anti-APP CTD | Hui Zheng lab (Xia et al, 2002) | N/A |
| Mouse anti-γ-tubulin (clone GTU-88) | Sigma-Aldrich | Cat# T6557 (RRID: AB_477584) |
| Mouse anti-ubiquitin (clone P4D1) | Santa Cruz | Cat# sc-8017 (RRID: AB_2762364) |
| Rabbit anti-LC3B | Sigma-Aldrich | Cat# L7543 (RRID: AB_796155) |
| Rat anti-Cd11b-FITC | BD Biosciences | Cat# 553310 (RRID: AB_394774) |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Mouse anti-IFNAR1 (clone MAR1-5A3) | Bio X Cell | Cat# BE0241 (RRID: AB_2687723) |
| Mouse IgG1 isotype (clone MOPC-21) | Bio X Cell | Cat# BE0083 (RRID: AB_1107784) |
| Tamoxifen | Sigma-Aldrich | Cat# T5648 |
| Recombinant mouse IFNβ | R&D Systems | Cat# #8234-MB-010 |
| Deposited Data | ||
| Mount Sanai Brain Bank (MSBB) | AMP-AD Knowledge Portal (https://adknowledgeportal.synapse.org) | Synapse ID: syn8484987 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J mice | NIA | N/A |
| 5XFAD mice | The Jackson Laboratories | Strain #034840-JAX |
| Ifnar1fl mice | The Jackson Laboratories | Strain #028256 |
| Cx3Cr1-CreERT2 mice | The Jackson Laboratories | Strain #021160 |
| Nestin-Cre mice | The Jackson Laboratories | Strain #003771 |
| ROSAmT/mG mice | The Jackson Laboratories | Strain #026862 |
| Mx1-Cre mice | The Jackson Laboratories | Strain #003556 |
| Thy1-eGFP-M mice | The Jackson Laboratories | Strain #007788 |
| Oligonucleotides | ||
| RT-qPCR primers | See Table S1 | N/A |
| Software and Algorithms | ||
| Leica confocal suite software | Leica (https://www.leica-microsystems.com) | RRID:SCR_013673 |
| Imaris (version 9.1) | Bitplane (www.bitplane.com) | RRID:SCR_007370 |
| ImageJ (version 1.47) | NIH (https://imagej.nih.gov/ij/) | RRID:SCR_003070 |
| GraphPad Prism (version 9.1.0) | GraphPad Software (www.graphpad.com) | RRID:SCR_002798 |
| R (version 3.5.1) | R Core Team (https://www.r-project.org/) | RRID:SCR_001905 |
| nSolver Advanced Analysis | Nanostring (www.nanostring.com) | RRID:SCR_003420 |
| InnateDB | Breuer et al, 2013 | N/A |
| NetworkAnalyst | Zhou et al, 2019 | N/A |
| ANY-maze Behavioral tracking software | Stoelting Co. (https://www.any-maze.com) | RRID:SCR_014289 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
C57BL/6J mice were obtained from the National Institute on Aging (NIA). 5XFAD (B6SJL-Tg(APPSwFlLon,PSEN1*M146L*L286V)6799Vas/Mmjax), Ifnar1fl (B6(Cg)-Ifnar1tm1.1Ees/J), Cx3Cr1-CreERT2 (B6.129P2(Cg)-Cx3cr1tm2.1(cre/ERT2)Litt/WganJ), Nestin-Cre (B6.Cg-Tg(Nes-cre)1Kln/J), ROSA-mT/mG (B6.129(Cg)-Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J), Mx1-Cre (B6.Cg-Tg(Mx1-cre)1Cgn/J), and Thy1-eGFP-M (Tg(Thy1-EGFP)MJrs/J) mice were obtained from Jackson Laboratories. 5XFAD mice were generated by breeding a heterozygous Tg parent with a homozygous non-Tg parent while maintaining the original background (Oakley et al., 2006). 5XFAD mice expressing the MXG reporter system were generated by crossing 5XFAD mice with the ROSAmT/mG reporter, then crossing 5XFAD;mT/mG+/+ mice with mT/mG+/+;Mx1-Cre mice to obtain mT/mG+/+;Mx1-Cre mice with or without the 5XFAD transgene. 5XFAD mice expressing the Thy1-eGFP reporter were generated by crossing 5XFAD mice with Thy1-eGFP mice. 5XFAD mice with conditional deletion of Ifnar1 were obtained by crossing 5XFAD;Ifnar1fl/fl mice with Ifnar1fl/fl;Cx3Cr1-CreERT2 (5XFAD;MKO) or Ifnar1fl/fl;Nestin-Cre (5XFAD;NKO) mice to obtain single mutant, double mutant and control (Ifnar1fl/fl) littermates. Mice of both sexes were used for all experiments throughout the study.
For Cre recombination in Cx3Cr1-CreERT2-containing mice, tamoxifen was administered to all mice (both Cre and non-Cre) at 6 weeks of age, as described (Parkhurst et al., 2013), with modifications. Briefly, 3 mg/ml tamoxifen (Sigma) was dissolved in ethanol and sunflower oil and delivered intraperitoneally to mice (after isopropanol sterilization of the abdomen) daily for five days, with a one-day gap after day 3 to allow animal recovery.
Mice with ad libitum access to food and water and were housed in mixed-genotype groups of 3–4 per cage under specific pathogen-free conditions and standard light/dark cycle. Both male and female mice were used in experiments, unless otherwise stated. Mice were analyzed between 3 and 11 months in the study, and precise ages of all animals used in experiments are listed in respective figure legends. All animal procedures were performed in accordance with NIH guidelines and with the approval of the Baylor College of Medicine Institutional Animal Care and Use Committee.
Primary cultures
Primary neurons were harvested and cultured as previously described (Lian et al., 2015). Briefly, P0 pups of mixed sexes were decapitated into ice-cold PBS and forebrain hemispheres were dissected and stripped of meninges. Tissues were digested in trypsin at 37°C for 15 minutes with gentle swirling. Digested tissues were triturated in multiple layers and dissociated cells were passed through a 70 μm filter. The cells were plated into poly D-lysine-coated culture plates (1× 105 cells/cm2) containing neuronal culture medium (Neurobasal medium supplemented with 2% B27, 1X GlutaMAX, and 1% Pen-Strep). Cells were maintained in incubators at 37°C, 95% O2, 5% CO2, and half the culture medium was replaced every 5–7 days.
For treatments, cultured neurons were stimulated at DIV21 with doses of IFNβ indicated in figures for the indicated time points. After the treatments, cells were rinsed with PBS and lysed with protein extraction buffer.
