

Abstract
Chromogranin A (CHGA) is an index granin protein critical for biogenesis and exocytotic release of catecholamine storage granules. It is elevated in plasma of patients with sympathetic over-activity and kidney dysfunction. Several CHGA polymorphisms are associated with hypertensive kidney disease. Previously, we unraveled the molecular mechanism by which CHGA expression is regulated in African Americans carrying a genetic variation associated with hypertensive chronic kidney disease (CKD). Experimental CKD mouse model were created by 5/6th nephrectomy (Npx) using wild-type (WT) and Chga−/− (KO) mouse strains to delineate the role of CHGA in CKD. WT-Npx mice expressing Chga developed exacerbated azotemia and fibrosis as compared to their KO-Npx counterparts. Gene expression profiling revealed downregulation of mitochondrial respiratory complexes genes consistent with maladaptive mitochondria in WT-Npx mice, contrasted to KO-Npx. In healthy individuals, an inverse relationship between circulating CHGA levels and glomerular function was observed. In vitro, mesangial cells treated with CHGA triggered nitric oxide release by a signaling mechanism involving scavenger receptor SR-A. The CHGA-treated and untreated mesangial cells displayed differential expression of cytokine, chemokine, complement, acute phase inflammatory and apoptotic pathway genes. Thus, build-up of plasma CHGA due to kidney injury served as an insult to the mesangial cells resulting in expression of genes promoting inflammation, fibrosis, and progression of CKD. These findings improve understanding of the role of elevated CHGA in the progression of CKD and reveal novel pathways that could be exploited for therapeutic strategies in hypertensive kidney disease.
Keywords: chromogranin A, hypertensive kidney disease, CKD, nephrectomy, mesangial cell
CONDENSED ABSTRACT
Epidemiological studies have demonstrated an excessive risk of CKD progression to ESRD among African Americans, even after controlling for confounding factors. In this population, several potential disease-susceptibility loci for hypertensive-CKD leading to ESRD have been identified including chromogranin A (CHGA) the adrenergic catecholamine storage protein. This study supports observations in humans that loss of kidney function leads to build-up of plasma CHGA and triggers mesangial cell activation via scavenger receptor, mounting an oxidative stress response potentiating inflammation and fibrosis eventuating in the disease phenotype. These findings should provide new inroads into therapeutic approaches for a currently intractable health disparity.
INTRODUCTION
The growing prevalence of CKD is a major public health challenge, associated with high morbidity and is risk of cardiovascular disease [1]. Catecholamine a measure of the sympathetic nerve over-activity, is associated with mortality and cardiovascular outcomes in ESRD [2]. Chromogranin A (CHGA) is an index member of the granin family co-stored and released into circulation with catecholamine from secretory granules of chromaffin cells and post-ganglionic sympathetic neurons [3]. A relationship has been established between CHGA, hypertension and kidney disease. Kidney dysfunction contributes to increasing levels of plasma CHGA [4, 5] and several CHGA gene polymorphisms contributing to traits of autonomic blood pressure control, hypertension, and hypertensive kidney disease have been identified [6–10]. In the African American Study of Kidney Disease and Hypertension (AASK) cohort, glomerular filtration rate (GFR) decline in CKD was associated with a polygenic trait with contributions from several genes involved in catecholamine biosynthesis, storage and catabolism including CHGA [8]. We previously unraveled the molecular mechanism by which CHGA expression is regulated in the African American carriers of a genetic variation in the CHGA 3’-UTR region that is associated with hypertensive nephropathy [11]. In this study, we seek to understand the role of CHGA in CKD progression.
The nephrectomy (Npx) mouse model which represents progressive kidney dysfunction due to reduced renal parenchyma was used to investigate the influence of CHGA on kidney function [12]. In this study it was observed that mice expressing Chga that underwent Npx surgery developed more severe azotemia and fibrosis compared to their Chga−/− (knockout, KO) counterparts. We profiled by microarray differential gene expression in both stains of mice with or without surgery and observed downregulation of mitochondrial respiratory complexes genes. In healthy individuals, a negative inverse correlation between circulating CHGA levels and glomerular filtration was observed. Loss of kidney function led to a significant elevation of plasma CHGA and to understand its consequence, mesangial cells in culture were treated in vitro with CHGA protein. This triggered NO release and activated genes involved in its biosynthesis. The signal transduction mechanism leading to NO production involved activation of scavenger receptor and gene expression of genes leading to kidney disease progression.
METHODS
Animal husbandry, nephrectomy and eGFR measurements.
Mice were housed in a 12-h light (0600–1800 h) and 12-h dark cycle under pathogen-free conditions, maintained on a normal chow-diet and allowed to have water ad libitum. Experiments were carried out in accordance with the IACUC at UCSD.
Progressive HT- kidney disease model, mimicked features of human CKD and was created sequentially: briefly, the upper and lower poles of the left kidney (two thirds) were resected. After one week, the remaining right kidney was removed through a right paramedian incision after ligation of the right renal artery, vein, and ureter [13]. Sham surgery was performed by manipulation of the renal pedicle in age and sex-matched mice used as controls. The mice were anesthetized with ketamine/xylazine (100 mg/kg and 10 mg/kg, respectively) and via precision vaporizer isoflurane (1.25 – 1.5% in 100% oxygen). Buprenorphine (0.1 mg/kg) was given 2–3 times per day, for 3 days, to mice during the recovery. To avoid overdosing of ketamine/xylazine, the strategy of half dosage of standard ketamine/xylazine (50 mg/kg and 5 mg/kg, respectively) and isoflurane (1.25 – 1.5% in 100% oxygen) was administered prior to operation. Standard processes for pre-operational and post-operational animal care was applied. The surgery when done in 2-stages it improves the survival chances of the mouse. This is in accordance with published literature.
The animals are then followed sequentially for 10 weeks, with weekly assessments of BP and HR using tail-cuff apparatus, as described previously [14]. Weekly blood sampling of operated mice was done by tail vein incision to obtain plasma for creatinine measurements [15]. Measurements of eGFR were estimated by renal clearance of creatinine by a method not confounded by circulating non-creatinine Jaffe chromogens in rodents. Plasma creatinine was measured using electrospray mass-spectrometry LC-MS and 10 μl sample volumes, at the UAB-O’Brien core.
At the terminal stage of the experiment, mice were euthanized by deep anesthesia in isoflurane and blood was collected by a transthoracic ventricular puncture in EDTA-tubes. The remnant kidney tissue samples were dissected and suspended in RNA later (Life Technologies, California, USA) for subsequent RNA extraction or in formaldehyde for histological analysis. The kidneys were fixed in 10% formalin and embedded in paraffin wax. Kidney sections on slides were stained with three color Masson’s Trichrome stain. To quantify fibrosis, digitized images were analyzed using ImageJ software (NIH) to calculate collagen volume fraction as percent area of collagen divided by total section area. At least 15 images from each group were analyzed. Plasma CHGA was measured using a mouse ELISA kit kindly provided by Angelo Corti[16].
Microarray analysis.
Kidney tissue 10 weeks post- Npx/ sham surgery in 4 cohorts: Chga +/+ sham/ Npx; Chga −/− sham/ Npx were harvested and their transcriptome analyzed in quadruplicates. Total RNA was isolated using a RNeasy Kit (QIAGEN). In a parallel experiment, RNA from CRL-1927 cells (4 replicates, with or without CHGA treatment in vitro) was isolated. The RNA quality was assessed by using an Agilent Model 2100 Bioanalyzer (Agilent Technologies) and labeled and hybridized to a mouse 12-plex array (100718_MM9_EXP_HX12, Nimblegene/Roche, Madison, WI). The array was scanned on a GenePix 4000B scanner (Molecular Devices, CA) and data extracted using Arraystar 4 (DNAStar Inc., WI). Raw intensity data were analyzed using a Bayesian variance modeling approach in VAMPIRE using median normalization and an FDR of 0.05. Principal component analysis and unsupervised hierarchical clustering using Euclidean distance were performed, and violin plots were generated using GeneSpring 14.5 (Agilent, CA). The list of genes with altered expression between the WT Npx and KO Npx, or with CHGA treatment in vitro, was subjected to gene ontology and pathway mapping using GeneGo’s Metacore software (St. Joseph, MI). Microarray data are available at GEO Accession Number (GSE106300).
Cell culture and NO assay.
