

Abstract
In CASP14, 39 research groups submitted more than 2,500 3D models on 22 protein complexes. In general, the community performed well in predicting the fold of the assemblies (for 80% of the targets), though it faced significant challenges in reproducing the native contacts. This is especially the case for the complexes without whole-assembly templates. The leading predictor, BAKER-experimental, used a methodology combining classical techniques (template-based modeling, protein docking) with deep learning-based contact predictions and a fold-and-dock approach. The Venclovas team achieved the runner-up position with template-based modeling and docking. By analyzing the target interfaces, we showed that the complexes with depleted charged contacts or dominating hydrophobic interactions were the most challenging ones to predict. We also demonstrated that if AlphaFold2 predictions were at hand, the interface prediction challenge could be alleviated for most of the targets. All in all, it is evident that new approaches are needed for the accurate prediction of assemblies, which undoubtedly will expand on the significant improvements in the tertiary structure prediction field.
Keywords: CASP, quaternary structure prediction, protein assembly, contact prediction, template-based modeling
INTRODUCTION
Proteins do not act alone; they carry out their functions as a part of protein communities. Various “omics” technologies have generated a wealth of information on these communities, waiting to be interpreted at the structural level. The recent developments in the Electron Microscopy (EM) field have helped accelerate the structure determination of protein complexes (Benjin & Ling, 2020; Ognjenovíc et al., 2019). In parallel, template-based modeling and diverse docking strategies have been developed and applied to produce reliable protein complex models (Lensink et al., 2019; Muratcioglu et al., 2015; Park et al., 2015; Porter et al., 2019; Vakser, 2020). CAPRI (Critical Assessment of PRedicted Interactions) has been gauging the accuracy of these computational approaches since 2002 (Janin et al., 2003; Wodak et al., 2020). In the meantime, CASP has seen substantial developments in the field of protein structure prediction (Kryshtafovych et al., 2019). Since 2016, CASP has been offering an independent assessment of protein assembly prediction, where the community has been focused on pushing the limits of the computational structural biology field forward in the area of protein assembly prediction (Guzenko et al., 2019; Lafita et al., 2018). As a reflection of this, in CASP14, we covered several challenging complexes, which were large, heterogeneous, and cannot be resolved with the classical assembly prediction methods. More and more groups have started to implement deep learning-based inter-residue distance/contact predictions into their assembly modeling to address the assembly-related challenges (Elofsson, 2021; Hopf et al., 2014; Ovchinnikov et al., 2014; Yan & Huang, 2021). Here we examine the state of the art in assembly prediction methods and identify the best performing CASP14 prediction groups.
METHODOLOGY
Classification of the CASP14 Assembly Targets
For all CASP14 assembly targets, except H1047, the symmetry and stoichiometry information were shared with the predictors (Table 1). We validated the provided symmetry information with AnAnaS (Analytical Analyzer of Symmetries) (Pagès et al., 2018; Pagès & Grudinin, 2018) and the biological interface definition with EPPIC (Bliven et al., 2018). For only one target, T1034o, we had a mismatch between the experimentally determined stoichiometry (tetramer) and the one predicted by EPPIC (dimer). The assembly targets were categorized according to the availability of templates for the individual subunits and the interface between them. The template search was performed with HHPred (https://toolkit.tuebingen.mpg.de/tools/hhpred, (Gabler et al., 2020; Söding et al., 2005)). As in the previous rounds, we classified a target as easy, if there is a structural template covering the whole assembly; medium, if there is a partial template for the subunits or their interaction; or difficult, if there is no template neither for the subunits nor the assembly. In CASP14, we had five targets (H1036, H1047, H1060, H1081, T1099o), which were challenging to assess with the standard CASP metrics. To assess these cases, we divided them into sub-targets and treated each sub-target as an independent evaluation unit. While identifying the sub-targets, we concentrated on isolating the minimum repeating unit(s) of the global assembly, as explained below (Figure S1):
Table 1.
The CASP14 assembly target list, involving each target’s stoichiometry, symmetry definition, assembly classification, prediction difficulty, taxonomy, the experimental technique used to resolve its structure, and the best predictor for that target ranked according to Eq. 3.
| Target ID | PDB ID | Stoichiometry | Symmetry | Assembly Classification | Difficulty | Taxonomy | Exp. Tech. | 1st ranking Group |
|---|---|---|---|---|---|---|---|---|
| H1036* | 6vn1(Oliver et al., 2020) | A3B3C3 | C3 | Heteromer | Medium | Virus | EM | BAKER-experimental |
| H1036v0* | 6vn1(Oliver et al., 2020) | A1B1C1 | - | Heteromer | Medium | Virus | EM | htjcadd |
| H1045* | A1B1 | - | Heteromer | Medium | Plant | X-Ray | Huang | |
| H1047 | A26B26 | - | Heteromer | Difficult | Bacteria | EM | E2E | |
| H1060v1* | 7CGO (Tan, et al.) | A2B1C1D1 | - | Heteromer | Medium | Virus | EM | BAKER-experimental |
| H1060v2* | 7CGO (Tan, et al.) | A3 | C3 | Homomer | Medium | Virus | EM | Venclovas |
| H1060v3* | 7CGO (Tan, et al.) | A3 | C3 | Homomer | Medium | Virus | EM | Venclovas |
| H1060v4* | 7CGO (Tan, et al.) | A12 | C12 | Homomer | Medium | Virus | EM | Takeda-Shitaka-Lab |
| H1060v5* | 7CGO (Tan, et al.) | A6 | C6 | Homomer | Medium | Virus | EM | BAKER-experimental |
| H1065 | A1B1 | - | Heteromer | Difficult | Bacteria | X-Ray | AILON | |
| H1072 | 6r17(Dunce et al., 2020) | A2B2 | C2 | Coiled Coil | Medium | Mammalian | X-Ray | Bates |
| H1081v0* | A5 | - | Homomer | Medium | Bacteria | EM | Zou | |
| H1097 | A1B1C1D1E1 | - | Intertwined | Medium | Bacteria | EM | BAKER-experimental | |
| T1032o* | 6n64(Chen et al., 2020) | A2 | C2 | Homomer | Easy | Mammalian | X-Ray | Zou |
| T1034o | 6tmm (Chiarini, et al., 2021) | A4 | D2 | Intertwined | Medium | Extremophile bacteria | X-Ray | Ornate-Select |
| T1038o | 6ya2(Bahat et al., 2020) | A2 | C2 | Homomer | Difficult | Virus | X-Ray | Bates |
| T1048o | 6un9 (Vorobiev, et al., 2020) | A4 | D2 | Coiled Coil | Medium | Bacteria | X-Ray | BAKER-experimental |
| T1052o* | A3 | C3 | Homomer | Easy | Virus | X-Ray | Vakser | |
| T1054o* | A2 | C2 | Homomer | Difficult | Bacteria | X-Ray | Venclovas | |
| T1061o | A3 | C3 | Homomer | Difficult | Virus | EM | BAKER-experimental | |
| T1070o* | A3 | C3 | Intertwined | Difficult | Virus | X-Ray | CAPRI-Shen | |
| T1078o* | A2 | C2 | Homomer | Medium | Fungi | X-Ray | Zou | |
| T1080o | A3 | C3 | Intertwined | Difficult | Bacteria | X-Ray | BAKER-experimental | |
| T1083o* | A2 | C2 | Coiled Coil | Medium | Bacteria | X-Ray | Venclovas | |
| T1084o | A2 | C2 | Coiled coil | Medium | Extremophile bacteria | X-Ray | BAKER-experimental | |
| T1087o* | A2 | C2 | Coiled Coil | Medium | Extremophile bacteria | X-Ray | BAKER-experimental | |
| T1099ov0* | 6ygh(Makbul et al., 2020) | A4 | - | Homomer | Medium | Virus | EM | Seok |
| T1099ov1* | 6ygh(Makbul et al., 2020) | A2 | C2 | Homomer | Medium | Virus | EM | Seok |
| T1099ov2* | 6ygh(Makbul et al., 2020) | A2 | - | Homomer | Medium | Virus | EM | Zou |
The CASP/CAPRI targets are indicated with *. The groups, which submitted a model having above 0.5 score for all the evaluation metrics are underscored.
H1036: is an antibody complex with a viral particle with A3B3C3 stoichiometry and C3 symmetry. The viral component had very good templates (pdb ids: 6esc(Vallbracht et al., 2017), 5ys6(Li et al., 2017)). On the other hand, there was no template for the virus antibody interface. As the viral protein has nearly five times more amino acids than the antibodies, its successful predictions were dominating our assessment. We, therefore, analyzed only the viral particle-antibody interface as the evaluation unit H1036v0 (Figure S1A).
H1047: is a bacterial assembly, formed by the 26 repeats of an A1B1 dimer. Here, we concentrated on the A1B1 dimer predictions only. As this complex has pseudosymmetry, we calculated the number of contacts between each A1B1, falling within 5Å. Then, we selected the A1B1 pair having the largest number of contacts as the reference. The same procedure was followed to isolate the relevant A1B1 evaluation unit from the models (Figure S1B).
H1060: is a bacteriophage T5 tail complex, composed of five stacked rings, involving four distinct monomers. The unique symmetric rings were offered as four separate targets (H1060v2–5). To assess the H1060 models, we manually isolated these four unique rings in PyMOL (The PyMOL Molecular Graphics System). The asymmetric repeating unit was defined as the vertical cross-section of the assembly (H106v1). This evaluation unit was defined upon selecting the mostly contacting vertical subunits (within 5 Å). The same protocol was applied to the submitted models (Figure S1C).