Expression analysis on human AD samples
The expression profiles of post-mortem human AD brain samples (Mount Sinai School of Medicine, AMP-AD RNAseq project, Synapse ID: syn8484987, download date: 13 Nov. 2019) were extracted for analysis. Data from parahippocampal gyrus (PHG; Brodmann area BM-36) of 195 subjects were used (n = 122 females, n = 73 males; median age at death of 85). Clinical covariates and sample information for all 195 subjects are included in Table S3. In all analyses, samples with RIN score less than 5.0 were excluded. For CDR, the three groups were defined based on score: CDR=0 is ‘NCI’ (no cognitive impairment), CDR=0.5 is ‘MCI’ (mild cognitive impairment), CDR≥1 is ‘Dementia’.
For pathway analysis, genes upregulated in ‘Dementia’ group compared to ‘NCI’ group with fold change >1.15 and P < 0.01 as determined by two-sided t-test (458 genes) were queried using the Pathway Analysis tool in InnateDB (Breuer et al., 2013) To visualize the relationships between pathways enriched in Dementia, upregulated genes were submitted to the NetworkAnalyst tool (Zhou et al., 2019) and significant REACTOME pathways were visualized using “List Enrichment Network” feature (Figure S6H).
For pathways correlated with IRF7 expression, all genes were subjected to Pearson correlations with IRF7 transcript levels, and two gene lists were constructed: all genes with Pearson r > 0.6 (positive correlates, 739 genes), and all genes with r < −0.6 (negative correlates, 832 genes). These lists were queried using InnateDB, as above, and the top ten REACTOME terms by P value were plotted in Figure S6I.
Pearson correlation for IRF7 and AXL association was computed, and simple linear regression was used to generate fit line in the plot (Figure 6H). Data for CDR plots were analyzed by one-way ANOVA with Bonferroni’s correction (Figure 6H).
METHOD DETAILS
Intracranial osmotic pump implants
For long-term i.c.v. administration of blocking antibodies, osmotic pumps delivering agents to the i.c.v. space were used according to manufacturer instructions (Alzet) and similarly to previously published protocols and studies (DeVos and Miller, 2013, Sanchez-Mendoza et al., 2016, DeVos et al., 2013). Briefly, 2 mg/ml of αIFNAR (clone MAR1–5A3) or IgG1 isotype control (clone MOPC-21) was loaded into osmotic pumps with mouse-specific steel brain infusion cannulae attached (Alzet #2004 with Brain Infusion Kit #3) and pre-soaked for ~40 hours in PBS at 37°C under aseptic conditions. For the surgeries, animals received subcutaneous Buprenex (1 mg/kg buprenorphine hydrochloride, Indivior, Richmond VA) pre-operatively, and were then anesthetized with inhalable isoflurane (2% Isothesia, Henry Schein, Dublin, OH). Anesthetized animals were placed in a Kopf stereotaxic instrument (David Kopf Instruments, Tujunga, CA), and cannulae were placed using coordinates to target lateral ventricles (−0.4 mm anteroposterior, ± 1.0 mm mediolateral, and −2.0 mm dorsoventral from the surface of the skull at bregma). Brain cannulae were cemented in place, pumps were inserted subcutaneously above the spine, and the wounds were sutured (Coated Vicryl Suture, RB-1 17 mm ½c Taper, Ethicon, Cincinnati, OH) and glued. Mice were closely monitored post-operatively and throughout the experiment, which lasted ~30 days after implantation. Animals were subjected to blinded behavioral assays (detailed below) and, within 1–2 days, brain tissues were collected for downstream analyses. Tissues were inspected upon microdissection to verify injection sites.
Behavioral assays
Similar to previously described (Chiu et al., 2018, Chiu et al., 2012, Chiu et al., 2017), novel object recognition (NOR) and Y maze tests were performed to assess cognitive function. All behavioral tests were performed and analyzed by a blinded researcher, and ANY-maze Behavioral tracking software (Stoelting Co., Wood Dale, IL) was used for quantification. In brief, for NOR, mice were transferred from the home cage to a novel testing arena containing two identical objects or 5 minutes (training phase) then returned to their home cage. After a one-hour inter-trial interval, the mice were transferred back to the arena containing one familiar object and one novel object (testing phase). Discrimination index was determined by the equation (TNovel − TFamiliar)/(TNovel + TFamiliar).
For the Y maze, spontaneous alternations were measured. In brief, mice were placed in a symmetrical three-arm, gray plastic Y-maze (35 cm length × 5 cm width × 15.5 cm height per arm, with an arm angle of 120°) with external spatial room cues. Mice were randomly placed in one of the arms. Movement was recorded for 5 minutes, and mouse exploration was evaluated. Perfect alternations were defined as exploration of all three arms sequentially before reentering a previously visited arm. All four paws must have been within the arm to be counted as an entrance. Results are represented as the ratio of the number of perfect alternations to the total number of possible alternations.
FACS sorting of microglia and non-microglia
FACS sorting of microglia was performed as previously described (Swartzlander et al., 2018), with slight modifications. Briefly, adult mice were perfused with PBS, brain tissues were gently minced with sterile razor blades, digested in papain (Worthington Biochemical, cat# LK003172) containing DNase (Worthington Biochemical, cat# LK003178), and triturated 3–4 times using a fire-polished glass Pasteur pipette. After incubation, papain digestion was neutralized with HBSS+ and the suspensions were pelleted at 310 × g for 5 minutes at 4°C. The pellets were resuspended in 1 mL of HBSS+, transferred to an ice-cold 1.7 mL Eppendorf tube and further triturated 3 times, and the supernatants were collected after a brief, low-speed centrifugation. The resulting supernatants were filtered through a pre-wetted 40 μm cell strainer (BD Biosciences, 352340) into a chilled 50-mL conical tube and centrifuged at 310 × g for 5 minutes at 4°C. The resulting pellets were depleted of myelin and other debris using a 20% isotonic Percoll PLUS (MilliporeSigma, cat# E0414–250ML) separation. The resulting pellets contained dissociated single cells. For isolation of microglial and non-microglial cells, the cell suspensions were incubated in 500 μL HBSS+ containing 1:100 Mouse BD Fc Block (BD Biosciences, cat# 553141) and 1:500 rat anti-CD11b–FITC (BD Biosciences, cat# 553310) on ice for 15–20 minutes. Cells were washed twice with HBSS+ and resuspended in 500 μL of HBSS+ prior to FACS separation using a BD Biosciences LSR Fortessa, whereupon CD11b+ and CD11b− cells were sorted into collection tubes, pelleted, and lysed with RNA extraction buffer for downstream qPCR analysis.