Mouse kidney glomerular mesangial cells (ATCC ® CRL-1927™) were grown in 3:1 mixture of DMEM high glucose and Ham’s F12 (Corning Cellgro) with 14 mM Hepes, 5 % fetal bovine serum and penicillin/streptomycin/glutamine at 37°C in 5 % CO2. Cells were treated with various stimuli in fresh culture media and incubated for up to 24 hours. Tissue culture supernatant was collected, diluted 1:1 with reaction diluent, filtered through Amicon Ultra-0.5 mL centrifugal filters (Ultracel, 10K, Millipore) and the flow through media used to determine total NO according to the manufacturer’s instruction (KGE001, R&D Systems, Inc., MN). Experiments were performed on at least two unique cell preparations. Data are representative of a typical experiment and are given as a mean of triplicates ± SEM.
CHGA and estimated GFR measurements in subjects.
Subjects were volunteers from Southern California and gave informed written consent; the protocol was approved by the UCSD Human Research Protection Program. CHGA and estimated GFR were measured in this large cohort of European ancestry as previously described [17]. None of these healthy individuals had a history of kidney dysfunction, GFR was estimated from plasma creatinine, age, sex and body size, using the simplified NIDDK Modification of Diet in Renal Disease algorithm: GFR (ml/min) =(186)*(pCr1.154)*(Age0.203)*(0.742 [if female])*(1.21 [if black]).
Statistical methods.
Data were analyzed by Student’s t-test and ANOVA and expressed as mean± SEM where applicable using Kaleidagraph (Synergy Software, Reading, PA) and or SPSS-23 (Chicago, IL) software. Results from at least 3 independent experiments were considered for analysis of in vitro data. Data with a ‘p’ value <0.05 were considered significant.
RESULTS
Chga+/+-Npx model mimics clinical observation of inverse correlation between circulating CHGA levels and GFR.
To evaluate the effect of circulating CHGA on glomerular function, both the WT and KO strains of mice were subjected to either Npx or sham surgery and assessed weekly for their plasma creatinine and BP levels for up to week 10 post-surgery. Both strains of mice with Npx displayed decreased renal function with the progression of time compared to mice that underwent sham surgery. Both strains did not display a significant difference in the build-up of plasma creatinine at 4 weeks post-Npx. After 4 weeks, the creatinine levels in the WT-Npx continued to increase whereas the KO-Npx did not display further deterioration of kidney function, potentially indicating a detrimental effect of CHGA in the progression of CKD (Fig.1A). WT-Npx mice displayed elevated plasma levels of the CHGA, contributed in part due to increased sympathetic activity and also in part failure of kidneys to filter CHGA (Fig.1B). High BP is a hemodynamic characteristic of CKD and accelerates the progression of kidney dysfunction by worsening glomerular injury and proteinuria. The phenotype of KO mice is hypertensive prior to surgery [18]. Kidney dysfunction due to Npx elevated BP in both strains of Npx mice and week 5 post-Npx, no significant difference in BP between strains was observed (Fig.1C), like the KO-Npx mice, the WT-Npx were hypertensive (BP ~150 mm Hg). To investigate whether CHGA levels are predictive of kidney disease progression in humans, we measured plasma CHGA in subjects with known GFR (Fig.1D) and a significant negative correlation was observed. Therefore, in both mice and humans, an inverse correlation suggested a decrease in GFR with increasing plasma CHGA concentration.
Figure 1. Impact of CHGA on glomerular function.
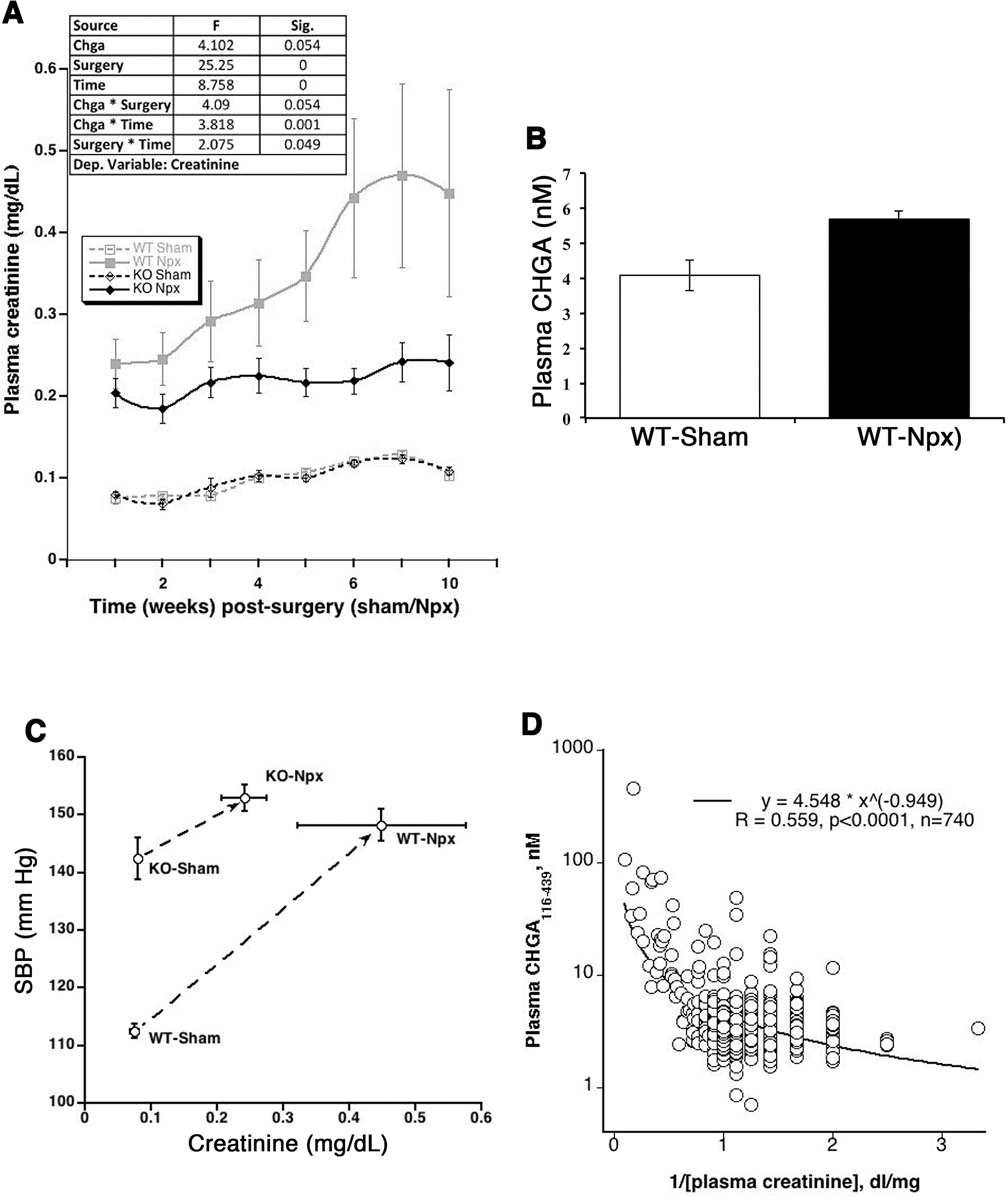
(A) Time course on the effect of CHGA accumulation on eGFR. WT and KO mice (N=12 each of the 4 groups), 8 weeks of age, were subjected to sham or Npx surgery and their weekly eGFR was estimated by measuring the plasma creatinine levels. The data was analyzed by repeated measure ANOVA using linear mixed model and showed significant differences in creatinine measurements between the two mouse strains subjected to Npx. (B) Elevated CHGA in the CKD mouse model. The WT-Npx mice showed 1.4-fold elevated plasma CHGA level as compared to sham operated mice. (C) Circulating CHGA concentration and human eGFR. A large cohort of healthy individuals of European ancestry were measured for plasma CHGA and eGFR. A significant negative correlation was observed suggesting decrease in glomerular function with increasing plasma CHGA concentration. (D) Ten weeks-post-Npx the BP did not differ significantly between Npx strains however, WT-Npx have significantly augmented azotemia compared to KO-Npx suggesting BP independent mechanism of CHGA pathogenesis.
Renal fibrosis augmented by CHGA.
Renal fibrosis causing stiffness and loss in structural integrity of the kidney greatly affects survival outcomes [19]. To assess fibrosis, sections of kidneys from WT and KO mice 10 weeks post sham/ Npx operation were stained with Masson’s trichrome. Both KO and WT Npx showed excess deposition of extracellular collagenous tissue, whereas staining was not present in kidneys from sham-operated mice. Strikingly, matrix deposition in the WT kidney was much more prominent with almost no empty extracellular space in kidney parenchyma as compared to the KO-Npx (Fig.2) consistent with worsened kidney function associated with WT-Npx mice. Collagen volume fraction (CVF) as calculated from Masson Trichrome Stained kidney tissue sections (Fig 2) was significantly increased in WT-Npx group compared to WT-sham (Supplementary Data 3).
