

Abstract
Guanine deaminase (GDA) deaminates guanine to xanthine. Despite its significance, the study of human GDA remains limited compared to other metabolic deaminases. As a result, its substrate and inhibitor repertoire are limited, and effective real-time activity, inhibitory, and discovery assays are missing. Herein, we explore two emissive heterocyclic cores, based on thieno[3,4-d]pyrimidine (thN) and isothiazole[4,3-d]pyrimidine (tzN), as surrogate GDA substrates. We demonstrate that, unlike the thieno analog, thGN, the isothiazolo guanine surrogate, tzGN, does undergo effective enzymatic deamination by GDA and yields the spectroscopically distinct xanthine analog, tzXN. Further, we showcase the potential of this fluorescent nucleobase surrogate to provide a visible spectral window for a real-time study of GDA and its inhibition.
Graphical Abstract
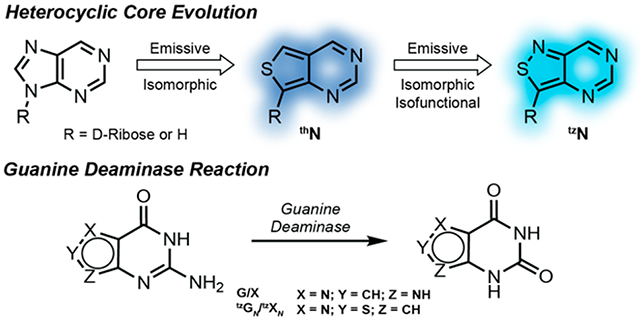
INTRODUCTION
Nucleobase and nucleoside deaminases play key roles in cellular pathways. Being responsible for the cellular levels of purine- and pyrimidine-based heterocycles, their over (or under) expression is frequently related to pathological conditions.1–4 Additionally, differences between human and bacterial enzymes provide intriguing opportunities for new therapeutic approaches, exploiting their distinct fidelity and inherent activities.1
Guanine deaminase (GDA), a hydrolytic zinc-based enzyme, converts guanine to xanthine.5,6 Recent observations have shown it impacts neuronal morphology7,8 and implicated GDA in traumatic brain injury,9 memory dysfunction, and psychiatric diseases.10 The study of human GDA, from the aminohydrolase superfamily, has remained somewhat sparse compared to other deaminases, including adenosine deaminase (ADA)11,12 and cytidine deaminase (CDA),13 which convert adenosine to inosine and cytidine to uridine, respectively. As a result, real-time activity and inhibitory assays for GDA, especially the human isoform, have yet to be advanced, and its substrate scope remains somewhat elusive.14
Diverse methods have been utilized for assessing the enzymatic activity and inhibition of deaminases. Most protocols capable of directly measuring GDA activity rely on the distinct chromatographic behavior or the UV absorption signature of the substrate/product pair.15 The former techniques are accurate but can be time-consuming, while the latter tools frequently suffer from spectral interference as many inhibitors share similar chromophoric properties with the substrate or product. To facilitate the biochemical study of deaminases, define their substrate scope, and identify inhibitors, innovative methods for monitoring their activity are needed. Although fluorescence spectroscopy might a priori appear most attractive due to its high sensitivity, it cannot be applied in such contexts due to the lack of any useful emissive features of the native purine nucleobases.
Over the past two decades our laboratory has designed, synthesized, and implemented minimally perturbing and responsive fluorescent nucleoside analogs.15–29 The guiding criterion for their design has been the diminution of structural and functional perturbations, which are inevitable consequences of modifying any native building block. Nucleosides that fulfill such critical constraints are thus coined isomorphic. If they function identically to their native counterparts, we define them as being isofunctional. We have advanced two emissive RNA alphabets, based on thieno[3,4-d]pyrimidine (1st generation; thN)30 and isothiazole[4,3-d]pyrimidine (2nd generation; tzN)31 heterocyclic cores (Figure 1a). The emissive adenosine analogs have been successfully used to advance real-time activity and inhibition assays of adenosine deaminase, exploiting the distinct emission profiles of the substrates (thA and tzA), and their corresponding deamination products (thI and tzI, respectively), with the latter (tzA) being essentially isofunctional (Figure 1b).32,33 The red-shifted absorption and visible-range emission of such fluorescent substrate surrogates opens a spectral window not accessible by other means, which facilitates the real-time monitoring of these reactions even in the presence of potentially interfering chromophores.34
Figure 1.
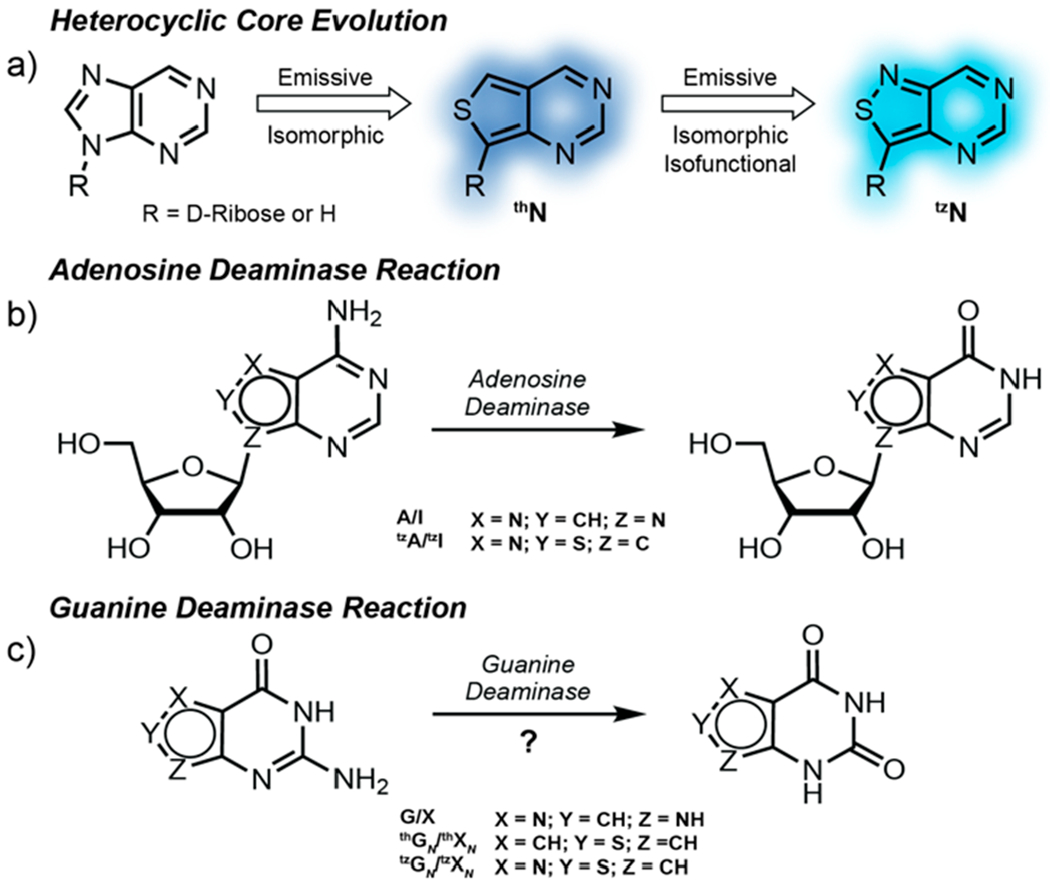
a) Heterocycle evolution pertaining to this study starting from the native purine core to thN to tzN. b) Adenosine deaminase conversion of A to I and isofunctional nucleoside analog tzA to tzI. c) Guanine deaminase conversion of G to X, isomorphic nucleobase analog thGN to thXN, and isofunctional nucleobase analog tzGN to tzXN.
