

Abstract
Down syndrome (DS), also known as trisomy 21 (T21), is the most common human chromosomal anomaly. Although DS can affect many organ systems, lung and heart disease are the leading causes of death. An abundance of existing data suggests that lung abnormalities originate postnatally in DS. However, a single report of branching insufficiency in DS has inferred a potential prenatal origin. The histology of T21 fetal lungs (n= 15) was assessed by an experienced pathologist. Spatial differences in cellular phenotypes were examined using immunohistochemistry (IHC). Comprehensive gene expression in prenatal T21 lungs (n= 19), and age-matched controls (n= 19), was performed using high-throughput RNA sequencing (RNAseq) and validated by RT-qPCR. Histopathological abnormalities were observed in approximately half of T21 prenatal lung samples analyzed, which included dilated terminal airways/acinar tubules, dilated lymphatics, and arterial wall thickening. IHC for Ki67 revealed significant reductions in epithelial and mesenchymal cell proliferation, predominantly in tissues displaying pathology. IHC demonstrated that airway smooth muscle was reduced and discontinuous in the proximal airway in conjunction with reduced SOX2. RNAseq identified 118 genes significantly dysregulated (FDR < 0.05) in T21 lung when unadjusted and 316 genes when adjusted for age. Ontology analysis showed that IFN pathway genes were appreciably upregulated, whereas complement and coagulation cascades and extracellular matrix pathway genes were downregulated. RT-qPCR confirmed the changes in genes associated with these pathways in prenatal T21 lungs. Our data demonstrate that specific histological, cellular, and molecular abnormalities occur prenatally in different compartments of human T21 lung, which could be representative of premature stage progression.
Keywords: lung development, Down syndrome, histopathology, type I interferon, extracellular matrix, RNAseq, trisomy 21
Introduction
Down syndrome (DS), commonly referred to as trisomy 21 (T21) due to the presence of a partial or complete third copy of human chromosome 21, is the most prevalent chromosomal abnormality worldwide, with an incidence of approximately 1 in 700 live births [1,2]. It is a multisystem syndrome, demonstrating a range of medical complications, including, but not limited to, craniofacial abnormalities, gastrointestinal disorders, and autoimmune conditions. However, approximately 75% of the mortality observed in DS is due to cardiovascular and pulmonary disorders [2]. Furthermore, more than 54% of the hospitalizations of individuals with DS are due to lung disease, with respiratory complications being the most common cause of hospital admissions in children with DS less than 3 years of age [3].
Lung manifestations in DS are broad and can affect the whole respiratory system. In the upper airways, individuals with DS may present with tracheal stenosis, bronchomalacia, reduced upper airway muscle tone, and airway obstruction [2,4–6]. Lower airways are also affected, with recurrent infections being the main manifestation [7–9]. Deficient alveolarization is also described in the postnatal lung of individuals with DS, manifesting with a reduced number of alveoli (between 58% and 83% the expected number) and enlarged alveolar airspaces [10,11]. The persistence of a doublecapillary network system within the alveoli of the postnatal lung of individuals with DS suggests an arrest or delay in vascular remodeling, as the double capillary network is characteristic of the saccular stage of lung development [11,12]. Pulmonary hypoplasia with a 25% decrease in the number of airway branch generations in post-mortem lungs of individuals with DS has also been reported [13,14]. As airway branching primarily occurs during the pseudoglandular stage of development, this suggests that these lung defects occur prenatally. Moreover, a recent study on a small cohort of prenatal T21 lungs demonstrated the presence of reduced vessel density and increased vessel wall thickness, hypothesized to result from increased expression of anti-angiogenic factors such as endostatin and tumstatin [15].
Lung development is orchestrated by a wide array of cellular and molecular events. Human airway branching in particular occurs between 6 and 16 weeks of gestation and is regulated by cell proliferation, differentiation, migration, and cell–cell interactions, as well as extracellular matrix (ECM) regulation. Chromosome 21, the smallest autosome, represents approximately 1–1.5% of the human genome [16] and contains 225 genes, of which 170 are conserved protein coding genes, as well as 59 pseudogenes [16,17]. Many of the unidentified/ novel genes require further investigation and may code for such factors as microRNAs and long non-coding RNAs. Several of these genes have been shown to regulate organogenesis in many organ systems such as the brain and bone. For example, DYRK1A, located on human chromosome 21, is important for skeletal development, and inhibiting its overexpression in DS mouse models rescues observed bone defects [18]. Other genes encoded on chromosome 21 include ECM and ECM-associated genes. ECM is critical for providing adequate tissue architecture in addition to appropriate mechanical stability to support the structure and function of the lung. Recent studies showed that deletion of Col6a1 in the mouse results in histopathologic alveolar and airway abnormalities [19], whereas a deficiency in the metalloproteinase ADAMTS18 causes pulmonary hypoplasia [20]. Although there are no data directly linking DS to impaired lung ECM, several studies have implicated dysregulated ECM components in the pathogenesis of other organs of individuals with DS. For example, increased COLVI in T21 fetal skin results in nuchal edema [21]. Another study showed abnormal histology in the thymus due to aberrant fibronectin, collagen I, and collagen IV [22]. Furthermore, a microarray on prenatal T21 hearts demonstrated differential expression of genes encoding cell adhesion and extracellular matrix proteins [23]. Additionally, many MMPs (matrix metalloproteases), ADAM (a disintegrin and metalloproteinase domain), and TIMP (tissue inhibitor of metalloproteinases) genes are dysregulated in DS [24–26]. Many of these ECM and ECM-associated proteins are expressed in the lung, suggesting that dysregulated expression in T21 lungs may contribute to developmental lung defects. Yet studies are limited in understanding the effects that dysregulated gene expression of Chr21 has on lung development.
In order to understand the origin of respiratory complications in DS, it is important to study the ontogeny of T21 lung development. As modeling DS in experimental systems has proven extremely challenging, we have used early second trimester lung samples to characterize defects in T21 lungs and their time of onset. Our data herein demonstrate that histological, cellular, and molecular abnormalities occur prenatally in multiple compartments of the human T21 lung, which has not been previously described.