Figure 2. CHGA accumulation due to injury augments renal fibrosis.
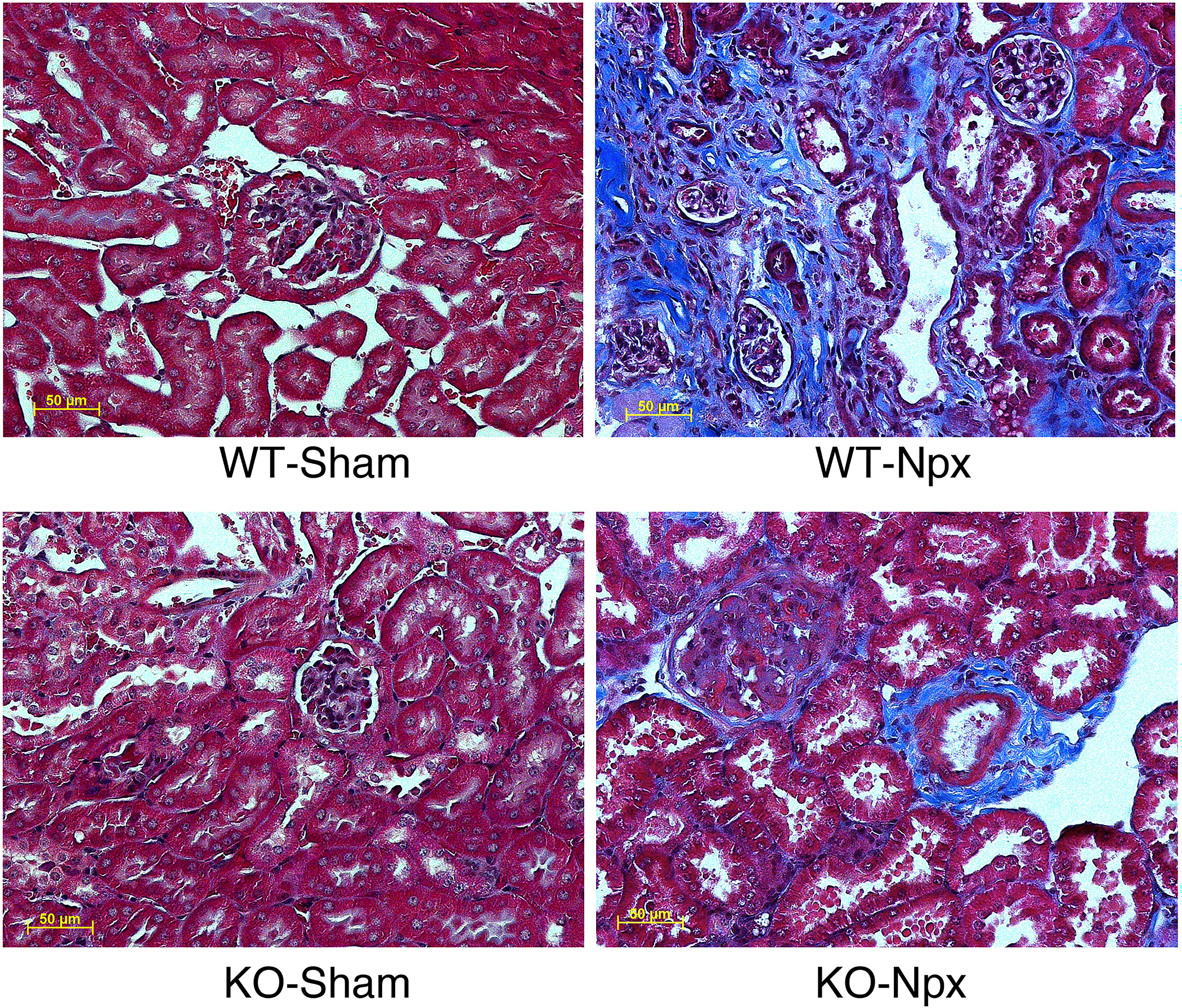
The kidney tissue was harvested 10-weeks post-surgery (sham/Npx) and histology sections were stained by Masson’s trichome. All pictographs have the same magnification.
CHGA imparts differential gene expression in kidneys subjected to injury.
Gene expression profiling by microarray was performed on kidney tissue at week 10 to assess the effects of strain and operation. Each group (Chga+/+sham, Chga+/+Npx, Chga−/− sham, and Chga−/− Npx) comprised of 4 animals. The analysis focused on genes differentially expressed between the WT-Npx and KO-Npx as we found little difference between WT and KO mice in the sham state. Fig. 3A is the Principal Component Analysis plot of the samples; a Venn diagram shows the number of genes altered by Npx in WT or KO mice (Fig.3B), as well as the genes altered by genotype in sham or Npx mice (Supplementary Data 1). The heat map provides pictograph of all the altered genes (Fig.3C). The GO enrichment reports for the 612 genes altered in WT-Npx vs KO-Npx indicate that genes involved in extracellular matrix remodeling are significantly enriched (Supplementary Data 2). The data also suggested that genes with oxidoreductase activity or associated with the mitochondrion are also enriched. These families of genes were inspected individually, and those representing ECM, transport, and mitochondrial genes are shown as violin plots (Fig.4A). Globally, genes in the extracellular matrix, transporters and mitochondrial families are elevated by Npx in both WT and KO mice. These findings are in agreement with the histological analysis (Fig. 2) showing fibrosis due to Npx. Several mitochondrial protein mRNAs (Lyrm1, Hsd3b1, Mrps33, Gm6181, Gm11810, Hsd3b5, Amacr, Lypla1, Ndufaf1, Auh, Immp2l, Gm4952, Agxt2l1, Nme1, Mpv17l, 8430408g22rik, Cox6a2, Acot13, Gm6462, Gm7180, Mrpl33, Gm2382) are overexpressed in WT-Npx, however the plot depicting expression of the electron transport chain and ATP synthase genes, showed decreased expression selectively in WT-Npx but not KO-Npx. This might suggest that mitochondrial efficiency is reduced in WT-Npx but somehow protected in KO-Npx. The expression data for the gene family members that were significantly altered in the Npx samples are shown separately (Fig. 4B), highlighting the different response of the WT and KO mice to kidney injury. ECM genes are more highly expressed in WT-Npx than KO-Npx consistent with the histology. Transporter and mitochondrial genes show greater variance in the expression for the WT Npx than the KO. However respiratory complex genes are significantly downregulated in WT-Npx vs. KO-Npx consistent with the entire gene family data (Fig. 4A) suggesting impaired mitochondria in WT- Npx mice but protection in the KO. Strikingly, the transcriptome analysis also showed that renin synthesis was suppressed and Klkb1 expression was upregulated in WT-Npx mice (Fig.4C).
Figure 3. Transcriptome analysis of the remnant kidney.