H1081: is a bacterial arginine decarboxylase made of 20 repeating units assembled in a D5 symmetry. This complex is made by two stacked decameric rings (2 x A10), where there is a reliable template for each (pdb id: 2vyc (Andréll et al., 2009)). For this target, we defined the evaluation unit as the pentamer as presented in H1081v0 (Figure S1D). H1081v0 contains all the unique interfaces (intra- and inter-decamer), which is repeated four times within the complex.
T1099o: is a 240-subunit viral capsid of the duck hepatitis B virus, having a T=4 icosahedral symmetry. This target was divided into three evaluation units, defining the core C3 and C5 symmetric interactions (T1099v0–2). As it was not possible to assign any rule to isolate the relevant dimers and tetramers from the models, we performed the annotation of the evaluation units manually, upon inspecting each model in PyMOL (The PyMOL Molecular Graphics System) (Figure S1E).
The dissection of H1036, H1060, H1081, and T1099o was carried out together with the CAPRI organizers.
To analyze the interface characteristics of the CASP14 assembly targets, we calculated the percentage of the polar, hydrophobic and charged residues residing within 7Å contact range. To calculate the interface RMSD (i-RMSD) between the targets and the AlphaFold2 monomer models (Jumper et al., 2021), we focused on the residues falling within 5Å contact range. The i-RMSDs were calculated over the interfacial backbone atoms (C, CA, N, O) with McLachlan algorithm (McLachlan, 1982) as implemented in the program ProFit (http://www.bioinf.org.uk/software/profit/ (Martin, 2021)).
CASP14 Assembly Scoring
The CASP14 assembly predictors were ranked according to the cumulative Z-score distributions of oligomeric lDDT (local Distance Difference Test, (Mariani et al., 2013)), TM (Template Modelling, (Xu & Zhang, 2010; Zhang & Skolnick, 2004)), ICS (Interface Contact, (Lafita et al., 2018)), and IPS (Interface Patch, (Lafita et al., 2018)) scores. lDDT and TM measure the global similarity, whereas ICS and IPS define the interface accuracy. The definitions of these metrics are:
lDDT-oligo measures the difference in the interatomic distances within a model and the reference structure. For convenience, throughout the paper, we will refer to this metric as lDDT.
TM assesses the topological similarity of the model with respect to the reference.
-
ICS is calculated as an F1-score, measuring the relationship between the precision (P) and recall (R) of predicted inter-chain contacts:
(Eq.1) In Eq. 1, M and T represent the model and target. P is the fraction of the correct inter-chain contacts among all model inter-chain contacts. R is the fraction of correctly reproduced native inter-chain contacts. Two residues are considered in contact when at least two of their non-hydrogen atoms (one from each residue) are within 5Å from each other.
IPS is calculated as a Jaccard coefficient (Jc) over the interface amino acids (I) predicted by the model (IM) compared to the target (IT):
| (Eq.2) |
To obtain the final ranking, we followed the below-given protocol:
Given a target, the Z-score of each metric was calculated either over the first or the best model submitted.
Then, the cases with Z-score < -2 were removed from the model list on a per-case basis.
Afterwards, a new Z-score set was calculated on the reduced case-based set.
Finally, the models having a Z-score > 0 were kept.
The final cumulative Z-scores were calculated according to Equation 3.
| (Eq.3) |
RESULTS and DISCUSSION
Twenty-two different protein complexes were offered for the CASP14 assembly prediction category. Among these, five were dissected into sub-targets, so that they can be assessed by using the standard CASP metrics (Methods, Figure S2A). In the end, the assembly predictions were assessed over 29 evaluation units, half of which came from the structures determined by EM techniques (Table 1). The CASP14 assemblies mostly came from bacterial (38%) and viral (48%) systems (Figure S2B). 38% of these targets were dimers and the remaining had three or more chains. These were mostly homomeric protein assemblies arranged in a symmetric fashion. For all the targets, except for H1047, the assembly stoichiometry was shared with the predictors. After the evaluation unit definitions, the stoichiometrically largest complex contained 12 monomers. In this round, we had 39 prediction programs (30 research groups) participating. A parallel joint CASP14-CAPRI round was organized on a subset of 12 targets (19 evaluation units), and additional eight groups took on CAPRI-only targets (Table S1–S2). The assessment of these targets was carried out in close collaboration with the CAPRI organizers [ref: CASP14-CAPRI50 paper]. On average, the assembly prediction community calculated 110 models per target, where the number of models submitted inversely correlated with the target difficulty (Figure S2C). Five groups (BAKER-experimental, Venclovas, Takeda-Shitaka-Lab, Bates_BMM, Risoluto) submitted a qualified prediction for all targets, 18 groups more than 20 targets, and 25 groups more than ten targets (Table S1–2).
Overall Evaluation
For scoring the CASP14 assembly models, we used a function made by the equal contributions of the Z-scores of ICS, IPS, lDDT, and TM metrics (Methods, Eq. 3). According to this function, BAKER-Experimental is the top-performing group, followed by Venclovas, Takeda-Shitaka, and Seok groups (Figure 1). The ranking remains the same irrespectively of whether we considered the best or the first submitted model. This ranking was also preserved when CASP/CAPRI targets were taken into account. According to CAPRI criteria (Lensink et al., 2007; Méndez et al., 2005), BAKER-experimental, Venclovas, and Seok performed equally well as the top predictors [ref: CASP14-CAPRI50 paper]. Our ranking difference stems from different approaches used by both communities; CASP ranking uses a combined Z-score-based function, while CAPRI measures the accuracy with a star-based system. Following the criteria of the previous rounds, the assembly targets were assessed separately in three difficulty categories: easy, medium, and difficult (see Methods). CASP14 target set contained two easy, 20 medium, and seven difficult cases (Figure S2A, Table 1). The BAKER-experimental method performed distinctively well in the medium category.
Figure 1.
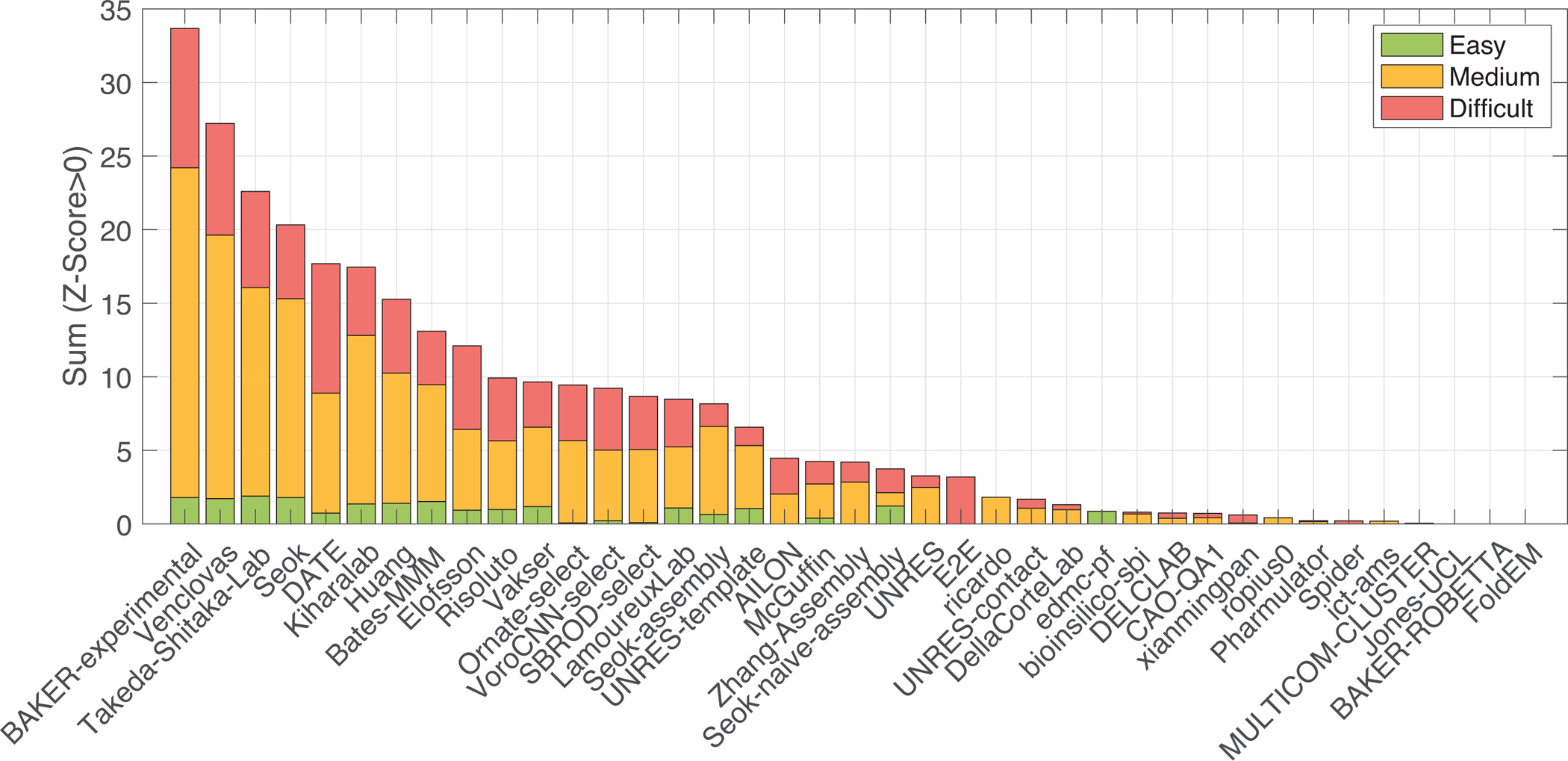
The ranking of the CASP14 Assembly Groups according to the Z-score-based ranking (Eq. 3).