Materials and methods
Human lung tissue
De-identified human fetal lungs obtained from second trimester pregnancies were collected under IRB approval (USC-HS-13-0399 and CHLA-14-2211) and provided to the lab by the USC fetal tissue biobank, the University of Washington Birth Defects Research Laboratory, and Advanced Biosciences Resources (seven lungs, ages 13–15 weeks; 18 lungs, ages 16+ weeks). The only information collected was gestational age and whether there were any known genetic conditions [chromosomal trisomy 21 (T21), T18, T13] or gestational complications such as low amniotic fluid. Informed consent was provided for each lung collected and used in this study. T21 diagnosis was made at the different collection centers by routine prenatal diagnostic methods. Lungs were received as fresh whole lobes and fixed overnight in 4% PFA. Lungs were then gradually dehydrated and embedded in paraffin wax.
Histologic review and morphometry
Histologic review of T21 lungs (n = 15) and age-matched controls (n = 15) was carried out by an experienced pediatric pathologist. All slides were imaged and measured together. Images were captured with a digital camera mounted on a Nikon Eclipse 80i microscope and analyzed using NIS-Elements Advanced Research Software v4.13 (Nikon Instruments Inc, Melville, NY, USA). For morphometry, measurements were made on three random muscular arteries accompanying proximal bronchioles with complete muscular walls. Wall thickness was measured in micrometers across the shorter axis of the vessel, from the inner to the outer aspect of the medial wall, and averaged per vessel.
Immunofluorescence staining
Immunohistochemistry was performed on formalin-fixed, paraffin-embedded human fetal lungs (T21 and non-T21) as previously described [27]. In brief, paraffin-embedded lungs were sectioned at 5 μm, rehydrated, and then boiled in a sodium citrate antigen retrieval solution for 12 min. A 15-min step in 3% H2O2 was performed to quench endogenous peroxidases to decrease background. Sections were incubated with the following primary antibodies overnight at 4 °C: CD31 (RB-10333-P0, 1:200; Neomarkers, Fremont, CA, USA), CDH1 (610181, 1:200; BD Biosciences, San José, CA, USA), Ki67 (RM-9106-S1, 1:200; Thermo Fisher Scientific, Fremont, CA, USA), LYVE1 (ab14917, 1:100; Abcam, Cambridge, UK), SOX2 (sc-17320, 1:100; Santa Cruz, Dallas, TX, USA), and SOX9 (AB5535, 1:500; Millipore, Burlington, MA, USA). Sections were then stained with appropriate fluorochrome-conjugated secondary antibodies in conjunction with Cy3-ACTA2 conjugated antibody (C6198, 1:200; Sigma-Aldrich, St Louis, MO, USA) when necessary. Slides were counterstained with DAPI (DE571; Life Technologies, Grand Island, NY, USA) and mounted using ProLong Diamond Antifade Mountant (Life Technologies).
Quantitative analysis of proliferation
T21 and non-T21 lungs immunostained for Ki67 and E-cadherin (CDH1) were imaged using a 20× objective. Nine images were captured and quantified per sample. An average of approximately 1900 cells was counted per image. Cells were counted using a custom macro in Fiji ImageJ software [28]. The green channel was subtracted from blue (nuclei) to better delineate nuclei. Nuclei were threshold educing the Bernsen local thresholding algorithm (radius 10), median-filtered (radius 1), and watershed-segmented. Nuclei of 50 pixels and larger were counted and the corresponding intensity in the red channel was measured using the Analyze Particles function; those with an average red intensity above an arbitrary threshold (set by visual examination) were considered positive.
Quantification of airway ACTA2
ACTA2 positive signal was measured in regions of interest (ROIs) of 0.05 mm2 using a pixel threshold on 8-bit converted images (ImageJ) and expressed as the area fraction of positive signal per ROI. One representative image per sample in each group (n = 8 per group) was selected for measurements. Fluorescent images of DAPI staining of the same samples were used to differentiate vascular ACTA2 signal from airways. The number of gaps in airway ACTA2 cell layers was counted manually and expressed as the average number of gaps per sample. The length of these gaps was measured as the distance between the closest ACTA2-positive cells bordering the gap and expressed as the average number of gaps per sample.
RNA extraction and sequencing
RNA was extracted from pieces of fetal human lung (T21 and non-T21) using a hand-held homogenizer and following the instructions of the iNtRon Biotechnology, Inc Easy-Spin™ Total RNA Extraction Kit (iNtRon Biotechnology, Inc, Burlington, MA, USA). Samples were treated with DNAse (Qiagen, Hilden, Germany). For RNAseq, cDNA libraries were generated using 100 ng of RNA isolated from lungs of DS samples (n = 19) and age-matched controls (n = 19). Library construction was performed using the TruSeq Stranded mRNA library kit (Illumina, San Diego, CA, USA). cDNA quantity was determined with the Qubit Fluorometer (Life Technologies, Grand Island, NY, USA) and RNA quality was assessed using the Agilent Bioanalyzer 2100 (Agilent, Santa Clara, CA, USA). Libraries were sequenced (single end reads) on the Illumina NovaSeq6000 (Illumina) to generate 20 million reads per sample.
Sequences were aligned against the human genome version of hg38 using the Splice Transcript Alignment to a Reference (STAR) algorithm [29], counted with HTSeq [30], and normalized for total counts (counts per million, CPM). We used a non-specific filtering strategy to remove genes with low expression values. Samples were excluded based on poor read count/mapped read numbers, or if there were extreme outliers following hierarchical clustering and PCA. Differential expression was assessed by paired DESeq [31] to identify genes with significant differences in mean expression [false discovery rate (FDR) < 0.05]. Genes identified as differentially expressed in individual comparisons were used for pathway analysis using the ToppGene Functional annotation tool or ToppFun [32] and ENRICHR [33].
RT-qPCR analyses
cDNA was synthesized from 250 ng of RNA using an iScript cDNA synthesis kit and quantitative PCR was performed as previously described [34] using non-commercial assays (http://pga.mgh.harvard.edu/primerbank). Differences in gene expression were tested using the Wilcoxon rank test (p < 0.05).
