

Abstract
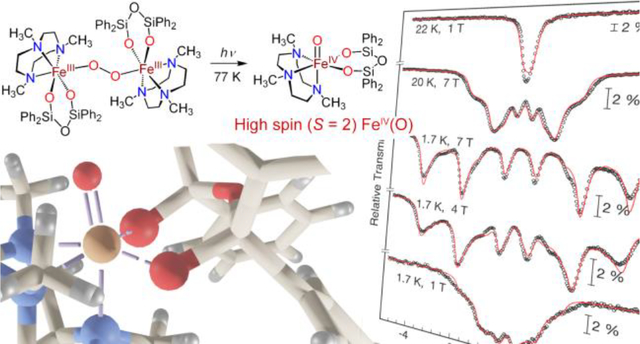
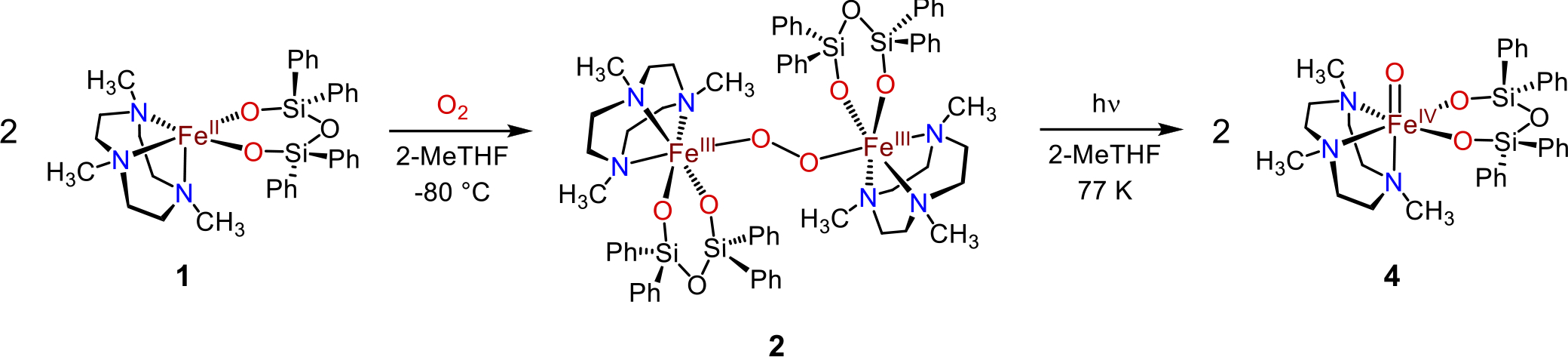
Addition of dioxygen at low temperature to the nonheme ferrous complex, FeII(Me3TACN)(OSiPh2)2O) (1), in 2-MeTHF produces a peroxo-bridged diferric complex, Fe2III(μ-O2)(Me3TACN)2((OSiPh2)2O)2 (2), which was characterized by UV-vis, resonance Raman, and variable field Mössbauer spectroscopies. Illumination of a frozen solution of 2 in THF with white light leads to homolytic O–O bond cleavage and generation of a FeIV(O) complex 4 (ν(FeO) = 818 cm−1; δ = 0.22 mm s−1, ΔEQ = 0.23 mm s−1). Variable field Mössbauer spectroscopy measurements show that 4 is a rare example of a high-spin, S = 2 FeIV(O) complex, and the first synthetic example to be generated directly from O2. Complex 4 is highly reactive, as expected for a high-spin ferryl, and decays rapidly in fluid solution at cryogenic temperatures. This decay process in 2-MeTHF involves C–H cleavage of the solvent. However, the controlled photolysis of 2 in situ with visible light and excess phenol substrate leads to competitive phenol oxidation, via the proposed transient generation of 4 as the active oxidant.
Graphical Abstract
INTRODUCTION
The controlled activation of dioxygen to generate strong oxidants capable of selectively oxidizing organic substrates is a significant chemical challenge. Despite the favorable thermodynamics for the complete four-electron reduction of O2, the initial one electron reduction step required to activate O2 is kinetically challenging.1–2 Redox-active transition metal centers are capable of binding O2 to promote reduction, but require appropriately tuned redox potentials and/or metal–oxygen bond thermodynamics in order to activate O2 via inner sphere mechanisms.3 Following O2 binding, a series of metal–oxygen intermediates can be generated, including metal –superoxo –peroxo, –oxo, and –hydroxo species, each of which has the potential to react with organic substrates.4–10 Furthermore, uncontrolled decay of these species can lead to the unproductive generation of chemically inert metal-oxides. It is a significant challenge to control the formation and decay of these reactive intermediates following the initial O2 binding to a transition metal center.
Metalloenzymes, in contrast, have evolved to activate O2 through highly choreographed mechanisms, leading to remarkably potent and selective catalytic oxidation chemistry. Nonheme iron oxidases and oxygenases are a major sub-class of these enzymes.11–12 Examples of mononuclear nonheme iron enzymes include α-ketoglutarate-dependent dioxygenases and the thiol dioxygenases. The proposed mechanisms for these enzymes include formation of an initial superoxo species, followed by a peroxo-bridged intermediate formed between the iron center and redox-active cofactor or substrate (Figure 1).
Figure 1.
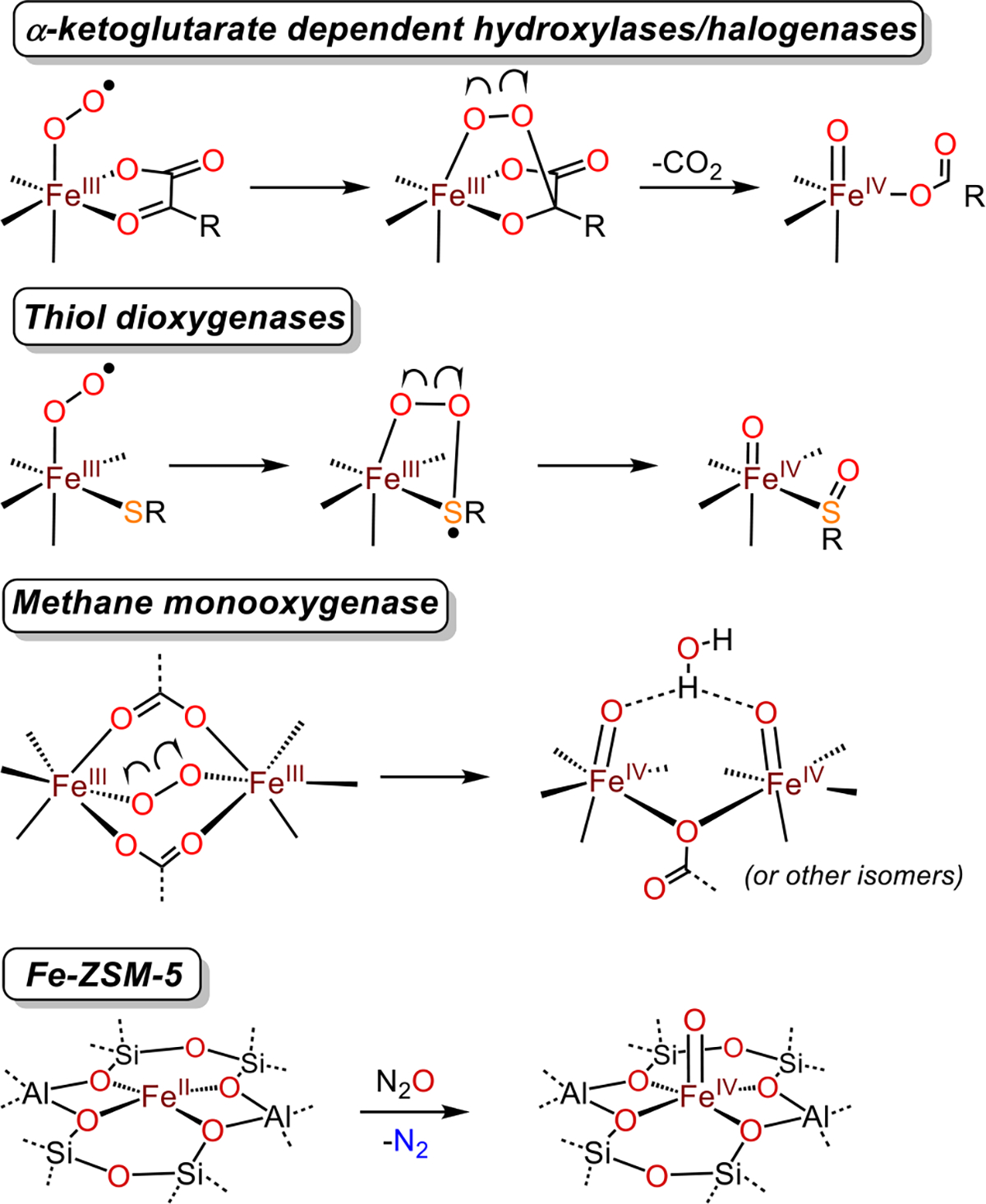
Cleavage of peroxo O–O bonds to give high-spin FeIV(O) intermediates in select nonheme iron enzymes and formation of a high-spin FeIV(O) intermediate in Fe-ZSM-5.
Homolytic cleavage of the O–O bond in the peroxo intermediate leads to a ferryl species (FeIV(O)) that serves as a potent oxidant, which, in the α-KG enzymes, can functionalize strong C–H bonds.11–14 The nonheme diiron enzyme methane monooxygenase is thought to use a similar strategy, in which a diferrous core reacts with O2 to generate a peroxo-bridged diferric intermediate. This intermediate can then undergo homolytic O-O cleavage to give a bis-μ-oxo diiron(IV) species,15 or alternatively, two terminal FeIV(O) units as shown in Scheme 1, that is capable of hydroxylating methane.16 The FeIV(O) intermediates in the aforementioned examples are thought to be high-spin (S = 2) FeIV(O) species. These quintet FeIV(O) species are, in general, predicted to be much more powerful oxidants than triplet (S = 1) FeIV(O) species.12, 17, 18
Scheme 1.
Generation of Complexes 2 and 4
The vast majority of well-characterized, synthetic FeIV(O) species exhibit intermediate-spin (S = 1) ground states and generally muted reactivity as opposed to what is seen for enzymatic FeIV(O) intermediates.19 The few examples of synthetic, high-spin FeIV(O) species that are reported take advantage of weak-field ligands, or a trigonal coordination environment, or both.20 These species also typically employ sterically bulky or H-bonding ligand sets, which help stabilize the ferryl core, but attenuate reactivity with exogenous organic substrates. A few examples of high-spin FeIV(O) species have been generated in the absence of sterically bulky ligands, and in these cases high reactivity toward C-H and O-H bonds is observed.21–22 A highly reactive high-spin FeIV(O) intermediate was characterized in the Fe-doped zeolite ZSM-5 (ZSM = Zeolite Socony Mobil), which can convert methane to methanol in the presence of N2O.23–24 Spectroscopic studies indicate that a tetragonal, silicate-ligated, high-spin FeIV(O) species is the active oxidant responsible for methane C–H bond hydroxylation (Figure 1). A high-spin FeIV(O) intermediate was also implicated, although not directly detected, in the oxidation of ethane to ethanol with N2O as the O-atom source in an Fe-containing metal-organic framework.25 In both of these heterogeneous systems, the S = 2 ground state was attributed to the relatively weak-field oxygen donors.