CASP14 Assembly Scoring
Like in CASP13, we considered a model to be successful if it scored above 0.5 according to all scoring measures (Guzenko et al., 2019). The community managed to submit at least one successful model for nine out of 29 targets, constituting the ~31% success rate. Among these, one is easy (T1052), one is difficult (H1065), and the rest (T1084o, H1045, H1036, T1083o, T1048o, H1081v0, H1060v5) are medium targets (Figure S3). The top five successfully predicted targets were T1084o (symmetric homodimer), H1045 (heterodimer), H1081v0 (heteropentamer), H1036 (heterononamer), and H1060v5 (symmetric hexamer) (Figure 2). To understand the impact of the scoring terms used, we plotted the distributions of ICS, IPS, lDDT, TM for each target, calculated over the submitted models (Figure 3). This analysis revealed that the least strict scoring metric is TM, while the strictest one is ICS. According to TM, 24 targets had a score greater than 0.5 (~80% success rate), whereas this number dropped to 10 for ICS (~34% success rate). So, given the stoichiometry, the community performs very well in predicting the assembly topology. On the other hand, the prediction of interacting residue pairs remains to be a challenge. The pairwise correlations of ICS, IPS, lDDT, TM scores for the best models illustrated that the shape-related terms (lDDT, TM) are almost always of higher values compared to the interface-related terms (ICS, IPS) (Figure 4). These metrics generally follow ICS < IPS < lDDT < TM order, which breaks for T1032o. The best model of T1032o has a higher ICS than lDDT. For this case, the experimentally determined structure covers 60% of the sequence provided to the predictors, which could be the reason for the unusual lDDT behavior (Figure S4A).
Figure 2.
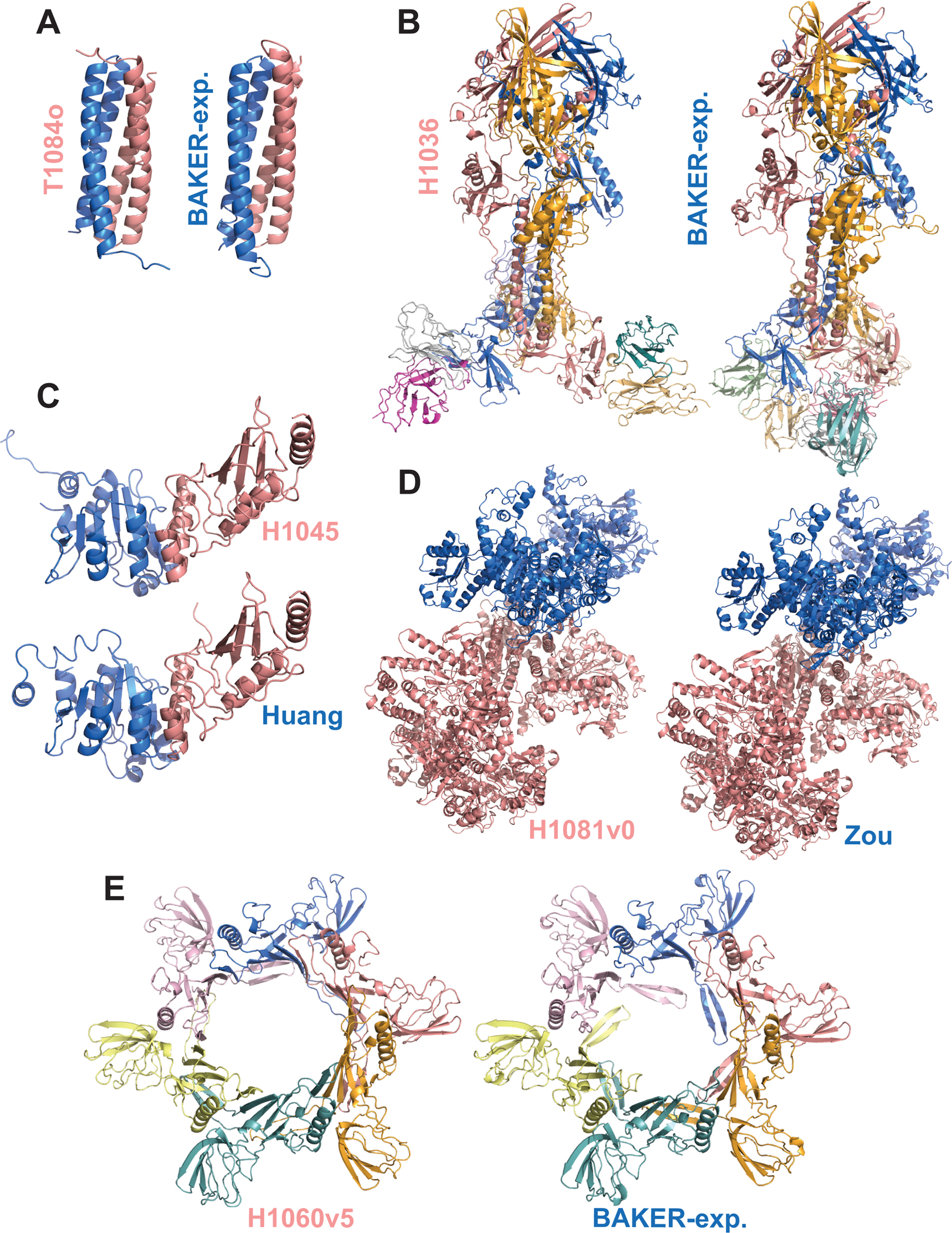
The top five successfully predicted targets and their best predicted models. (A) T1084o, (B) H1036, (C) H1045, (D) H1081v0, (E) H1060v5, where each monomer is represented with a different color.
Figure 3.
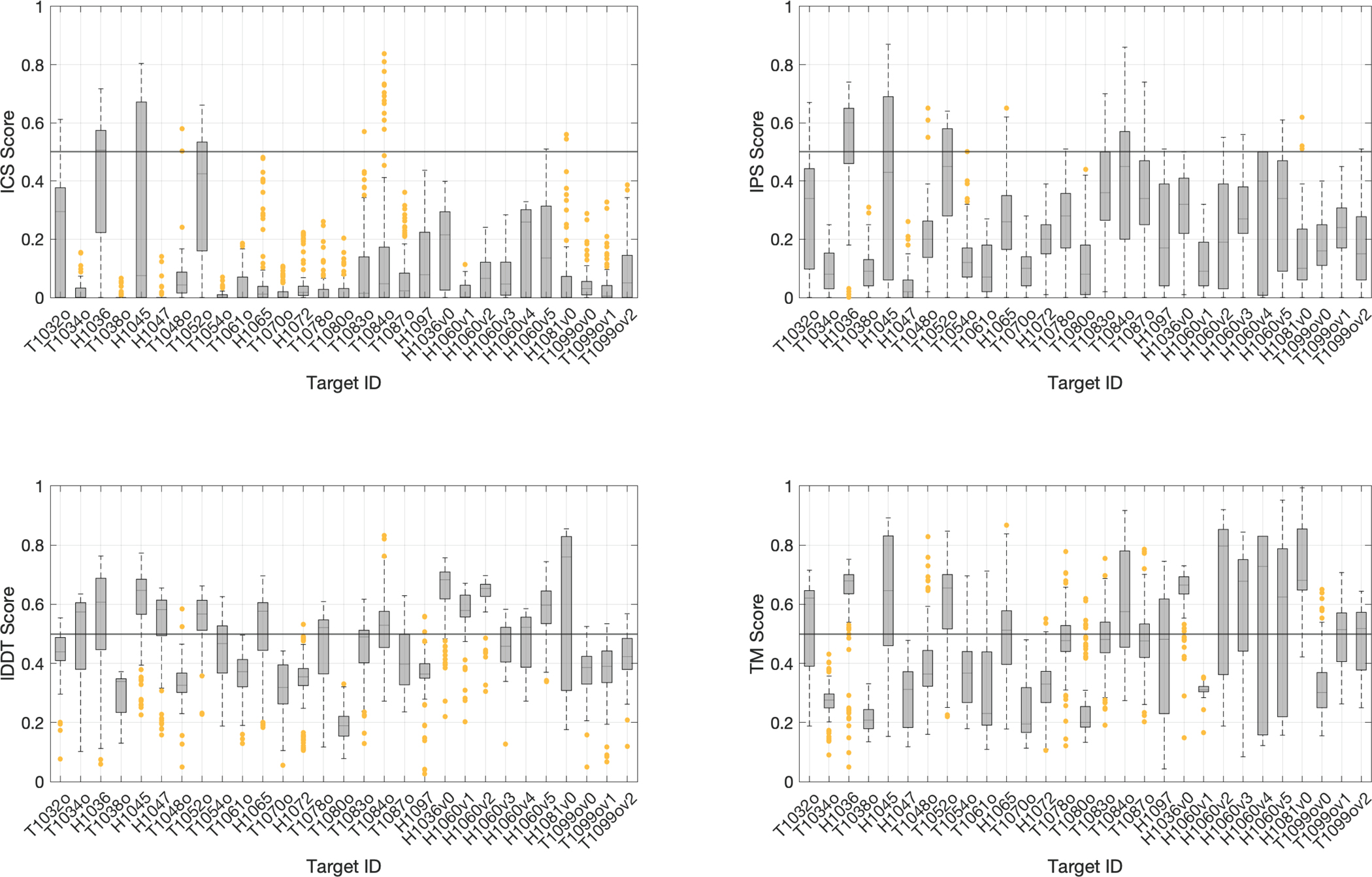
The per-target distribution of each scoring term (ICS, IPS, lDDT, TM). The successful model criterion 0.5 is marked with a solid gray line.
Figure 4.