Applying such approaches to nucleobase-processing enzymes such as human GDA represents, however, a minimally explored territory, as highly emissive nucleobase analogs have not been broadly tested and scrutinized as substrates.6,13 The lack of the rather large and contact forming d-ribose residue presents the challenge of altering guanine without disturbing contacts at the active site of such a potentially fastidious nucleobase-processing enzyme.35 We therefore set out to investigate two emissive nucleobase analogs, thieno[3,4-d]pyrimidine (thGN) and isothiazole[4,3-d]pyrimidine (tzGN) (Figure 1c), and demonstrate that while the former is not a viable substrate, tzGN does undergo facile GDA-mediated deamination to yield the fluorescently distinct xanthine analog, tzXN. The spectral differences displayed by the substrate and product are exploited for a real-time monitoring of the enzymatic reaction. Moreover, the potential of tzGN as a tool for GDA screenings is showcased by measuring GDA inhibition by a previously reported inhibitor and identifying two new inhibitors. Taken together, these findings highlight the necessity of the N7 for substrate recognition by GDA and provide information regarding its substrate and inhibitor scope.
RESULTS AND DISCUSSION
The four nucleobases, containing either a thienopyrimidine-core, thGN and thXN, or an isothiazolepyrimidine-core, tzGN and tzXN, were prepared according to established procedures (Figure 2a, Schemes S1–S3).30,31,35,36 Briefly, treatment of commercially available methyl 4-aminothiophene 3-carboxylate hydrochloride with chloroformamidine hydrochloride in DMSO2 at 125 °C yields thGN in one step. The corresponding xanthine analog, thXN, was synthesized in two steps by treating the same starting material with KOCN to yield the corresponding urea, which was then cyclized under basic conditions with sodium methoxide in methanol (Scheme S1). For the isothiazole-based nucleobases tzGN and tzXN, methyl 4-aminothiazole 3-carboxylate hydrochloride, a key precursor, was prepared from methyl thioglycolate and N-tosylated Oxyma, followed by decarboxylation and esterification (Scheme S2).37 Conversion of the isothiazole-based precursor to tzGN and tzXN followed the same cyclization procedures described above for the thiophene derivatives (Scheme S3). Crystal structures have been determined for all nucleobases (Figure 3a–d, Tables S1–S4).
Figure 2.
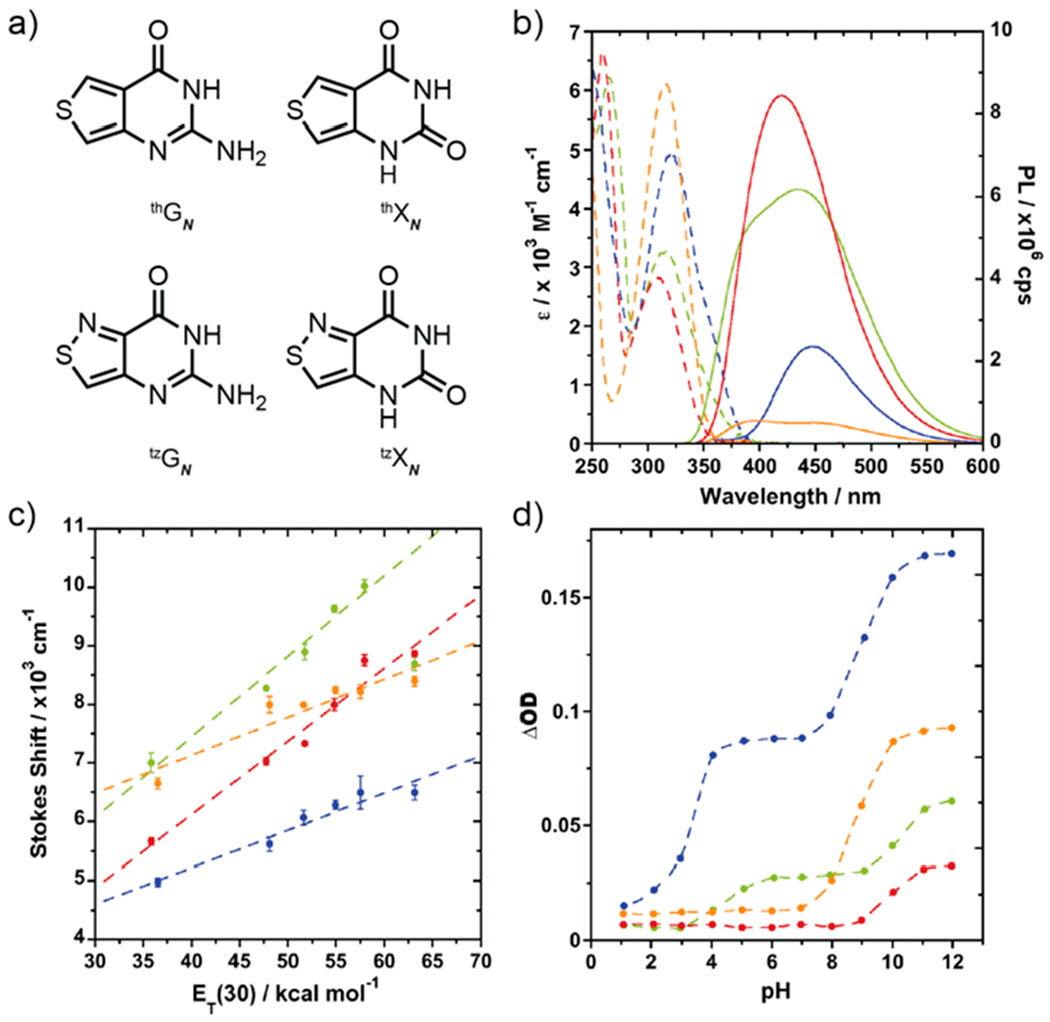
a) Structures of thGN, thXN, tzGN, and tzXN nucleobases. b) Absorption (dashed lines) and emission (solid lines) of thGN (green), thXN (red), tzGN (blue), and tzXN (orange) in water. c) Stokes shift correlations versus solvent polarity [ET(30) of water/dioxane mixtures] for thGN (green), thXN (red), tzGN (blue), and tzXN (orange). d) Change in optical density versus pH for thGN (green), thXN (red), tzGN (blue), and tzXN (orange).
Figure 3.