Our lab has focused on understanding how nonheme iron centers activate O2, particularly in the presence of sulfur donors.26 We recently reported a peroxo-bridged diferric complex, derived from the reaction of O2 with the thiolate-ligated FeII(Me3TACN)(S2SiMe2), and showed that controlled, homolytic O–O bond cleavage can be initiated by photolysis or thermolysis, producing FeIV(O)(Me3TACN)(S2SiMe2).27–28 This FeIV(O) species abstracts hydrogen from relatively weak O–H bonds, but is inert toward C–H bond substrates. Spectroscopic and computational studies pointed to an S = 1 ground state for this ferryl species. In the work presented here, we have replaced the sulfur donors in the former species with weaker-field silanolate oxygen donors, with the hypothesis that this substitution could lead to a much more reactive, high-spin FeIV(O) species. A similar strategy has been attempted by Limberg to model the ferryl intermediate in Fe-ZSM-5.29–30 Although these compounds have the same tetragonal symmetry and all-oxygen coordination environment at Fe as seen in Fe-ZSM-5, an FeIV(O) species has not yet been observed in these systems. Herein, we report the reactivity of the FeII complex, FeII(Me3TACN)((OSiPh2)2O) (1),31 an analog of FeII(Me3TACN)(S2SiMe2) in which the sulfur donors are replaced with anionic siloxide oxygen donors. Reaction of 1 with dioxygen produces a metastable, peroxo-bridged, diferric species (2) which can be trapped at −80 °C. Upon warming, this species rapidly decays to give a stable FeIII2(μ-O) complex (3), but illumination of 2 with a white light source prior to warming instead generates a FeIV(O) complex (4). This ferryl species is extremely reactive, rapidly decomposing in solution at cryogenic temperatures, but amenable to spectroscopic characterization in frozen solvent matrix. Spectroscopic and computational studies, including high-field Mössbauer, show unambiguously that 4 is a high-spin (S = 2) ferryl complex, and is strikingly similar to the ferryl intermediate in the nonheme iron enzyme TauD. Complex 4 is also, to our knowledge, the only high-spin FeIV(O) complex that is prepared directly from O2 and a ferrous precursor. Decay of 4 likely proceeds via H-atom abstraction from solvent. However, if 4 is generated via photolysis of 2 in the presence of excess phenol substrate, the H-atom transfer (HAT) from phenol to 4 appears to be competitive with the solvent-induced decay process. Thus we can harness the oxidizing power of a high-spin ferryl with light at low temperature.
RESULTS AND DISCUSSION
Addition of excess, dry, dioxygen gas to a solution of 131 in THF at 23 °C results in a rapid color change to purple that persists for about 1 s then gradually turns pale yellow. In contrast, when the same reaction was carried out at − 80 °C, the purple species 2 was stabilized, exhibiting features at 350 nm (ε ~ 5000 M−1 cm−1 per Fe) and 560 nm (ε ~ 3200 M−1 cm−1 per Fe) in the UV-vis spectrum (Figure 2). Complex 2 can be generated in THF, 2-MeTHF, CS2, acetone, and butyronitrile and is stable over one month at −80 °C. Attempts to obtain crystals of 2 have thus far been unsuccessful. EPR spectroscopy indicates that 2 is EPR silent, which is consistent with an integer spin ground state for 2. Mössbauer spectra of complex 2(57Fe) at 80 K in THF reveal a quadrupole doublet with δ = 0.50 mm s−1 and │ΔEQ│= 0.63 mm s−1 (Figure 3).
Figure 2.
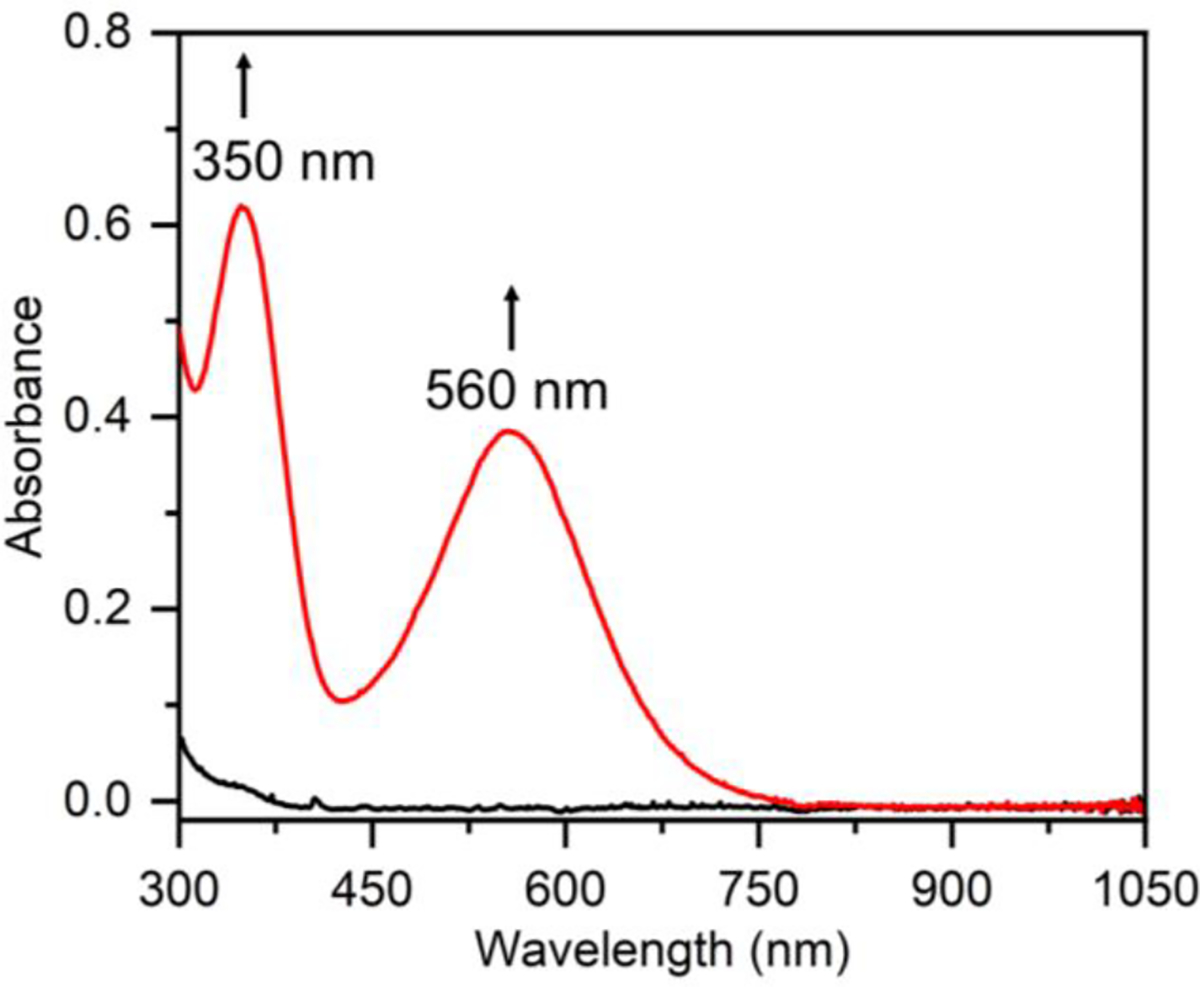
UV-vis spectra showing conversion of 1 (0.12 mM, black line) to 2 (red line) immediately following exposure to excess O2(g) in THF at −80 °C.
Figure 3.
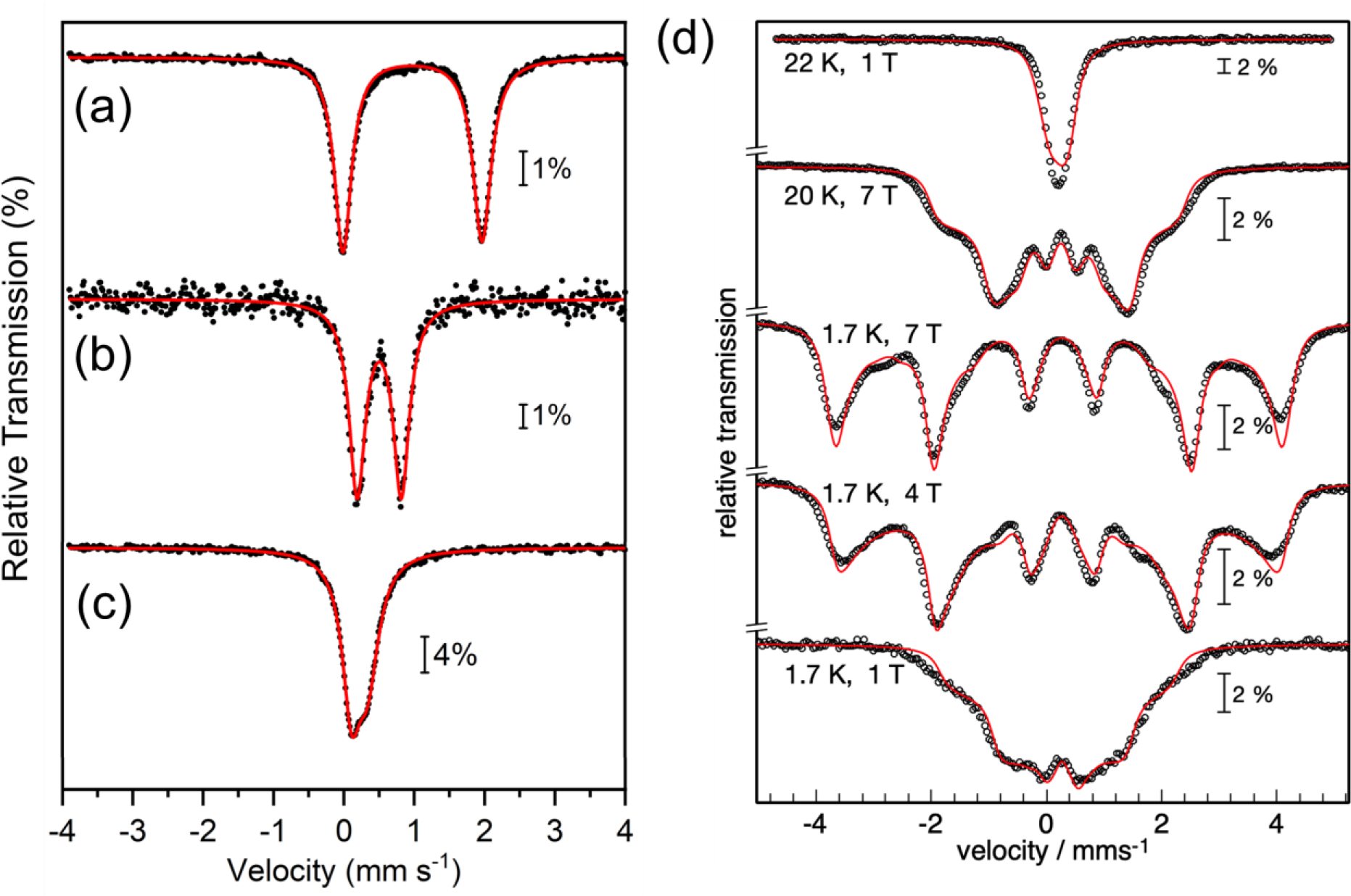
Zero-field Mössbauer spectra (80 K) of (a) 1, (b) 2, and (c) 4. (d) Variable field Mössbauer spectra of 4 recorded at 1.7 – 22 K with 1 – 7 T field applied perpendicular to the γ rays. Data shown in black. Best global fits shown as red lines.