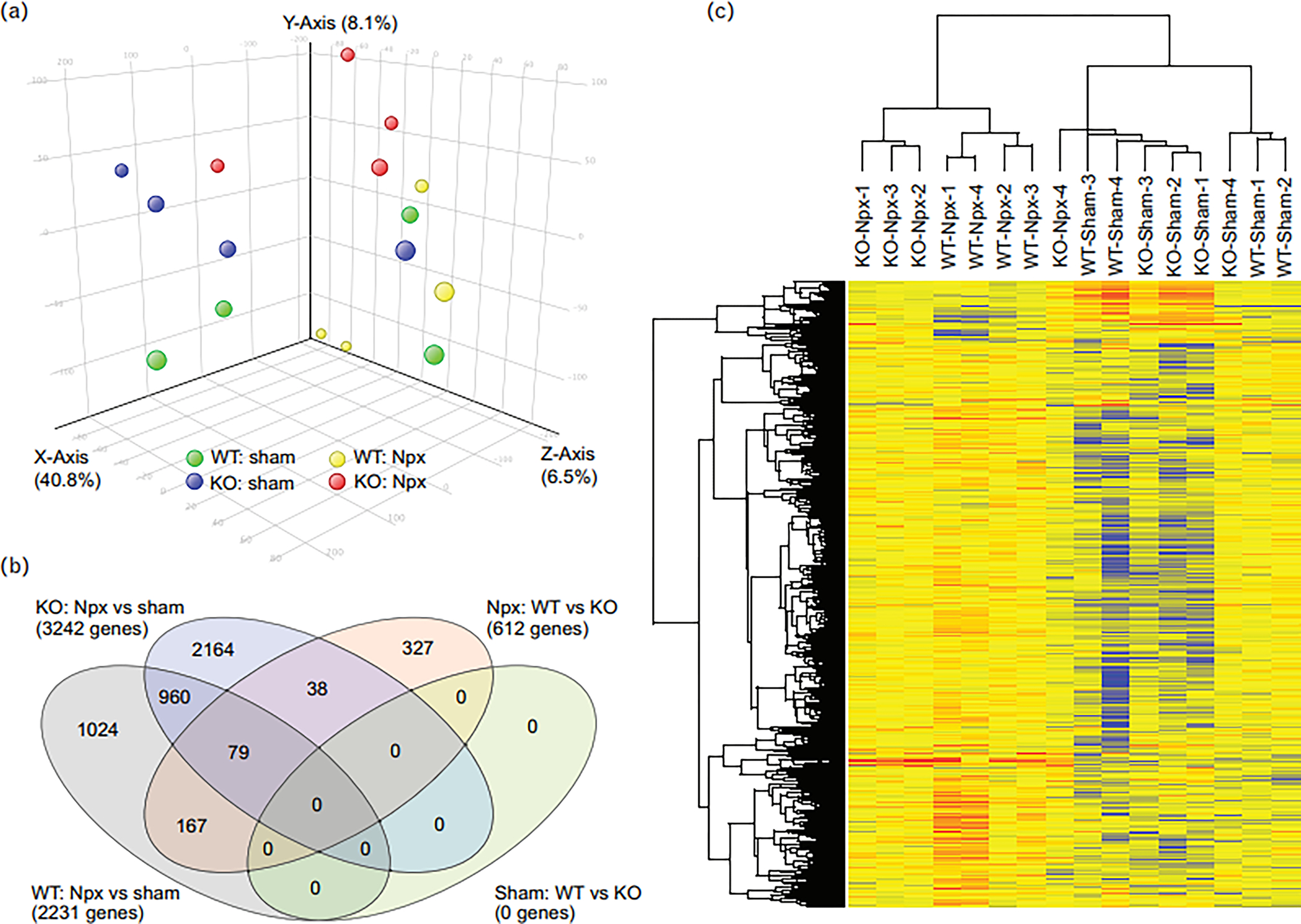
(A) PCA plot of the kidney tissue of mice subjected to sham and Npx surgery. (B) Venn diagram showing the number of genes altered by Npx in WT or KO mice, as well as the genes altered by genotype in sham or Npx mice. (C) The heatmap depicts all the altered genes. All the sham samples cluster together, as do most Npx samples.
Figure 4. Differential expression of genes by strains in response to nephrectomy.
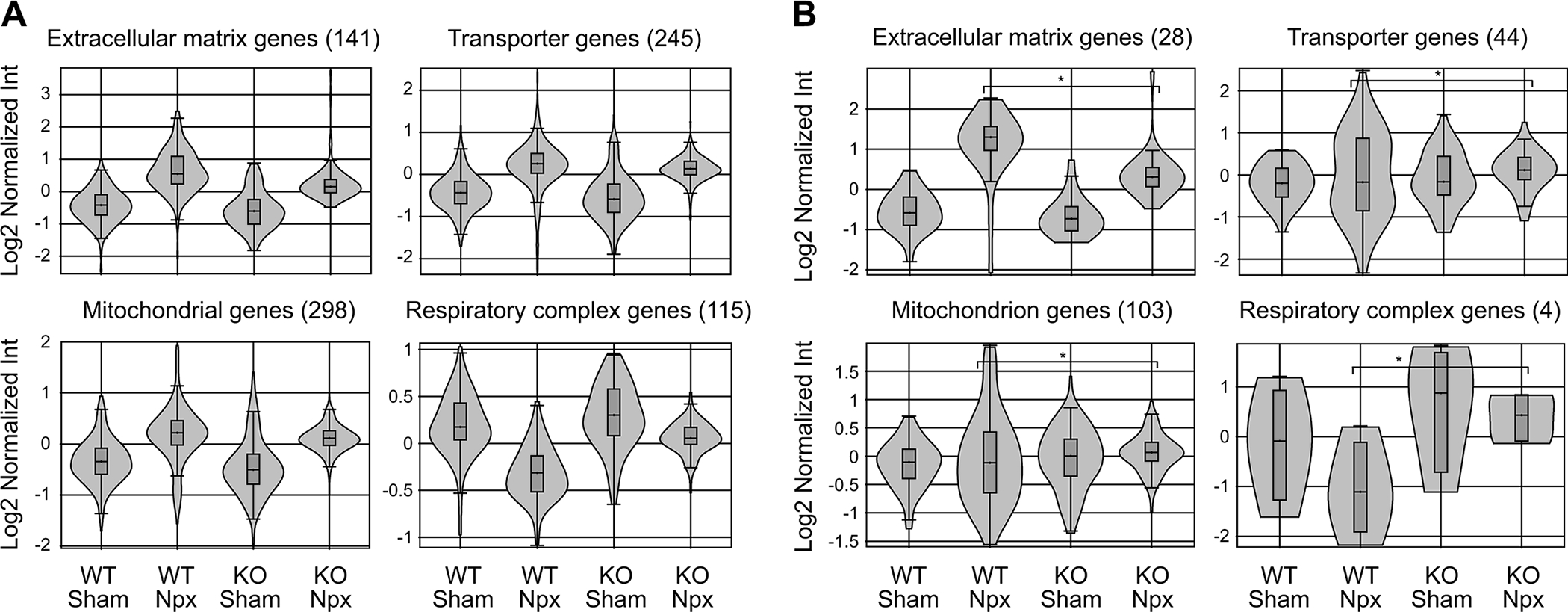
(A) Transcriptome analysis of ECM, transport, and mitochondrial gene families significantly altered in the complete dataset shown as violin plots. Most were up due to Npx in both WT and KO strains. The plot showing electron transport chain and ATP synthase genes were down in WT-Npx but not KO-Npx suggesting reduced mitochondrial efficiency in WT-Npx but somehow protected mitochondrial function in KO-Npx. (B) Gene families differentially expressed between WT-Npx and KO-Npx. There was no difference between WT and KO mice in the sham state. Expression of ECM genes were higher in WT than KO. Expression of transporter and mitochondrial genes showed greater variance in WT-Npx. Respiratory complex genes were downregulated in WT-Npx vs KO-Npx similar to D, again suggesting impaired mitochondria in WT-Npx.
CHGA treatment of mesangial cells triggers NO synthesis.
Since CHGA builds up in the plasma due to loss of kidney function (Figs. 2 & 3) we investigated the response of kidney cells to elevated CHGA. CRL-1927 mouse mesangial kidney cells were incubated with recombinant CHGA protein and the release of NO measured over time (Fig. 5A). In the presence of CHGA, the NO secretion was 2.9-fold and 2.1-fold higher than untreated cells at 8 and 24 hours respectively. LPS, a potent activator of the immune system resulted in 5.9 and 3.8-fold stimulation over untreated cells at the same time points. The stimulation of NO release by mesangial cells treated with CHGA was by upregulation of nitric oxide synthase (iNOS) transcription (Fig.5B) and was abolished by a specific iNOS pharmacological inhibitor L-NIL. The signal transduction mechanism leading to NO release involved protein tyrosine kinase and de novo protein synthesis, as it was blocked by genistein and cycloheximide (Fig.5C). The CHGA induced NO release was completely blocked by pre-incubating cells in the presence of the scavenger receptor (SR-A) ligand fucoidan. Poly-inosinic acid the ligand for both toll-like receptor 3 ligand (TLR3), and SR-A also blocked NO production to some extent (Fig.5D). The activation of iNOS expression was specific to full–length CHGA protein; CHGA’s peptide derivatives- catestatin, vasostatin hydrophobic loop, and WE-14 failed to stimulate NO release (data not shown). Tropomyosin, a protein that has a coiled-coil structure like full-length CHGA, also stimulated NO release whereas poly(Glu)n peptide with a random coil form was ineffective (Fig.5E).
Figure 5. CHGA triggers NO release from mouse glomerular mesangial cells.