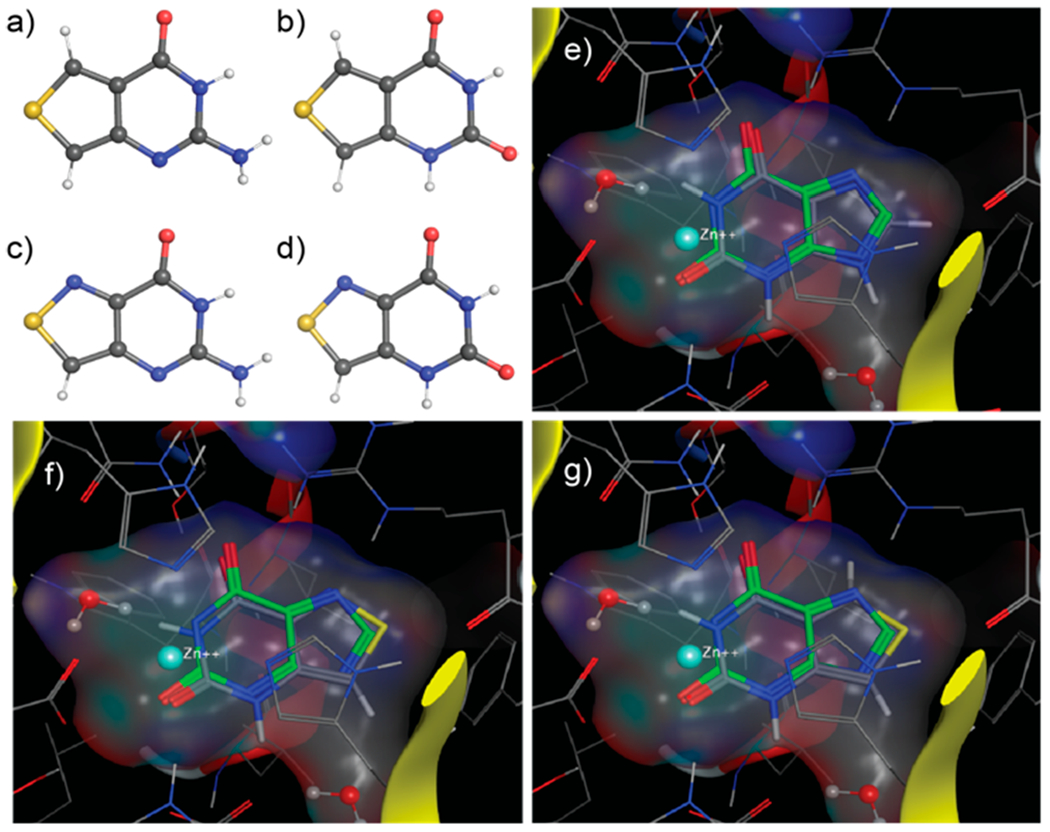
X-ray crystal structures of nucleobases. a) thGN, b) thXN, c) tzGN, and d) tzXN. MOE docking of e) xanthine as reference (ΔΔG 0), f) tzXN (ΔΔG 0.28), and g) thXN (ΔΔG 1.89) in GDA active site (PDB ID 2UZ9, see Methods).
To assess the likelihood of spectroscopically distinguishing between the substrates and their corresponding products, the fundamental spectroscopic properties of the nucleobases were determined (Tables 1 and S5). Absorption spectra in water displayed red-shifted maxima of the nucleobase analogs compared to their native counterparts, showing maxima at 315, 308, 320, and 315 nm for thGN, thXN, tzGN, and tzXN, respectively. Excitation at their absorption maxima gave rise to visible emission, which peaked at 439, 420, 446, and 394 nm for thGN, thXN, tzGN, and tzXN, respectively (Figure 2b). As expected, the emission quantum yields of the thiophene analogs, thGN and thXN (0.40 and 0.46, respectively) were higher than their isothiazole counterparts, tzGN and tzXN (0.07 and 0.02, respectively.)
Table 1.
Photophysical Data for Nucleobase Analogs
| λ abs,max a | ε b | λ em,max a | ϕ | |
|---|---|---|---|---|
| thGN | 315 | 3.0 | 439 | 0.40 |
| thXN | 308 | 3.2 | 420 | 0.46 |
| tzGN | 320 | 5.4 | 446 | 0.07 |
| tzXN | 315 | 6.1 | 394 | 0.02 |
λabs,max and λem,max are in nm.
ε is in 103 M−1 cm−1. Measured in triplicate in water.
Additionally, the sensitivity of photophysical parameters to environmental polarity was evaluated, and spectroscopically derived pKa values were determined (Table S5). The absorption and emission parameters of each nucleobase were measured in dioxane, water, and mixtures thereof. By linearly correlating the calculated Stokes shift against the solvent polarity of each sample, the chromophore’s responsiveness was defined. The four nucleobases showed differing levels of sensitivity to polarity, with the thiophene derivatives showing substantial impact on Stokes shift compared to the isothiazole derivatives (Figure 2c, Figure S1). The nucleobases also showed responsiveness to pH changes. pKa values were extrapolated by plotting the change in optical density versus pH (Figure 2d, Figure S2). The guanine derivatives, thGN and tzGN, show two pKa values (pKa = 4.41, 10.19 and pKa = 3.28, 8.96, respectively), while the xanthine derivatives, thXN and tzXN, show one pKa value (pKa = 9.92 and pKa = 8.8, respectively), as expected.31,38,39
To compare the susceptibility of thGN and tzGN to GDA-mediated deamination, enzymatic reactions were first analyzed by HPLC and then monitored in real-time by absorption and emission spectroscopy. HPLC analysis confirmed the complete transformation of native guanine to xanthine by GDA within 500 s (Figure 4a and S3a). The reaction of tzGN with GDA showed conversion to the corresponding product, tzXN (Figures 4a and S3b). However, the reaction of thGN showed no conversion of substrate across all time points studied (Figure S3c). By plotting the area under the curve at different reaction times (Figure 4b), comparable reaction half-life values for guanine deamination (G t1/2 = 27 s) and tzGN deamination (tzGN t1/2 = 21 s) were calculated assuming pseudo-first-order reaction conditions (Table 2). Kinetic parameters were also calculated by fitting HPLC-monitored deamination reactions to a set of ordinary differential equations consistent with Michaelis-Menten kinetics (see Methods). Similar values were obtained for guanine (KM = 12 ± 9 μM; k2 = 36 ± 27 s−1; k2/KM = 3.0 ± 0.3 μM−1 s−1) and its isothiazolo analog tzGN (KM = 8 ± 3 μM; k2 = 32 ± 7 s−1; k2/KM = 4.2 ± 1.0 μM−1 s−1; see Table 2). We note that the KM value obtained for the deamination of guanine is comparable to previously published figures for rabbit liver GDA (KM of 12.5 μM),40 and the k2/KM is comparable to k1/[GDA] (G k1/[GDA] = 2.6 μM−1 s−1) from the pseudo-first-order kinetic curves (Table 2).
Figure 4.