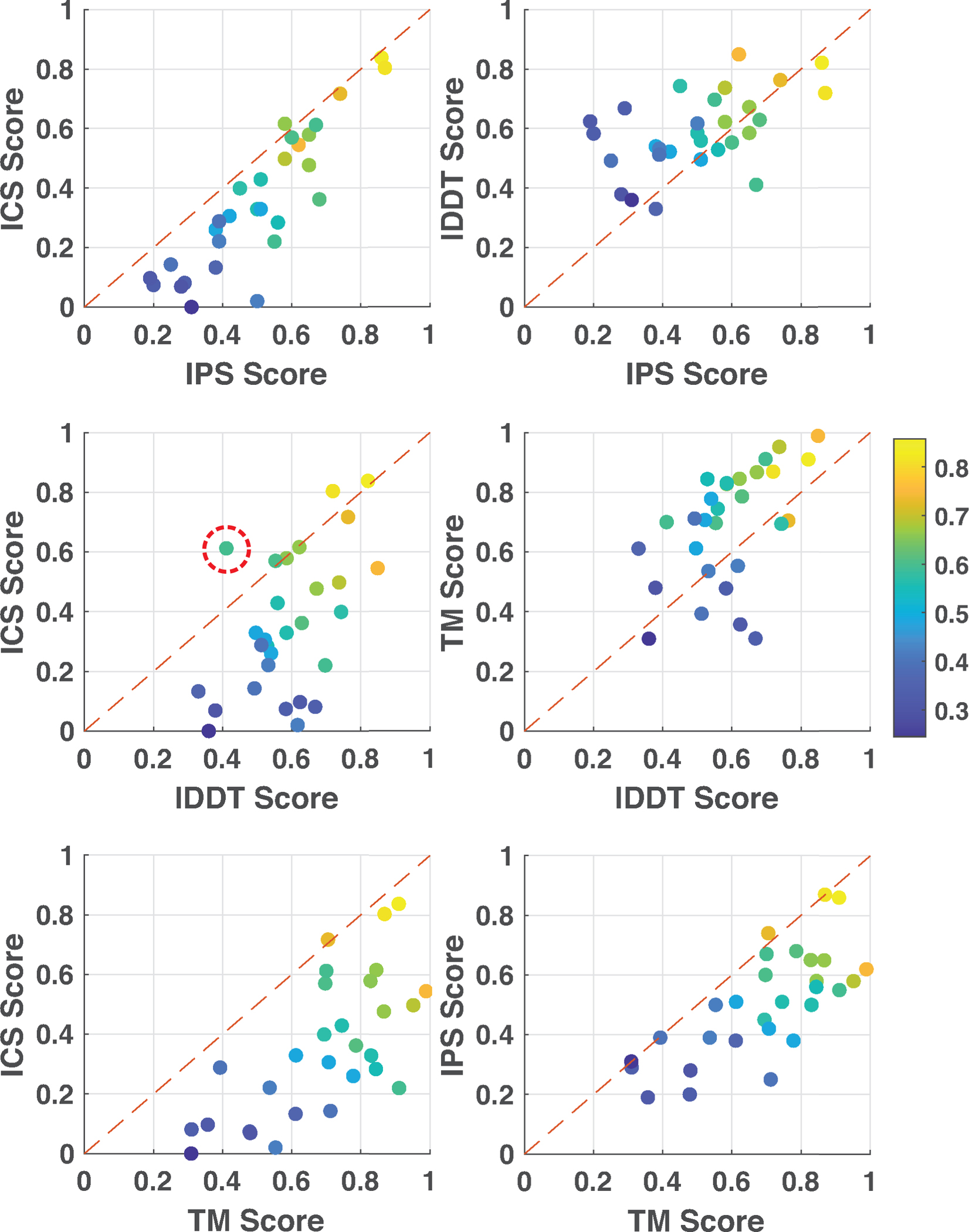
The pairwise relationship of each scoring term belonging to the best model (of each target). The scattered data points are colored according to their mean scores in all plots. The mean score is defined by the average of the four assessment metrics as given in mean-score=(ICS+IPS+TM+lDDT)/4). For only one target, T1032o, shape-related metric lDDT has a worse score than the interface-related ICS (encircled in red in middle-left figure).
CASP14 in Comparison with the Previous Assembly Rounds
In CASP14, there were fewer assembly targets than in previous rounds of CASP (22 in CASP14, 42 in CASP13, and 30 in CASP12). Even so, the technological advancements in the EM field were reflected in this assembly round. In the previous editions, up to 15% of the assembly targets were resolved by EM, whereas this number increased to 32% in CASP14 (calculated over the original 22 target set, Table 1). Accordingly, the complexity of assembly stoichiometries increased as well. In CASP13, the largest two assemblies were composed of 18 and 10 subunits, whereas in CASP14, four targets had more than 15 monomers, i.e., H1060, H1081, T1099o, H1047 (Figure S5). The increase in subunit size not only impacted the prediction difficulty but also posed a significant challenge on the assessment due to the combinatorial chain mapping problem (which we overcame by dissecting these big complexes into their sub-complexes). To provide a quantitative comparison across all CASP assembly rounds, we gathered the best five models submitted for each case and plotted their mean score distribution (Figure 5). This depiction showed that CASP13 has more models in the high score bins (>0.8). On the other hand, when we calculated the percentage of the models having a mean score > 0.5, we observed that with 44.2%, CASP14 has the highest success rate, followed by 32.3% in CASP13 and 30.6% in CASP12. So, even though the target difficulty increased in CASP14, the community posted a ~15% improvement in the assembly prediction.
Figure 5.
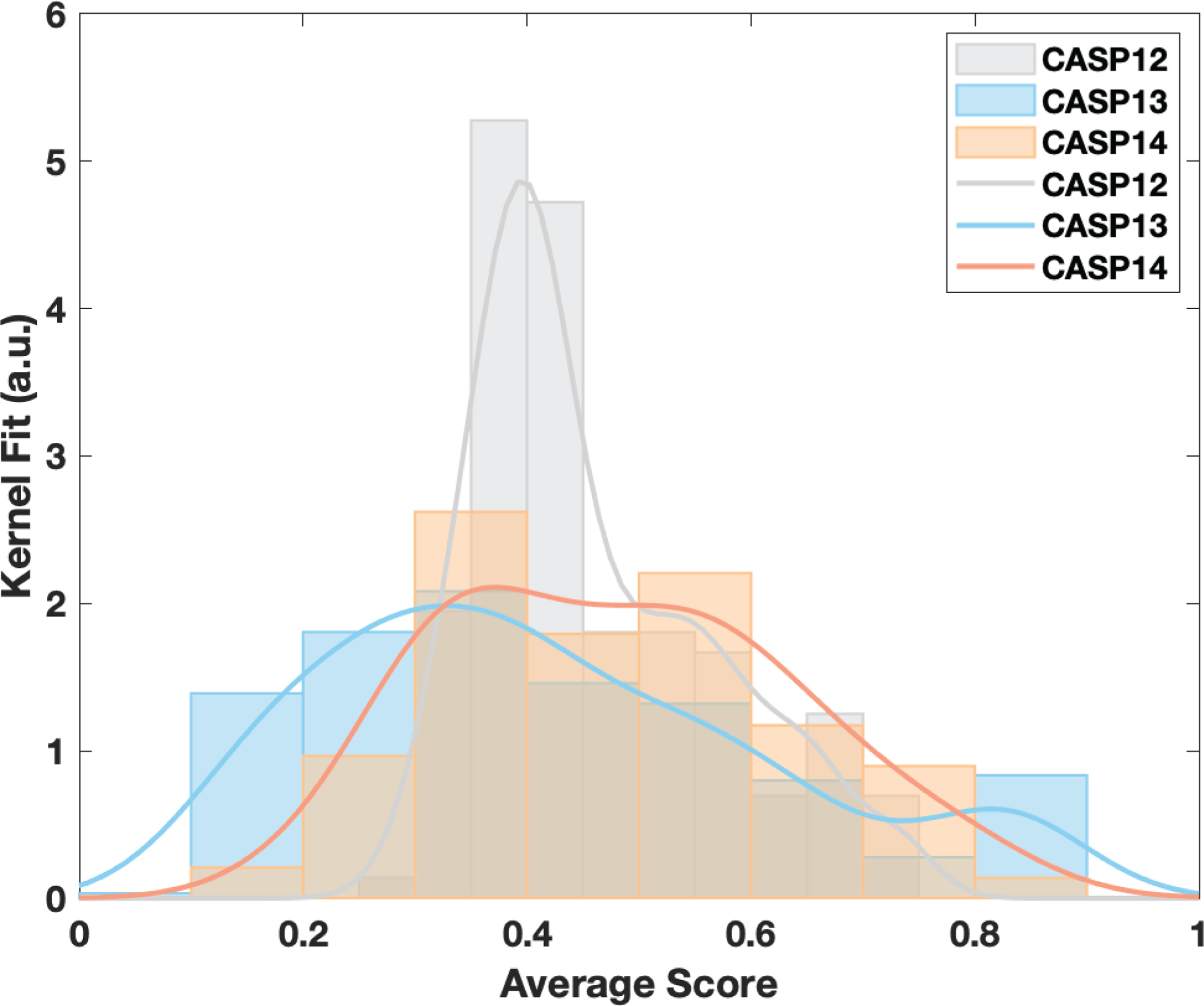
The mean-score distributions of CASP12, CASP13 and CASP14 assembly models, when the top five models of each target were considered.
A Close Look into the Target Characteristics
We investigated the CASP14 assembly targets under four assembly-based categories: homomers (48%), heteromers (21%), coiled-coils (17%), and assemblies having intertwined monomers (14%) (Table 1). For the assessment of homomeric targets, Seok-naïve-assembly predictions were used as a baseline indicator (Lensink et al., 2018). These predictions came from a classical template-based approach (established by the consecutive use of HHPred (Gabler et al., 2020; Söding et al., 2005) and Modeller (Webb & Sali, 2016)). The Seok-naïve-assembly predictions have been used as a reference to assess the homomeric predictions since the earlier CASP rounds (Guzenko et al., 2019). In CASP14, all the top-performing groups produced better quality models than the Seok-naïve-assembly (Figure S6).