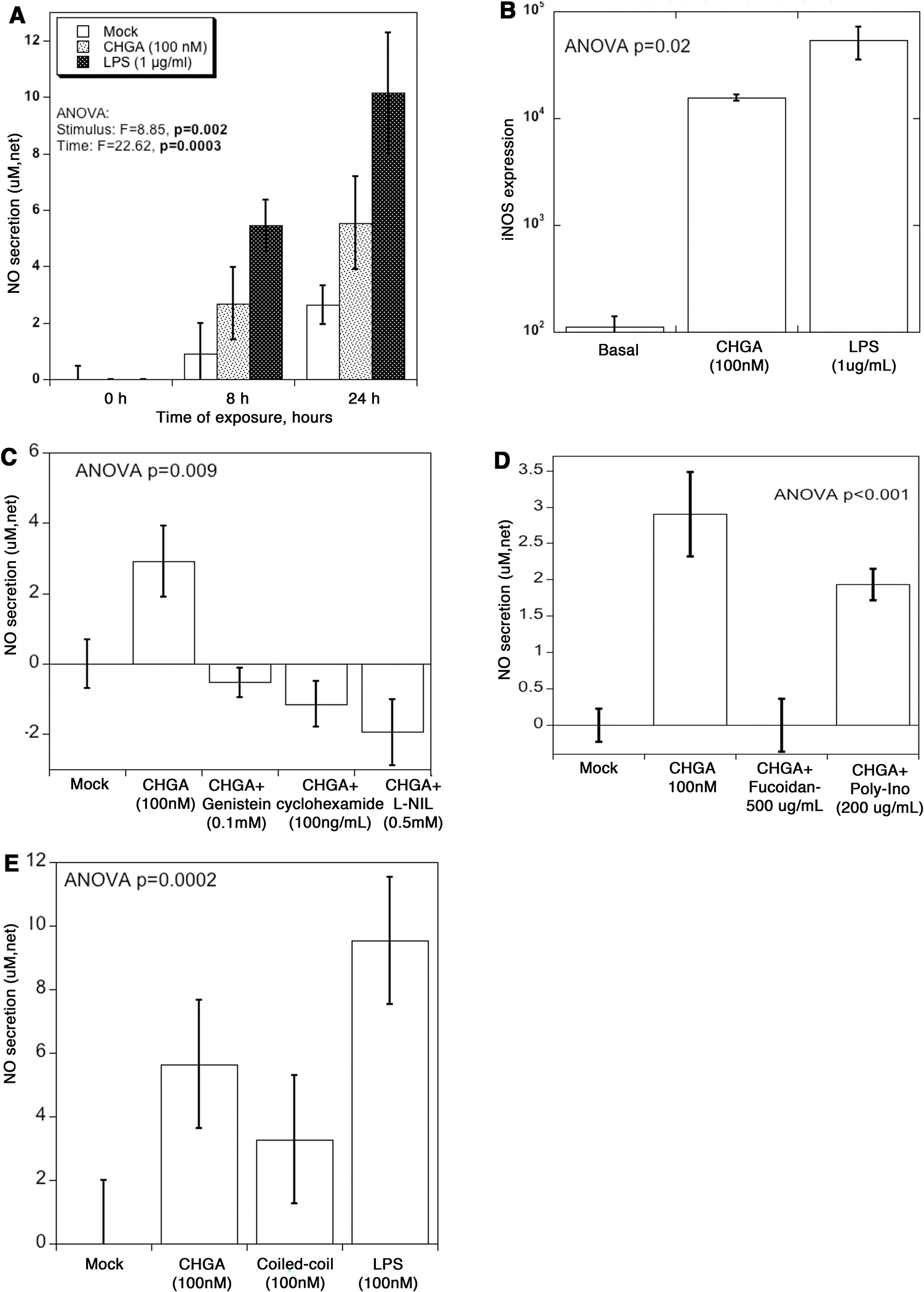
(A) CRL-1927 cells were treated with CHGA (100 nM) or LPS (1 μg/mL) for 8 and 24 hours, and the tissue culture media collected to measure total NO. The assay determined NO concentration based on the enzymatic conversion of nitrate to nitrite by nitrate reductase, followed by a colorimetric detection by Griess reagent. Control media was from cells left untreated. Net secretion was calculated by subtracting zero-hour basal secretion from all other time points. (B) Cells were treated with CHGA or LPS for 24 hours, and the total RNA extracted and processed for real-time PCR. Relative amount of the iNOS mRNA was quantified as iNOS/beta actin ratio. (C) Specificity of CHGA induced NO secretion. Cells were treated with CHGA in presence and absence of L-NIL, cycloheximide and genistein for 24 hours, and net NO secretion was measured from culture media. (D) Cells were pre-treated with SR-A blocker fucoidan or poly-ionosine for 1 hour prior to the addition of CHGA. After 24 hours tissue culture supernatant was harvested and the released NO measured. (E) Cells were treated with full length CHGA, a coiled-coil protein tropomyosin, poly-L-glutamic acid or LPS for 24-hour and the tissue culture supernatant collected to measure total NO.
Concerted transcription of genes involved in NO production and pro-inflammatory markers in mesangial cells treated with CHGA.
Following CHGA or mock treatment of CRL-1927 cells, gene expression was analyzed by microarray. A two-stage amplification of NO synthesis was brought about by an increase in transcription of both the arginine transporter Slc7a7 and iNOS (Fig.6A). L-arginine transported by Slc7a7 serves as a substrate for iNOS to catalyze the production of NO. Expression of several solute carrier family genes was also upregulated in CHGA-treated cells (Fig.6B). Differential expression of pro-inflammatory genes was also observed in mesangial cells including the upregulation of several chemokine and cytokine genes (Fig.6C). Three of the four complement family members were over-expressed in CHGA-treated cells, and the fourth was under-expressed (Fig.6D). Acute phase response genes (Saa3 and Orm1) are upregulated in response to CHGA treatment as in case of serious injury and inflammation (Fig.6E). Oxidative stress is a major cause of damage associated with elevated NO that leads to apoptosis and Fas expression plays a central role in this process. Fas expression was upregulated significantly due to CHGA treatment (Fig.6F).
Figure 6. CHGA impacts transcription of genes involved in NO synthesis and pro inflammatory markers.
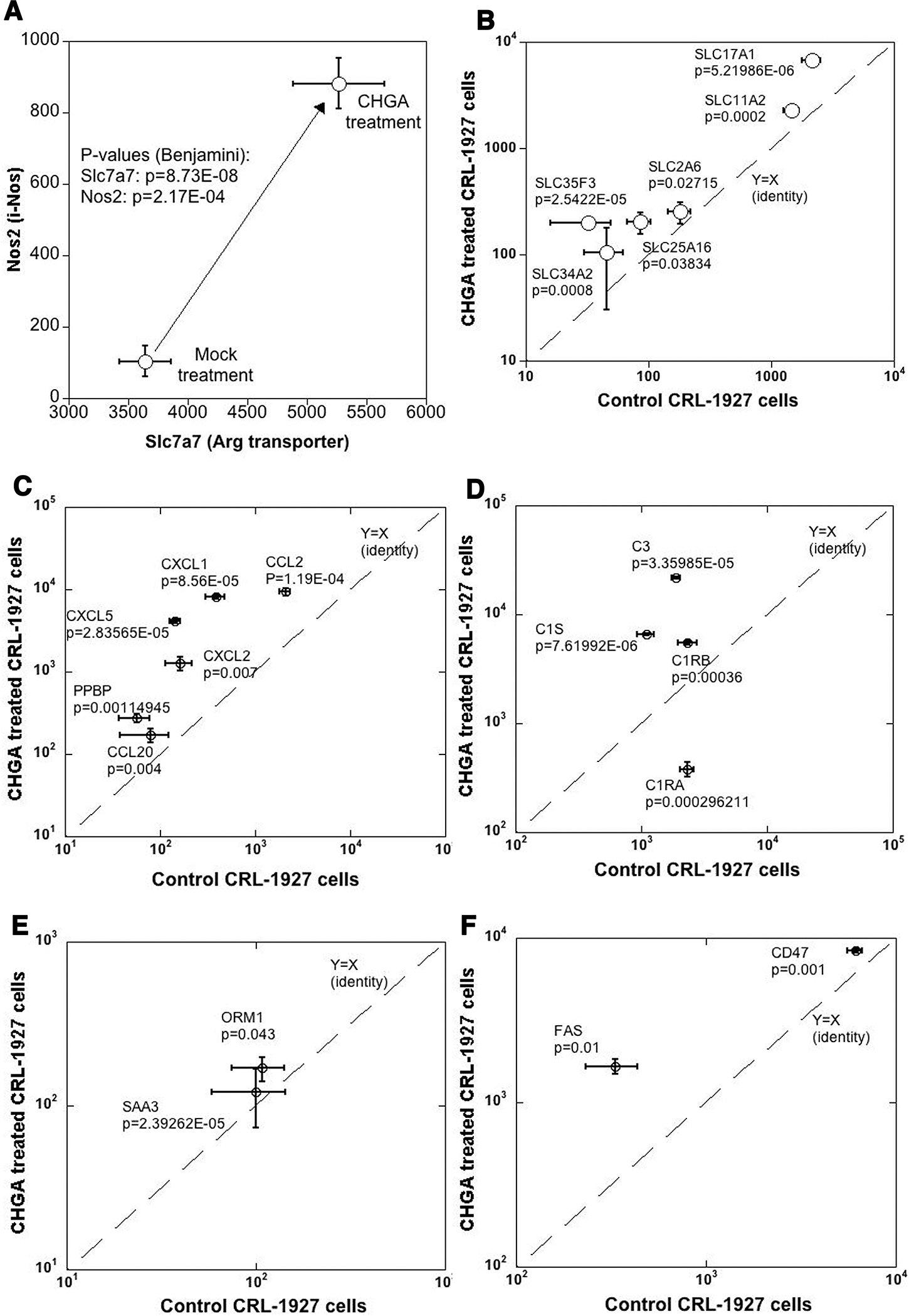
The mesangial cells were treated with CHGA (100 nM) or untreated for 24 hours, harvested and transcriptomes compared (A) A 2-stage amplification of transcripts involved in NO synthesis. Both the precursor Arg transporter and NO biosynthesis enzyme iNOS showed concomitant over-expression. (B) Several solute carrier transport family genes are over-expressed. Similarly, CHGA treated cells overexpress: (C) Cytokine and chemokines family transcripts (D) Complement family transcripts (E) Acute phase inflammatory markers (F) Transcripts related to cellular apoptosis.