Immunoblotting
Total protein from brain tissues and cell cultures were extracted using RIPA buffer, separated by SDS-PAGE, and immunoblotted using antibodies against full-length APP (APP-C), ubiquitin, LC3, Ifitm3, and γ-tubulin as a loading control. Blots were visualized by fluorescent of fluor-conjugated secondary antibodies on a LI-COR Odyssey blot imager.
Mouse tissue immunofluorescence
Mice were perfused with ice-cold saline (0.9% NaCl) after deep anesthesia with ketamine/xylazine, and brains were extracted, fixed overnight at 4°C in 4% paraformaldehyde (Santa Cruz, cat# sc-281692), and dehydrated in 30% sucrose until sectioning. Brains were sectioned into 30-μm tissue sections using a freezing microtome, and tissue was stored in cryoprotectant at −20°C. For staining, floating sections were washed in phosphate-buffered saline (PBS) and blocked for 1 hour at room temperature in a blocking buffer of 10% normal donkey serum (Millipore, cat# S30–100ML) and 1% Triton X-100 in TBS. Primary antibodies were diluted in blocking buffer and applied to sections overnight at 4°C. Tissues were washed with PBS-T (PBS with 0.1% Tween-20) three times, then incubated with fluorescent secondary antibodies diluted in blocking buffer for 1 hour at room temperature. After final washing in PBS-T, sections were mounted on glass slides, allowed to dry, and coverslipped with ProLong Glass Antifade mountant (Life Technologies, cat# P36982). In one experiment, perineuronal nets were visualized with fluorescently labelled Wisteria floribunda agglutinin (Vector Labs, cat# B-1355–2), which was used in conjunction with primary antibody incubation. Tissues from the MXG reporter animals were stained with anti-GFP antibody to amplify the signal.
For antibody validation experiments (such as for pStat proteins, synaptic markers and TAM-related molecules, as presented in Figures S5 and S6, respectively), primary antibodies were omitted from the overnight incubation in a subset of parallel sections, and all sections were then incubated with secondary antibodies. Multiple sections with and without each primary antibody validated were stained simultaneously and were imaged side-by-side using the same microscope settings to confirm specific signals.
In a subset of experiments, dense core plaques were visualized using thioflavin S (Sigma, cat# T1892) or methoxy-X04 dye (Tocris, cat# 4920), as described. For thioflavin S staining (using a modified protocol optimized by the Joanna Jankowsky laboratory), sections were rinsed following IF in PBS, then transferred to solution containing 0.0002% thioflavin S in PBS for 8 mins, rinsed in 40% ethanol in PBS twice for 2 mins, rinsed in PBS, then mounted on slides. For X04, sections were dried overnight following IF, then rehydrated in PBS for 30 s, transferred to 40% ethanol in PBS for 30 s, incubated in 40% ethanol containing 1 μM methoxy X04 dye for 3 min, rinsed in 40% ethanol for 30 s, rinsed in 95% ethanol for 30 s, dipped in tap water three times, and then air dried before mounting.
For GFP sequestration into plaques in the MxG and Thy1-GFP models, brain sections were stained with Iba1 antibody (Wako) and methoxy-X04. Endogenous GFP was imaged without antibody amplification. Individual plaques were imaged at high magnification (63X) and multiple single-plane images in the centers of plaque X04+ signals were assessed for GFP signals.
Image quantification procedures
Neuropathological parameter quantification
Basic assessments of plaque load and extent of gliosis were performed by scanning and stitching entire brain sections on an EVOS Cell Imaging system (Thermo) with a 4X lens. Images were loaded into ImageJ (NIH), where regions of interest (ROIs) were drawn around relevant brain regions. The fluorescent signal thresholds were adjusted to accurately overlap the signals in each ROI, and the percent area within the ROIs were recorded. Multiple sections were analyzed as technical replicates for each animal.
Other neuropathological parameters, such as LAMP1+ DNs, pNF+ DAs, and X04+ plaque volumes, were imaged using confocal microscopy on a Leica SPE system. Briefly, Z stacks (>5 μm total thickness, 0.5 μm step size) were captured using a 40X oil immersion lens. Images were quantified on Imaris 9.1 software by creating 3D surfaces of each signal. The overall DN and DA volumes in each image were divided by the overall plaque volumes in each image, yielding a ratio. Multiple images were analyzed as technical replicates from each animal.
Quantification of cell density
Histological measurement of cFos+ neuron density was performed as described (Richetin et al., 2020). Briefly, fixed brain sections were stained as described above using antibodies against NeuN and cFos and imaged at 40X on the confocal microscope. Three sections were imaged per animal at the dentate gyrus, and 25 μm Z-stacks were flattened before cFos+ neurons were manually counted in each image by an investigator blinded to genotypes and treatments. Means of the three values were plotted and analyzed using ANOVA.
Similarly, microglia were counted in a subset of experiments using specific nuclear marker PU.1, in which multiple stained sections per animal were imaged and manual counting yielded relative microglial densities, or the number of microglia associated with individual plaques.
Plaque/microglial barrier quantification
For quantification of plaque barrier formed by microglial processes, sections were stained with Iba1 antibody and X04, and 150–300 plaques per experiment were imaged on the confocal at 40X magnification. Single-plane images through plaque centers were captured for analysis. The free-hand tracing tool in ImageJ (NIH) was used to carefully trace the X04 plaque border with and without microglial process contact, and measurements were taken of each segment to yield a % coverage for each plaque, which were then pooled into groups for analysis.
Analyses of marker occupancy and uptake
Expression of various markers by specific cell types (e.g. microglial Axl, microglial CD68, microglial Clec7a, Ifitm3 or APP within astrocytes or DNs, tau in DNs), as well as engulfed signals (e.g. PSD95 or synaptophysin by microglia, Aβ within microglial CD68+ vesicles) were quantified on Imaris 9.1 software (Bitplane) using the “Co-loc” feature. Briefly, Z-stacks (>5 μm total thickness, <1 μm step size, channel averaging as-needed for weaker signals) were captured by confocal microscopy, and fluorescence channels for cellular markers (e.g. Iba1, GFAP, and LAMP1) or subcellular markers (e.g. CD68) were used to create a mask within which the occupancy of the target marker was calculated as a percentage of the 3D space. Similarly, nuclear Stat1 signal was quantified by 3D cropping around individual microglial nuclei and using the “Co-loc” feature of Imaris to determine % occupancy in the 3-dimensional nuclei. Within all co-localization experiments, consistent thresholds were used for each signal to prevent biased measurements. Multiple images (3 to 6) were analyzed as technical replicates from each animal. In some experiments, individual cells were digitally isolated for analysis.