Among the homomeric predictions, T1061o was particularly interesting. Being the only difficult homomeric target, T1061o forms a bacteriophage tail together with H1060. The best monomeric structure prediction for this target was modeled by AlphaFold2, with 61.78 GDT-TS. In this case, AlphaFold2 could not predict the fold of the final 220 residues. Even so, by using template-based modeling, the Baker group could obtain a T1061o model with an impressive TM score (0.70) but with a very low ICS (0.16) (Figure 6A). Another interesting homomer is 1099v0, for which we asked the predictors to submit a minimal sub-complex, which would encompass the integral symmetry to construct the T=4 icosahedral viral capsid (Figure S1E). Here, we expected to receive at least a tetramer. Even though there was a template for 1099v0 with 19.5% sequence identity within 2 Å RMSD (pdb id:3j2v (Yu et al., 2013)), the best average score generated for this target remained low (0.29 ICS and 0.39 TM, as submitted by Seok). Our analysis showed that CAPRI-Shen utilized the available template the best in terms of the shape prediction with a TM-score of 0.65 (Figure 6B).
Figure 6.
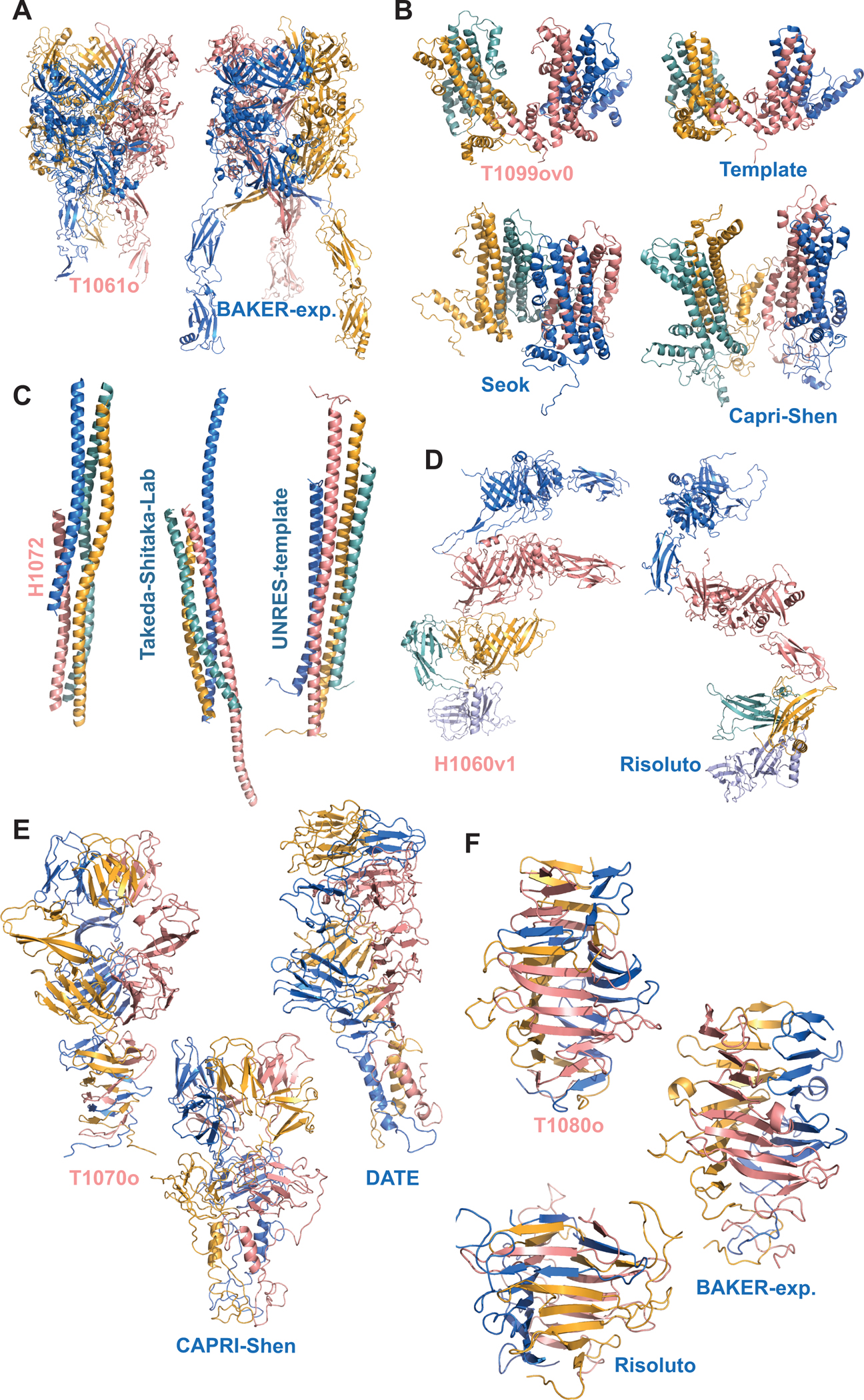
The target and prediction highlights of the CASP14 assembly round. (A) T1061o, (B) T1099o (also with the template (pdb id:3j2v(Yu et al., 2013))), (C) H1072, (D) H1060v1, (E) T1070o, (F) T1080o, where each monomer is represented with a different color.
Among the coiled-coil complexes, the most challenging target was the heteromeric H1072 (Figure 6C). As there was no template for its interface, the best interface scores produced for H1072 were 0.22 ICS and 0.32 IPS (Takeda-Shitaka-Lab). For this one, the best TM got as high as 0.55 (UNRES-template). Under the category of heteromers, as indicated above, H1060 forms the bacteriophage tail subcomplex with T1061. This complex comprises five stacked rings made by four different monomers, where their rings were assessed as different evaluation units. Among these, the most interesting evaluation unit was the heteromeric H1060v1, the asymmetric vertical slice of H1060. None of the groups could predict H1060v1 successfully. The best score to note in this case was generated with 0.35 TM by Risoluto (Figure 6D). Another noteworthy prediction came for H1047, which is a bacterial flagellar complex. H1047 is the only target for which we did not provide the stoichiometry information. Among all the groups, only Seok and KiharaLab could predict the H1047 complex stoichiometry correctly. Although the interface predictions for these targets were off, E2E and Baker could produce models with 0.48 and 0.45 TM scores, respectively (Figure S4B).
From the intertwined cases, T1070o and T1080o involved a high degree of domain swap. As a result, none of the groups could predict these targets successfully. The monomeric AlphaFold2 predictions for these targets had 43.12 GDT-TS for T1070o and 82.3 GDT-TS for T1080o. The challenge in the monomer modeling was reflected in the assembly category with very low ICS and IPS scores. Even so, for T1070o, CAPRI-Shen and DATE could produce models with 0.48 and 0.46 TM scores, respectively (Figure 6E). In the case of T1080o, Risoluto and Baker generated good TM scores at the extent of 0.60 (Figure 6F).
How do the interface characteristics impact the prediction difficulty?
We calculated the biophysical characteristics of the interface for each target, i.e., the percentage of hydrophobic, charged, and polar interface residues (Methods). Here, we examined the relationship between these interface properties and the best ICS and IPS scores produced for the targets. As a result, we observed that if the interface is deprived of charged contacts or dominated by hydrophobic interactions, its prediction becomes more challenging (Figure 7A). Five targets with the least charged interfaces are H1047, T1078o, T1080o, T1038o, H1036v0 (Figure 7B). Their best ICS scores are 0.07, 0.26, 0.13, 0.00, and 0.40, and their best IPS scores are 0.20, 0.38, 0.38, 0.31, and 0.45, respectively. It is also interesting to see that three targets among the five least-charged ones fall into the difficult category. Alongside, five targets with the most hydrophobic interfaces are T1080o, H1072, T1070o, T1034o, and T1078o (Figure 7B). Their best ICS scores are 0.13, 0.22, 0.07, 0.10, and 0.26, and their best IPS scores are 0.38, 0.39, 0.28, 0.19 and 0.38, respectively (Figure 7A, middle). Here, three of the five most hydrophobic interfaces came from the intertwined targets (T1080o, T1070o, T1034o). When we ranked the targets according to their best ICS scores in descending order, seven of them had either a charged-depleted interaction or a hydrophobically-dominating one. We should note that the correlation between ICS/IPS and the charge/hydrophobicity characteristics percentages of the interface is not high (R~0.5), but still significant (p-value<0.05, Figure 7A). Such a correlation was not observed for the ICS/IPS vs. polarity relationship.
Figure 7.