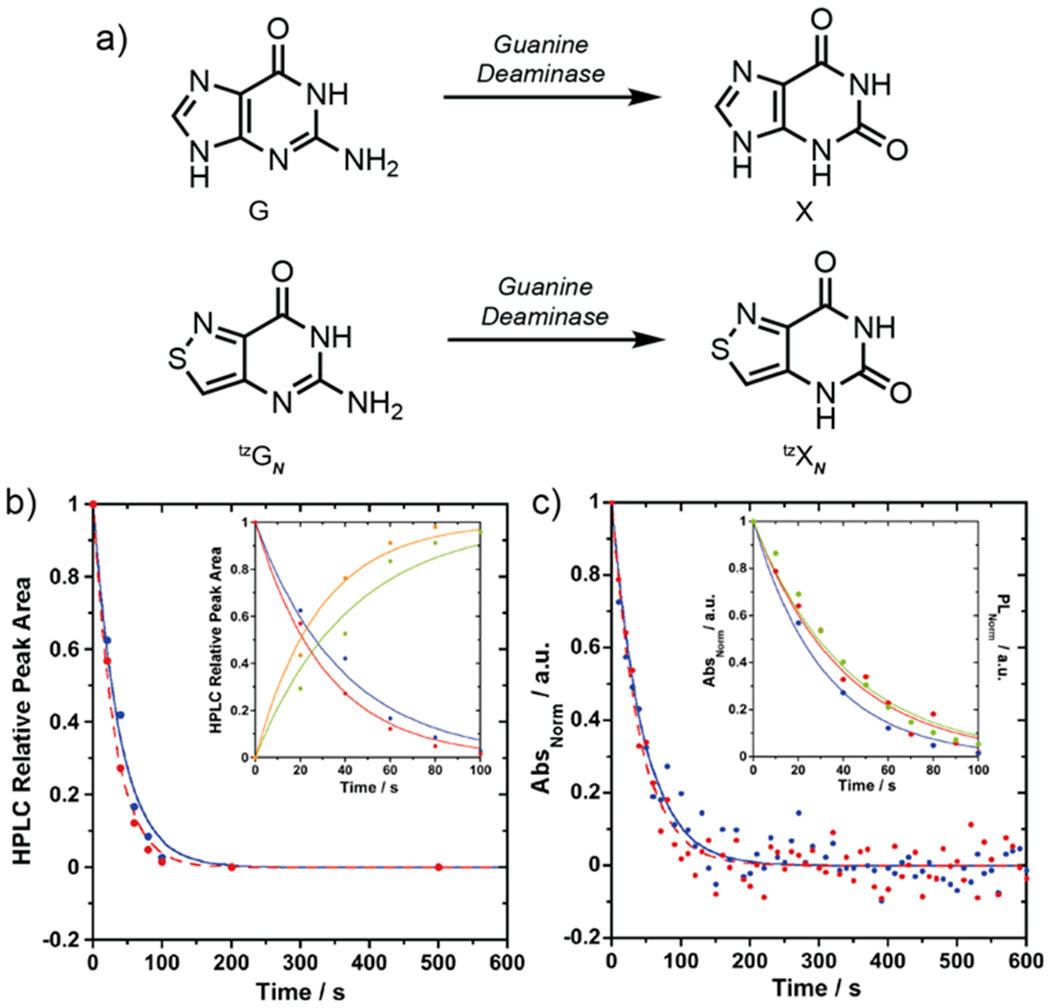
a) Conversion of guanine to xanthine and conversion of isofunctional tzGN to tzXN by GDA. b) HPLC relative peak area versus time for consumption of guanine to xanthine at 260 nm (blue) and consumption of tzGN to tzXN at 320 nm (red). Inset: HPLC relative peak area versus time for the consumption of guanine (blue) and formation of xanthine (green) at 260 nm and the consumption of tzGN (red) and formation of tzXN (orange) at 320 nm from 0 to 100 s. c) Absorption changes upon enzymatic deamination of guanine to xanthine at 270 nm (blue) and tzGN to tzXN at 355 nm (red). Inset: Enzymatic deamination of tzGN to tzXN monitored by absorbance (at 355 nm, red), fluorescence (450 nm, excitation at 328 nm, green), and HPLC relative peak area over time (at 320 nm, blue).
Table 2.
GDA Reaction Rates
| HPLC |
absorbance |
emission |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| λ mntr a | k 1 b | t 1/2 c | K M d | k 2 d | k2/KMd | λ mntr a | k 1 b | t 1/2 c | λ mntr a | k 1 b | t 1/2 c | |
| G to X | 260 | 26 ± 2 | 27 | 12 ± 9 | 36 ± 27 | 3.0 ± 0.3 | 270 | 22 ± 1 | 31 | |||
| thGN to thXN | 320 | NR | NR | NR | NR | NR | 355 | NR | NR | 450e | NR | NR |
| tzGN to tzXN | 260 | 32 ± 1 | 21 | 8 ± 3 | 32 ± 7 | 4.2 ± 1.0 | 355 | 25 ± 1 | 27 | 450f | 24 ± 1 | 29 |
λmntr reported in nm and represents wavelength monitored.
Pseudo-first-order reaction kinetics slope of the exponential approximation in 10−3 s−1.
t1/2 is reaction half-life calculated assuming pseudo-first-order kinetics.
KM, k2, and k2/KM are reported in μM, s−1, and 106 M−1 s−1, respectively.
λexc at 360 nm for thGN reactions.
λexc at 328 nm for tzGN reactions. Experiments done in triplicate.
Rewardingly, spectroscopic analyses agree with the chromatographic analysis (Figure 4c). When monitoring the enzymatic deamination of tzGN spectroscopically, an increase in absorbance at 355 nm and an emission decrease at 450 nm when exciting at 328 nm, the isosbestic point, were observed. The reaction half-life for tzGN was calculated using the same assumptions mentioned previously (tzGN abs t1/2 = 27 s; t1/2 em = 29 s) and was found to be comparable to the native substrate (G abs t1/2 = 31 s), as shown in Table 2. The transformation of both native guanine to xanthine and tzGN to tzXN by GDA is also illustrated by the absorption maxima shift (Figure S4). Thienopyrimidine thGN showed no change in absorption or emission in the presence of GDA over 500 s.
Reactions monitored by HPLC as well as real-time absorption and emission spectroscopy suggest that tzGN, the isothiazolepyrimidine substrate, is equally susceptible to GDA-mediated deamination as native guanine. Intriguingly, thGN, the thienopyrimidine guanine analog, is unreactive and does not undergo GDA-mediated deamination. These findings showcase the functionality and increased isomorphicity of the isothiazolepyrimidine-based nucleobase alphabet, which reinstated the N7 moiety, compared to the thienopyrimidine-based nucleobases, even without the large and contact forming d-ribose residue the nucleoside counterparts possess. Further support is obtained by Molecular Operating Environment (MOE) molecular docking experiments (Figure 3e–g, Figures S5 and S6) using the published crystallographic data for xanthine-bound human GDA (PDB ID 2UZ9). The enzyme recognition preferences for tzXN over thXN are illustrated by the markedly higher positive ΔΔG values obtained for the latter vs the former (ΔΔG 1.89 and 0.28, respectively). A similar trend was observed for the substrates Figure S6). Docking also shows a plausible structure deformation induced by Arg235, which is projected toward the N7 position at the native substrate. This basic site, present in native G/X and the isothiazolo analogs, tzGN/tzXN, likely hydrogen bonds to Arg235. Clashing of this arginine side chain with the CH group of thGN at the same position likely renders the thiophenopyrimidine-based substrate unrecognizable by GDA.