These parameters are typical of high spin ferric ions. Accordingly, magnetic Mössbauer spectra recorded with 7 T applied field in the temperature range 1.7 – 80 K reveal the presence of a ferric dimer complex with total spin St = 0 ground state. The corresponding antiferromagnetic super-exchange coupling was found to be J = −23(±5) cm−1, which is relatively weak compared to other diferric peroxo complexes32 (we use the notation Hex = −2JS1·S2; details of the simulations with local spins S1 = S2 = 5/2 are given in the Supporting Information, Figure S4).
The low J value of 2 is typical of an unsupported peroxo bridge,33–34 and resembles that of Kitajima’s oxygen adduct of FeII(OBz)(HB(3,5-iPr2pz)3) with J = −33 cm−1.35 In contrast, diferric peroxo complexes with an additional oxo- or hydroxo bridge usually show stronger antiferromagnetic coupling.33 It should be noted that the exchange pathway through the peroxo bridge depends on the dihedral angle of the iron magnetic axes.36 For example, dicopper peroxo complexes can show substantial ferromagnetic coupling that is dependent on the dihedral angle of the magnetic axes.37
The electric field gradient (efg) for the two indistinguishable iron sites of 2 was found to be negative (ΔEQ = −0.63 mm s−1), but the asymmetry parameter, η = 0.93, is close to its conventional upper limit (η = 1), rendering two efg components, Vzz and Vyy, with opposite signs but of about the same size. This situation, with weak ΔEQ and large η, suggests an absence of asymmetry in the coordination environment, which may be expected for iron-peroxo groups with typical Fe-O bond lengths, in contrast to the short Fe-O bonds seen for terminal FeIV(O) groups.38
Resonance Raman (RR) spectroscopy on 2 was performed at 110 K with 647 nm laser excitation. The spectrum of 2 clearly displays two isotope-sensitive bands at 896 cm−1 (Δ18O = −50 cm−1) and 415 cm−1 (Δ18O = −17 cm−1) (Figure 4). These bands are assigned to ν(O–O) and ν(Fe–O) modes, respectively, and are consistent with an Fe(peroxo) species.32 Taken together, the Mössbauer, EPR, and RR spectroscopic data are consistent with the assignment of 2 as a (peroxo-bridged)diferric complex, which results from the reaction between 1 and O2 at low temperature (Scheme 1).
Figure 4.
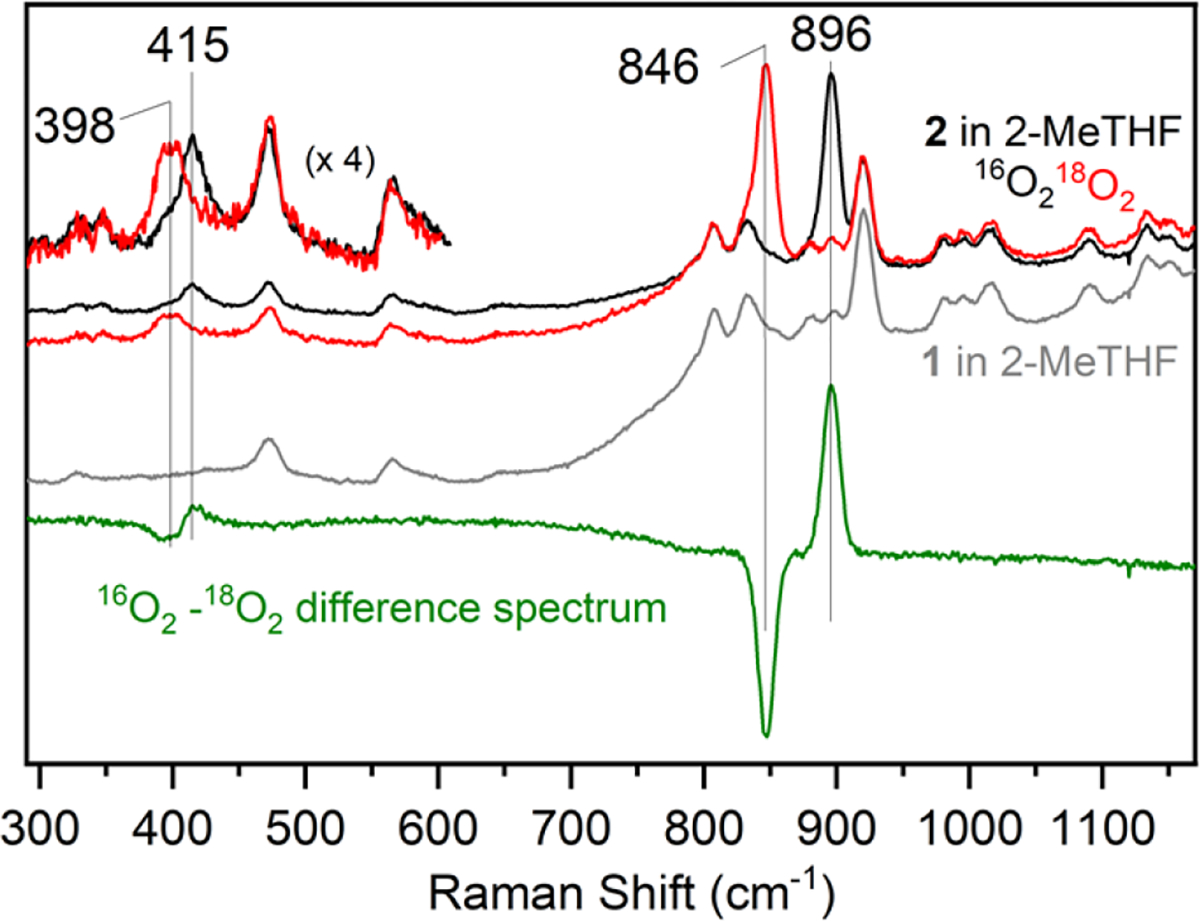
RR spectra of 2 in frozen 2-MeTHF at 110 K (λexc = 647 nm). Black and red spectra show 2(16O) and 2(18O) data, respectively. The grey spectrum shows the spectrum for 1 in 2-MeTHF. The green spectrum is the difference spectrum for 2(16O) – 2(18O).
We recently reported a related (peroxo)diiron complex bearing the silanedithiolate ligand, (S2SiMe2)2−.27 This complex exhibited a ν(O–O) of 849 cm−1, which is more activated than that seen for 2. The less activated O–O bond of 2 is surprising given the more negative E1/2(FeIII/FeII) potential seen for the starting ferrous complex 1 (E1/2 = −0.90 vs Fc+/Fc)31 compared with that of the thiolate-ligated ferrous precursor FeII(Me3TACN)(S2SiMe2) (E1/2 = −0.60 V vs Fc+/Fc).27 It has been proposed that the Fe–O–O angle in peroxo(diiron) complexes can influence the ν(O–O) and ν(Fe–O) stretching frequencies, with more obtuse angles favoring a larger ν(O–O) and smaller ν(Fe–O).39 Given the increased steric demands of the ((OSiPh2)2O)2− ligand compared to the (S2SiMe2)2− ligand, it is expected that the Fe---Fe distance, and thus the Fe–O–O angle will be larger in 2, which could give rise to the observed trends for the vibrational frequencies. This hypothesis is also consistent with the relatively low ν(Fe–O) stretching frequency in 2 compared to that of other peroxo(diiron) complexes.32, 39 Differences in covalency within the complexes due to the siloxide donors compared to thiolate donors may also explain differences in the degree of O–O bond activation.
Calculations using density functional theory (DFT) were performed to aid in the assignment of 2. The structure of 2 was optimized with a BP86-D3/def2-tzvp/def2-SVP(C,H) functional/basis set combination using broken symmetry (BS), assuming coupling between the two S = 5/2 iron centers. The Mössbauer parameters were calculated as δ = 0.57 mm s−1 and │ΔEQ│= 0.75 mm s−1, in good agreement with the experimental data. The DFT-calculated O–O bond distance of 1.383 Å is nearly identical to that calculated for FeIII2(O2)(Me3TACN)2(S2SiMe2)2 (d(O–O) = 1.392 Å); however, the optimized Fe---Fe distance for 2 of 4.797 Å is significantly longer than the Fe---Fe distance of 4.612 Å calculated for the S-ligated complex. The significant elongation in the Fe---Fe distance for 2 is consistent with the increased steric bulk of the siloxide ligand compared to the silanedithiolate ligand. The calculated Fe–O–O angles of 2 at 134.1° and 132.9° are larger than the average Fe–O–O angle of 119.3° calculated for FeIII2(O2)(Me3TACN)2(S2SiMe2)2.27 These computational data support the hypothesis of a relatively weak activation of the O–O bond in 2 related to a long Fe---Fe distance and obtuse Fe–O–O angles.