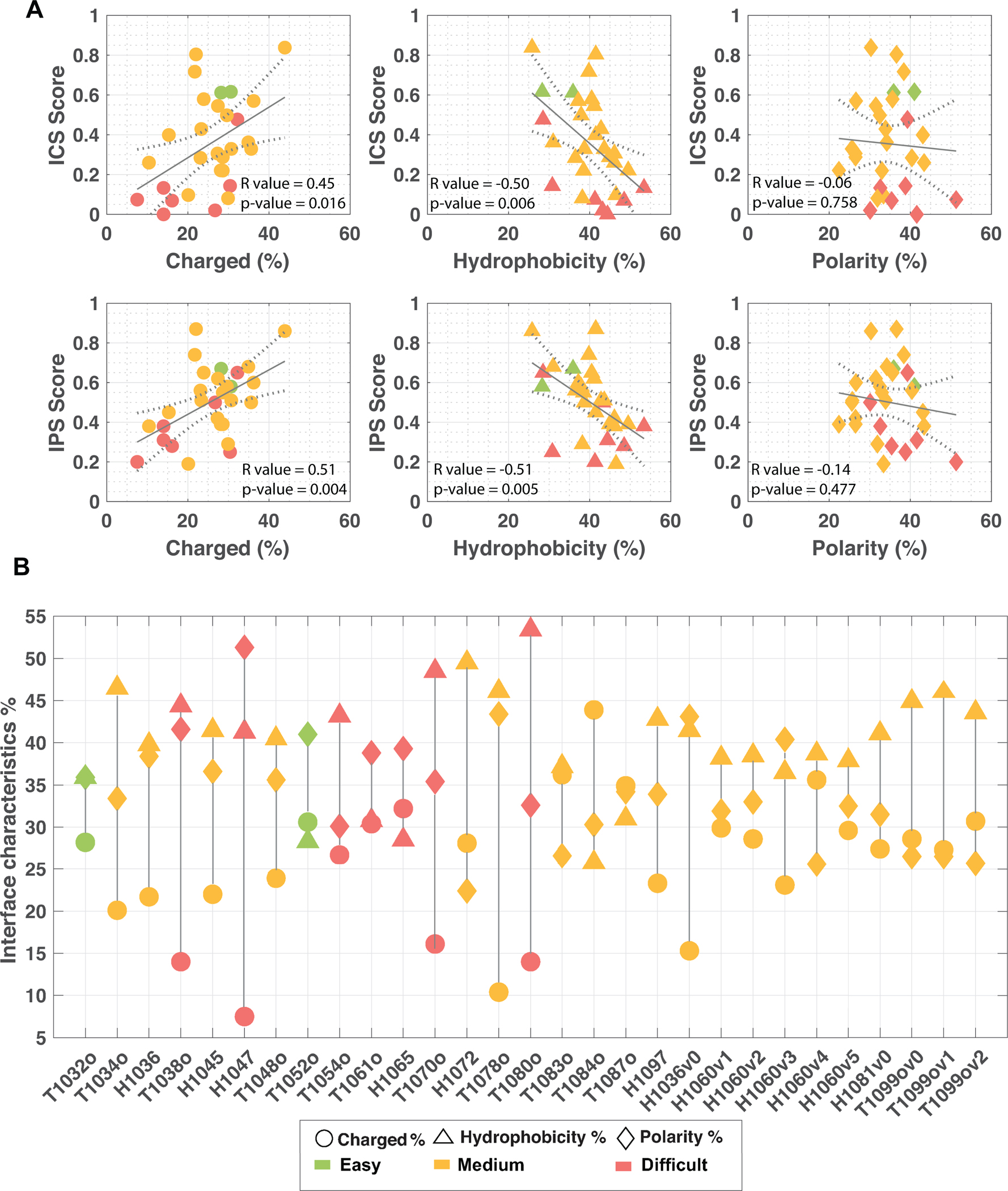
(A) The relationship between the interface characteristics and the interface-related scores (ICS and IPS): the charged interface percent vs. ICS/IPS score (left), the hydrophobic interface percent vs. ICS/IPS score (middle), and the polar interface percent vs ICS/IPS score (right). Each data point was colored according to its target difficulty (easy: green, medium: orange and difficult: red). The fitting was performed with the least square fitting option of Matlab (MATLAB and Statistics Toolbox Release 2020b). (B) The biophysical interface characteristics of each target. The charged interface percent was marked with circles, hydrophobicity with triangles and polarity with diamonds. Each data point was colored according to its target difficulty as in panel A of this figure.
The impact of using contact predictions on the prediction performance
The distribution of the assembly scoring metrics, as presented in Figure 3, showed that there is no group performing distinctively better than the rest of the community. When we counted the occurrence of the top-raking groups over the best three models, BAKER-experimental came up 14, Venclovas 16, Takeda-Shitaka-Lab seven, and Seok four times. This observation indicated that Venclovas produced consistently good models for multiple targets, whereas BAKER-experimental performed significantly better for specific targets. For the assembly prediction, Venclovas, Takeda-Shitaka-Lab, and Seok followed a classical approach: they used template-based modeling or protein-protein docking, where relevant. The leading group BAKER-experimental, besides these approaches, utilized contact predictions, together with a fold-and-dock approach for ten targets (H1047, H1060v5, H1065, H1072, H1097, T1048o, T1080o, T1083o, T1084o, T1087o [Baker’s Team CASP paper]. Here, we investigated the contribution of BAKER-experimental-specific approaches to Baker Group’s success. For this, we studied the relationship between TM and ICS of the best models submitted by BAKER-experimental and Venclovas. As presented in Figure 8, the BAKER-experimental models had a slightly better TM and ICS correlation than the Venclovas models (0.78 vs. 0.64). So, BAKER-experimental performed slightly better in predicting the correct interfacial contacts. BAKER-experimental submitted a top-ranking model for nine targets (H1036, H1060v1, H1060v5, H1097, 1048o, T1061o, T1080o, T1084o, T1087o). Here, only T1048o and T1084o submissions were successful. These targets were modeled with a contact incorporated fold-and-dock approach. For T1048o, BAKER-experimental produced a top model with an ICS of 0.58, where the second-ranking group, Risoluto, had an ICS of 0.17. For T1084o, BAKER-experimental produced a 0.83 ICS, followed by Kiharalab with 0.69 ICS. Both of these targets were homomeric coiled coil assemblies, indicating that BAKER-experimental’s fold-and-dock worked distinctively well on them. Finally, as introduced above, even though BAKER-experimental could not produce a successful model for the intertwined T1080o, together with Risoluto, they submitted models with good TM scores (Figure 6F).
Figure 8.
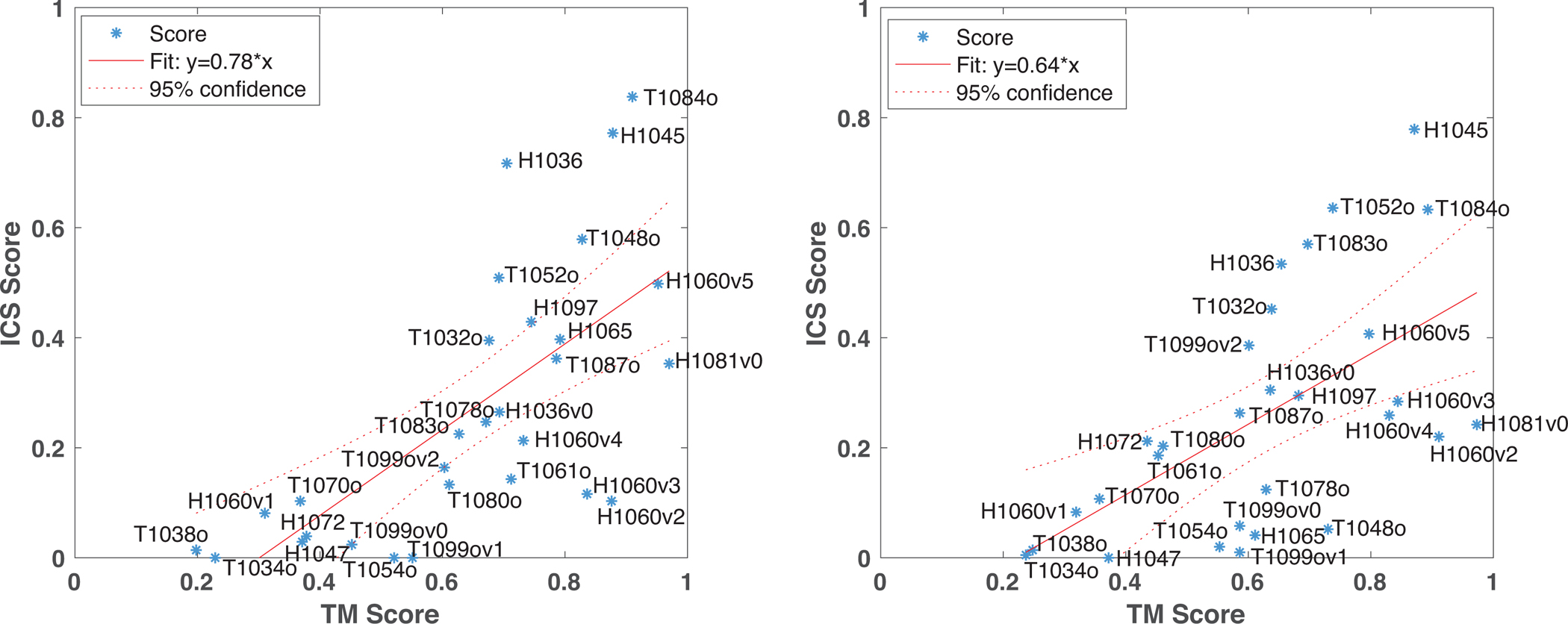
The ICS vs. TM score correlations for the best models submitted by BAKER-experimental (left) and Venclovas groups (right). Each data point was labeled with its corresponding target id. The fitting was performed with the least square fitting option of Matlab (MATLAB and Statistics Toolbox Release 2020b).
According to CASP abstracts, except BAKER-experimental, ten groups used a contact-based approach for at least one target: Huang, Elofsson, Risoluto, LamoureuxLab, Ailon, Zhang-assembly, edmc-pf, UNRES-contact, DellaCorteLab, bioinsilico-sbi (Figure S7). Two of these methods performed very well for H1047 (medium) and H1065 (difficult). In the case of H1047, even though the interface prediction was off, H1047 topology could still be modeled by E2E with a TM score of 0.5. E2E’s approach is based on TrRosetta (Yang et al., 2020) (Figure S4B). For H1065, Elofsson generated a 0.5 ICS score. For this target, Elofsson used trRosetta, as well as template-based docking. Strikingly, their best H1065 model emanated from using the CASP server models as an input to TMdock (Anishchenko et al., 2017; Pozzati et al., 2021), personal communication). So, although the contact-based sophisticated algorithms make a difference for specific targets, the classical assembly methods still prevail for most of the cases.