Thus, this study describes the association between hypertensive CKD and CHGA, a gene known to play critical roles within the sympathoadrenal response.
DISCUSSION
Epidemiological studies have demonstrated an excessive risk of CKD progression to ESRD among African Americans, even after controlling for confounding factors[20, 21]. In this population, several potential disease-susceptibility loci and gene polymorphisms for hypertensive-CKD leading to ESRD have been identified including CHGA, the adrenergic catecholamine storage protein[8–10, 22–25]. This study supports observations in humans that loss of kidney function leads to build-up of plasma CHGA and triggers mesangial cell activation via scavenger receptor, mounting an oxidative stress response potentiating inflammation and fibrosis ensuing in the disease phenotype.
Sympathetic activation due to kidney failure results in hypertension and cardiovascular complications in many of these patients[26, 27]. CHGA and its hypotensive peptide derivative catestatin have been implicated in both the heritability[28]and pathogenesis of essential hypertension[29–31]. CHGA catalyzes the biogenesis and regulated exocytosis of catechaolamine storage granules in neuroendocrine cells and thus contributes to blood pressure homeostasis[32, 33]. Acetylcholine stimulation results in the simultaneous release of catecholamine and CHGA and the secreted catecholamine goes on to triggers cardiovascular target cells to increase blood pressure[3]. In this study, we explored the role of the CHGA in kidney disease. Kidneys have an essential role in clearing proteins including CHGA from blood; however due to loss of kidney function, there is build-up of CHGA in plasma of patients[34] and in WT-Npx mice (Fig. 2). Our data indicate that post-kidney injury, elevated levels of CHGA in plasma is detrimental to recovery of kidney function (Fig.1). Also stress induced by nephrectomy augmented sympathetic response contributing to CHGA release from adrenergic stores which along with loss of GFR elevated circulating CHGA in mice.
Studies in other rodent models have also shown association between sympathetic nervous system activity and rate of progression of kidney disease. In obese spontaneously hypertensive rats, progressive increase in blood pressure, cardiac remodeling and renal injury were mediated by renal sympathetic activation as they were attenuated by renal sympathetic denervation[35]. In 5/6 nephrectomized rats sympathectomy attenuated hypertension however, the level of proteinuria in in these rats was reduced to a greater extent than what would be expected solely on the basis of lower blood pressure confirming involvement of sympathetic nervous system[36]. Campese’s study[37] indicates that increased sympathetic nervous system activity contributes to the pathogenesis of hypertension and progressive glomerulosclerosis in rats with the renal ablation model of chronic renal failure. In the mouse AKI model elevated neurohormonal signaling of the sympathetic nervous system and the endothelin system play a driving force in kidney and heart damage involving GPCR-Gβγ signaling[38].
Our findings suggest the probable chain of events involves increased sympathetic response, diminished kidney filtration leading to accumulation of CHGA in plasma, triggering augmented expression of pro-fibrotic and inflammation genes resulting in progression of CKD. These findings are in line with the patient cohort confirming the inverse correlation between GFR and plasma CHGA levels (Fig. 4).
Previous studies have reported that plasma CHGA is elevated in CKD patients[4] and several CHGA gene, promoter and 3’UTR polymorphisms have been associated with HT kidney disease[8, 9]. CHGA acts via glomerular endothelium to regulate kidney function, and plasma CHGA correlates positively with endothelin-1 and negatively with GFR[17]. In African Americans with HT-ESRD, CHGA has been identified as a disease-susceptiblity locus, and these patients also show an “intermediate” biological phenotype of diminished circulating levels of the catestatin (the hypotensive peptide derived from CHGA that serves as a nicotinic cholinergic receptor antagonist)[9]. Our study suggests that while injured kidneys contribute to sympathetic activation and elevated BP, the disease outcome is not entirely a result of sympathetic activation. In both strains of Npx mice the sympathetic response is elevated however, loss of kidney function is dramatically accelerated in mice expressing Chga. Similar findings have been observed previously in 5/6th nephrectomy rats whose sympathetic overactivity in early phase after nephrectomy is closely related to progression of CKD, however is independent of changes in blood pressure[39]. In the hypertensive and hyperadrenergic Chga−/− mice, glomerular hyperfiltration is observed[11].
The differential response by both strains to surgery is parsed by mitochondrial dysfunction in mice expressing Chga and as a result expression of the electron transport chain and ATP synthase genes are down in WT-Npx but not KO-Npx (Fig.4). Overall, in WT-Npx as compared to KO-Npx mitochondrial genes are overexpressed, and this generates superoxide radicals leading to oxidative stress and inflammation. These CHGA-induced mitochondrial genes’ transcriptional response was not observed in vitro potentially due to the absence of regulatory factors present in vivo. The kidney receives 20% of cardiac output and consumes 10% of the body’s oxygen to perform its functions, and mitochondrial dysfunction is recognized as a leading factor to many chronic and acute renal diseases[40]. CHGA triggers oxidative stress injury and inflammation leading to energy shortage from either impaired mitochondrial biogenesis or ATP energetics. This could result in the recruitment of immune cells, inflammatory cytokines accumulation, apoptosis, and tissue injury. CHGA triggers mesangial cells to release NO by activating iNOS (Nos2) expression (Fig.5). Under normal physiological condition, the kidneys generate ROS such as NO in amounts that are beneficial to maintain electrolyte homeostasis and extracellular fluid volume[41]. However, inflammatory conditions promote the induction of iNOS expression and the generation of much larger amounts of NO, which participates in modulating aspects of inflammatory processes. NO now binds to ferrous heme sites and reacts with biological molecules to disrupt iron-sulfur centers and reacts with catecholamine and generates other reactive oxygen and nitrogen molecules resulting in pathophysiological processes [40].
The activation of Toll-like receptor 4 (TLR4 or human ortholog SR-A) plays a fundamental role in pathogen recognition and activation of innate immunity[42],[43]. Anders proposed that some inflammation also accompanies non-pathogen injury to the kidney because damaged renal tissues activate innate immunity by releasing immune-stimulatory “danger signaling” molecules. The resulting non-pathogen signaling turns maladaptive causing renal tissue scarring. This happens because intrarenal inflammation is unable to attenuate the metabolic, hemodynamic, toxic, or autoimmune drivers of kidney injury[44]. Our findings (Fig.5) suggest CHGA might be such a signaling molecule activating TLR 4/SR-A which is known to bind to endotoxin LPS. LPS similarly via TLR triggers an inflammatory response of chemokines and cytokines[45]. TLRs on or in kidney cells transduce the recognition of such danger signals into the secretion of cytokines, chemokines, acute phase inflammatory makers and apoptosis genes as is observed in CRL-1927 cells treated with CHGA. TLR4 has an important role in renal interstitial fibrosis and CKD progression[46].
CONCLUSION
Our results are consistent with the model (Fig.7) depicting the chain of events from kidney injury to the development of CKD. Data from mice support observations in humans that after loss of kidney function, build-up of plasma CHGA activates via SR-A mesangial cells, mounting an oxidative stress response potentiating inflammation and fibrosis eventuating in CKD phenotype. These findings should provide new inroads into therapeutic approaches for a currently intractable health disparity.