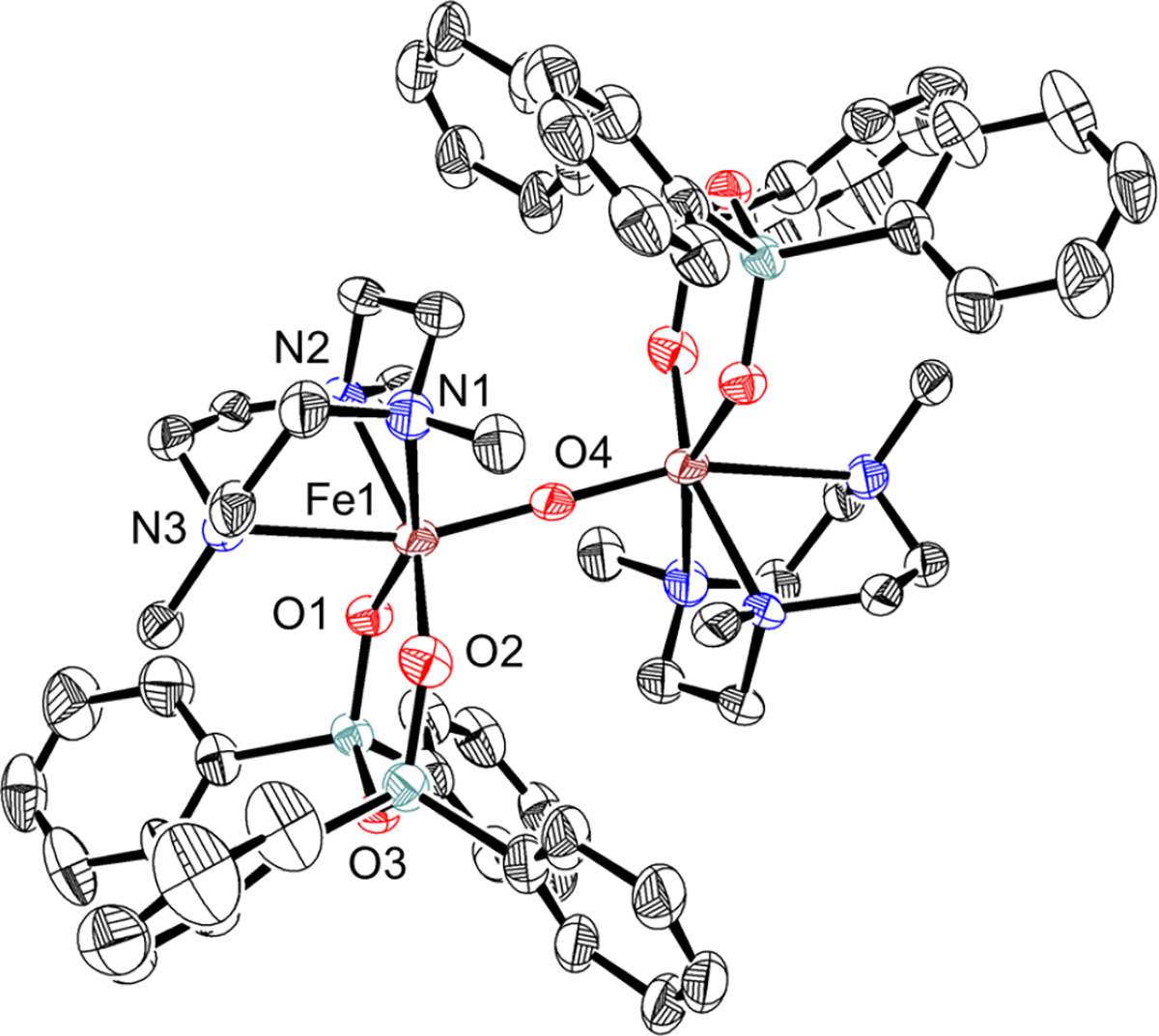
Warming a solution of 2 from −80 °C to 23 °C results in the loss of the purple color associated with 2 and formation of a new pale yellow species, 3. Analysis by Mössbauer spectroscopy reveals a new major quadrupole doublet with parameters δ = 0.49 mm s−1,│ΔEQ│= 1.03 mm s−1 that makes up 75% of the fit. A second minor component (15%) with δ = 0.40 mm s−1,│ΔEQ│= 1.48 mm s−1 is also observed. The isomer shifts are typical of high-spin ferric ions, whereas the relatively large quadrupole splittings in both subspecies show substantial anisotropy in the iron coordination. These features are known for μ-oxo bridged dimers.38 Analysis by EPR spectroscopy reveals that the thermal decay products are EPR-silent. Crystallization of the solution following warm-up produced pale yellow blocks that were suitable for single crystal X-ray diffraction (XRD). The crystal structure for 3 is shown in Figure 5 and reveals an FeIII2(μ-O) complex with an unsupported, linear core (∠Fe–O–Fe = 180.0°). The Fe–oxo bond length is 1.8343(5) Å, and the d(Fe---Fe) distance is 3.669 Å. The NTACN donor trans to the oxo ligand exhibits a weakened interaction with the Fe center, with an Fe–N distance of 2.494(4) Å. The trans N distance is the longest Fe–NTACN distance documented in the Cambridge Structural Database40 and likely is due to the steric crowding caused by the diphenyl siloxide groups. Geometry optimization of 3, using the same computational method as for 2, reproduces the XRD structure with the long Fe–NTACN distance (calcd: 1.515 Å) and the linear Fe–O–Fe angle (calcd: 178.4°). Prediction of the Mössbauer parameters of 3 by DFT gave δ = 0.50 mm s−1 and │ΔEQ│= 1.07 mm s−1, which are in excellent agreement with the major component seen in the experimental data.
Figure 5.
Displacement ellipsoid plot (50% probability level) for 3 at 110(2) K. Selected bond distances (Å): Fe1–O1: 1.932(3), Fe1–O2: 1.941(2), Fe1–N1: 2.273(4), Fe1–N2: 2.250(4), Fe1–N3: 2.494(4), Fe1–O4: 1.8343(5).
Previously we showed that irradiation of FeIII2(O2)(Me3TACN)2(S2SiMe2)2 at −135 °C led to O–O bond cleavage to give a metastable FeIV(O) complex.27 However, irradiation of 2 under similar low temperature conditions results in bleaching of the solution and an intractable mixture of products. In contrast, initial freezing of a solution of 2 at 77 K followed by irradiation with visible light leads to the formation of a pale, yellow-green species. Analysis by Mössbauer spectroscopy revealed a single quadrupole doublet with δ = 0.22 mm s−1 and │ΔEQ│= 0.23 mm s−1 (Figure 3c), indicating quantitative formation of the new species, 4. The low isomer shift for 4 is in the range typically observed for FeIV(O) centers (−0.19 mm s−1 − 0.35 mm s−1).7, 19, 27 The │ΔEQ│ value is significantly lower than that for other S = 1 FeIV(O) complexes, and is more comparable with S = 2 FeIV(O) species,19 including the silicate-ligated S = 2 FeIV(O) species in Fe-doped zeolites23–24 (δ = 0.30 mm s−1, │ΔEQ│= 0.50 mm s−1).
Variable field Mössbauer studies allowed us to assign the ground spin state of 4. The magnetic Mössbauer spectra of 4, recorded at liquid helium temperatures with fields of 1 – 7 T applied perpendicular to the γ rays, are governed by wide and strongly temperature- and field dependent magnetic hyperfine splitting (Figure 3d). Particularly the field dependence at 1.7 K (middle traces) is typical of an integer spin system with substantial, apparently positive zero-field splitting (zfs), for which the ground level of the electronic spin manifold would be essentially “mS=0” in nature. The collapse of the magnetic splitting at 22 K, in spite of the presence of a 1 T field (top trace), suggests fast paramagnetic relaxation. In contrast, strong fields of 4 and 7 T induced symmetric six-line spectra without significant quadrupole perturbation (relative shift of inner four vs. the outer two lines), in accord with the small quadrupole splitting of the zero-field spectrum (Figure 3c). The intensity ratio of the six-line pattern is close to 3:4:1:1:4:3 which shows strong polarization of the internal fields along the applied-fields direction perpendicular to the γ rays. Although this spectral behavior for powders or frozen solutions is in principle consistent with an easy (x/y) plane of magnetic anisotropy, as expected for a low lying “mS=0” ground level, and as encountered for most other Fe(IV) oxo complexes with positive D and E/D ≈ 0,7, 54 the asymmetry of the individual Mössbauer hyperfine lines of 2 suggests deviations from axial symmetry. The ‘intensity smearing’ towards lower magnetic splitting, particularly of the outer Mössbauer lines at 4 and 7 T, indicates partial misalignment of applied and internal fields due to higher magnetic anisotropy of the ground level than expected for an easy plane of magnetization. In terms of a spin Hamiltonian description, this means finite rhombicity, E/D > 0, and eventually also anisotropic magnetic hyperfine tensor components, Axx ≠ Ayy.
Moreover, the strong-field spectra of 4 resemble remarkably well the 8T Mössbauer spectra of the high-spin Fe(IV)-oxo intermediate J of taurine/α-ketoglutarate dioxygenase (TauD)41, 55 (except for details of the line shape and the different intensity ratio due to different field orientation parallel to the γ rays). The overall magnetic splitting of about 8 mm s−1 is similar for both and reveals a similarly strong internal hyperfine field of more than 30 T. This value is the most significant indication of a high-spin state for the Fe(IV)oxo complex 4 with S = 2, like for TauD41, 55 and other examples (Table 1).21, 42, 46, 48, 50–51 The corresponding wide spectral splitting clearly exceeds the splitting of corresponding spectra from low-spin Fe(IV)-oxo species with S = 1 ground states.7, 56
Table 1.
Spectroscopic properties of selected FeIV(O) centers
| FeIV(O) species | S | λmax | d(Fe-O) (cm−1) | ν(Fe=O) (cm−1) | δ, ΔEQ (mm s−1) | D (cm−1) | Axx, Ayy, Azz/gNβN (T) | Ref |
|---|---|---|---|---|---|---|---|---|
| 4 | 2 | − | − | 818 | 0.22, −0.23 | 13.2 | −20.4, −16.9, −30.4 | This work |
| TauD J | 2 | 318 | 1.62 | 821 | 0.31. −0.88 | 10.5 | −18.4,−17.6, −31 | 41–43 |
| Fe-ZSM-5 | 2 | 498 | 1.63 | 885 | 0.30, 0.50a | 8.0 | − | 23–24 |
| [FeIV(O)(TQA)(MeCN)]2+ | 2 | 400, 650, 900 | − | 838 | 0.24, −1.05 | −18.2, −16.6, − | 21 | |
| [FeIV(O)(H2O)5]2+ | 2 | 320 | 0.30, −0.33 | 9.7 | −20.3, −20.3, − | 44 | ||
| [FeIV(O)(TMG3tren)]2+ | 2 | 400, 825, 866 | 1.661 | 843 | 0.24, −0.29 | 5.0 | −15.5, −14.8, −28.0 | 45–46 |
| [FeIV(O)(H3buea)]− | 2 | 440, 550, 808 | 1.68 | 799 | 0.02, +0.43 | 4.7 | −14.7, −14.9, −26.7 | 47–48 |
| [FeIV(O)(tpaPh)]− | 2 | 400, ~900 | 1.62 | 850 | 0.09, 0.51a | − | 49 | |
| [FeIV(O)(TMG2dien)(CH3CN)]2+ | 2 | 380, 805 | 1.65 | 807 | 0.08, +0.58 | 4.5 | −13.9, 15.8, −26.0 | 50 |
| [FeIV(O)(tBu3TACN)]2+ | 2 | 356 | 1.66 | 802 | 0.11, +0.96 | −10.1, −3.3, −36.1 | 51 | |
| FeIV(O)(TMC)(CH3CN)]2+ | 1 | 824 | 1.646 | 839 | 0.17, +1.24 | 29 | −23, −18, −3 | 52–53 |
Sign not determined.
According to the arguments above, spin Hamiltonian simulations with the assumption of S=2 for the paramagnetic center of 4 yielded a nice global fit with D = 13.2 cm−1, E/D = 0.19 with g = 2 (fixed), ΔEQ = −0.2 mms−1, η = 0.4, and a non-axial A-tensor with Axx/gNβN = −20.42 T, Ayy/gNβN = −16.87 T and Azz/gNβN = −30.36 T (Figure 3d, red lines). In contrast, attempts to fit the spectra of 4 with S = 1 failed to reproduce the distinct field- and temperature dependence of the data set (due to the limited number of magnetic substates), or yielded unrealistic parameters. Particularly, the large overall splitting of the strong-field spectra would require unreasonably high A values (Figure S6). Hence, we conclude that 4 is a Fe(IV)-oxo complex with S = 2 ground state.