How much could the use of AlphaFold2 tertiary structure predictions improve the assembly predictions?
In CASP14, the AlphaFold2 team provided impressive predictions for the monomeric protein targets. During CASP14, AlphaFold2 did not participate in the assembly round and their predictions were not made available to the community. Therefore, we analyzed how much the use of AlphaFold2 tertiary structure predictions could have improved the assembly predictions. For this, we measured the r.m.s. difference between the interfaces of the assembly monomers and the AlphaFold2 models (i-RMSD, Methods). This i-RMSD calculation could be performed over 16 complexes, for which there was a readily AlphaFold2 tertiary structure prediction available (either on the whole complex or on one of the subunits) (Table 2). As a result, except for T1061o and T1070o (both difficult targets), all the i-RMSDs values were turned out to be smaller than 2.5–3.0 Å (a threshold to categorize an interfacial change as small-to-medium (Karaca & Bonvin, 2013)). Though, only five of these rather small i-RMSD cases could be successfully modeled in the assembly round (H1045, H1065, T1052o, T1083o, and T1084o). These small(er) i-RMSD values indicate that these targets are mainly obligate and for them, if AlphaFold2 predictions were at hand, the prediction of the conformational changes upon binding would not pose a barrier during the assembly prediction. This assumption is only challenged by a small i-RMSD case, T1054o (its monomers are of 1.5 Å i-RMSD), where the presence of AlphaFold2 model would not ease the prediction difficulty. In this case, the 1.5 Å change emanates from the conformational difference of the first twenty amino acids, which directly blocks the dimerization interface (Figure S4C). All in all, if AlphaFold2 predictions were available, the interface prediction challenge could be alleviated for most of the “unsuccessful” targets, even for the challenging intertwined T1080o (as discussed above).
Table 2.
The interface RMSD (i-RMSD) values measured between the assembly monomer and its related best CASP14 tertiary prediction. Target name and i-RMSD of subunits are tabulated, respectively. The cases experiencing a small or medium scale conformational changes are represented in bold. The cases, for which there was at least one successfully predicted assembly model, are underlined.
| Target Name | i-RMSD of subunit1 (Å) | i-RMSD of subunit2 (Å) | i-RMSD of subunit3 (Å) | i-RMSD of subunit4 (Å) |
|---|---|---|---|---|
| H1045 | 0.7 | 0.5 | ||
| H1047 | 1.4 | 0.4 | ||
| H1060 | 2.0 (v1) | 2.8 (v3) | 2.6 (v1) | 0.9 (v4) |
| H1065 | 0.7 | 0.9 | ||
| T1034o | 1.9 | |||
| T1038o | 1.9 | |||
| T1052o | 1.4 | |||
| T1054o | 1.5 | |||
| T1061o | 20.2 | 8.0 | ||
| T1070o | 10.6 | 6.0 | ||
| T1078o | 1.0 | 0.9 | ||
| T1080o | 1.4 | |||
| T1083o | 1.5 | |||
| T1084o | 1.3 | |||
| T1087o | 0.4 | |||
| T1099o | 1.2 | 1.6 | 1.5 | 2.1 |
CONCLUSIONS and PERSPECTIVES
The accurate prediction of protein-protein interactions is of vital importance to understand protein function. To this end, many algorithms have been deployed to accurately model protein assemblies. To contribute to the development of the assembly prediction field, here, we portrayed the state of the CASP14 assembly prediction round. Given the stoichiometry of the complex, for 80% of the cases, the community could predict the topology of the assembly. Though, the accurate prediction of inter-chain contacts remained an issue (for 70% of the cases). Strikingly, this was even the case for contact-based approaches, which helped to predict a better interface only for a few cases.
When compared to CASP13, we observed a 15% improvement in the quality of the models submitted. However, by analyzing the available CASP14 tertiary structure models, we showed that most of the CASP14 assembly targets were obligate. So, the assessed methods are yet to be probed with more “flexible” cases. Upon examining the CASP14 target interfaces, we presented that the interfaces deprived of charge or extremely hydrophobic were the most difficult ones to predict. By using this observation as a clue, we speculate that it might be possible to predict the limits of the available methods, for example, by analyzing the solvent-accessible residues of the tertiary structure predictions.
Thanks to the technological advancements in the EM field, in this round, we could offer intricate protein complexes as targets. To assess such difficult cases with the standard CASP metrics, we had to dissect them into sub-targets. For larger and more intricate complexes, new methodologies, expanding on the available ones should be developed (Lafita, 2017). A way to incorporate the resolution of EM map into the assessment strategy should also be considered within this context.
All in all, while assessing the assembly prediction community, we outlay that new approaches are needed for the accurate prediction of assembly interfaces, especially for utilizing MSA-based distance/contact predictions. Towards addressing this need, we are sure that the significant improvements in tertiary structure prediction field will pave the way for the community.
Supplementary Material
Acknowledgements
We wholeheartedly thank CASP and CAPRI organizing committees for assisting us in all the steps during the assembly assessment round. We specially thank Shoshana Wodak and Marck F. Lensink for the fruitful discussions on the common CASP/CAPRI targets, and Baker, Venclovas, Seok, and Elofsson groups for case-based discussions. We thank Ayşe Berçin Barlas and Büşra Savaş for helping us in dissecting the evaluation units of H1081, H1060 and H1047 models. Finally, we would express our gratitude to all the experimentalists, offering their complexes to the CASP14 assembly round.
Funding information
EK and BO are supported by the EMBO Installation Grant, grant number 4421. AK is supported by the US National Institute of General Medical Sciences (NIGMS/NIH), grant number GM100482.
Footnotes
Conflict of interest disclosure
The authors declare no conflicts of interest.
Data availability statement
The raw data, which is analyzed in this paper can be accessed at https://predictioncenter.org/casp14/multimer_results.cgi
REFERENCES
- Andréll J, Hicks MG, Palmer T, Carpenter EP, Iwata S, & Maher MJ (2009). Crystal structure of the acid-induced arginine decarboxylase from Escherichia coli: reversible decamer assembly controls enzyme activity. Biochemistry, 48(18), 3915–3927. [DOI] [PubMed] [Google Scholar]
- Anishchenko I, Kundrotas PJ, & Vakser IA (2017). Modeling complexes of modeled proteins. Proteins: Structure, Function and Bioinformatics, 85(3), 470–478. 10.1002/prot.25183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahat Y, Alter J, & Dessau M (2020). Crystal structure of tomato spotted wilt virus G N reveals a dimer complex formation and evolutionary link to animal-infecting viruses. 10.1073/pnas.2004657117/-/DCSupplemental [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjin X, & Ling L (2020). Developments, applications, and prospects of cryo-electron microscopy. In Protein Science (Vol. 29, Issue 4, pp. 872–882). Blackwell Publishing Ltd. 10.1002/pro.3805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliven S, Lafita A, Parker A, Capitani G, & Duarte JM (2018). Automated evaluation of quaternary structures from protein crystals. PLoS Computational Biology, 14(4). 10.1371/journal.pcbi.1006104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Birkinshaw RW, Gurzau AD, Wanigasuriya I, Wang R, Iminitoff M, Sandow JJ, Young SN, Hennessy PJ, Willson TA, Heckmann DA, Webb AI, Blewitt ME, Czabotar PE, & Murphy JM (2020). Crystal structure of the hinge domain of Smchd1 reveals its dimerization mode and nucleic acid-binding residues. In Sci. Signal (Vol. 13). http://stke.sciencemag.org/ [DOI] [PubMed] [Google Scholar]
- Chiarini V, Fiorillo A, Camerini S, Crescenzi M, Nakamura S, Battista T, … Ilari A (2021). Structural basis of ubiquitination mediated by protein splicing in early Eukarya. Biochimica et Biophysica Acta - General Subjects. [DOI] [PubMed] [Google Scholar]
- Dunce JM, Salmon LJ, & Davies OR (2020). Structural basis of meiotic chromosome synaptic elongation through hierarchical fibrous assembly of SYCE2-TEX12. In bioRxiv. bioRxiv. 10.1101/2020.12.30.424799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elofsson A (2021). Toward Characterising the Cellular 3D-Proteome. Frontiers in Bioinformatics, 1. 10.3389/fbinf.2021.598878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabler F, Nam SZ, Till S, Mirdita M, Steinegger M, Söding J, Lupas AN, & Alva V (2020). Protein Sequence Analysis Using the MPI Bioinformatics Toolkit. Current Protocols in Bioinformatics, 72(1). 10.1002/cpbi.108 [DOI] [PubMed] [Google Scholar]