Puncta co-localization studies
For synaptic, pStat1, and TAM marker co-localization studies, published protocols for analysis of high-magnification punctate signals were modified. For synaptic co-localization studies, brain sections were stained using antibodies recognizing synaptophysin and PSD95, marking pre- and post-synaptic terminals, respectively. For synaptic localization with pStat1 or TAM-related molecules (i.e., Axl, Mer, and Gas6), synapse markers were used in conjunction with antibodies for target molecules. Antibodies were validated as described in the immunofluorescence section. Confocal imaging was performed using a 63X oil objective with a 3.0–6.0 digital zoom. Z-stacks of 5 μm thickness from the middle of the tissues (to avoid staining artifacts) were obtained with a 0.2 μm step-size, and 2 frame-averaged 512×512 images for each channel. Due to variation in synaptic puncta densities by brain region, care was taken to image from the same area (i.e., the plaque-laden subiculum) consistently across all samples and experiments.
Z-stacks with pre- and post- synaptic puncta and signals for other molecules were analyzed using the “Spots” feature of Imaris 9.1 software (Bitplane). Spots were generated automatically (with manual adjustment for accurate puncta representation) for each channel separately, and total numbers of spots were recorded. Subsequently, spots were analyzed by the “Co-localize Spots” MATLAB plugin. Pre- and post-synaptic puncta were defined as co-localized if their centers were within 200 nm. Co-localization between TAM molecules and synapses also used a max distance of 200 nm between Spots. In a set of validation experiments, one of the two fluorescent channels was flipped along the X axis before spot detection to determine the specificity or randomness of co-localization between markers. Percent co-localized puncta were calculated for both the original and mismatched channel orientations, and co-localized puncta were deemed nonrandom if higher values were observed using original channel orientations.
For quantification of Axl+ puncta localization to dendritic spines in Thy1-eGFP mice, GFP+ cortical dendrites were imaged by confocal microscopy using a 63X oil lens to generate Z-stacks with a step-size of 0.2 μm. Images were then analyzed on Imaris 9.1 software (Bitplane) by creating a 3D surface of dendritic spines and a spots object for Axl+ puncta. Puncta within 100 nm of the dendrite surface were isolated and counted using the “Find Spots Close to Surface” MATLAB plugin. Dendrite segments of at least 10 μm in length were included in analysis, and data are represented as the relative frequency of Axl+ puncta in contact with dendrites per unit of dendrite length.
Gene expression profiling
Quantitative Real-Time PCR
Total RNA was isolated from brain tissues and FACS-sorted cell pellets by lysing them in Qiagen RLT buffer with 1% β-mercaptoethanol and processing with RNeasy Mini isolation kit (Qiagen) according to manufacturer protocols, including DNase digestion steps. About 500ng–1ug of RNA was used to generate cDNA by reverse transcription using iScript Reverse Transcription Supermix reagent (BioRad, #170–8840). qRT-PCR was performed using iTaq Universal SYBR Green Supermix (BioRad, #172–5124) on a CFX384 Touch Real-Time PCR Detection System with primer sequences listed in Table S1.
Nanostring
Nanostring gene expression data was generated from hippocampal tissues using the nCounter® Mouse Neuroinflammation panel. nCounts of mRNA transcripts were normalized using the geometric means of 10 housekeeping genes (Csnk2a2, Ccdc127, Xpnpep1, Lars, Supt7l, Tada2b, Aars, Mto1, Tbp, and Fam104a) and analyzed using nSolver Advanced Analysis software. Results are provided in Table S2.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data in bar plots are presented as means ± s.e.m. Violin plots are presented as medians (solid lines) and quartiles (dashed lines). Unless otherwise noted, differences between two groups were analyzed by two-tailed Student’s t-tests, and differences between three or more groups were analyzed by one-way ANOVA with Bonferroni’s post-hoc multiple-comparisons tests, as indicated in figure legends. Human transcriptome data were statistically analyzed as described in “Expression analysis on human AD samples” section, above. GraphPad Prism (v9.1.0) was used for all statistical analyses. P values less than 0.05 were considered significant (noted as *P < 0.05, **P < 0.01, ***P < 0.001 in plots), and those over 0.05 were considered non-significant (“ns”, or numerical P values listed in certain plots). All n values are listed in figure legends for each respective plot. Female and male mice were used for all experiments. All micrographs shown are images representative of multiple replicates as indicated. No statistical methods were used to predetermine sample sizes, but similar publications were used as guidelines. Data was assumed to have a normal distribution, though this was not formally determined. Experimental animals were randomly assigned to treatment groups. In vivo experiments, such as osmotic pump implantations and behavioral studies, were performed by a researcher blinded to the genotypes and treatment groups.
Supplementary Material
Table S1. Oligonucleotides, Related to STAR Methods section “Gene expression profiling”
Table S2. Nanostring data, Related to Figure 3 and Figures S2, S3, and S6
Highlights.
IFN-I-activated microglia and other brain cells arise and expand with amyloidosis
Blockade of brain IFN-I restores cognition, neuroinflammation, and synaptic loss
Pre- and post-synaptic loss are IFN-I-dependent and mediated by different cell types
IFN-I signaling in neural cells promotes plaque accumulation
Acknowledgements
The study was funded by NIH grants AG057587 (W.C. and H.Z.); AG020670 (H.Z.), AG062257 (H.Z.), NS093652 (H.Z.), BrightFocus ADR A20183775 (W.C.), Brown Foundation 2020 Healthy Aging Initiative (W.C.) and Cure Alzheimer’s Fund (H.Z.). R.K. and C.C. were partially supported by The Cancer Prevention Institute of Texas (CPRIT) RP170005, RP210227, RP200504, NIH P30 shared resource grant CA125123, and NIEHS grants P30 ES030285 and P42 ES027725, and NIMHD P50MD015496. We acknowledge technical assistance from Haiying Liu, Nadia Aithmitti, and Bianca Contreras, and generosity from Michele Comerota. Our gratitude goes to Dr. Andre Catic for providing MxG founder mice; to Lita Duraine at NRI TEM Core of Baylor College of Medicine for technical consultation of electron microscopy; and to Dr. Joanna Jankowsky for critical reading and comments. We thank the Neurobehavioral Core at Baylor College of Medicine, which is supported by IDDRC grant number U54HD083092 from the Eunice Kennedy Shriver National Institute of Child Health & Human Development. Some graphics were created with BioRender.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
W.C. received honorarium as an invited speaker in a symposium sponsored by AstroZeneca. H.Z. is a member of the Scientific Advisory Board of the Tau Consortium, Rainwater Charitable Foundation.