As stated, the presence of the nitrogen at a position equivalent to N7 in the purine skeleton endows tzGN, the isothiazole purine surrogate, with substrate recognition features that appear lacking in thGN. While comprehensive investigations of the GDA substrate scope have not been pursued, previous substrate and inhibitor analyses of aminohydrolase isoforms suggest that the O6, N3, and N7 points of contact are important for substrate recognition.6,17,40,41 Intriguingly, while tzGN retains these contact points, it is lacking the NH group found in the purine’s 9 position. Previously reported inhibitors, such as valciclovir and derivatives of azepinomycin, containing substituents at the N9 position suggest GDA can tolerate diverse groups at that position, which may explain the high tolerance and the native deamination rate displayed by tzGN.14,42
The change in emission signal intensity upon GDA-mediated deamination of tzGN to tzXN facilitates real-time monitoring of the enzymatic reaction and its inhibition. To demonstrate its potential, three inhibitors were tested, 5-aminoimidazole-4-carboxamide (AICA), 4-aminoisothiazole-3-carboxamide (ATCA), and 4-imidazolecarboxylic acid (ICA, Figure 5a). AICA is an established competitive inhibitor of GDA.40 We synthesized ATCA, the isothiazole counterpart (Scheme S4). Reaction conditions used for previous experiments were sustained with the exception of inhibitor addition at various concentrations. Percent inhibition plots were fitted with sigmoidal Hill curves to obtain IC50 values of the tested inhibitors. AICA (IC50 = 100 μM) showed comparable inhibition to ATCA (IC50 = 80 μM), while ICA is comparatively a less potent inhibitor (IC50 = 2 mM, Figure 5b).
Figure 5.
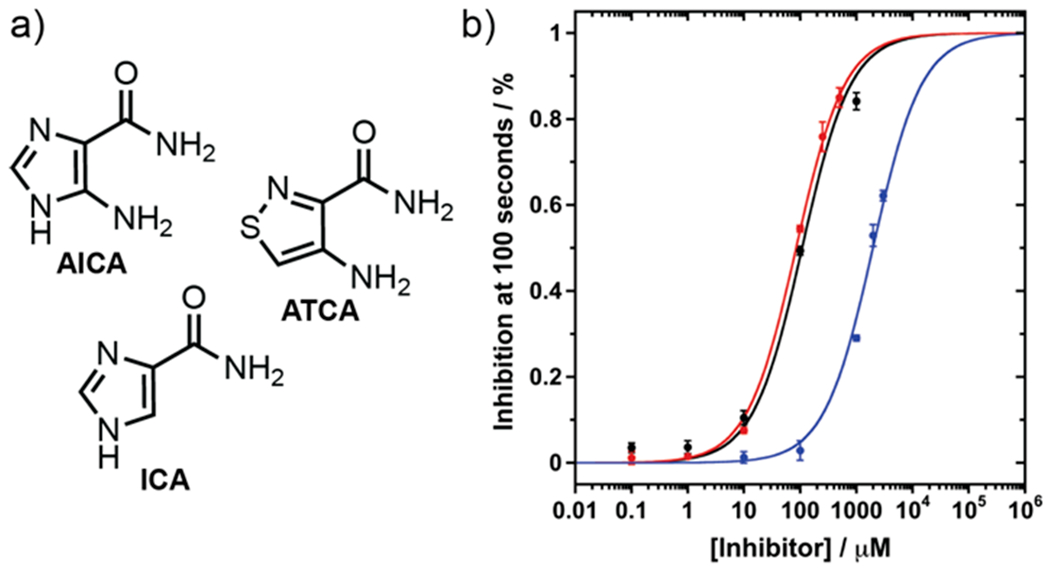
a) GDA inhibitor structures of AICA, ATCA, and ICA. b) Overlay of % inhibition at 100 s and sigmoidal Hill plot for AICA (black), ATCA (red), and ICA (blue).
CONCLUSION
This study set out to assess the utility of two emissive nucleobase analogs, thGN and tzGN, as substrates for the enzymatic reaction of GDA. We demonstrate that isothiazole-[4,3-d]pyrimidine, tzGN, behaves as an isomorphic and isofunctional emissive surrogate for guanine. The interconversion of tzGN to tzXN can successfully be tracked in real-time as the reaction substrate and product display distinct photophysical properties. The strategic replacement of the GDA substrate with emissive counterparts from both previously published emissive alphabets demonstrate the significance of N7 for GDA substrate recognition. Moreover, this discovery provides a biochemical tool to study the activity of human GDA and offers a spectral window for fabricating real-time high throughput discovery assays of GDA. We illustrate this through a small pilot study of inhibitor effects on GDA activity, which includes one previously reported inhibitor and two new inhibitors.
METHODS
Expression and Purification of Human GDA.
Wild-type human GDA gene, cloned into pET-28B vector (Genescript), was expressed with an N-terminal hexahistidine tag. The construct was sequence-verified (GENEWIZ). The plasmid (4 μg) was dissolved in water to a concentration of 28.8 ng/μL. E. coli BL21 (DE3) cells were transformed with the GDA containing plasmid. Overnight starter cultures were grown in Lubria Broth (LB) medium at 37 °C with 50 μg/mL kanamycin. The overnight culture was used to inoculate 1 L of LB medium with 50 μg/mL kanamycin. Cells were grown while shaking at 37 °C until an OD600 of 0.6 was reached, and the culture was down tempered to 18 °C over a period of 1 h. To induce target protein production, 0.5 mM IPTG was added, and the medium was left to shake at 18 °C overnight. Cells were harvested after overnight growth by centrifugation at 3500 rpm for 35 min at 4 °C.
Cells were resuspended in lysis buffer [20 mM HEPES, 500 mM NaCl, 10% glycerol, 0.5 mM TCEP (pH 7.5), supplemented with one tablet of Complete EDTA-free protease inhibitor (Roche Applied Science)] and lysed by sonication. Cell lysate was centrifuged at 10,000 rpm for 1 h at 4 °C before the supernatant was decanted and filtered through a 0.45 μm syringe filter. GDA was purified by immobilized metal affinity chromatography. An IMAC Ni-charged column [2 mL HisPur Ni-NTA Resin (Thermo Scientific)] was equilibrated with lysis buffer to which the lysate was then added. The column was washed with 50 mL of wash buffer supplemented with 20 mM imidazole. The bound protein was eluted from the column with elution buffer (wash buffer supplemented with 200 mM imidazole). Fractions were analyzed by SDS-PAGE, and those containing the target protein were pooled, subsequently concentrated, and buffer exchanged into storage buffer [20 mM Tris-HCl buffer (pH 8.0), 10% glycerol, 1 mM DTT] using a centrifugal filter device with a 10 kDa molecular weight cutoff. After protein expression and purification, the final protein concentration was 5.3 mg mL−1 (0.1 mM) in a total volume of 1.7 mL. Protein aliquots were snap-frozen and stored at −80 °C until further use. Before being used in enzymatic reactions, the GDA protein stock was diluted 1:100 in sodium phosphate buffer [50 mM (pH 7.4)].