Figure 7.
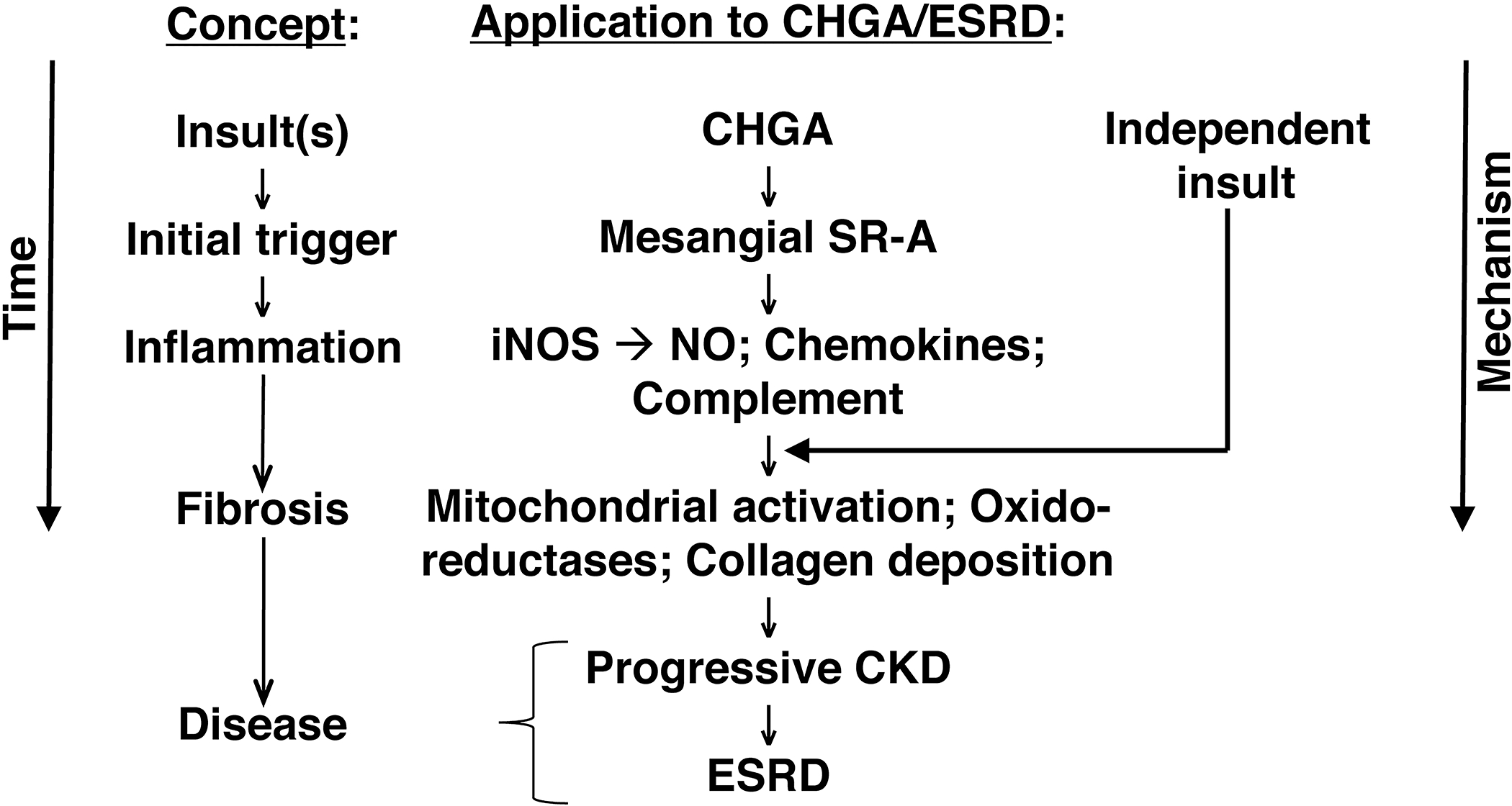
CKD/ESRD and the CHGA pathway: From pathogenic molecule to disease.
Supplementary Material
Supplementary data 1. Gene expression raw data with FDR correction
Supplementary data 2. Enrichment analysis report by GO molecular functions
Supplementary data 3. Collagen volume fraction calculated from Masson Trichrome stained kidney tissue sections
Support:
Funding for this research was with grants R01HL108629 (NHLBI) and R01DK094894 (NIDDK) awarded to SMV. This work was also supported by the UAB-UCSD O’Brien Core Center for Acute Kidney Injury Research (NIH P30-DK079337).
ABBREVIATIONS:
- CHGA
Chromogranin A
- CKD
hypertensive chronic kidney disease
- Npx
5/6th nephrectomy
- WT
wild-type (Chga+/+)
- KO
knockout (Chga−/−)
- SR-A
scavenger receptor
- ESRD
end stage renal disease
- AASK
African American Study of Kidney Disease and Hypertension
- GFR
glomerular filtration rate
- NO
nitric oxide
- BP
blood pressure
- HR
heart rate
- iNOS
nitric oxide synthase
- CRL-1927
mouse mesangial cells
Footnotes
The authors have declared that no conflict of interest exists.
REFERENCES
- 1.Webster AC, Nagler EV, Morton RL, Masson P. Chronic Kidney Disease. Lancet. 2017; 389 (10075):1238–52. [DOI] [PubMed] [Google Scholar]
- 2.Zoccali C, Mallamaci F, Parlongo S, Cutrupi S, Benedetto FA, Tripepi G, et al. Plasma norepinephrine predicts survival and incident cardiovascular events in patients with end-stage renal disease. Circulation. 2002; 105 (11):1354–9. [DOI] [PubMed] [Google Scholar]
- 3.Taupenot L, Harper KL, O’Connor DT. The chromogranin-secretogranin family. N Engl J Med. 2003; 348 (12):1134–49. [DOI] [PubMed] [Google Scholar]
- 4.O’Connor DT, Pandlan MR, Carlton E, Cervenka JH, Hslao RJ. Rapid radioimmunoassay of circulating chromogranin A: in vitro stability, exploration of the neuroendocrine character of neoplasia, and assessment of the effects of organ failure. Clin Chem. 1989; 35 (8):1631–7. [PubMed] [Google Scholar]
- 5.Tramonti G, Ferdeghini M, Annichiarico C, Norpoth M, Donadio C, Bianchi R, et al. Relationship between renal function and blood level of chromogranin A. Ren Fail. 2001; 23 (3–4):449–57. [DOI] [PubMed] [Google Scholar]
- 6.Chen Y, Rao F, Rodriguez-Flores JL, Mahapatra NR, Mahata M, Wen G, et al. Common genetic variants in the chromogranin A promoter alter autonomic activity and blood pressure. Kidney Int. 2008; 74 (1):115–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen Y, Rao F, Rodriguez-Flores JL, Mahata M, Fung MM, Stridsberg M, et al. Naturally occurring human genetic variation in the 3’-untranslated region of the secretory protein chromogranin A is associated with autonomic blood pressure regulation and hypertension in a sex-dependent fashion. J Am Coll Cardiol. 2008; 52 (18):1468–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Y, Rao F, Wen G, Gayen JR, Zhang K, Vaingankar SM, et al. Naturally occurring genetic variants in human chromogranin A (CHGA) associated with hypertension as well as hypertensive renal disease. Cell Mol Neurobiol. 2010; 30 (8):1395–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Salem RM, Cadman PE, Chen Y, Rao F, Wen G, Hamilton BA, et al. Chromogranin A polymorphisms are associated with hypertensive renal disease. J Am Soc Nephrol. 2008; 19:600–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu L, Jiang L, Zhou XJ, Zhu L, Zhang H. Common genetic variants in the chromogranin a promoter are associated with renal injury in IgA nephropathy patients with malignant hypertension. Ren Fail. 2010; 32 (1):41–6. [DOI] [PubMed] [Google Scholar]
- 11.Zhang K, Mir SA, Hightower CM, Miramontes-Gonzalez JP, Maihofer AX, Chen Y, et al. Molecular Mechanism for Hypertensive Renal Disease: Differential Regulation of Chromogranin A Expression at 3’-Untranslated Region Polymorphism C+87T by MicroRNA-107. J Am Soc Nephrol. 2015; 26:1816–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gagnon RF, Gallimore B. Characterization of a mouse model of chronic uremia. Urol Res. 1988; 16 (2):119–26. [DOI] [PubMed] [Google Scholar]
- 13.Hayakawa S, Ohashi K, Shibata R, Kataoka Y, Miyabe M, Enomoto T, et al. Cardiac myocyte-derived follistatin-like 1 prevents renal injury in a subtotal nephrectomy model. J Am Soc Nephrol. 2015; 26 (3):636–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mir SA, Zhang K, Milic M, Gu Y, Rieg T, Ziegler M, et al. Analysis and validation of traits associated with a single nucleotide polymorphism Gly364Ser in catestatin using humanized chromogranin A mouse models. J Hypertens. 2016; 34:68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Durschlag M, Wurbel H, Stauffacher M, Von Holst D. Repeated blood collection in the laboratory mouse by tail incision--modification of an old technique. Physiol Behav. 1996; 60 (6):1565–8. [DOI] [PubMed] [Google Scholar]
- 16.Corti A, Gasparri A, Chen FX, Pelagi M, Brandazza A, Sidoli A, et al. Characterisation of circulating chromogranin A in human cancer patients. Br J Cancer. 1996; 73 (8):924–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Y, Mahata M, Rao F, Khandrika S, Courel M, Fung MM, et al. Chromogranin A regulates renal function by triggering Weibel-Palade body exocytosis. J Am Soc Nephrol. 2009; 20 (7):1623–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mahapatra NR, O’Connor DT, Vaingankar SM, Hikim AP, Mahata M, Ray S, et al. Hypertension from targeted ablation of chromogranin A can be rescued by the human ortholog. J Clin Invest. 2005; 115 (7):1942–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eddy AA. Overview of the cellular and molecular basis of kidney fibrosis. Kidney Int. supplements. 2014; 4 (1):2–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Freedman BI, Limou S, Ma L, Kopp JB. APOL1-Associated Nephropathy: A Key Contributor to Racial Disparities in CKD. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2018; 72 (5s1):S8–s16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moxey-Mims M Kidney Disease in African American Children: Biological and Nonbiological Disparities. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2018; 72 (5s1):S17–s21. [DOI] [PubMed] [Google Scholar]