References
- Altman JB, Taft J, Wedeking T, Gruber CN, Holtmannspotter M, Piehler J & Bogunovic D 2020. Type I IFN is siloed in endosomes. Proc Natl Acad Sci U S A, 117, 17510–17512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asnis GM & De La Garza R, 2nd 2005. Interferon-induced depression: strategies in treatment. Prog Neuropsychopharmacol Biol Psychiatry, 29, 808–18. [DOI] [PubMed] [Google Scholar]
- Baik SH, Kang S, Son SM & Mook-Jung I 2016. Microglia contributes to plaque growth by cell death due to uptake of amyloid beta in the brain of Alzheimer’s disease mouse model. Glia, 64, 2274–2290. [DOI] [PubMed] [Google Scholar]
- Breuer K, Foroushani AK, Laird MR, Chen C, Sribnaia A, Lo R, Winsor GL, Hancock RE, Brinkman FS & Lynn DJ 2013. InnateDB: systems biology of innate immunity and beyond--recent updates and continuing curation. Nucleic Acids Res, 41, D1228–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheadle L, Rivera SA, Phelps JS, Ennis KA, Stevens B, Burkly LC, Lee WA & Greenberg ME 2020. Sensory Experience Engages Microglia to Shape Neural Connectivity through a Non-Phagocytic Mechanism. Neuron, 108, 451–468 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WT, Lu A, Craessaerts K, Pavie B, Sala Frigerio C, Corthout N, Qian X, Lalakova J, Kuhnemund M, Voytyuk I, et al. 2020. Spatial Transcriptomics and In Situ Sequencing to Study Alzheimer’s Disease. Cell, 182, 976–991 e19. [DOI] [PubMed] [Google Scholar]
- Cheon H & Stark GR 2009. Unphosphorylated STAT1 prolongs the expression of interferon-induced immune regulatory genes. Proc Natl Acad Sci U S A, 106, 9373–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu GS, Boukelmoune N, Chiang ACA, Peng B, Rao V, Kingsley C, Liu HL, Kavelaars A, Kesler SR & Heijnen CJ 2018. Nasal administration of mesenchymal stem cells restores cisplatin-induced cognitive impairment and brain damage in mice. Oncotarget, 9, 35581–35597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu GS, Chatterjee D, Darmody PT, Walsh JP, Meling DD, Johnson RW & Freund GG 2012. Hypoxia/reoxygenation impairs memory formation via adenosine-dependent activation of caspase 1. J Neurosci, 32, 13945–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu GS, Maj MA, Rizvi S, Dantzer R, Vichaya EG, Laumet G, Kavelaars A & Heijnen CJ 2017. Pifithrin-mu Prevents Cisplatin-Induced Chemobrain by Preserving Neuronal Mitochondrial Function. Cancer Res, 77, 742–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H, Proll SC, Szretter KJ, Katze MG, Gale M Jr. & Diamond MS 2013. Differential innate immune response programs in neuronal subtypes determine susceptibility to infection in the brain by positive-stranded RNA viruses. Nat Med, 19, 458–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho HJ, Jin SM, Son SM, Kim YW, Hwang JY, Hong HS & Mook-Jung I 2009. Constitutive JAK2/STAT1 activation regulates endogenous BACE1 expression in neurons. Biochem Biophys Res Commun, 386, 175–80. [DOI] [PubMed] [Google Scholar]
- Cho HJ, Kim SK, Jin SM, Hwang EM, Kim YS, Huh K & Mook-Jung I 2007. IFN-gamma-induced BACE1 expression is mediated by activation of JAK2 and ERK1/2 signaling pathways and direct binding of STAT1 to BACE1 promoter in astrocytes. Glia, 55, 253–62. [DOI] [PubMed] [Google Scholar]
- Chung W-S, Clarke LE, Wang GX, Stafford BK, Sher A, Chakraborty C, Joung J, Foo LC, Thompson A, Chen C, et al. 2013. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature, 504, 394–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crapser JD, Spangenberg EE, Barahona RA, Arreola MA, Hohsfield LA & Green KN 2020. Microglia facilitate loss of perineuronal nets in the Alzheimer’s disease brain. EBioMedicine, 58, 102919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’errico P, Ziegler-Waldkirch S, Aires V, Hoffmann P, Mezo C, Erny D, Monasor LS, Liebscher S, Ravi VM, Joseph K, et al. 2022. Microglia contribute to the propagation of Abeta into unaffected brain tissue. Nat Neurosci, 25, 20–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B & Karran E 2016. The Cellular Phase of Alzheimer’s Disease. Cell, 164, 603–15. [DOI] [PubMed] [Google Scholar]
- Devos SL, Goncharoff DK, Chen G, Kebodeaux CS, Yamada K, Stewart FR, Schuler DR, Maloney SE, Wozniak DF, Rigo F, et al. 2013. Antisense reduction of tau in adult mice protects against seizures. J Neurosci, 33, 12887–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devos SL & Miller TM 2013. Direct intraventricular delivery of drugs to the rodent central nervous system. J Vis Exp, e50326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Domizio J, Dorta-Estremera S, Gagea M, Ganguly D, Meller S, Li P, Zhao B, Tan FK, Bi L, Gilliet M, et al. 2012. Nucleic acid-containing amyloid fibrils potently induce type I interferon and stimulate systemic autoimmunity. Proc Natl Acad Sci U S A, 109, 14550–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dipasquale O, Cooper EA, Tibble J, Voon V, Baglio F, Baselli G, Cercignani M & Harrison NA 2016. Interferon-alpha acutely impairs whole-brain functional connectivity network architecture - A preliminary study. Brain Behav Immun, 58, 31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ejlerskov P, Hultberg JG, Wang J, Carlsson R, Ambjorn M, Kuss M, Liu Y, Porcu G, Kolkova K, Friis Rundsten C, et al. 2015. Lack of Neuronal IFN-beta-IFNAR Causes Lewy Body- and Parkinson’s Disease-like Dementia. Cell, 163, 324–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipello F, Morini R, Corradini I, Zerbi V, Canzi A, Michalski B, Erreni M, Markicevic M, Starvaggi-Cucuzza C, Otero K, et al. 2018. The Microglial Innate Immune Receptor TREM2 Is Required for Synapse Elimination and Normal Brain Connectivity. Immunity, 48, 979–991 e8. [DOI] [PubMed] [Google Scholar]
- Fritz-French C & Tyor W 2012. Interferon-alpha (IFNalpha) neurotoxicity. Cytokine Growth Factor Rev, 23, 7–14. [DOI] [PubMed] [Google Scholar]
- Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, Walker AJ, Gergits F, Segel M, Nemesh J, et al. 2019. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity, 50, 253–271 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasel P, Rose IVL, Sadick JS, Kim RD & Liddelow SA 2021. Neuroinflammatory astrocyte subtypes in the mouse brain. Nat Neurosci, 24, 1475–1487. [DOI] [PubMed] [Google Scholar]
- Hayley S, Scharf J & Anisman H 2013. Central administration of murine interferon-alpha induces depressive-like behavioral, brain cytokine and neurochemical alterations in mice: a mini-review and original experiments. Brain Behav Immun, 31, 115–27. [DOI] [PubMed] [Google Scholar]
- Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, et al. 2016. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science, 352, 712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu WL, Ma YL, Hsieh DY, Liu YC & Lee EH 2014. STAT1 negatively regulates spatial memory formation and mediates the memory-impairing effect of Abeta. Neuropsychopharmacology, 39, 746–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang KL, Marcora E, Pimenova AA, Di Narzo AF, Kapoor M, Jin SC, Harari O, Bertelsen S, Fairfax BP, Czajkowski J, et al. 2017. A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer’s disease. Nat Neurosci, 20, 1052–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Happonen KE, Burrola PG, O’connor C, Hah N, Huang L, Nimmerjahn A & Lemke G 2021. Microglia use TAM receptors to detect and engulf amyloid beta plaques. Nat Immunol, 22, 586–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur JY, Frost GR, Wu X, Crump C, Pan SJ, Wong E, Barros M, Li T, Nie P, Zhai Y, et al. 2020. The innate immunity protein IFITM3 modulates gamma-secretase in Alzheimer’s disease. Nature, 586, 735–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, et al. 2017. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell, 169, 1276–1290 e17. [DOI] [PubMed] [Google Scholar]
- Knopman DS, Amieva H, Petersen RC, Chetelat G, Holtzman DM, Hyman BT, Nixon RA & Jones DT 2021. Alzheimer disease. Nat Rev Dis Primers, 7, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koffie RM, Hyman BT & Spires-Jones TL 2011. Alzheimer’s disease: synapses gone cold. Mol Neurodegener, 6, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korvatska O, Kiianitsa K, Ratushny A, Matsushita M, Beeman N, Chien WM, Satoh JI, Dorschner MO, Keene CD, Bammler TK, et al. 2020. Triggering Receptor Expressed on Myeloid Cell 2 R47H Exacerbates Immune Response in Alzheimer’s Disease Brain. Front Immunol, 11, 559342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, Beckers L, O’loughlin E, Xu Y, Fanek Z, et al. 2017. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity, 47, 566–581 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemke G 2019. How macrophages deal with death. Nat Rev Immunol, 19, 539–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Chiou B, Gilman CK, Luo R, Koshi T, Yu D, Oak HC, Giera S, Johnson-Venkatesh E, Muthukumar AK, et al. 2020. A splicing isoform of GPR56 mediates microglial synaptic refinement via phosphatidylserine binding. EMBO J, 39, e104136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Viengkhou B, Denyer G, West PK, Campbell IL & Hofer MJ 2018. Microglia have a more extensive and divergent response to interferon-alpha compared with astrocytes. Glia, 66, 2058–2078. [DOI] [PubMed] [Google Scholar]
- Lian H, Yang L, Cole A, Sun L, Chiang AC, Fowler SW, Shim DJ, Rodriguez-Rivera J, Taglialatela G, Jankowsky JL, et al. 2015. NFkappaB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron, 85, 101–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Munch AE, Chung WS, Peterson TC, et al. 2017. Neurotoxic reactive astrocytes are induced by activated microglia. Nature, 541, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieb K, Engelbrecht MA, Gut O, Fiebich BL, Bauer J, Janssen G & Schaefer M 2006. Cognitive impairment in patients with chronic hepatitis treated with interferon alpha (IFNalpha): results from a prospective study. Eur Psychiatry, 21, 204–10. [DOI] [PubMed] [Google Scholar]
- Mattsson N, Insel P, Nosheny R, Zetterberg H, Trojanowski JQ, Shaw LM, Tosun D, Weiner M & Alzheimer’s Disease Neuroimaging I 2013. CSF protein biomarkers predicting longitudinal reduction of CSF beta-amyloid42 in cognitively healthy elders. Transl Psychiatry, 3, e293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minter MR, Moore Z, Zhang M, Brody KM, Jones NC, Shultz SR, Taylor JM & Crack PJ 2016. Deletion of the type-1 interferon receptor in APPSWE/PS1DeltaE9 mice preserves cognitive function and alters glial phenotype. Acta Neuropathol Commun, 4, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, et al. 2006. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci, 26, 10129–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olah M, Menon V, Habib N, Taga MF, Ma Y, Yung CJ, Cimpean M, Khairallah A, Coronas-Samano G, Sankowski R, et al. 2020. Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer’s disease. Nat Commun, 11, 6129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owlett LD, Karaahmet B, Le L, Belcher EK, Dionisio-Santos D, Olschowka JA, Elliott MR & O’banion MK 2022. Gas6 induces inflammation and reduces plaque burden but worsens behavior in a sex-dependent manner in the APP/PS1 model of Alzheimer’s disease. J Neuroinflammation, 19, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR 3rd, Lafaille JJ, Hempstead BL, Littman DR & Gan WB 2013. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell, 155, 1596–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peet G, Bennett FC & Bennett ML 2020. Please eat (only part) of me: synaptic phosphatidylserine cues microglia to feast: Two new studies identify how a common apoptotic cell flag is used to sculpt neural circuits. EMBO J, 39, e105924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podvin S, Jones A, Liu Q, Aulston B, Ransom L, Ames J, Shen G, Lietz CB, Jiang Z, O’donoghue AJ, et al. 2020. Dysregulation of Exosome Cargo by Mutant Tau Expressed in Human-induced Pluripotent Stem Cell (iPSC) Neurons Revealed by Proteomics Analyses * . Molecular & Cellular Proteomics, 19, 1017–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz M, Masuda T, Wheeler MA & Quintana FJ 2021. Microglia and Central Nervous System-Associated Macrophages-From Origin to Disease Modulation. Annu Rev Immunol, 39, 251–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Propson NE, Roy ER, Litvinchuk A, Kohl J & Zheng H 2021. Endothelial C3a receptor mediates vascular inflammation and blood-brain barrier permeability during aging. J Clin Invest, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramdhani S, Navarro E, Udine E, Efthymiou AG, Schilder BM, Parks M, Goate A & Raj T 2020. Tensor decomposition of stimulated monocyte and macrophage gene expression profiles identifies neurodegenerative disease-specific trans-eQTLs. PLoS Genet, 16, e1008549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richetin K, Steullet P, Pachoud M, Perbet R, Parietti E, Maheswaran M, Eddarkaoui S, Begard S, Pythoud C, Rey M, et al. 2020. Tau accumulation in astrocytes of the dentate gyrus induces neuronal dysfunction and memory deficits in Alzheimer’s disease. Nat Neurosci, 23, 1567–1579. [DOI] [PubMed] [Google Scholar]