Initial attempts to characterize the new ferryl complex by RR spectroscopy in 2-MeTHF were unsuccessful, likely due to the prominent solvent bands7 between 800–900 cm−1 obscuring the expected ν(Fe-O) stretching mode. However, changing the solvent to THF eliminated this problem. Mössbauer spectroscopy confirmed formation of the ferryl complex 4 in THF (Figure S8). RR data collected on 4 in THF with 350 nm laser excitation (Figure 6) shows a prominent vibrational band at 818 cm−1 that shifts to 788 cm−1 upon substitution with 18O. The observed downshift (Δ18O = −30 cm−1) is close to the value expected for an isolated Fe–O harmonic oscillator (−36 cm−1) and is in a typical range seen for other FeIV(O) complexes (Table 1).7
Figure 6.
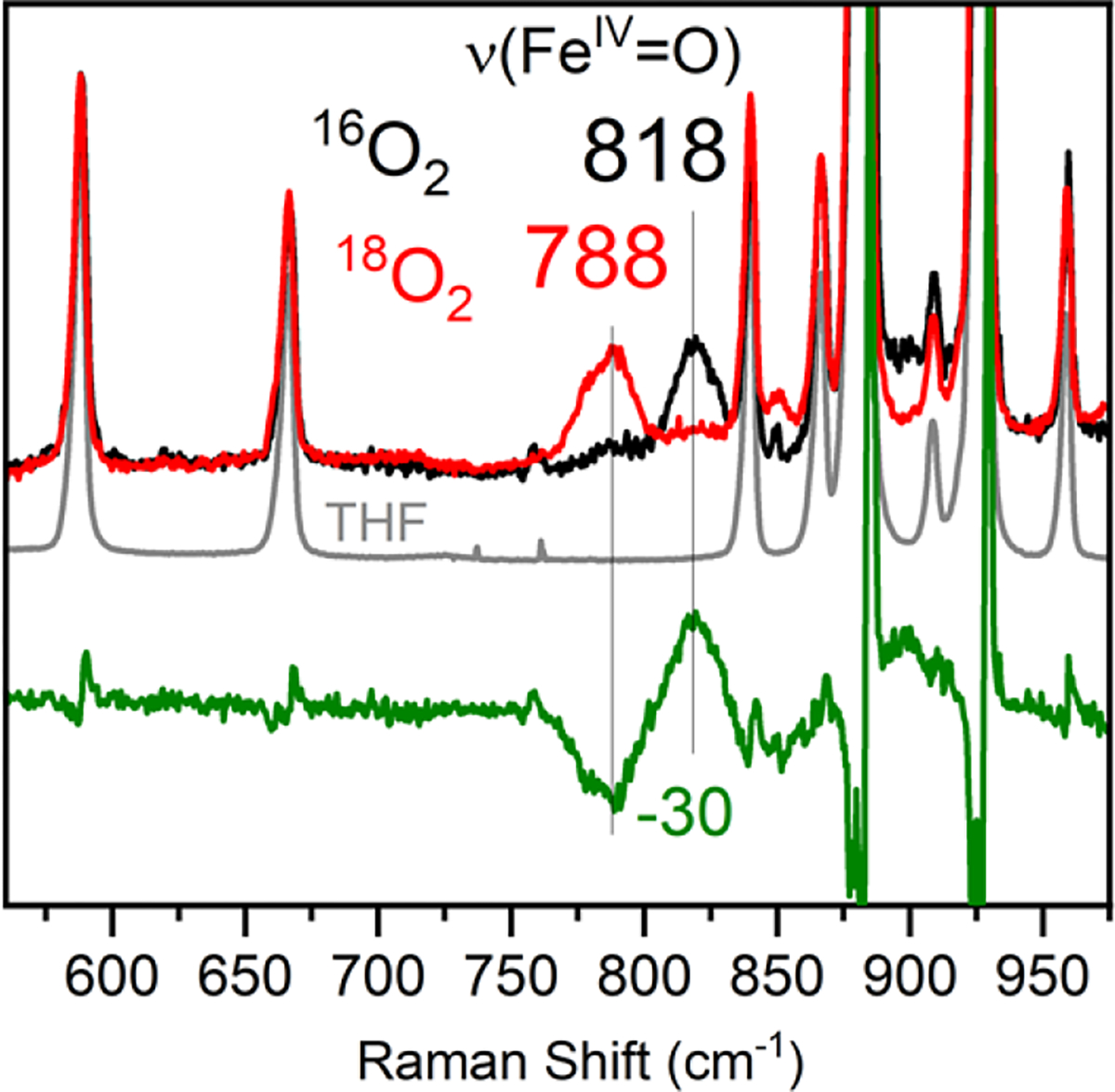
RR spectra of 4 in frozen THF at 110 K (λexc = 350 nm). Black and red spectra show 4(16O) and 4(18O) data, respectively. The grey spectrum shows the spectrum for pure THF. The green spectrum is the difference spectrum for 4(16O) – 4(18O).
DFT calculations for FeIV(O)(Me3TACN)((OSiPh2)2O) were performed for both S = 1 and S = 2 spin states, and several functional/basis set combinations were tested. Calculations using the BP86 functional, which is known to be biased towards low spin configurations,57 gave single point energies for both S = 1 and S = 2 within 2 kcal mol−1 of each other, which is within error for this functional. The B3LYP/def2-tzvp/def2-svp(C,H) functional/basis set combination calculates the S = 2 spin state to be 4 kcal mol−1 lower in energy. Mössbauer parameters for both spin states using the B3LYP/def2-tzvp/def2-svp(C,H) level of theory were calculated, and gave δ = 0.02 mm s−1 and │ΔEQ│= 1.93 mm s−1 for S = 1, and δ = 0.12 mm s−1 and │ΔEQ│= 0.30 mm s−1 for S = 2. While the predicted S = 1 parameters are a poor match with the experimental data, the computed S = 2 parameters fit well, in accordance with the assignment of an S = 2 spin ground state for 4. The optimized geometry for 4 (Figure 7) reveals a non-linear Ntrans–Fe=O angle of 167°. It has been proposed that tilting of the Fe=O core correlates with increased HAT reactivity,58 in line with the observed instability of 4 in solution. Taken together, the data show that a high spin FeIV(O) complex, 4, is generated via O–O cleavage following irradiation of 2 in frozen 2-MeTHF (Scheme 1).
Figure 7.
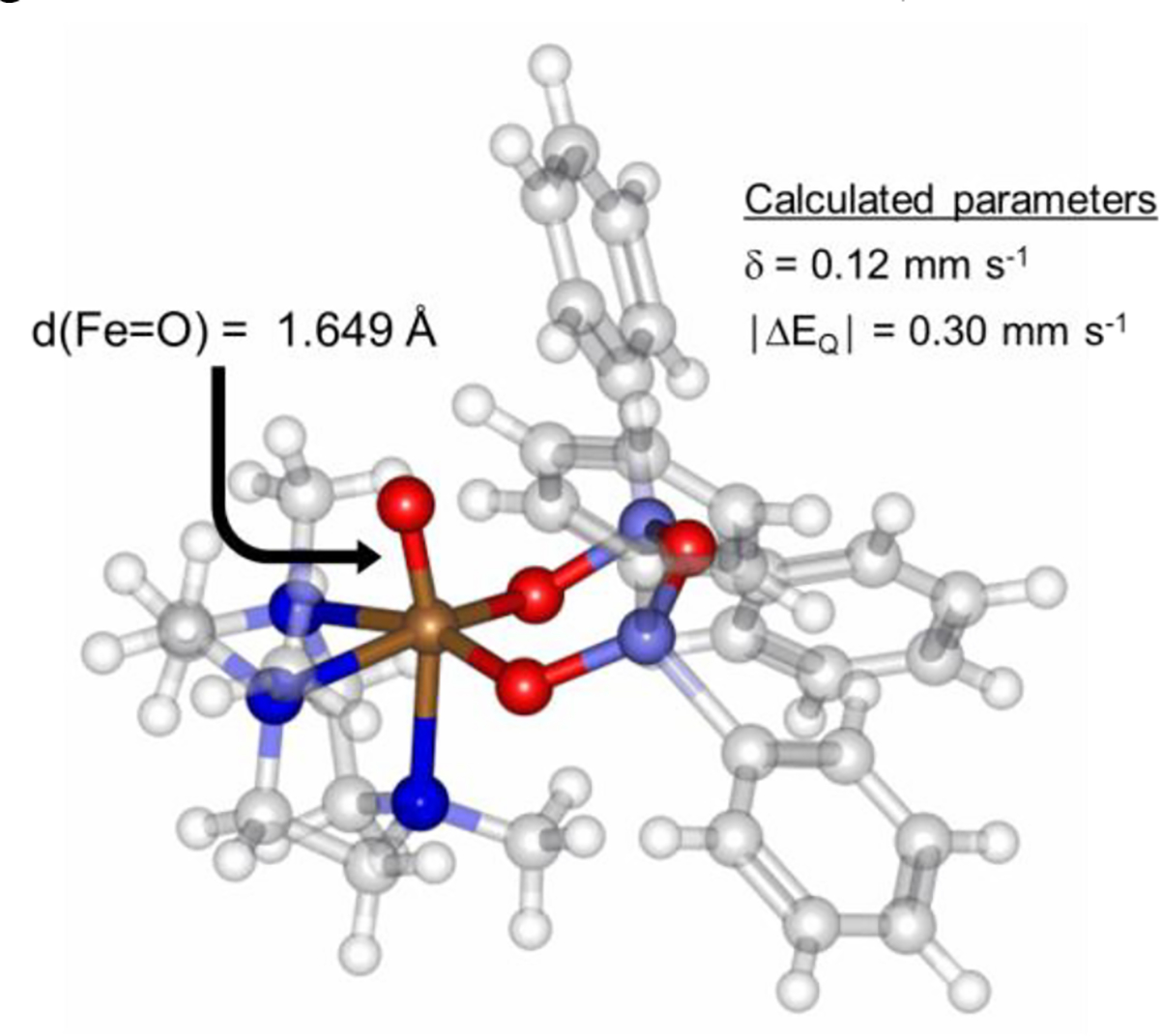
DFT geometry-optimized structure of 4.