- 22.Hayek SS, Koh KH, Grams ME, Wei C, Ko YA, Li J, et al. A tripartite complex of suPAR, APOL1 risk variants and alphavbeta3 integrin on podocytes mediates chronic kidney disease. Nat Med. 2017; 23 (8):945–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Foster MC, Coresh J, Fornage M, Astor BC, Grams M, Franceschini N, et al. APOL1 variants associate with increased risk of CKD among African Americans. J Am Soc Nephrol. 2013; 24 (9):1484–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boger CA, Heid IM. Chronic kidney disease: novel insights from genome-wide association studies. Kidney Blood Press Res. 2011; 34 (4):225–34. [DOI] [PubMed] [Google Scholar]
- 25.Hsu CC, Bray MS, Kao WH, Pankow JS, Boerwinkle E, Coresh J. Genetic variation of the renin-angiotensin system and chronic kidney disease progression in black individuals in the atherosclerosis risk in communities study. J Am Soc Nephrol. 2006; 17 (2):504–12. [DOI] [PubMed] [Google Scholar]
- 26.Klag MJ, Whelton PK, Randall BL, Neaton JD, Brancati FL, Ford CE, et al. Blood pressure and end-stage renal disease in men. N Engl J Med. 1996; 334 (1):13–8. [DOI] [PubMed] [Google Scholar]
- 27.Schlaich MP, Socratous F, Hennebry S, Eikelis N, Lambert EA, Straznicky N, et al. Sympathetic activation in chronic renal failure. J Am Soc Nephrol. 2009; 20 (5):933–9. [DOI] [PubMed] [Google Scholar]
- 28.Takiyyuddin MA, Parmer RJ, Kailasam MT, Cervenka JH, Kennedy B, Ziegler MG, et al. Chromogranin A in human hypertension. Influence of heredity. Hypertension. 1995; 26 (1):213–20. [DOI] [PubMed] [Google Scholar]
- 29.Sahu BS, Sonawane PJ, Mahapatra NR. Chromogranin A: a novel susceptibility gene for essential hypertension. Cellular and molecular life sciences : CMLS. 2010; 67 (6):861–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mazza R, Tota B, Gattuso A. Cardio-vascular activity of catestatin: interlocking the puzzle pieces. Curr Med Chem. 2015; 22:292–304. [DOI] [PubMed] [Google Scholar]
- 31.Troger J, Theurl M, Kirchmair R, Pasqua T, Tota B, Angelone T, et al. Granin-derived peptides. Prog Neurobiol. 2017; 154:37–61. [DOI] [PubMed] [Google Scholar]
- 32.Friese RS, Altshuler AE, Zhang K, Miramontes-Gonzalez JP, Hightower CM, Jirout ML, et al. MicroRNA-22 and promoter motif polymorphisms at the Chga locus in genetic hypertension: functional and therapeutic implications for gene expression and the pathogenesis of hypertension. Hum Mol Genet. 2013; 22:3624–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Loh YP, Cheng Y, Mahata SK, Corti A, Tota B. Chromogranin A and derived peptides in health and disease. J Mol Neurosci. 2012; 48 (2):347–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hsiao RJ, Mezger MS, O’Connor DT. Chromogranin A in uremia: progressive retention of immunoreactive fragments. Kidney Int. 1990; 37 (3):955–64. [DOI] [PubMed] [Google Scholar]
- 35.Linz D, Hohl M, Schutze J, Mahfoud F, Speer T, Linz B, et al. Progression of kidney injury and cardiac remodeling in obese spontaneously hypertensive rats: the role of renal sympathetic innervation. Am J Hypertens. 2015; 28 (2):256–65. [DOI] [PubMed] [Google Scholar]
- 36.Amann K, Rump LC, Simonaviciene A, Oberhauser V, Wessels S, Orth SR, et al. Effects of low dose sympathetic inhibition on glomerulosclerosis and albuminuria in subtotally nephrectomized rats. J Am Soc Nephrol. 2000; 11 (8):1469–78. [DOI] [PubMed] [Google Scholar]
- 37.Campese VM. The kidney and the neurogenic control of blood pressure in renal disease. J Nephrol. 2000; 13 (3):221–4. [PubMed] [Google Scholar]
- 38.Kamal FA, Travers JG, Schafer AE, Ma Q, Devarajan P, Blaxall BC. G Protein-Coupled Receptor-G-Protein betagamma-Subunit Signaling Mediates Renal Dysfunction and Fibrosis in Heart Failure. J Am Soc Nephrol. 2017; 28 (1):197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kobuchi S, Tanaka R, Funai A, Suzuki R, Yazawa M, Tsutsui H, et al. Involvement of Renal Sympathetic Nerve Overactivation in the Progression of Chronic Kidney Disease in Rats. J Cardiovasc Pharmacol. 2014; 63 (1):9–15. [DOI] [PubMed] [Google Scholar]
- 40.Ratliff BB, Abdulmahdi W, Pawar R, Wolin MS. Oxidant Mechanisms in Renal Injury and Disease. Antioxid Redox Signal 2016; 25 (3):119–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gonzalez-Vicente A, Garvin JL. Effects of Reactive Oxygen Species on Tubular Transport along the Nephron. Antioxidants (Basel, Switzerland). 2017; 6 (2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, et al. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol. (Baltimore, Md : 1950). 1999; 162 (7):3749–52. [PubMed] [Google Scholar]
- 43.Inferences Beutler B., questions and possibilities in Toll-like receptor signalling. Nature. 2004; 430 (6996):257–63. [DOI] [PubMed] [Google Scholar]
- 44.Anders HJ. Toll-like receptors and danger signaling in kidney injury. J Am Soc Nephrol. 2010; 21 (8):1270–4. [DOI] [PubMed] [Google Scholar]
- 45.Lund S, Christensen KV, Hedtjarn M, Mortensen AL, Hagberg H, Falsig J, et al. The dynamics of the LPS triggered inflammatory response of murine microglia under different culture and in vivo conditions. J Neuroimmunol. 2006; 180 (1–2):71–87. [DOI] [PubMed] [Google Scholar]
- 46.Souza AC, Tsuji T, Baranova IN, Bocharov AV, Wilkins KJ, Street JM, et al. TLR4 mutant mice are protected from renal fibrosis and chronic kidney disease progression. Physiol Rep. 2015; 3(9): e12558. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data 1. Gene expression raw data with FDR correction
Supplementary data 2. Enrichment analysis report by GO molecular functions
Supplementary data 3. Collagen volume fraction calculated from Masson Trichrome stained kidney tissue sections