Synthetic Procedures.
All synthetic procedures and characterizations of compounds are reported in the Supporting Information.
Photophysical Properties: General Methods.
Absorption spectra were measured on a Shimadzu UV-2450 spectrometer with 0.5 mm resolution and setting the slit at 1 nm. Emission spectra were measured on a Horiba Fluoromax-4 equipped with a cuvette holder and stirring system. Emission measurements were taken with resolution at 1 nm and setting both the excitation and emission slits at 3 nm.
All measurements were carried out in a 10 mm four-sided quartz cuvette purchased from Helma. All spectra were corrected for the blank. Both instruments were equipped with a thermostat-controlled ethylene glycol-water bath, and all measurements were taken at 37 °C. Measurements were recorded after a 3 min temperature equilibration period.
Concentrated stock solutions for xanthine, thGN thXN tzGN and tzXN were prepared in DMSO, and a stock solution of guanine was prepared in water basified to pH 12 with sodium hydroxide. Samples for experiments were prepared with stock nucleobase diluted to a total sample volume of 3 mL in deionized water, mixed with a pipet for 10 s, and placed in the cuvette holder. All samples contain 0.3 v/v % DMSO, except guanine samples.
Quantum Yield Measurements.
All sample concentrations were adjusted to optical density lower than 0.1 at the excitation wavelength (λex). The fluorescence quantum yield (ϕ) of each nucleobase was evaluated based on 2-aminopurine (0.68 in water, λex 320 nm) as an external standard by using the following equation
| (1) |
where ϕSTD is the fluorescence quantum yield of the standard, I and ISTD are the integrated area of the emission band of the sample and the standard, respectively, OD and ODSTD are the optical density at the excitation wavelength for the sample and standard, respectively, and n and nSTD are the solvent refractive index of the sample and the standard solutions, respectively.
Sensitivity to pH.
Sodium phosphate buffers with a final concentration of 50 mM were prepared and adjusted to the desired experimental pH values using HCl or NaOH prior to spectral measurements. Changes in optical density, at 310 and 350 nm, were plotted versus pH. The pKa values were determined by interpolation of the fitting curves.
Sensitivity to Polarity.
Experiments evaluating the effect of polarity were performed in water, dioxane, and mixtures of 20, 40, 60, and 80 v/v % water in dioxane. The sample ET(30) values were determined by dissolving Reichardt’s dye in an aliquot of the same solvent used to dilute the nucleobase DMSO sample. The observed wavelength absorption maximum () was then converted to the ET(30) values (Table S6) using the following equation
| (2) |
Enzymatic Deamination: General Methods.
Reaction conditions were the same for all GDA reactions monitored by spectroscopy or chromatography. Concentrated stock solutions for xanthine, thGN thXN tzGN and tzXN were prepared in DMSO, and the stock solution of guanine was prepared in water basified to pH 12 with sodium hydroxide. Samples were prepared in a 10.00 mm four-sided quartz cuvette from Helma. Reactions had a total reaction volume of 3 mL with nucleobase and enzyme concentrations of 3 μM and 10 nM in sodium phosphate buffer [50 mM (pH 7.4)]. All measurements were taken at 37 °C, and GDA was introduced after a 3 min temperature equilibration period.
Real-Time Monitoring of GDA Reactions via Absorption and Emission.
GDA-mediated conversion of guanine and its analogs (thGN and tzGN was monitored by absorption and emission spectroscopy. Absorbance measurements of the enzymatic conversion of guanine (and its analogs) were performed on a Shimadzu UV-2459 spectrometer taking a point every 10 s for 600 s after the addition of GDA with a slit setting of 1 nm. The conversion was monitored at 270 nm for guanine to xanthine and at 355 nm for the analogs (thGN and tzGN to tzXN). Emission measurements of the conversion of guanine (and analogs) were performed on a Horiba Fluoromax-4 with a cuvette holder and a built-in stirring system with excitation and emission slits set to 3 nm and taking points every 10 s for 600 s after the addition of GDA.
Emission was monitored at 450 nm with excitation at 360 and 328 nm, respectively, for thGN and tzGN. The thGN reaction showed no change in absorption or emission intensity in the presence of GDA over 600 s. Each experiment was done in triplicate. There is a 6 s lag time after GDA is added to the cuvette after the time 0 measurement.
Steady State Absorption Measurements in the Presence of GDA.
Steady state absorption spectra over time were performed on a Shimadzu UV-2450 spectrophotometer setting the slit at 5 nm, using a resolution of 0.5 nm, taking a measurement every 20 s. All spectra were corrected for the blank. Smoothing of data was done on spectra for plotting.
HPLC Analysis of Enzymatic Conversion of Native G to X and of tzGN to tzXN.
GDA-mediated conversion was monitored by chromatography. HPLC was carried out with an Agilent 1200 system with a Polaris 5 C18-A 250 × 4.9 mm column. Solutions of 0.1% formic acid (Honeywell Fluka) were prepared by dissolving 1 mL of formic acid in 1 L total volume of acetonitrile (J. T. Baker) or water. Solutions were filtered using Millipore type GNWP 0.2 μm filters before use. Each injection (75 μL) for guanine or tzGN experiments was subjected to a linear gradient of 0.5% to 10% acetonitrile in water with 0.1% formic acid for 20 min, followed by a flush and equilibration for 10 min. thGN injections were subjected to a linear gradient of 0.5% to 10% acetonitrile in water with 0.1% formic acid for 30 min, followed by a flush and equilibration for 10 min. Each run was monitored at 260, 280, and 320 nm with a calibrated reference at 650 nm and slit set at 1 nm.
A concentrated stock solution of guanine, thGN, and tzGN was diluted in phosphate buffer. The solution was warmed to 37 °C for 3 min with stirring before addition of a GDA stock solution. After the addition of GDA, the enzymatic conversion was quenched in aliquots (after 20, 40, 60, 80, 100, 200, and 500 s) by adding formic acid (0.55 M) and placing the aliquots on ice. Each 100 μL aliquot was filtered and analyzed by HPLC.
HPLC traces were corrected for the blank, and the relative areas were plotted as a function of time. Trend lines represent a loss of substrate and product apparition over time for a pseudo-first-order kinetic reaction.
A set of ordinary differential equations (ODEs) consistent with Michaelis–Menten kinetics (eqs 3–6) was solved using the Runge–Kutta method with a method with a variable time step in MatLab (function ode45). Initial concentrations used for enzyme and substrate were 10 nM and 3 μM, respectively. The product and enzyme substrate complex were assumed to have initial concentrations of 0 μM. The resulting fitted curves for each species were optimized by iteratively testing k values that maximized R.2 This yielded k1, k−1, and k2 values from which KM and k2/KM values were derived.
| (3) |
| (4) |
| (5) |
| (6) |
Inhibition Studies.