Results
Histopathological abnormalities observed in prenatal T21 lungs
As limited data have demonstrated a decrease in airway branching in a proportion of individuals with DS, we hypothesized that the onset of this abnormality occurred during the stages of lung development when branching is established. We first sought to determine whether any gross morphological abnormalities are present in T21 fetal lungs. The histology of prenatal T21 lungs (n = 15), ranging in age from 13 to 20 weeks of gestation, was assessed by hematoxylin and eosin stain for histopathologic alterations and compared with age-and sex-matched non-DS lungs (n = 15). The presence of dilated respiratory bronchioles/acinar spaces, lymphatic dilatation, and capillary congestion was compared with the age-matched controls for each sample. The presence of one or more of these parameters was assigned a score of 0 (no difference), 1+ (rare foci), 2+ (scattered foci), or 3+ (many foci). T21 lungs frequently exhibited abnormalities in the distal airways, lymphatics, and vasculature. In eight (53.3%) of the cases, there was mild to moderate dilatation of the respiratory bronchioles and acinar spaces, some containing edema (yellow arrows in Figure 1F versus 1E; Table 1). Concurrent with these findings was dilatation of lymphatics (lymphangiectasia) (blue arrows in Figure 1F versus 1E) and/or congested capillaries in alveolar septa (Table 1). In three additional cases, only vascular changes were present (Table 1). Airway and vascular alterations were not seen in lungs that were less than 16 weeks of gestation (Figure 1B versus 1A) and generally became more pronounced with older gestational age. Thus, histopathologic assessment demonstrated a variable degree and number of lung defects, with some lungs displaying only one and others displaying a combination of the anomalies listed above (Table 1 and Figure 1E versus 1D). While many lungs had prominent terminal airway dilatation and dilated lymphatics, a subset of T21 lungs from a similar developmental stage (16 and 17 weeks, representative of the late pseudoglandular/early canalicular stages of lung development based on the accepted human lung staging [35,36]) had no distinguishable alterations from corresponding non-T21 lungs (Figure 1D versus 1C).
Figure 1.
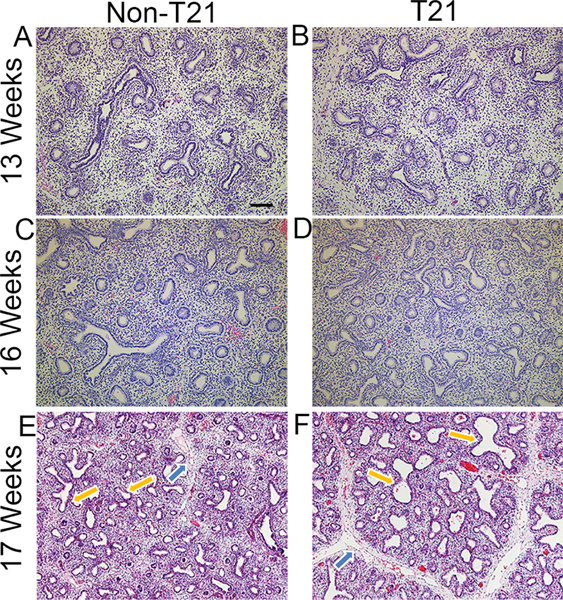
Fetal T21 lungs present with dilated airways and lymphatics. Hematoxylin and eosin staining shows histopathological defects in fetal T21 lungs compared with age- and sex-matched control non-T21 lungs. T21 lungs in the pseudoglandular stage (13 weeks’ gestation) show development comparable to non-T21 lungs (B versus A). The same is observed in a subset of lungs in the late pseudoglandular/early canalicular stages (16 weeks’ gestation) (D versus C). However, approximately 50% of T21 lungs at 16 weeks’ gestation and onward have dilated airways and lymphatics compared with age-and sex-matched non-T21 lungs (F versus E); Yellow arrows delineate airways and blue arrows lymphatics. Scale bar: 100 μm. n = 15 for each group (T21 and non-T21).
Table 1.
Morphometric analyses of T21 samples at different gestational ages.
| T21 sample | Compared with age-matched controls | Dilated radial branches and acinar tubules | Lymphatics | Muscularized arteries | Congested vessels |
|---|---|---|---|---|---|
|
| |||||
| T21–1 | 13 weeks | 0 | 0 | 1+ | 0 |
| T21–2 | 15.4 weeks | 0 | 1+ | 0 | 0 |
| T21–3 | 15.4 weeks | 0 | 0 | 0 | 0 |
| T21–4 | 16.1 weeks | 2+ some contain edema | 1+ | 2+ | 1+ |
| T21–5 | 16.2 weeks | 0 | 0 | 0 | 0 |
| T21–6 | 16.3 weeks | 1 + | 0 | 1 + | 1+ |
| T21–7 | 17 weeks | 0 | 1+ | 0 | 1 + |
| T21–8 | 17.2 weeks | 1 + | 1+ | 0 | 0 |
| T21–9 | 17.6 weeks | 2+ contain edema | 2+ | 0 | 1 + |
| T21–10 | 18 weeks | 2+ | 1+ | 2+ | 0 |
| T21–11 | 18.1 weeks | 0 | 0 | 0 | 0 |
| T21–12 | 18.2 weeks | 1+ | 0 | 1 + | 1+ |
| T21–13 | 19.3 weeks | 0 | 0 | 0 | 0 |
| T21–14 | 19 weeks | 2+ (some variability) | 1+ | 1+ | 0 |
| T21–15 | 20 weeks | 2+ (some variability) | 1+ | 0 | 0 |
Morphometric analyses were performed on H&E-stained lung sections of 15 different T21 samples and compared with age- and sex-matched non-T21 samples. Distal bronchiolar/acinar tubule dilatation, lymphatic dilatation, capillary congestion, and muscularization of arteries with measurements were assessed.
Decreased proliferation in T21 lungs with abnormal histopathology
Cell proliferation, differentiation, and a well-established proximal–distal axis are essential for proper lung branching and development. Since several abnormalities were observed in T21 lungs during development, we sought to assess whether these events are accompanied by defects in cell proliferation. We evaluated cell proliferation using immunostaining for Ki67 on each sample that had undergone histopathological assessment. Quantification of the total number of proliferating cells in either the epithelium or the mesenchyme of T21 lungs displaying histological defects demonstrated a significant percent decrease in proliferation in both compartments compared with age- and sex-matched non-T21 lungs with epithelium (8.8 ± 1.0% in T21 versus 14.4 ± 1.3% in non-T21, n = 7; p = 0.0044) and mesenchyme (8.3 ± 0.7% in T21 versus 20.2 ± 2.6% in non-T21, n = 7; p = 0.0121) (Figure 2D versus 2C and 2F). In contrast, T21 fetal lungs without histological defects did not have significantly altered proliferation in either the epithelium (10.4 ± 1.9% in T21 versus 13.1 1.6% in non-T21, n = 7, p = 0.07) or the mesenchyme (11.2 ± 1.7% in T21 versus 12.2 ± 1.5% in non-T21, n = 7, p = 0.5854) (Figure 2B versus 2A and 2E). This suggests that decreased cell proliferation is associated with the structural lung abnormalities reported [37]. Altered cell proliferation with abnormal lung structures then led to the investigation of cellular differentiation.