Having determined that the FeIV(O) complex 4 could be generated in frozen matrix from irradiation of 2, we reasoned that it might be possible to transiently form 4 in fluid solution at low temperature, and intercept it with an appropriate, pre-loaded substrate. Irradiation of a solution of 2 pre-loaded with excess 2,4,6-tri-tert-butylphenol (ttbp) (63 equiv) at −80 °C leads to the rapid, isosbestic decay of 2 and concomittant formation of phenoxyl radical (λmax = 386, 400, 630 nm) (Figure S9). Similarly, irradiation of a frozen matrix (77 K) of 2 and excess ttbp in 2-MeTHF, followed by thawing to −130 °C, resulted in the formation of a sharp g = 2 signal in the EPR spectrum indicative of a phenoxyl radical (44(2)% based on Fe). The EPR spectrum of this reaction mixture also shows peaks typical of a high-spin (S = 5/2) FeIII product. Taken together, these data are consistent with transient formation of 4 in fluid solution followed by hydrogen atom abstraction from ttbp to give the expected FeIII(OH) product (Scheme 2).
Scheme 2.
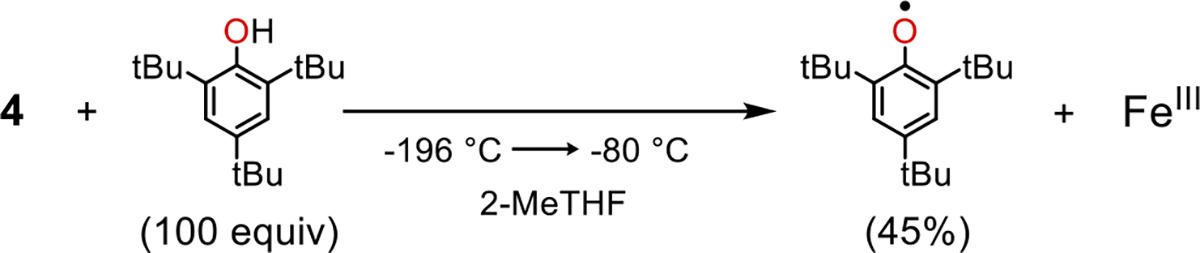
Phenol Oxidation by Complex 4
In the absence of phenol substrate, we hypothesized that the decay of 4 upon thawing from frozen matrix in 2-MeTHF might arise from H-atom abstraction from the C-H bond of the solvent. High-spin FeIV(O) complexes are known to be highly reactive H-atom abstractors, cleaving C–H bonds with bond dissociation free energy (BDFE) values as high as 99 kcal mol−1.20 The weakest C-H bond in 2-MeTHF can be expected to have a BDFE ~85 kcal mol−1.59–60 Thawing of 4 in frozen matrix (2-MeTHF) to just above the freezing point of the solvent (−136 °C) leads to Mössbauer (Figure S10) and EPR (Figure S11) spectra consistent with a high-spin (S = 5/2) FeIII species. Warming 4 to room temperature followed by removal of solvent in vacuo and analysis by electron impact mass spectrometry revealed peaks consistent with 2-MeTHF oxidation (Figure S12).
The expected product from H-atom abstraction would be a mononuclear FeIII(OH) species. We were not able to isolate this complex, and therefore turned to DFT calculations for further insight. Geometry optimization of FeIII(OH)(Me3TACN)((OSiPh2)2O) followed by a single point energy calculation to obtain the Mössbauer parameters gave calculated values of δ = 0.46 mm s−1,│ΔEQ│= 0.83 mm s−1, which are in good agreement with the experimental data for the decay product of 4. The BDFE(O–H) for this FeIII(OH) complex was also calculated by DFT (see Experimental Section for details), and gave a value of Fe(O-H) = 98 kcal mol−1, which is quite high compared to other O–H/C–H bonds,61 and is consistent with 4 being a powerful H-atom abstracting agent.
Conclusions
The siloxide-ligated ferrous complex FeII(Me3TACN)((OSiPh2)2O) (1) reacts with O2 to give a metastable peroxo-bridged diferric complex, Fe2III(μ-O2)(Me3TACN)2((OSiPh2)2O)2 (2), at −80 °C. Resonance Raman spectroscopy indicated a relatively high ν(O–O) and a low ν(Fe–O) of 896 and 417 cm−1, respectively, which can be correlated with the large Fe–O–O angle that would be expected for a differic, unsupported peroxo-bridged species.39 The assignment of 2 as an unsupported peroxo-bridged species is also consistent with the low magnetic coupling constant of J = −23(±5) cm−1, which was determined from variable-field Mössbauer experiments. Thermal decay of 2 leads to the μ-oxo-bridged, differic complex 3. Complex 2 is also photosensitive, and upon exposure to white light in fluid solution, decays to an intractable mixture of products. However, visible light illumination of 2 in a frozen solvent matrix leads to controlled homolytic O–O bond cleavage and production of the FeIV(O) complex, 4.
Complex 4 represents a rare example of an S = 2 FeIV(O) prepared directly from O2, and the first such species derived via photolytic O–O cleavage of a peroxo(diiron) complex. Interestingly, complex 4 shows striking similarities to the ferryl intermediate J in the catalytic cycle of TauD.41–42 The ν(Fe=O) of TauD-J (821 cm−1) is nearly identical to that for 4 (818 cm−1), and the measured hyperfine parameters for 4 (Azz = −30.4 T; Ayy = −20.4 T; Axx = −16.9 T) are very close to those for TauD-J (Azz = −31.0 T; Ayy = −17.6 T; Axx = −18.4 T).
Although it was necessary to keep 4 in a frozen matrix for characterization, we were able to harness the power of this potent oxidant through photorelease from 2 in fluid solution. Thawing a frozen solution of 4 results in rapid decay, likely via H-atom abstraction with solvent. However, when complex 2 is photolyzed in the presence of a phenol substrate, 4 is generated and intercepted to produce the expected phenoxyl radical product. Our studies show that the peroxo-bridged diiron species, 2, can be converted into the more reactive 4 in situ to perform H-atom abstraction. Thus we are able to use visible light to trigger the rapid production of a highly reactive FeIV(O) species. The ability to cleave inert C–H bonds using abundant and benign oxidants such as O2 remains a major challenge in synthetic chemistry. This work indicates that controlled, light-mediated O–O bond cleavage could be a possible strategy for producing other FeIV(O) species that are capable of cleaving C–H bonds. We hypothesize that with careful ligand design it will be possible to extend the lifetime of S = 2 FeIV(O) to promote more controlled substrate oxidation reactivity.
EXPERIMENTAL SECTION
General Considerations.
All syntheses and manipulations were conducted in an N2-filled drybox (Vacuum Atmospheres, O2 < 0.2 ppm, H2O < 0.5 ppm) or using standard Schlenk techniques under an atmosphere of Ar unless otherwise noted. Me3TACN was purchased from Matrix Scientific, degassed by three freeze-pump-thaw cycles, and stored over 3 Å molecular sieves prior to use. Fe(OTf)2•2MeCN and 57Fe(OTf)2•2MeCN were prepared according to a literature procedure.62 57Fe metal (95.93%) was purchased from Cambridge Isotope Laboratories. 18O2 (98 atom %) was purchased from ICON Isotopes (Summit, N.J.). Potassium hydride (KH) in paraffin was purchased from Sigma-Aldrich, washed with diethyl ether and pentane, and dried under vacuum prior to use. H2(OSiPh2)2O was synthesized according to a reported procedure.63 All other reagents were purchased from commercial vendors and used without further purification. Acetonitrile and acetonitrile-d3 were distilled from CaH2. Tetrahydrofuran, tetrahydrofuran-d8 (≥ 99.5% D), pentane, hexane, and 2-MeTHF were dried over Na/benzophenone and subsequently distilled. Butyronitrile was distilled from Na2CO3/KMnO4 according to a reported procedure.64 Diethyl ether was obtained from a PureSolv solvent purification system. All solvents were degassed by a minimum of three freeze-pump-thaw cycles and stored over freshly activated 3 Å molecular sieves in the drybox following distillation.
Instrumentation.
The 1H NMR spectra were measured on a Bruker 300 MHz or a Bruker 400 MHz spectrometer. Chemical shifts were referenced to reported solvent resonances.65 All photolysis experiments were carried out using a halogen lamp. UV–vis experiments were carried out on a Cary Bio-50 or Cary 60 UV-vis spectrophotometer equipped with a Unisoku USP-203A cryostat using a 1 cm modified Schlenk cuvette. Midwest Microlab LLC (Indianapolis, IN) conducted elemental analyses on samples prepared and shipped in ampules sealed under vacuum. EPR measurements were performed on a Bruker X-band EPR spectrometer in 5 mm quartz EPR tubes (Wilmad). Zero-field Mössbauer spectra were recorded on a spectrometer from SEE Co. (Edina, MN) operating in the constant acceleration mode in a transmission geometry. The sample was kept in an SVT - 400 cryostat from Janis Research Co. (Wilmington, MA), using liquid He as a cryogen for 5 K data collection and liquid N2 as a cryogen for 80 K measurements. Isomer shifts were determined relative to the centroid of the spectrum of a metallic foil of α–Fe collected at room temperature. Data analysis was performed using version F of the program WMOSS (www.wmoss.org) and quadrupole doublets were fit to Lorentzian lineshapes.66
Magnetic Mössbauer spectra were recorded on a conventional spectrometer with alternating constant acceleration of the γ-source (MPI-CEC, own construction). The sample temperature was maintained constant in a cryogen-free closed-cycle Mössbauer cryostat from Cryogenic Ltd, equipped with top-loading variable-temperature insert (vti) and a split-pair super-conducting magnet for fields up to 7 T, oriented vertical and perpendicular to the γ rays. The vti allows sample temperature settings in the range 1.7 K to 300 K. The 57Co/Rh source (1.8 GBq) was kept at room temperature and was positioned inside the gap of the magnet system by using a re-entrant bore tube. The source was adjusted horizontally to a zero-field position in ca. 9 cm distance from the sample. The minimum experimental line width was 0.3 mm s−1 (full width at half-height). The detector was a Si-Drift diode (150 mm2 SDD CUBE) of a AXAS-M1 system from Ketek GmbH, mounted at the tip of a vacuum-tight 200 mm stainless steel finger. This finger was also inserted horizontally into the cryostat in the gap of the magnet to approach the sample to increase the aperture of the γ beam. The magnetic Mössbauer spectra were simulated with the program mx.SL (by E.B.) by diagonalization of the usual spin Hamiltonian. More detail is given in the Supporting Information.
Computational Studies.