Fluorescence real-time monitoring of the enzymatic conversion of tzGN to tzXN with GDA in the presence of inhibitors was followed by emission spectroscopy by monitoring the intensity signal at 450 nm with excitation at 328 nm. 5-Aminoimidazole-4-carboxamide (AICA) was tested as an inhibitor in the enzymatic reaction for tzGN to tzXN using the same reaction conditions but supplemented with inhibitor concentrations of 0.1, 1, 10, 100, and 1000 μM. 4-Aminoisothiazole-3-carboxamide (ATCA) was also tested as an inhibitor in the reaction under the same conditions but supplemented with inhibitor concentrations of 0.1, 1, 10, 100, 250, and 500 μM. 4-Imidazolecarboxylic acid (ICA) was also tested as a potential inhibitor under the same reaction conditions with 10, 100, 1000, 2000, and 3000 μM concentrations. IC50 values were determined from plots of percent inhibition against respective inhibitor concentration on a logarithmic axis, fitted with sigmoidal Hill curves. All measurements were done in triplicate.
Docking of Guanine, Xanthine, and Analogs in GDA.
Docking simulations were performed with the Molecular Operating Environment (MOE) 2020 software suite. The crystal structure of human guanine deaminase (guaD) in complex with zinc and its product xanthine was obtained from the Protein Data Bank (PDB ID 2UZ9). The crystal structure was chosen because it contains the native product xanthine. The chosen ligands were placed by the Triangle Matcher method and ranked with the London dG values scoring function. A total of 30 poses were refined using the rigid receptor method. Then five poses were rescored using a GBVI/WSA dG scoring function. The pose with the ligand most closely aligned with the xanthine ligand was used for comparison, and the ΔΔG value was calculated for tzGN and thGN with native guanine as the reference and for tzXN and thXN with native xanthine as the reference. The equation below was used where ΔGref is the GBV1/WSA dG scoring of the reference native ligand and ΔGlig is the GBVI/WSA dG scoring of the ligand analogs (eq 7).
| (7) |
Supplementary Material
ACKNOWLEDGMENTS
We thank the National Institutes of Health (through grant no. GM 069773) for generous support, the Chemistry & Biochemistry MS facility, and the UCSD X-ray Crystallography Facility (especially M. Gembicki and C. Moore). We also thank the Burkhart Lab at UCSD for advice on protein expression and purification and R. Anand for helpful discussions and insight.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.1c00232.
Synthetic schemes, experimental methods, photophysical data, spectra, and X-ray crystallographic data for thGN, thXN, tzGN, and tzXN (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acschembio.1c00232
The authors declare no competing financial interest.
Contributor Information
Marcela S. Bucardo, Department of Chemistry and Biochemistry, University of California, San Diego, La Jolla, California 92093, United States
You Wu, Department of Chemistry and Biochemistry, University of California, San Diego, La Jolla, California 92093, United States.
Paul T. Ludford, III, Department of Chemistry and Biochemistry, University of California, San Diego, La Jolla, California 92093, United States.
Yao Li, Department of Chemistry and Biochemistry, University of California, San Diego, La Jolla, California 92093, United States.
Andrea Fin, Department of Chemistry and Biochemistry, University of California, San Diego, La Jolla, California 92093, United States; Present Address: Dipartimento di Scienza e Tecnologia del Farmaco, University of Turin, Via P. Giuria, 9, 10125 Turin, Italy.
Yitzhak Tor, Department of Chemistry and Biochemistry, University of California, San Diego, La Jolla, California 92093, United States.
REFERENCES
- (1).Gaded V, and Anand R (2018) Nucleobase Deaminases: A Potential Enzyme System for New Therapies. RSC Adv. 8 (42), 23567–23577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Wang J, Bing T, Zhang N, Shen L, He J, Liu X, Wang L, and Shangguan D (2019) The Mechanism of the Selective Antiproliferation Effect of Guanine-Based Biomolecules and Its Compensation. ACS Chem. Biol 14 (6), 1164–1173. [DOI] [PubMed] [Google Scholar]
- (3).Slater PG, Cammarata GM, Monahan C, Bowers JT, Yan O, Lee S, and Lowery LA (2019) Characterization of Xenopus Laevis Guanine Deaminase Reveals New Insights for Its Expression and Function in the Embryonic Kidney. Dev. Dyn 248 (4), 296–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Jung JM, Noh TK, Jo SY, Kim SY, Song Y, Kim Y-H, and Chang SE (2020) Guanine Deaminase in Human Epidermal Keratinocytes Contributes to Skin Pigmentation. Molecules 25 (11), 2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Paletzki RF (2002) Cloning and Characterization of Guanine Deaminase from Mouse and Rat Brain. Neuroscience 109 (1), 15–26. [DOI] [PubMed] [Google Scholar]
- (6).Shek R, Hilaire T, Sim J, and French JB (2019) Structural Determinants for Substrate Selectivity in Guanine Deaminase Enzymes of the Amidohydrolase Superfamily. Biochemistry 58 (30), 3280–3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Akum BF, Chen M, Gunderson SI, Riefler GM, Scerri-Hansen MM, and Firestein BL (2004) Cypin Regulates Dendrite Patterning in Hippocampal Neurons by Promoting Microtubule Assembly. Nat. Neurosci 7 (2), 145–152. [DOI] [PubMed] [Google Scholar]
- (8).Rodríguez AR, O’Neill KM, Swiatkowski P, Patel MV, and Firestein BL (2018) Overexpression of Cypin Alters Dendrite Morphology, Single Neuron Activity, and Network Properties via Distinct Mechanisms. J. Neural Eng 15 (1), No. 016020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Swiatkowski P, Sewell E, Sweet ES, Dickson S, Swanson RA, McEwan SA, Cuccolo N, McDonnell ME, Patel MV, Varghese N, Morrison B, Reitz AB, Meaney DF, and Firestein BL (2018) Cypin: A Novel Target for Traumatic Brain Injury. Neurobiol. Dis 119, 13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Zuccarini M, Giuliani P, Frinchi M, Mudò G, Serio RM, Belluardo N, Buccella S, Carluccio M, Condorelli DF, Caciagli F, Ciccarelli R, and Di Iorio P (2018) Uncovering the Signaling Pathway behind Extracellular Guanine-Induced Activation of NO System: New Perspectives in Memory-Related Disorders. Front. Pharmacol 9, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Wilson DK, Rudolph FB, and Quiocho FA (1991) Atomic Structure of Adenosine Deaminase Complexed with a Transition-State Analog: Understanding Catalysis and Immunodeficiency Mutations. Science 252 (5010), 1278–1284. [DOI] [PubMed] [Google Scholar]