Figure 2.

T21 fetal lungs with histopathologic defects have significantly decreased proliferation. T21 lungs that do not present with histopathologic abnormalities have comparable proliferation to age- and sex-matched non-T21 lungs (B versus A, E; n = 7 of each group). T21 lungs with defects have significantly decreased proliferation in both the epithelium and the mesenchyme (D versus C, F) (*p = 0.0044; **p = 0.0121; n = 7 of each group); 16-week lungs. IF staining: red = Ki67; green = CDH1 (E-cadherin). Scale bar: 50 μm.
Prenatal T21 lungs present with altered epithelial differentiation
During the late pseudoglandular to early canalicular stages of development, SOX9 is expressed in the distal epithelial tips, where as airway smooth muscle cells(SMCs)surround SOX2+ progenitor cells of the proximal airways [27]. During lung development, epithelial SOX9 progenitors differentiate into SOX2-expressing airway cells [38,39], whereas SOX2 is known to regulate epithelial cell differentiation [40]. Immunofluorescence (IF) staining demonstrated a notable decrease in SOX2-positive cells compared with age-matched non-T21 controls (Figure 3B versus 3A) but displayed no change in SOX9-positive cells (Figure 3B versus 3A). RT-qPCR analysis confirmed a significant decrease in SOX2 expression in T21 versus non-T21 lungs (p = 0.0051, n = 19, Figure 3C) and no significant change in SOX9 expression (Figure 3C). Furthermore, whereas in a normally developing human fetal lung the airway SMCs are in close proximity to the epithelium, uniformly underlining it until reaching the distal epithelial tips, ACTA2 staining of T21 fetal lungs showed a thinner and interrupted layer of SMCs around the dilated airways, as shown in Figure 3B. This was confirmed via quantification of the airway SMCs, demonstrating that the % of ACTA2+ signal within a comparable area was significantly decreased in the T21 lungs compared with the nonT21 (Figure 3D; n = 8; p = 0.0014), and the gaps within the SMC layer were greater in number (Figure 3D; n = 8; p = 0.0006) and larger in size (Figure 3D; n = 8; p < 0.0001) in the T21 lungs.
Figure 3.

T21 fetal lungs have fewer SOX2-positive cells and compromised airway SMC differentiation. (A, B) Compared with age-matched non-T21 lungs (A), fewer SOX2+cells (green) were observed in the proximal airways of T21 lungs (B), while the number of SOX9+ cells (red) in the distal airways did not change. SMC differentiation is also compromised in T21 lungs compared with age-matched non-T21 lungs, noted by the disrupted SMC layer surrounding T21 airways, shown by ACTA2 IF (white) and indicated by white arrows (B versus A). Scale bar: 50 μm; n = 11 for each group. (C) RT-qPCR for SOX2 demonstrated decreased expression in fetal T21 lungs, whereas there was no change for SOX9 expression (*p = 0.0051; n = 19). (D) Airway smooth muscle quantification shows more (*p = 0.0006, n = 8) and larger (*p < 0.0001, n = 8) intermittent gaps within the airway SMC layer of T21 lungs compared with non-T21 lungs. Furthermore, the percentage of ACTA2 within a comparable region of interest is significantly less in the T21 airways than in the non-T21 airways (*p = 0.0014, n = 8).
Prenatal T21 lungs display defects in vascular and lymphatic endothelium
In addition to compromised epithelial structure, histopathological assessment determined that a subset of fetal T21 lungs have lymphangiectasia, capillary congestion, and increased muscularization of pulmonary arteries (Table 1). To further investigate the vascular and lymphatic defects in prenatal T21 lungs, IF staining was performed. LYVE1, a lymphatic endothelial marker, clearly demonstrated that there was dilatation of the lymphatics in T21 lungs compared with their non-T21 counterparts (Figure 4D versus 4C). However, no disruptions within the lymphatic structures themselves were noted, such as interruptions or a thickening of the endothelial layer. To assess the vascular endothelium, we used the endothelial marker CD31. In addition to decreased CD31 staining, the T21 lungs demonstrated a disorganized vascular endothelial cell distribution (Figure 4B versus 4A). While the vascular network is well organized in the nonT21 fetal lung throughout the mesenchyme, in close proximity to the epithelium (Figure 4A), in the T21 lungs it is disrupted and patchy throughout the mesenchyme, maintaining a distance from the epithelium (Figure 4B). Furthermore, RT-qPCR analysis confirmed significantly increased expression of the anti-angiogenic factors amyloid-beta precursor protein (APP) (p = 0.037; n = 12) and COL4A3 (p = 0.041; n = 12) in T21 versus non-T21 lungs (Figure 4E). This corroborates previous findings demonstrating diminished vascular growth and increased anti-angiogenic factors in fetal T21 lungs [15].
Figure 4.