- Roy ER, Wang B, Wan YW, Chiu G, Cole A, Yin Z, Propson NE, Xu Y, Jankowsky JL, Liu Z, et al. 2020. Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J Clin Invest, 130, 1912–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadleir KR, Kandalepas PC, Buggia-Prevot V, Nicholson DA, Thinakaran G & Vassar R 2016. Presynaptic dystrophic neurites surrounding amyloid plaques are sites of microtubule disruption, BACE1 elevation, and increased Abeta generation in Alzheimer’s disease. Acta Neuropathol, 132, 235–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sainaghi PP, Bellan M, Lombino F, Alciato F, Carecchio M, Galimberti D, Fenoglio C, Scarpini E, Cantello R, Pirisi M, et al. 2017. Growth Arrest Specific 6 Concentration is Increased in the Cerebrospinal Fluid of Patients with Alzheimer’s Disease. J Alzheimers Dis, 55, 59–65. [DOI] [PubMed] [Google Scholar]
- Sala Frigerio C, Wolfs L, Fattorelli N, Thrupp N, Voytyuk I, Schmidt I, Mancuso R, Chen WT, Woodbury ME, Srivastava G, et al. 2019. The Major Risk Factors for Alzheimer’s Disease: Age, Sex, and Genes Modulate the Microglia Response to Abeta Plaques. Cell Rep, 27, 1293–1306 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salih DA, Bayram S, Guelfi S, Reynolds RH, Shoai M, Ryten M, Brenton JW, Zhang D, Matarin M, Botia JA, et al. 2019. Genetic variability in response to amyloid beta deposition influences Alzheimer’s disease risk. Brain Commun, 1, fcz022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Mendoza EH, Carballo J, Longart M, Hermann DM & Doeppner TR 2016. Implantation of Miniosmotic Pumps and Delivery of Tract Tracers to Study Brain Reorganization in Pathophysiological Conditions. J Vis Exp, e52932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Varo R, Trujillo-Estrada L, Sanchez-Mejias E, Torres M, Baglietto-Vargas D, Moreno-Gonzalez I, De Castro V, Jimenez S, Ruano D, Vizuete M, et al. 2012. Abnormal accumulation of autophagic vesicles correlates with axonal and synaptic pathology in young Alzheimer’s mice hippocampus. Acta Neuropathol, 123, 53–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott-Hewitt N, Perrucci F, Morini R, Erreni M, Mahoney M, Witkowska A, Carey A, Faggiani E, Schuetz LT, Mason S, et al. 2020. Local externalization of phosphatidylserine mediates developmental synaptic pruning by microglia. EMBO J, 39, e105380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastian Monasor L, Muller SA, Colombo AV, Tanrioever G, Konig J, Roth S, Liesz A, Berghofer A, Piechotta A, Prestel M, et al. 2020. Fibrillar Abeta triggers microglial proteome alterations and dysfunction in Alzheimer mouse models. Elife, 9, doi: 10.7554/eLife.54083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Q, Chowdhury S, Ma R, Le KX, Hong S, Caldarone BJ, Stevens B & Lemere CA 2017. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci Transl Med, 9, eaaf6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangenberg E, Severson PL, Hohsfield LA, Crapser J, Zhang J, Burton EA, Zhang Y, Spevak W, Lin J, Phan NY, et al. 2019. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat Commun, 10, 3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartzlander DB, Propson NE, Roy ER, Saito T, Saido T, Wang B & Zheng H 2018. Concurrent cell type-specific isolation and profiling of mouse brains in inflammation and Alzheimer’s disease. JCI Insight, 3, e121109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai DJ, Hsu WL, Liu YC, Ma YL & Lee EH 2011. Novel role and mechanism of protein inhibitor of activated STAT1 in spatial learning. EMBO J, 30, 205–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentine AD, Meyers CA, Kling MA, Richelson E & Hauser P 1998. Mood and cognitive side effects of interferon-alpha therapy. Semin Oncol, 25, 39–47. [PubMed] [Google Scholar]
- Venegas C, Kumar S, Franklin BS, Dierkes T, Brinkschulte R, Tejera D, Vieira-Saecker A, Schwartz S, Santarelli F, Kummer MP, et al. 2017. Microglia-derived ASC specks cross-seed amyloid-beta in Alzheimer’s disease. Nature, 552, 355–361. [DOI] [PubMed] [Google Scholar]
- Wachholz S, Esslinger M, Plumper J, Manitz MP, Juckel G & Friebe A 2016. Microglia activation is associated with IFN-alpha induced depressive-like behavior. Brain Behav Immun, 55, 105–113. [DOI] [PubMed] [Google Scholar]
- Wilton DK, Dissing-Olesen L & Stevens B 2019. Neuron-Glia Signaling in Synapse Elimination. Annu Rev Neurosci, 42, 107–127. [DOI] [PubMed] [Google Scholar]
- Wu T, Dejanovic B, Gandham VD, Gogineni A, Edmonds R, Schauer S, Srinivasan K, Huntley MA, Wang Y, Wang TM, et al. 2019. Complement C3 Is Activated in Human AD Brain and Is Required for Neurodegeneration in Mouse Models of Amyloidosis and Tauopathy. Cell Rep, 28, 2111–2123 e6. [DOI] [PubMed] [Google Scholar]
- Xia X, Wang P, Sun X, Soriano S, Shum WK, Yamaguchi H, Trumbauer ME, Takashima A, Koo EH & Zheng H 2002. The aspartate-257 of presenilin 1 is indispensable for mouse development and production of beta-amyloid peptides through beta-catenin-independent mechanisms. Proc Natl Acad Sci U S A, 99, 8760–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda M, Nagappan-Chettiar S, Johnson-Venkatesh EM & Umemori H 2021. An activity-dependent determinant of synapse elimination in the mammalian brain. Neuron, 109, 1333–1349 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Gaiteri C, Bodea LG, Wang Z, Mcelwee J, Podtelezhnikov AA, Zhang C, Xie T, Tran L, Dobrin R, et al. 2013. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell, 153, 707–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng LS, Hitoshi S, Kaneko N, Takao K, Miyakawa T, Tanaka Y, Xia H, Kalinke U, Kudo K, Kanba S, et al. 2014. Mechanisms for interferon-alpha-induced depression and neural stem cell dysfunction. Stem Cell Reports, 3, 73–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G, Soufan O, Ewald J, Hancock REW, Basu N & Xia J 2019. NetworkAnalyst 3.0: a visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res, 47, W234–W241. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Oligonucleotides, Related to STAR Methods section “Gene expression profiling”
Table S2. Nanostring data, Related to Figure 3 and Figures S2, S3, and S6
Data Availability Statement
All data including behavior data and microscopy data generated in this study are available from the lead contact on request. Nanostring data are provided in Table S2. Human transcriptomic data (MSBB) is publicly available from AMP-AD (see Key Resources Table).