- (12).Kinoshita T, Nakanishi I, Terasaka T, Kuno M, Seki N, Warizaya M, Matsumura H, Inoue T, Takano K, Adachi H, Mori Y, and Fujii T (2005) Structural Basis of Compound Recognition by Adenosine Deaminase. Biochemistry 44 (31), 10562–10569. [DOI] [PubMed] [Google Scholar]
- (13).Bitra A, Biswas A, and Anand R (2013) Structural Basis of the Substrate Specificity of Cytidine Deaminase Superfamily Guanine Deaminase. Biochemistry 52 (45), 8106–8114. [DOI] [PubMed] [Google Scholar]
- (14).Egeblad L, Welin M, Flodin S, Gräslund S, Wang L, Balzarini J, Eriksson S, and Nordlund P (2012) Pan-Pathway Based Interaction Profiling of FDA-Approved Nucleoside and Nucleobase Analogs with Enzymes of the Human Nucleotide Metabolism. PLoS One 7 (5), e37724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Wojczewski C, Stolze K, and Engels JW (1999) Fluorescent Oligonucleotides - Versatile Tools as Probes and Primers for DNA and RNA Analysis. Synlett 1999 (10), 1667–1678. [Google Scholar]
- (16).Hawkins M (2001) Fluorescent Pteridine Nucleoside Analogs: A Window on DNA Interactions. Cell Biochem. Biophys 34, 257–281. [DOI] [PubMed] [Google Scholar]
- (17).Rist MJ, and Marino JP (2002) Fluorescent Nucleotide Base Analogs as Probes of Nucleic Acid Structure, Dynamics and Interactions. Curr. Org. Chem 6 (9), 775–793. [Google Scholar]
- (18).Millar DP (1996) Fluorescence Studies of DNA and RNA Structure and Dynamics. Curr. Opin. Struct. Biol 6 (3), 322–326. [DOI] [PubMed] [Google Scholar]
- (19).Okamoto A, Saito Y, and Saito I (2005) Design of Base-Discriminating Fluorescent Nucleosides. J. Photochem. Photobiol., C 6, 108–122. [Google Scholar]
- (20).Ranasinghe RT, and Brown T (2005) Fluorescence Based Strategies for Genetic Analysis. Chem. Commun 44, 5487–5502. [DOI] [PubMed] [Google Scholar]
- (21).Silverman AP, and Kool ET (2006) Detecting RNA and DNA with Templated Chemical Reactions. Chem. Rev 106 (9), 3775–3789. [DOI] [PubMed] [Google Scholar]
- (22).Wilson JN, and Kool ET (2006) Fluorescent DNA Base Replacements: Reporters and Sensors for Biological Systems. Org. Biomol. Chem 4 (23), 4265–4274. [DOI] [PubMed] [Google Scholar]
- (23).Dodd D, and Hudson R (2009) Intrinsically Fluorescent Base-Discriminating Nucleoside Analogs. Mini-Rev. Org. Chem 6, 378–391. [Google Scholar]
- (24).Wilhelmsson LM (2010) Fluorescent Nucleic Acid Base Analogues. Q. Rev. Biophys 43 (2), 159–183. [DOI] [PubMed] [Google Scholar]
- (25).Wierzchowski J, Antosiewicz JM, and Shugar D (2014) 8-Azapurines as Isosteric Purine Fluorescent Probes for Nucleic Acid and Enzymatic Research. Mol. BioSyst 10 (11), 2756–2774. [DOI] [PubMed] [Google Scholar]
- (26).Jones AC, and Neely RK (2015) 2-Aminopurine as a Fluorescent Probe of DNA Conformation and the DNA-Enzyme Interface. Q. Rev. Biophys 48 (2), 244–279. [DOI] [PubMed] [Google Scholar]
- (27).Wilhelmsson M Fluorescent Analogs of Biomolecular Building Blocks: Design and Applications; 2016; DOI: 10.1002/9781119179320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Xu W, Chan KM, and Kool ET (2017) Fluorescent Nucleobases as Tools for Studying DNA and RNA. Nat. Chem 9 (11), 1043–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Saito Y, and Hudson RHE (2018) Base-Modified Fluorescent Purine Nucleosides and Nucleotides for Use in Oligonucleotide Probes. J. Photochem. Photobiol., C 36, 48–73. [Google Scholar]
- (30).Shin D, Sinkeldam RW, and Tor Y (2011) Emissive RNA Alphabet. J. Am. Chem. Soc 133 (38), 14912–14915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Rovira AR, Fin A, and Tor Y (2015) Chemical Mutagenesis of an Emissive RNA Alphabet. J. Am. Chem. Soc 137 (46), 14602–14605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Sinkeldam RW, McCoy LS, Shin D, and Tor Y (2013) Enzymatic Interconversion of Isomorphic Fluorescent Nucleosides: Adenosine Deaminase Transforms an Adenosine Analogue into an Inosine Analogue. Angew. Chem. Int. Ed 52 (52), 14026–14030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Ludford PT, Rovira AR, Fin A, and Tor Y (2019) Fluorescing Isofunctional Ribonucleosides: Assessing Adenosine Deaminase Activity and Inhibition. ChemBioChem 20 (5), 718–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Adamek RN, Ludford P, Duggan SM, Tor Y, and Cohen SM (2020) Identification of Adenosine Deaminase Inhibitors by Metal-Binding Pharmacophore Screening. ChemMedChem 15 (22), 2151–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Srivatsan SG, Weizman H, and Tor Y (2008) A Highly Fluorescent Nucleoside Analog Based on Thieno[3,4-d]Pyrimidine Senses Mismatched Pairing. Org. Biomol. Chem 6 (8), 1334–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Rovira AR, Fin A, and Tor Y (2017) Expanding a Fluorescent RNA Alphabet: Synthesis, Photophysics and Utility of Isothiazole-Derived Purine Nucleoside Surrogates. Chem. Sci 8 (4), 2983–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Gewald K, and Bellmann P (1979) Synthese und Reaktionen von 4-Aminoisothiazolen. Liebigs Ann. Chem 1979 (10), 1534–1546. [Google Scholar]
- (38).Rogstad KN, Jang YH, Sowers LC, and Goddard WA (2003) First Principles Calculations of the PKa Values and Tautomers of Isoguanine and Xanthine. Chem. Res. Toxicol 16 (11), 1455–1462. [DOI] [PubMed] [Google Scholar]
- (39).Didier P, Kuchlyan J, Martinez-Fernandez L, Gosset P, Léonard J, Tor Y, Improta R, and Mely Y (2020) Deciphering the PH-Dependence of Ground- and Excited-State Equilibria of Thienoguanine. Phys. Chem. Chem. Phys 22 (14), 7381–7391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Lewis AS, and Glantz MD (1974) Rabbit Liver Guanine Deaminase. Chemical, Physical, and Kinetic Properties. J. Biol. Chem 249 (12), 3862–3866. [PubMed] [Google Scholar]
- (41).Bergstrom JD, and Bieber AL (1979) Characterization of Purified Guanine Aminohydrolase. Arch. Biochem. Biophys 194 (1), 107–116. [DOI] [PubMed] [Google Scholar]
- (42).Chakraborty S, Shah NH, Fishbein JC, and Hosmane RS (2011) A Novel Transition State Analogue Inhibitor of Guanase Based on Azepinomycin Ring Structure: Synthesis and Biochemical Assessment of Enzyme Inhibition. Bioorg. Med. Chem. Lett 21 (2), 756–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.