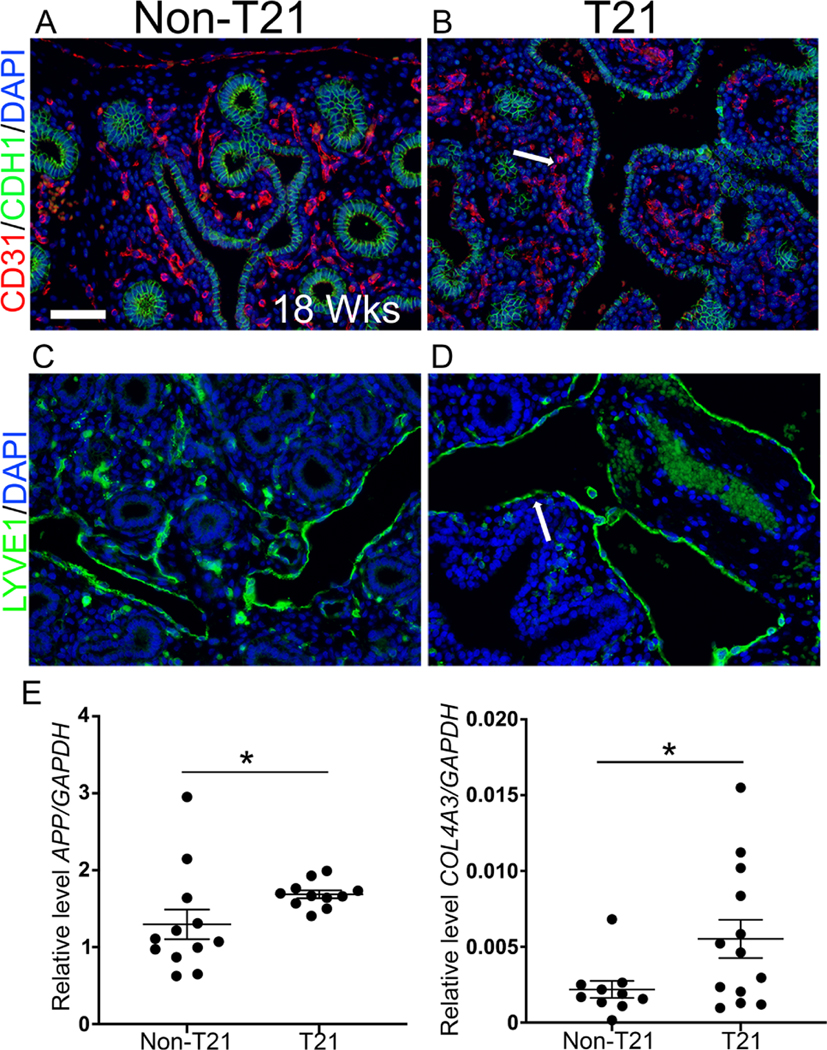
T21 fetal lungs have compromised endothelial structures. (A, B) Vascular endothelium stained for CD31 (red) showed that the vascular network of T21 fetal lungs was minimal and disrupted (indicated by white arrow) (B) compared with age-matched non-T21 fetal lungs (A). (C, D) Lymphatic endothelium stained for LYVE1 (green) demonstrated that T21 fetal lungs often had dilatation of lymphatics (lymphangiectasia) (D), compared with sex-matched control non-T21 lungs (C), but that the endothelium itself was not disrupted. Scale bar: 50 μm; n = 11 for each group; 18-week gestation lungs pictured. (E) RT-qPCR for anti-angiogenic factor mRNAs APP and COL4A3 (encoding tumstatin) had increased expression in fetal T21 lungs (*p = 0.037, *p = 0.041, respectively; n = 12 for each group).
Gene expression is altered in prenatal T21 lungs
A comprehensive gene expression profile was generated from prenatal T21 lungs (n = 19) and age-matched non-T21 controls (n = 19) using high-throughput RNA sequencing (RNAseq). The analytical dataset includes values from 40 902 genes for 38 samples, post filtering. The average number of sequence reads in the samples was high (50 ± 8 million sequence reads). Overall, approximately 68% of possible genes showed detectable transcript. DESeq2 (at FDR < 0.05) identified 118 genes to be differentially expressed in T21 lungs, when compared with age-matched controls, of which 99 were upregulated and 19 downregulated in T21 lungs. When adjusted for gender, DESeq2 (at FDR < 0.05) identified 124 genes to be differentially expressed in T21 lungs, of which 82 were upregulated and 42 downregulated. Similarly, age-adjusted DESeq2 analysis (FDR < 0.05) identified 316 genes to be differentially expressed in T21 lungs, of which 112 were upregulated and 204 downregulated in T21 lungs. Genes identified as differentially expressed by DESeq2 on transcriptomic data adjusted for gestational age are shown in supplementary material, Table S1. When adjusted for both age and gender, 98 genes were differentially expressed in T21 lungs, of which 82 were upregulated and 16 downregulated in T21 lungs. Fifty-four genes were common across all the comparisons (Figure 5A). Interestingly, many of these genes were not localized on chromosome 21. This is likely because some of the dysregulated genes on chromosome 21 may be chromosome modifiers [41,42]. Since age-adjusted analysis had the biggest effect on gene expression changes, it might indicate an early initiation of lung defects associated with DS.
Figure 5.

Pathway analysis from sequencing data of fetal T21 lungs. (A) Bulk RNAseq of 19 T21 and 19 non-T21 fetal lungs identified hundreds of differentially expressed genes (FDR < 0.05). Different gene sets were significantly differentially expressed when adjusting for gender (98 genes), age (316 genes), or both (124). (B) Circos plot of select pathways that were dysregulated in T21 (left) and the genes contributing to that enrichment (right) arranged in decreasing order of magnitude of change. Direction of change is indicated by the color of the bar, with red indicating increased expression and green indicating reduced expression in T21 lungs. (C) Pathway analysis showed an upregulation of genes associated with IFN α/β signaling in T21 lungs and downregulation of ECM enzymes/regulators and ECM-associated genes. The greatest changes were observed with age adjustment analyses.
Pathway analysis was performed in ENRICHR and ToppFun, using individual sets of differentially expressed genes identified by each of the aforementioned analysis (Figure 5C). When restricted to the differentially expressed genes located only on chromosome 21, no additional or novel pathways were detected. The significantly inhibited pathways included lipoprotein metabolism, complement and coagulation cascades, and ECM-associated proteins, enzymes, and regulators, while the interferon (IFN) signaling pathway (Figure 5B) was consistently activated.
Pathways associated with genes differentially expressed in T21 lungs compared with non-T21 are listed in supplementary material, Table S2.
We validated significant gene expression changes (p < 0.05) between T21 lungs and age-matched controls using RT-qPCR for six genes (IFI27, MX1, FABP4, COL6A1, PCNT, and SOX2) out of a total of 22 genes to validate the expression of specific genes. Interferon alpha-inducible protein 27 (IFI27), which is located on chromosome 14 and is involved in type I IFN signaling, and MX dynamin-like GTPase 1 (MX1), which is an IFN-induced GTP binding protein located on chromosome 21, were both significantly upregulated in T21 samples compared with their corresponding non-T21 samples (n = 18, p = 0.0038; n = 18, p = 0.0035, respectively; Figure 6). This confirms that not only is the IFN signaling pathway upregulated but also that genes not located on chromosome 21 are differentially expressed. We further analyzed the expression of ECM-associated genes (Figure 6). RT-qPCR analysis demonstrated that COL6A1, located on chromosome 21, was significantly upregulated in T21 lungs compared with non-T21 (n = 18, p = 0.0115; Figure 6C). This was further confirmed at the protein level, where there was greater staining for COL6A1 in T21 lungs compared with non-T21 (Figure 6B versus 6A). However, RT-qPCR measurement of FGF23, which is located on chromosome 12 and necessary for regulating mineralization of the ECM, demonstrated a trend towards decreased expression in the T21 fetal lungs compared with the non-T21 lungs, corroborating the pathway analysis (Figure 6C). RT-qPCR analysis showed that expression of PCNT, localized on chromosome 21 (encoding pericentrin), and FABP4, localized on chromosome 8 (encoding fatty acid-binding protein 4), were both significantly upregulated in T21 lungs compared with non-T21 lungs, demonstrating that differential gene expression affects genes on other chromosomes, not only chromosome 21 (n = 18 for each group, p = 0.025, p = 0.011, respectively; Figure 6).