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-Aβ (clone 126E10 | Novus Biologicals | Cat# NBP2-62566 |
| Chicken anti-GFP | Abcam | Cat# ab13970 (RRID: AB_300798) |
| Goat anti-Iba1 | Novus Biologicals | Cat# NB100-1028 (RRID: AB_521594) |
| Rabbit anti-Iba1 | Wako | Cat# 019-19741 (RRID: AB_839504) |
| Rat anti-PU.1 | Novus Biologicals | Cat# MAB7124 |
| Rat anti-Clec7a (clone R1-8g7) | Invivogen | Cat# mabg-mdect (RRID: AB_2753143) |
| Goat anti-Axl | R&D Systems | Cat# AF854 (RRID: AB_355663) |
| Rat anti-CD68 (clone FA-11) | BioRad | Cat# MCA1957GA (RRID: AB_324217) |
| Rabbit anti-Olig2 | Millipore | Cat# AB9610 (RRID: AB_570666) |
| Rat anti-LAMP1 (clone 1D4B) | BD Biosciences | Cat# 553792 (RRID: AB_2134499) |
| Mouse anti-GFAP (clone GA5) | Millipore | Cat# MAB360 (RRID: AB_11212597) |
| Mouse anti-PSD95 (clone 6G6-1C9) | Millipore | Cat# MAB1596 (RRID: AB_2092365) |
| Rabbit anti-PSD95 | Invitrogen | Cat# 51-6900 (RRID: AB_2533914) |
| Rabbit anti-synaptophysin | Abcam | Cat# MA5-14532 (RRID: AB_10983675) |
| Goat anti-synaptophysin | R&D Systems | Cat# AF5555 (RRID: AB_2198864) |
| anti-Vglut1 | Synaptic Systems | Cat# 135302 (RRID: AB_887877) |
| Rabbit anti-GluR2/3/4 | Thermo | Cat# PA5-17314 (RRID: AB_10985891) |
| Anti-VGAT | Synaptic Systems | Cat# 131011 (RRID: AB_887872) |
| Mouse anti-GAD65 | BD Pharmingen | Cat# 559931 (RRID: AB_397380) |
| Rabbit anti-cFos | Millipore | Cat# ABE457 (RRID: AB_2631318) |
| Rabbit anti-Fragilis (anti-Ifitm3) | Abcam | Cat# ab15592 (RRID: AB_2122095) |
| Rabbit anti-Stat1 (clone D1K9Y) | Cell Signaling | Cat# 14994 (RRID: AB_2799965) |
| Rabbit anti-Stat1, pTyr701 (clone 58D6) | Cell Signaling | Cat# 9167 (RRID: AB_561284) |
| Rabbit anti-Stat3, pTyr705 (clone D3A7) | Cell Signaling | Cat# 9145 (RRID: AB_2491009) |
| Mouse anti-NeuN (clone A60) | Millipore | Cat# MAB377 (RRID: AB_2298772) |
| Mouse anti-pNF (clone SMI31) | Biolegend | Cat# 801601 (RRID: AB_2564641) |
| Mouse anti-p-Tau (CP13) | Peter Davies lab (Polydoro et al., 2009) | N/A |
| Rabbit anti-human Tau (K9JA) | Agilent (Dako) | Cat# A0024 (RRID: AB_10013724) |
| Rat anti-C3 (clone 11H9) | Hycult | Cat# HM1045 (RRID: AB_10129042) |
| Goat anti-Mer | R&D Systems | Cat# AF591 (RRID: AB_2098565) |
| Goat anti-Gas6 | R&D Systems | Cat# AF986 (RRID: AB_2263130) |
| Anti-APP CTD | Hui Zheng lab (Xia et al, 2002) | N/A |
| Mouse anti-γ-tubulin (clone GTU-88) | Sigma-Aldrich | Cat# T6557 (RRID: AB_477584) |
| Mouse anti-ubiquitin (clone P4D1) | Santa Cruz | Cat# sc-8017 (RRID: AB_2762364) |
| Rabbit anti-LC3B | Sigma-Aldrich | Cat# L7543 (RRID: AB_796155) |
| Rat anti-Cd11b-FITC | BD Biosciences | Cat# 553310 (RRID: AB_394774) |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Mouse anti-IFNAR1 (clone MAR1-5A3) | Bio X Cell | Cat# BE0241 (RRID: AB_2687723) |
| Mouse IgG1 isotype (clone MOPC-21) | Bio X Cell | Cat# BE0083 (RRID: AB_1107784) |
| Tamoxifen | Sigma-Aldrich | Cat# T5648 |
| Recombinant mouse IFNβ | R&D Systems | Cat# #8234-MB-010 |
| Deposited Data | ||
| Mount Sanai Brain Bank (MSBB) | AMP-AD Knowledge Portal (https://adknowledgeportal.synapse.org) | Synapse ID: syn8484987 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J mice | NIA | N/A |
| 5XFAD mice | The Jackson Laboratories | Strain #034840-JAX |
| Ifnar1fl mice | The Jackson Laboratories | Strain #028256 |
| Cx3Cr1-CreERT2 mice | The Jackson Laboratories | Strain #021160 |
| Nestin-Cre mice | The Jackson Laboratories | Strain #003771 |
| ROSAmT/mG mice | The Jackson Laboratories | Strain #026862 |
| Mx1-Cre mice | The Jackson Laboratories | Strain #003556 |
| Thy1-eGFP-M mice | The Jackson Laboratories | Strain #007788 |
| Oligonucleotides | ||
| RT-qPCR primers | See Table S1 | N/A |
| Software and Algorithms | ||
| Leica confocal suite software | Leica (https://www.leica-microsystems.com) | RRID:SCR_013673 |
| Imaris (version 9.1) | Bitplane (www.bitplane.com) | RRID:SCR_007370 |
| ImageJ (version 1.47) | NIH (https://imagej.nih.gov/ij/) | RRID:SCR_003070 |
| GraphPad Prism (version 9.1.0) | GraphPad Software (www.graphpad.com) | RRID:SCR_002798 |
| R (version 3.5.1) | R Core Team (https://www.r-project.org/) | RRID:SCR_001905 |
| nSolver Advanced Analysis | Nanostring (www.nanostring.com) | RRID:SCR_003420 |
| InnateDB | Breuer et al, 2013 | N/A |
| NetworkAnalyst | Zhou et al, 2019 | N/A |
| ANY-maze Behavioral tracking software | Stoelting Co. (https://www.any-maze.com) | RRID:SCR_014289 |