All geometry optimizations were performed in the ORCA-4.1.2 program package.67 Initial geometries were obtained from the X-ray crystallographic models of 1 or 3 and altered as needed. Optimized geometries were calculated using the BP86 functional68–69 or with B3LYP70 in combination with the D3 dispersion correction,71 which gave satisfactory results in reproducing the experimentally derived bond metrics and vibrational frequencies. The basis set def2-TZVP was used for all Fe, N, O, and Si atoms, and the def2-SVP basis set was used for all C and H atoms.72 To reduce computational costs, the Resolution of Identity (RI) approximations73 in tandem with the def2/J auxiliary basis set72, 74 were employed. Due to SCF convergence difficulties in some cases, damping parameters were altered using the slowconv function in ORCA. Broken symmetry calculations were carried out as previously described.75–77 Frequency calculations at the same level of theory confirmed that all optimizations had converged to true minima on the potential energy surface (i.e., no imaginary frequencies). The optimized structures were used for Mössbauer calculations. All Mössbauer calculations were performed in the ORCA-3.0.2 program package.78 Mössbauer parameters were computed using the B3LYP70 functional and a combination of CP(PPP)79 for Fe and def2-TZVP80–81 for all other atoms. The angular integration grid was set to Grid4 (NoFinalGrid), with increased radial accuracy for the Fe atom (IntAcc 7). To simulate solid state effects, a continuum solvation model was included (COSMO) with methanol designated as solvent, which has been shown to lead to accurate predictions of Mössbauer parameters.82 The isomer shift was obtained from the electron density at the Fe nucleus, using a previously reported linear fit function, δcalc = a(ρ(0) − C) + b where a = −0.424 mm s−1 a.u.3, b = 7.55 mm s−1, and C = 11800 a.u.3, which was derived by plotting ρ(0) versus the experimental isomer shift of a series of Fe complexes.82 Bond dissociation free energies were calculated at 1 atm and 25 °C using the geometry optimization and frequency calculations performed with B3LYP. Corrections for vibrational, zero-point energy, and contributions from translational, rotational, and vibrational modes to the energy and entropy of the H-atom transfer were accounted for. The electronic energy of H• used in the calculation of the BDFE values is 313.1 kcal mol−1.83
Synthesis of FeIII(μ-O)(Me3TACN)2((OSi2Ph)2O)2 (3).
A solution of 1 (38 mg, 0.059 mmol) in THF (10 mL) was cooled to − 78 °C and excess O2 was added to afford purple 2. The solution was then sparged with Ar gas for 30 min, warmed to 23 °C, and the solvent was removed under reduced pressure to produce a pale orange solid. Vapor diffusion of pentane to a solution of 3 in THF afforded pale yellow crystals after 12 h (9.8 mg, 26%). Anal. calcd for C66H82N6O7Si4Fe2: C, 61.19; H, 6.38; N, 6.49. Found: C, 61.55; H, 6.60; N, 6.13.
Preparation of 57FeIII(O2)(Me3TACN)2((OSi2Ph)2O)2 (2(57Fe)) for Mössbauer spectroscopy.
A solution of 57Fe-enriched 1(57Fe) (3.9 mM, 400 mL) in 2-MeTHF was transferred to a Delrin Mössbauer cup, and cooled to −80 °C. Excess O2 was bubbled through the solution of 1(57Fe) in the dark, resulting in the formation of 2 (dark purple). The solution of 2 was frozen in liquid nitrogen and stored at 77 K until it was loaded into the Mössbauer spectrometer.
Preparation of FeIV(O)(Me3TACN)((OSi2Ph)2O) (4(57Fe)) for Mössbauer spectroscopy.
A solution of 57Fe-enriched 1(57Fe) (5.5 mM, 400 mL) in 2-MeTHF was transferred to a Delrin Mössbauer cup, and cooled to −80 °C. Excess O2 was bubbled through the solution of 1(57Fe) in the dark, resulting in the formation of 2 (dark purple). The solution of 2 was frozen in liquid nitrogen, and covered with a glass petri dish to filter out UV light. Irradiation with a white light source for 15 min under liquid nitrogen resulted in a color change from purple to pale yellow-green. The frozen solution of 4 was stored at 77 K until it was loaded into the Mössbauer spectrometer.
Preparation of 2 and 4 for Resonance Raman Spectroscopy.
Complex 2.
A stock solution of 1 was prepared in 2-MeTHF (1.9 mM), and an aliquot was transferred to a 5 mm NMR tube and sealed with a septum in a drybox. The sealed NMR tube was removed from the drybox and cooled to −105 °C in an Et2O/N2(l) bath. Excess 16O2 (natural abundance) or 18O2 (98%) was added to the solution of 1 in the dark, yielding a color change from pale blue to purple. The reaction was allowed to proceed with frequent manual mixing for 4 min, and then the reaction mixture containing 2 was slowly annealed in liquid nitrogen and stored at 77 K until needed.
Complex 4.
Complex 2 was prepared in THF using either excess 16O2 (natural abundance) or 18O2 (98%) at −78 °C in a 5 mm NMR tube, and then frozen in liquid nitrogen. Irradiation for 20 min with a white light source resulted in a color change from purple to pale yellow-green of the outer layer of the frozen solution, indicative of the formation of 4. The inner core of the sample remained unchanged because THF does not form a good glass, and the white light source (150 W halogen lamp) only penetrates the outer layers of the frozen solution. However, this conversion was sufficient for RR analysis. Samples were stored at 77 K until needed.
Resonance Raman (RR) spectra were obtained using a custom McPherson 2061/207 spectrograph (set at 0.67 m with variable gratings) equipped with a liquid nitrogen-cooled CCD detector (LN-1100PB, Princeton Instruments). After attempts with different laser excitations, the 647- and 351-nm lines from a Kr laser (Innova 300, Coherent) were selected for the RR characterizations of complexes 2 and 4, respectively. Long-pass filters (RazorEdge, Semrock) were placed in front of the spectrograph entrance slit to attenuate the Rayleigh scattering. RR spectra were recorded using a 180o scattering geometry on samples maintained at 110 K inside a copper cold-finger and with continuous spinning. Frequencies were calibrated relative to aspirin and are accurate to ± 1 cm−1.
Phenol oxidation.
In a typical reaction, a stock solution of 1 was prepared in 2-MeTHF (1.6 mM), and an aliquot was transferred into a 5 mm EPR tube. An aliquot of a stock solution of ttbp (50 μL, 100 equiv) was added and the solution was cooled to −130 °C. Excess O2 was then bubbled directly into the solution, resulting in a color change from colorless to purple. The sample was then frozen at 77 K and irradiated with a white light source for 15 min resulting in a color change from purple to pale yellow-green. The reaction mixture was warmed to −130 °C, and a color change to pale blue was noted. The reaction mixture was manually mixed for 5 min, and then slowly annealed in liquid nitrogen and stored at 77 K until needed. Quantitation of the phenoxyl radical was accomplished by measuring the double integral value of the EPR first derivative spectrum and comparing this value against a calibration curve using (2,2,6,6-tetramethylpiperidin-1-yl)oxyl as a standard.
Supplementary Material
ACKNOWLEDGMENT
The NIH (R01GM119374 to D.P.G.) is gratefully acknowledged for financial support. J.B.G. would like to thank JHU for the Sonneborn Fellowship. Computer time was provided by the Maryland Advanced Research Computing Center (MARCC).
Footnotes
ASSOCIATED CONTENT
Supporting Information
Resonance Raman, Mössbauer, EPR, and UV–vis spectra, mass spectrometry data, crystallographic information, computational details, and DFT coordinates (PDF)
REFERENCES
- (1).Borden WT; Hoffmann R; Stuyver T; Chen B Dioxygen: What Makes This Triplet Diradical Kinetically Persistent? J. Am. Chem. Soc 2017, 139, 9010–9018. [DOI] [PubMed] [Google Scholar]
- (2).Sawyer DT; Williams RJP, Oxygen Chemistry. Oxford University Press: 1991; p 240. [Google Scholar]
- (3).Lacy DC Applications of the Marcus cross relation to inner sphere reduction of O2: implications in small-molecule activation. Inorg. Chem. Front 2019, 6, 2396–2403. [Google Scholar]
- (4).Chiang C-W; Kleespies ST; Stout HD; Meier KK; Li P-Y; Bominaar EL; Que L Jr.; Münck E; Lee W-Z Characterization of a Paramagnetic Mononuclear Nonheme Iron-Superoxo Complex. J. Am. Chem. Soc 2014, 136, 10846–10849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Nam W Synthetic Mononuclear Nonheme Iron–Oxygen Intermediates. Acc. Chem. Res 2015, 48, 2415–2423. [DOI] [PubMed] [Google Scholar]
- (6).Hong S; Lee Y-M; Ray K; Nam W Dioxygen activation chemistry by synthetic mononuclear nonheme iron, copper and chromium complexes. Coord. Chem. Rev 2017, 334, 25–42. [Google Scholar]
- (7).McDonald AR; Que L Jr. High-valent nonheme iron-oxo complexes: Synthesis, structure, and spectroscopy. Coord. Chem. Rev 2013, 257, 414–428. [Google Scholar]
- (8).Sahu S; Goldberg DP Activation of Dioxygen by Iron and Manganese Complexes: A Heme and Nonheme Perspective. J. Am. Chem. Soc 2016, 138, 11410–11428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Ching W-M; Zhou A; Klein JEMN; Fan R; Knizia G; Cramer CJ; Guo Y; Que L Jr. Characterization of the Fleeting Hydroxoiron(III) Complex of the Pentadentate TMC-py Ligand. Inorg. Chem 2017, 56, 11129–11140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Drummond MJ; Ford CL; Gray DL; Popescu CV; Fout AR Radical Rebound Hydroxylation Versus H-Atom Transfer in Non-Heme Iron(III)-Hydroxo Complexes: Reactivity and Structural Differentiation. J. Am. Chem. Soc 2019, 141, 6639–6650. [DOI] [PubMed] [Google Scholar]
- (11).Martinez S; Hausinger RP Catalytic Mechanisms of Fe(II)- and 2-Oxoglutarate-dependent Oxygenases. J. Biol. Chem 2015, 290, 20702–20711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Solomon EI; Goudarzi S; Sutherlin KD O2 Activation by Non-Heme Iron Enzymes. Biochemistry 2016, 55, 6363–6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Bollinger JM Jr.; Price JC; Hoffart LM; Barr EW; Krebs C Mechanism of Taurine: α-Ketoglutarate Dioxygenase (TauD) from Escherichia coli. Eur. J. Inorg. Chem 2005, 2005, 4245–4254. [DOI] [PubMed] [Google Scholar]