- Guzenko D, Lafita A, Monastyrskyy B, Kryshtafovych A, & Duarte JM (2019). Assessment of protein assembly prediction in CASP13. Proteins: Structure, Function and Bioinformatics, 87(12), 1190–1199. 10.1002/prot.25795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopf TA, Schärfe CPI, Rodrigues JPGLM, Green AG, Kohlbacher O, Sander C, Bonvin AMJJ, & Marks DS (2014). Sequence co-evolution gives 3D contacts and structures of protein complexes. ELife, 3. 10.7554/eLife.03430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janin JL, Henrick K, Moult J, Eyck L. ten, Sternberg MJE, Vajda S, Vakser I, & Wodak SJ (2003). CAPRI: A Critical Assessment of PRedicted Interactions. http://capri.ebi.ac.uk/ [DOI] [PubMed]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, … & Hassabis D (2021). Highly accurate protein structure prediction with AlphaFold. Nature, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaca E, & Bonvin AMJJ (2013). Advances in integrative modeling of biomolecular complexes. In Methods (Vol. 59, Issue 3, pp. 372–381). Academic Press Inc. 10.1016/j.ymeth.2012.12.004 [DOI] [PubMed] [Google Scholar]
- Kryshtafovych A, Schwede T, Topf M, Fidelis K, & Moult J (2019). Critical assessment of methods of protein structure prediction (CASP)—Round XIII. In Proteins: Structure, Function and Bioinformatics (Vol. 87, Issue 12, pp. 1011–1020). John Wiley and Sons Inc. 10.1002/prot.25823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafita A (2017). Assessment of protein assembly prediction in CASP12 & Conformational dynamics of integrin alpha-I domains. 10.3929/ethz-a-010863273 [DOI]
- Lafita A, Bliven S, Kryshtafovych A, Bertoni M, Monastyrskyy B, Duarte JM, Schwede T, & Capitani G (2018). Assessment of protein assembly prediction in CASP12. Proteins: Structure, Function and Bioinformatics, 86, 247–256. 10.1002/prot.25408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lensink MF, Brysbaert G, Nadzirin N, Velankar S, Chaleil RAG, Gerguri T, Bates PA, Laine E, Carbone A, Grudinin S, Kong R, Liu RR, Xu XM, Shi H, Chang S, Eisenstein M, Karczynska A, Czaplewski C, Lubecka E, … Wodak SJ (2019). Blind prediction of homo- and hetero-protein complexes: The CASP13-CAPRI experiment. Proteins: Structure, Function and Bioinformatics, 87(12), 1200–1221. 10.1002/prot.25838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lensink MF, Méndez R, & Wodak SJ (2007). Docking and scoring protein complexes: CAPRI 3rd Edition. Proteins: Structure, Function and Genetics, 69(4), 704–718. 10.1002/prot.21804 [DOI] [PubMed] [Google Scholar]
- Lensink MF, Velankar S, Baek M, Heo L, Seok C, & Wodak SJ (2018). The challenge of modeling protein assemblies: the CASP12-CAPRI experiment. Proteins: Structure, Function and Bioinformatics, 86, 257–273. 10.1002/prot.25419 [DOI] [PubMed] [Google Scholar]
- Li X, Yang F, Hu X, Tan F, Qi J, Peng R, Wang M, Chai Y, Hao L, Deng J, Bai C, Wang J, Song H, Tan S, Lu G, Gao GF, Shi Y, & Tian K (2017). Two classes of protective antibodies against Pseudorabies virus variant glycoprotein B: Implications for vaccine design. PLoS Pathogens, 13(12). 10.1371/journal.ppat.1006777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makbul C, Nassal M, & Böttcher B (2020). Slowly folding surface extension in the prototypic avian hepatitis B virus capsid governs stability. ELife, 9, 1–23. 10.7554/ELIFE.57277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariani V, Biasini M, Barbato A, & Schwede T (2013). IDDT: A local superposition-free score for comparing protein structures and models using distance difference tests. Bioinformatics, 29(21), 2722–2728. 10.1093/bioinformatics/btt473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MATLAB and Statistics Toolbox Release 2020b, The MathWorks, Inc., Natick, Massachusetts, United States. [Google Scholar]
- McLachlan A (1982). Rapid Comparison of Protein Structures'. Acta Cryst A38, 871–873. [Google Scholar]
- Méndez R, Leplae R, Lensink MF, & Wodak SJ (2005). Assessment of CAPRI predictions in Rounds 3–5 shows progress in docking procedures. Proteins: Structure, Function and Genetics, 60(2), 150–169. 10.1002/prot.20551 [DOI] [PubMed] [Google Scholar]
- Muratcioglu S, Guven-Maiorov E, Keskin O, & Gursoy A (2015). Advances in template-based protein docking by utilizing interfaces towards completing structural interactome. In Current Opinion in Structural Biology (Vol. 35, pp. 87–92). Elsevier Ltd. 10.1016/j.sbi.2015.10.001 [DOI] [PubMed] [Google Scholar]
- Ognjenovíc JO, Grisshammer R, & Subramaniam S (2019). Annual Review of Biomedical Engineering Frontiers in Cryo Electron Microscopy of Complex Macromolecular Assemblies. 10.1146/annurev-bioeng-060418 [DOI] [PubMed]
- Oliver SL, Xing Y, Chen DH, Roh SH, Pintilie GD, Bushnell DA, Sommer MH, Yang E, Carfi A, Chiu W, & Arvin AM (2020). A glycoprotein B-neutralizing antibody structure at 2.8 Å uncovers a critical domain for herpesvirus fusion initiation. Nature Communications, 11(1). 10.1038/s41467-020-17911-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovchinnikov S, Kamisetty H, & Baker D (2014). Robust and accurate prediction of residue-residue interactions across protein interfaces using evolutionary information. ELife, 2014(3). 10.7554/eLife.02030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagès G, & Grudinin S (2018). Analytical symmetry detection in protein assemblies. II. Dihedral and cubic symmetries. Journal of Structural Biology, 203(3), 185–194. 10.1016/j.jsb.2018.05.005 [DOI] [PubMed] [Google Scholar]
- Pagès G, Kinzina E, & Grudinin S (2018). Analytical symmetry detection in protein assemblies. I. Cyclic symmetries. Journal of Structural Biology, 203(2), 142–148. 10.1016/j.jsb.2018.04.004 [DOI] [PubMed] [Google Scholar]
- Park H, Lee H, & Seok C (2015). High-resolution protein-protein docking by global optimization: Recent advances and future challenges. In Current Opinion in Structural Biology (Vol. 35, pp. 24–31). Elsevier Ltd. 10.1016/j.sbi.2015.08.001 [DOI] [PubMed] [Google Scholar]
- Porter KA, Desta I, Kozakov D, & Vajda S (2019). What method to use for protein–protein docking? In Current Opinion in Structural Biology (Vol. 55, pp. 1–7). Elsevier Ltd. 10.1016/j.sbi.2018.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozzati G, Zhu W, Lamb J, Bassot C, Kundrotas P, & Elofsson A (2021). Limits and potential of combined folding and docking using PconsDock. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues JPGLM, Melquiond ASJ, Karaca E, Trellet M, van Dijk M, van Zundert GCP, Schmitz C, de Vries SJ, Bordogna A, Bonati L, Kastritis PL, & Bonvin AMJJ (2013). Defining the limits of homology modeling in information-driven protein docking. Proteins: Structure, Function and Bioinformatics, 81(12), 2119–2128. 10.1002/prot.24382 [DOI] [PubMed] [Google Scholar]
- Söding J, Biegert A, & Lupas AN (2005). The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Research, 33(SUPPL. 2). 10.1093/nar/gki408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J, Zhang X, Wang XX, Chang S, Wu H, Wang T, … Zu Y (to be published). Structural basis of assembly and torque transmission of the bacterial flagellar motor. Cell [DOI] [PubMed] [Google Scholar]
- The PyMOL Molecular Graphics System, V. 2. [Google Scholar]
- Vakser IA (2020). Challenges in protein docking. In Current Opinion in Structural Biology (Vol. 64, pp. 160–165). Elsevier Ltd. 10.1016/j.sbi.2020.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallbracht M, Brun D, Tassinari M, Vaney M-C, Pehau-Arnaudet G, Guardado-Calvo P, Haouz A, Klupp BG, Mettenleiter TC, Rey FA, & Backovic M (2017). Structure-Function Dissection of Pseudorabies Virus Glycoprotein B Fusion Loops. Journal of Virology, 92(1). 10.1128/jvi.01203-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorobiev S, Seetharaman JK, Xiao R, Everett J, Acton T, Montelione G, … Hunt J (2020). Crystal Structure of the Q7VLF5_HAEDU protein from Haemophilus ducreyi. Northeast Structural Genomics Consortium Target Hdr25. [Google Scholar]
- Webb B, & Sali A (2016). Comparative protein structure modeling using MODELLER. Current Protocols in Bioinformatics, 2016, 5.6.1–5.6.37. 10.1002/cpbi.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wodak SJ, Velankar S, & Sternberg MJE (2020). Modeling protein interactions and complexes in CAPRI: Seventh CAPRI evaluation meeting, April 3–5 EMBL-EBI, Hinxton, UK. In Proteins: Structure, Function and Bioinformatics (Vol. 88, Issue 8, pp. 913–915). John Wiley and Sons Inc. 10.1002/prot.25883 [DOI] [PubMed] [Google Scholar]
- Xu J, & Zhang Y (2010). How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics, 26(7), 889–895. 10.1093/bioinformatics/btq066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y, & Huang S-Y (2021). Accurate prediction of inter-protein residue–residue contacts for homo-oligomeric protein complexes. Briefings in Bioinformatics. 10.1093/bib/bbab038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Anishchenko I, Park H, Peng Z, Ovchinnikov S, Baker D, & analyzed data D (2020). Improved protein structure prediction using predicted interresidue orientations. 10.1073/pnas.1914677117/-/DCSupplemental [DOI] [PMC free article] [PubMed]
- Yu X, Jin L, Jih J, Shih C, & Hong Zhou Z (2013). 3.5Å cryoEM Structure of Hepatitis B Virus Core Assembled from Full-Length Core Protein. PLoS ONE, 8(9). 10.1371/journal.pone.0069729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, & Skolnick J (2004). Scoring function for automated assessment of protein structure template quality. Proteins: Structure, Function and Genetics, 57(4), 702–710. 10.1002/prot.20264 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data, which is analyzed in this paper can be accessed at https://predictioncenter.org/casp14/multimer_results.cgi