Figure 6.

Gene analysis of fetal T21 lungs. (A, B) Immunofluorescence staining for ECM-associated protein COL6A1 (red; gene localized on chromosome 21) demonstrated increased staining in T21 lungs (B) compared with non-T21 lungs (A). (C) This was confirmed by RT-qPCR, where COL6A1 was significantly upregulated in T21 fetal lungs compared with non-T21 (p = 0.0115). The ECM-associated gene FGF23 showed a trend towards downregulation in the T21 lungs compared with non-T21 (n = 18 for each group). RT-qPCR for IFN pathway-associated genes IFI27 and MX1 showed both to be significantly upregulated (n = 18 for each group; p = 0.0038, p = 0.0035, respectively) in T21 fetal lungs compared with non-T21. RT-qPCR showed that PCNT and FABP4 were also significantly upregulated in T21 fetal lungs compared with non-T21 (p = 0.025, p = 0.011, respectively; n = 18 for each group).
Discussion
Although many and diverse complications are associated with Down syndrome, pulmonary-related disorders comprise one of the largest percentages of morbidity and mortality within these individuals, with more than 54% of DS hospitalizations arising from lung disease [2,3]. Furthermore, respiratory complications are the most common cause of hospital admissions in children with DS 3 years of age or younger [3], suggesting that many pulmonary issues are initiated in utero. This was further implicated by a study reporting a 25% decrease in the number of airway branch generations in lungs of pediatric patients with DS [14], indicating that defects likely originate during the pseudoglandular stage of development. Our histopathological assessments of T21 human fetal lungs, demonstrating dilatations of the respiratory bronchioles and acinar spaces in over 50% of our samples, tentatively starting at 16 weeks of gestation (Figure 1E versus 1F), which is considered to be the late pseudoglandular to early canalicular stages of lung development, confirm that abnormal lung development is in fact initiated in utero. However, the limited number of samples below 16 weeks of gestation that were analyzed in our cohort limits our ability to determine whether these defects occur at a specific time during gestation. A larger cohort of samples will be necessary to conduct time-specific analyses in order to determine the exact gestational age at which these defects are first seen.
The pseudoglandular and canalicular stages of lung development are associated with increased cell proliferation and differentiation within airway epithelium and associated mesenchyme [27,43]. Given the altered branching observed in a subset of our T21 fetal lungs and noting that these findings were first seen during the pseudoglandular and canalicular stages of lung development, it was necessary to assess how proliferation and differentiation were being affected. Previous studies demonstrated a reduction in proliferation potency in fetal T21 neural precursor and fibroblasts compared with their non-T21 counterparts [44,45]. Interestingly, we noted a similar phenomenon in fetal T21 lungs displaying histological defects, demonstrating a significant decrease in epithelial and mesenchymal proliferation compared with the non-T21 control group and T21 group with no histological defects (Figure 2). This was accompanied by altered epithelial differentiation, where we observed decreased SOX2 expression within the developing proximal airways/lungs in T21, alongside abnormal airway smooth muscle patterning with disrupted ACTA2 localization (Figure 3). Proximal/distal patterning (proximal expression of SOX2 and distal expression of SOX9) is important for proper lung branching [27]. We previously reported that co-localization of SOX2/SOX9 in the distal buds and proper distribution of ACTA2+ cells are required for proper lung branching [27]. In addition, we demonstrated that a decrease in cell proliferation occurs concomitantly with the transition from a branching program to the canalicular stage of development. Furthermore, SOX2 is strongly expressed in the proximal airway during development and decreases significantly as the airway epithelial cells differentiate [27,46]. Taken together, these facts, along with our findings of decreased SOX2, ACTA2, and cell proliferation in addition to dilatation in T21 lungs, suggest that the lung is prematurely advancing into the canalicular stage. It is currently unclear why defects were seen only in a subset of lungs, but this may be related to genetic differences in T21 and/or concurrent anomalies in the fetus. As our analyses did not extend beyond 23 weeks of gestation, we cannot exclude variable time of onset as well.
One of the characteristic properties of the saccular stage of lung development is the establishment of a double capillary network system [12], which merges into a single capillary system postnatally. Postnatal T21 lungs often maintain this double capillary network, which may contribute to their increased risk for developing persistent pulmonary hypertension and pulmonary arterial hypertension [13]. Compromised angiogenesis in T21 lungs in utero is further supported by the disrupted CD31 staining (Figure 4). Of note, many of the lungs demonstrating vascular developmental issues also present with airway defects (Table 1). It has been previously shown that compromised angiogenesis impairs alveolar development [47]; therefore it would be important to further assess whether this phenomenon is a result of aberrant crosstalk amongst the compartments or is influenced by a set of dysregulated genes. In parallel with the vascular endothelial network, our histopathological assessment showed that the lymphatic endothelial network is also compromised in T21 lungs. Studies have previously shown that the pulmonary lymphatic endothelial cells are established between the canalicular and saccular stages of development [48]. Although there are no observed disruptions within the endothelium itself, we noted dilatation of the lymphatics, which is seen in pulmonary lymphangiectasia. Pulmonary lymphangiectasia is not uncommon in DS, either as a primary abnormality of the lymphatics or secondary to congenital heart disease. It is unclear what underlies lymphangiectasia in our samples, as we were not provided with information about the presence of cardiac or other anomalies.
A few hundreds of genes are encoded on chromosome 21, many of which have been shown to play important roles in neurological development, immune response, and homeostasis of several organs such as the brain and bone. In this study, we performed comprehensive RNA-sequencing analyses on T21 and non-T21 fetal lung samples. In addition to demonstrating significant upregulation of several genes encoded on chromosome 21 (e.g. COL6A1), T21 lungs also showed dysregulation of many genes not encoded on chromosome 21. Ontology analysis showed that complement and coagulation cascades and ECM pathway genes were significantly downregulated, whereas interferon (IFN) pathway genes were significantly upregulated (Figure 5). Consistent with our findings, a previous study showed increased anti-angiogenic factors in T21 fetal lungs (APP and tumstatin) accompanied by impaired vasculogenesis (Figure 4) [15]. Moreover, our data demonstrated dysregulation of ECM components important for proper lung development and patterning in T21 lungs (Figure 6). Comparable findings were reported in a study on T21 hearts, where several ECM components were modulated [23]. Interestingly, the type I IFN pathway, shown to be activated in our dataset (Figures 5 and 6), is one of the major signaling cascades consistently activated in DS [45]. Interstitial lung disease is often associated with interferonopathies, immune disorders caused by dysregulation of type I IFN [49]. Aberrant type I IFN signaling has also been associated with pulmonary arterial hypertension, one of the most common manifestations in DS [50]. Furthermore, the type I interferon receptors IFNAR1 and IFNAR2 are encoded on chromosome 21, suggesting an important role in DS pathophysiology [45]. While this pathway has been studied in the context of immune response, notably infections such with SARS-CoV-2 [51], it has not been studied in the context of lung development. In summary, we demonstrate that several lung defects in DS are initiated during fetal life, which are accompanied by modulation of several signaling pathways important in DS and lung development. More studies are needed to investigate the upregulated and downregulated pathways within our fetal T21 lung cohort and determine how they may contribute to the observed branching, vascular, and lymphatic abnormalities. Understanding the mechanisms associated with respiratory complications in DS can lead to therapeutic avenues that will improve the survival and quality of life of individuals with DS.
Supplementary Material
Acknowledgements
We thank Melissa L Wilson (Department of Preventive Medicine, University of Southern California) and Family Planning Associates for coordinating fetal tissue collection. We also thank Dr Brendan H Grubbs and Matthew E Thornton (Department of Obstetrics and Gynecology, Maternal Fetal Medicine Division, Keck School of Medicine, University of Southern California), the Birth Defects Research Lab at Washington University, and ABR for providing the fetal tissues. This work was funded by NIH/NHLBI R01HL141856 (to DA) and an ATS Unrestricted Grant: Pulmonary (to SD).
Footnotes
No conflicts of interest were declared.
Data availability statement
The metadata, raw data, and processed data of the RNAseq studies described have been uploaded into the GEO database, accession number GSE175539 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE175539).
References
- 1.Weijerman ME, van Furth AM, Vonk Noordegraaf A, et al. Prevalence, neonatal characteristics, and first-year mortality of Down syndrome: a national study. J Pediatr 2008; 152: 15–19. [DOI] [PubMed] [Google Scholar]
- 2.Colvin KL, Yeager ME. What people with Down Syndrome can teachus about cardiopulmonary disease. Eur Respir Rev 2017; 26: 160098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davidson MA. Primary care for children and adolescents with Downsyndrome. Pediatr Clin North Am 2008; 55: 1099–1111. [DOI] [PubMed] [Google Scholar]
- 4.Watts R, Vyas H. An overview of respiratory problems in childrenwith Down’s syndrome. Arch Dis Child 2013; 98: 812–817. [DOI] [PubMed] [Google Scholar]
- 5.Shott SR. Down syndrome: analysis of airway size and a guide forappropriate intubation. Laryngoscope 2000; 110: 585–592. [DOI] [PubMed] [Google Scholar]
- 6.Mitchell RB, Call E, Kelly J. Diagnosis and therapy for airwayobstruction in children with Down syndrome. Arch Otolaryngol Head Neck Surg 2003; 129: 642–645. [DOI] [PubMed] [Google Scholar]
- 7.Mitwalli M, Wahba Y, Shaltout A, et al. Lymphocyte subgroups and recurrent infections in children with Down syndrome – a prospective case control study. Cent Eur J Immunol 2018; 43: 248–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tenenbaum A, Hanna RN, Averbuch D, et al. Hospitalization of children with Down syndrome. Front Public Health 2014; 2: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan M, Park JJ, Shi T, et al. The burden of respiratory syncytial virus (RSV) associated acute lower respiratory infections in children with Down syndrome: a systematic review and meta-analysis. J Glob Health 2017; 7: 020413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cooney TP, Wentworth PJ, Thurlbeck WM. Diminished radial countis found only postnatally in Down’s syndrome. Pediatr Pulmonol 1988; 5: 204–209. [DOI] [PubMed] [Google Scholar]
- 11.Bush D, Abman SH, Galambos C. Prominent intrapulmonary bronchopulmonary anastomoses and abnormal lung development in infants and children with Down syndrome. J Pediatr 2017; 180: 156–162.e1. [DOI] [PubMed] [Google Scholar]
- 12.Cooney TP, Thurlbeck WM. Pulmonary hypoplasia in Down’s syndrome. N Engl J Med 1982; 307: 1170–1173. [DOI] [PubMed] [Google Scholar]
- 13.Saji T Clinical characteristics of pulmonary arterial hypertension associated with Down syndrome. Pediatr Int 2014; 56: 297–303. [DOI] [PubMed] [Google Scholar]
- 14.Schloo BL, Vawter GF, Reid LM. Down syndrome: patterns of disturbed lung growth. Hum Pathol 1991; 22: 919–923. [DOI] [PubMed] [Google Scholar]
- 15.Galambos C, Minic AD, Bush D, et al. Increased lung expression of anti-angiogenic factors in Down syndrome: potential role in abnormal lung vascular growth and the risk for pulmonary hypertension. PLoS One 2016; 11: e0159005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hattori M, Fujiyama A, Taylor TD, et al. The DNA sequence of human chromosome 21. Nature 2000; 405: 311–319. [DOI] [PubMed] [Google Scholar]
- 17.Gardiner K, Costa AC. The proteins of human chromosome 21.Am J Med Genet C Semin Med Genet 2006; 142C: 196–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blazek JD,AbeysekeraI,Li J,et al.Rescueoftheabnormalskeletalphenotype in Ts65Dn Down syndrome mice using genetic and therapeutic modulation of trisomic Dyrk1a. Hum Mol Genet 2015; 24: 5687–5696. [DOI] [PubMed] [Google Scholar]
- 19.Mereness JA, Bhattacharya S, Ren Y, et al. Collagen VI deficiency results in structural abnormalities in the mouse lung. Am J Pathol 2020; 190: 426–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu T, Lin X, Pan YH, et al. ADAMTS18 deficiency leads to pulmonary hypoplasia and bronchial microfibril accumulation. iScience 2020; 23: 101472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brand-Saberi B, Flöel H, Christ B, et al. Alterations of the fetal extracellular matrix in the nuchal oedema of Down’s syndrome. Ann Anat 1994; 176: 539–547. [DOI] [PubMed] [Google Scholar]
- 22.Fonseca EC, Lannes-Vieira J, Villa-Verde DM, et al. Thymic extracellular matrix in Down’s syndrome. Braz JMedBiol Res 1989; 22: 971–974. [PubMed] [Google Scholar]
- 23.Conti A, Fabbrini F, D’Agostino P, et al. Altered expression of mitochondrial and extracellular matrix genes in the heart of human fetuses with chromosome 21 trisomy. BMC Genomics 2007; 8: 268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsilingaridis G, Yucel-Lindberg T, Modéer T. Altered relationshipbetween MMP-8 and TIMP-2 in gingival crevicular fluid in adolescents with Down’s syndrome. J Periodontal Res 2013; 48: 553–562. [DOI] [PubMed] [Google Scholar]
- 25.Yamazaki-Kubota T, Miyamoto M, Sano Y, et al. Analysis of matrix metalloproteinase (MMP-8 and MMP-2) activity in gingival crevicular fluid from children with Down’s syndrome. J Periodontal Res 2010; 45: 170–176. [DOI] [PubMed] [Google Scholar]
- 26.Baviera G, Chimicata S, De Domenico R, et al. First- and secondtrimester ADAM12s in Down syndrome screening. Clin Chem 2010; 56: 1355–1357. [DOI] [PubMed] [Google Scholar]
- 27.Danopoulos S, Alonso I, Thornton ME, et al. Human lung branching morphogenesis is orchestrated by the spatiotemporal distribution of ACTA2, SOX2, and SOX9. Am J Physiol Lung Cell Mol Physiol 2018; 314: L144–L149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schindelin J,Arganda-CarrerasI,Frise E,et al.Fiji:anopen-sourceplatform for biological-image analysis. Nat Methods 2012; 9: 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013; 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anders S, Pyl PT, Huber W. HTSeq – a Python framework to work with high-throughput sequencing data. Bioinformatics 2015; 31: 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Love MI, Huber W, Anders S. Moderated estimation of fold changeand dispersion for RNA-seq data with DESeq2. Genome Biol 2014; 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen J, Bardes EE, Aronow BJ, et al. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res 2009; 37: W305–W311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuleshov MV, Jones MR, Rouillard AD, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 2016; 44: W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bhattacharya S, Go D, Krenitsky DL, et al. Genome-wide transcriptional profiling reveals connective tissue mast cell accumulation in bronchopulmonary dysplasia. Am J Respir Crit Care Med 2012; 186: 349–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Volckaert T, De Langhe SP. Wnt and FGF mediated epithelial– mesenchymal crosstalk during lung development. Dev Dyn 2015; 244: 342–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kitaoka H, Burri PH, Weibel ER. Development of the human fetal airway tree: analysis of the numerical density of airway endtips. Anat Rec 1996; 244: 207–213. [DOI] [PubMed] [Google Scholar]
- 37.Varner VD, NelsonCM. Cellular and physicalmechanisms of branching morphogenesis. Development 2014; 141: 2750–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Volckaert T, Campbell A, Dill E, et al. Localized Fgf10 expression is not required for lung branching morphogenesis but prevents differentiation of epithelial progenitors. Development 2013; 140: 3731–3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abler LL, Mansour SL, Sun X. Conditional gene inactivation revealsroles for Fgf10 and Fgfr2 in establishing a normal pattern of epithelial branching in the mouse lung. Dev Dyn 2009; 238: 1999–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tompkins DH, Besnard V, Lange AW, et al. Sox2 activates cell proliferation and differentiation in the respiratory epithelium. Am J Respir Cell Mol Biol 2011; 45: 101–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Letourneau A, Santoni FA, Bonilla X, et al. Domains of genome-wide gene expression dysregulation in Down’s syndrome. Nature 2014; 508: 345–350. [DOI] [PubMed] [Google Scholar]
- 42.Ruparelia A, Wiseman F, Sheppard O, et al. Down syndrome and the molecular pathogenesis resulting from trisomy of human chromosome 21. J Biomed Res 2010; 24: 87–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schittny JC. Development of the lung. Cell Tissue Res 2017; 367: 427–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stagni F, Giacomini A, Emili M, et al. Neurogenesis impairment: an early developmental defect in Down syndrome. Free Radic Biol Med 2018; 114: 15–32. [DOI] [PubMed] [Google Scholar]
- 45.Sullivan KD, Lewis HC, Hill AA, et al. Trisomy 21 consistently activates the interferon response. Elife 2016; 5: e16220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gontan C, de Munck A, Vermeij M, et al. Sox2 is important for two crucial processes in lung development: branching morphogenesis and epithelial cell differentiation. Dev Biol 2008; 317: 296–309. [DOI] [PubMed] [Google Scholar]
- 47.Jakkula M, Le Cras TD, Gebb S, et al. Inhibition of angiogenesis decreases alveolarization in the developing rat lung. Am J Physiol Lung Cell Mol Physiol 2000; 279: L600–L607. [DOI] [PubMed] [Google Scholar]
- 48.Norman TA Jr, Gower AC, Chen F, et al. Transcriptional landscape of pulmonary lymphatic endothelial cells during fetal gestation. PLoS One 2019; 14: e0216795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu ZX, Song HM. Toward a better understanding of type I interferonopathies: a brief summary, update and beyond. World J Pediatr 2020; 16: 44–51. [DOI] [PubMed] [Google Scholar]
- 50.George PM, Oliver E, Dorfmuller P, et al. Evidence for the involvement of type I interferon in pulmonary arterial hypertension. Circ Res 2014; 114: 677–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Espinosa JM Down syndrome and COVID-19: a perfect storm? Cell Rep Med 2020; 1: 100019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The metadata, raw data, and processed data of the RNAseq studies described have been uploaded into the GEO database, accession number GSE175539 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE175539).