- (14).Tamanaha E; Zhang B; Guo Y; Chang W.-c.; Barr EW; Xing G; St. Clair J; Ye S; Neese F; Bollinger JM Jr.; Krebs C Spectroscopic Evidence for the Two C–H-Cleaving Intermediates of Aspergillus nidulans Isopenicillin N Synthase. J. Am. Chem. Soc 2016, 138, 8862–8874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Jacobs AB; Banerjee R; Deweese DE; Braun A; Babicz JT; Gee LB; Sutherlin KD; Böttger LH; Yoda Y; Saito M; Kitao S; Kobayashi Y; Seto M; Tamasaku K; Lipscomb JD; Park K; Solomon EI Nuclear Resonance Vibrational Spectroscopic Definition of the Fe(IV)2 Intermediate Q in Methane Monooxygenase and Its Reactivity. J. Am. Chem. Soc 2021, 143, 16007–16029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Cutsail GE; Banerjee R; Zhou A; Que L Jr.; Lipscomb JD; DeBeer S High-Resolution Extended X-ray Absorption Fine Structure Analysis Provides Evidence for a Longer Fe···Fe Distance in the Q Intermediate of Methane Monooxygenase. J. Am. Chem. Soc 2018, 140, 16807–16820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Srnec M; Wong SD; England J; Que L Jr.; Solomon EI π-Frontier molecular orbitals in S = 2 ferryl species and elucidation of their contributions to reactivity. Proc. Natl. Acad. Sci. U. S. A 2012, 109, 14326–14331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Shaik S; Chen H; Janardanan D Exchange-enhanced reactivity in bond activation by metal–oxo enzymes and synthetic reagents. Nat. Chem 2011, 3, 19–27. [DOI] [PubMed] [Google Scholar]
- (19).Klein JEMN; Que L Jr., Biomimetic High-Valent Mononuclear Nonheme Iron-Oxo Chemistry. In Encyclopedia of Inorganic and Bioinorganic Chemistry, 2016; pp 1–22. [Google Scholar]
- (20).Puri M; Que L Jr. Toward the Synthesis of More Reactive S = 2 Non-Heme Oxoiron(IV) Complexes. Acc. Chem. Res 2015, 48, 2443–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Biswas AN; Puri M; Meier KK; Oloo WN; Rohde GT; Bominaar EL; Münck E; Que L Modeling TauD-J: A High-Spin Nonheme Oxoiron(IV) Complex with High Reactivity toward C–H Bonds. J. Am. Chem. Soc 2015, 137, 2428–2431. [DOI] [PubMed] [Google Scholar]
- (22).Pestovsky O; Bakac A Reactivity of Aqueous Fe(IV) in Hydride and Hydrogen Atom Transfer Reactions. J. Am. Chem. Soc 2004, 126, 13757–13764. [DOI] [PubMed] [Google Scholar]
- (23).Snyder BER; Böttger LH; Bols ML; Yan JJ; Rhoda HM; Jacobs AB; Hu MY; Zhao J; Alp EE; Hedman B; Hodgson KO; Schoonheydt RA; Sels BF; Solomon EI Structural characterization of a non-heme iron active site in zeolites that hydroxylates methane. Proc. Natl. Acad. Sci. U. S. A 2018, 115, 4565–4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Snyder BER; Vanelderen P; Bols ML; Hallaert SD; Böttger LH; Ungur L; Pierloot K; Schoonheydt RA; Sels BF; Solomon EI The active site of low-temperature methane hydroxylation in iron-containing zeolites. Nature 2016, 536, 317–321. [DOI] [PubMed] [Google Scholar]
- (25).Xiao DJ; Bloch ED; Mason JA; Queen WL; Hudson MR; Planas N; Borycz J; Dzubak AL; Verma P; Lee K; Bonino F; Crocellà V; Yano J; Bordiga S; Truhlar DG; Gagliardi L; Brown CM; Long JR Oxidation of ethane to ethanol by N2O in a metal–organic framework with coordinatively unsaturated iron(II) sites. Nat. Chem 2014, 6, 590–595. [DOI] [PubMed] [Google Scholar]
- (26).Gordon JB; Goldberg DP, Sulfur-Ligated, Oxidative Nonheme Iron Enzymes and Related Complexes. In Comprehensive Coordination Chemistry III, Constable EC; Parkin G; Que L Jr, Eds. Elsevier: 2021; Vol. 8, pp 333–377. [Google Scholar]
- (27).Gordon JB; Vilbert AC; DiMucci IM; MacMillan SN; Lancaster KM; Moënne-Loccoz P; Goldberg DP Activation of Dioxygen by a Mononuclear Nonheme Iron Complex: Sequential Peroxo, Oxo, and Hydroxo Intermediates. J. Am. Chem. Soc 2019, 141, 17533–17547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).A subsequent study described the formation of an FeIII(O2•-) species from FeII and O2, which was then proposed to decay through a similar peroxo-bridged, diferric intermediate to give a terminal FeIV(O) species. Kass D; Corona T; Warm K; Braun-Cula B; Kuhlmann U; Bill E; Mebs S; Swart M; Dau H; Haumann M; Hildebrandt P; Ray K Stoichiometric Formation of an Oxoiron(IV) Complex by a Soluble Methane Monooxygenase Type Activation of O2 at an Iron(II)-Cyclam Center. J. Am. Chem. Soc 2020, 142, 5924–5928. [DOI] [PubMed] [Google Scholar]
- (29).Beckmann F; Kass D; Keck M; Yelin S; Hoof S; Cula B; Herwig C; Krause KB; Ar D; Limberg C High-spin square planar iron(II) alkali metal siloxide complexes – influence of the alkali metal and reactivity towards O2 and NO. Z. Anorg. Allg. Chem 2021, 647, 960–967. [Google Scholar]
- (30).Pinkert D; Keck M; Tabrizi SG; Herwig C; Beckmann F; Braun-Cula B; Kaupp M; Limberg C A high-spin square planar iron(II)-siloxide and its tetrahedral allogon – structural and spectroscopic models of Fe-zeolite sites. Chem. Commun 2017, 53, 8081–8084. [DOI] [PubMed] [Google Scholar]
- (31).Dey A; Gordon JB; Albert T; Sabuncu S; Siegler MA; MacMillan SN; Lancaster KM; Moënne-Loccoz P; Goldberg DP A Nonheme Mononuclear {FeNO}7 Complex that Produces N2O in the Absence of an Exogenous Reductant. Angew. Chem. Int. Ed 2021, 60, 21558–21564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Jasniewski AJ; Que L Jr. Dioxygen Activation by Nonheme Diiron Enzymes: Diverse Dioxygen Adducts, High-Valent Intermediates, and Related Model Complexes. Chem. Rev 2018, 118, 2554–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Cranswick MA; Meier KK; Shan X; Stubna A; Kaizer J; Mehn MP; Münck E; Que L Jr. Protonation of a Peroxodiiron(III) Complex and Conversion to a Diiron(III/IV) Intermediate: Implications for Proton-Assisted O–O Bond Cleavage in Nonheme Diiron Enzymes. Inorg. Chem 2012, 51, 10417–10426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Frisch JR; Vu VV; Martinho M; Münck E; Que L Jr. Characterization of Two Distinct Adducts in the Reaction of a Nonheme Diiron(II) Complex with O2. Inorg. Chem 2009, 48, 8325–8336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Kitajima N; Tamura N; Amagai H; Fukui H; Moro-oka Y; Mizutani Y; Kitagawa T; Mathur R; Heerwegh K Monomeric Carboxylate Ferrous Complexes as Models for the Dioxygen Binding Sites in Non-Heme Iron Proteins. The Reversible Formation and Characterization of μ-Peroxo Diferric Complexes. J. Am. Chem. Soc 1994, 116, 9071–9085. [Google Scholar]
- (36).Du Bois J; Mizoguchi TJ; Lippard SJ Understanding the dioxygen reaction chemistry of diiron proteins through synthetic modeling studies. Coord. Chem. Rev 2000, 200–202, 443–485. [Google Scholar]
- (37).Kindermann N; Bill E; Dechert S; Demeshko S; Reijerse EJ; Meyer F A Ferromagnetically Coupled (S=1) Peroxodicopper(II) Complex. Angew. Chem. Int. Ed 2015, 54, 1738–1743. [DOI] [PubMed] [Google Scholar]
- (38).Kurtz DM Oxo- and hydroxo-bridged diiron complexes: a chemical perspective on a biological unit. Chem. Rev 1990, 90, 585–606. [Google Scholar]
- (39).Brunold TC; Tamura N; Kitajima N; Moro-oka Y; Solomon EI Spectroscopic Study of [Fe2(O2)(OBz)2{HB(pz’)3}]2: Nature of the μ–1,2 Peroxide–Fe(III) Bond and Its Possible Relevance to O2 Activation by Non-Heme Iron Enzymes. J. Am. Chem. Soc 1998, 120, 5674–5690. [Google Scholar]
- (40).Groom CR; Bruno IJ; Lightfoot MP; Ward SC The Cambridge Structural Database. Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater 2016, 72, 171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Price JC; Barr EW; Tirupati B; Bollinger JM; Krebs C The First Direct Characterization of a High-Valent Iron Intermediate in the Reaction of an α-Ketoglutarate-Dependent Dioxygenase: A High-Spin Fe(IV) Complex in Taurine/α-Ketoglutarate Dioxygenase (TauD) from Escherichia coli. Biochemistry 2003, 42, 7497–7508. [DOI] [PubMed] [Google Scholar]
- (42).Sinnecker S; Svensen N; Barr EW; Ye S; Bollinger JM; Neese F; Krebs C Spectroscopic and Computational Evaluation of the Structure of the High-Spin Fe(IV)-Oxo Intermediates in Taurine: α-Ketoglutarate Dioxygenase from Escherichia coli and Its His99Ala Ligand Variant. J. Am. Chem. Soc 2007, 129, 6168–6179. [DOI] [PubMed] [Google Scholar]
- (43).Proshlyakov DA; Henshaw TF; Monterosso GR; Ryle MJ; Hausinger RP Direct Detection of Oxygen Intermediates in the Non-Heme Fe Enzyme Taurine/α-Ketoglutarate Dioxygenase. J. Am. Chem. Soc 2004, 126, 1022–1023. [DOI] [PubMed] [Google Scholar]
- (44).Pestovsky O; Stoian S; Bominaar EL; Shan X; Münck E; Que L Jr; Bakac A Aqueous FeIV=O: Spectroscopic Identification and Oxo-Group Exchange. Angew. Chem. Int. Ed 2005, 44, 6871–6874. [DOI] [PubMed] [Google Scholar]
- (45).England J; Guo Y; Farquhar ER; Young VG Jr; Münck E; Que L Jr The Crystal Structure of a High-Spin Oxoiron(IV) Complex and Characterization of Its Self-Decay Pathway. J. Am. Chem. Soc 2010, 132, 8635–8644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).England J; Martinho M; Farquhar ER; Frisch JR; Bominaar EL; Münck E; Que L Jr. A Synthetic High-Spin Oxoiron(IV) Complex: Generation, Spectroscopic Characterization, and Reactivity. Angew. Chem. Int. Ed 2009, 48, 3622–3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Gupta R; Lacy DC; Bominaar EL; Borovik AS; Hendrich MP Electron Paramagnetic Resonance and Mössbauer Spectroscopy and Density Functional Theory Analysis of a High-Spin FeIV–Oxo Complex. J. Am. Chem. Soc 2012, 134, 9775–9784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Lacy DC; Gupta R; Stone KL; Greaves J; Ziller JW; Hendrich MP; Borovik AS Formation, Structure, and EPR Detection of a High Spin FeIV—Oxo Species Derived from Either an FeIII—Oxo or FeIII—OH Complex. J. Am. Chem. Soc 2010, 132, 12188–12190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Bigi JP; Harman WH; Lassalle-Kaiser B; Robles DM; Stich TA; Yano J; Britt RD; Chang CJ A High-Spin Iron(IV)–Oxo Complex Supported by a Trigonal Nonheme Pyrrolide Platform. J. Am. Chem. Soc 2012, 134, 1536–1542. [DOI] [PubMed] [Google Scholar]
- (50).England J; Guo Y; Van Heuvelen KM; Cranswick MA; Rohde GT; Bominaar EL; Münck E; Que L A More Reactive Trigonal-Bipyramidal High-Spin Oxoiron(IV) Complex with a cis-Labile Site. J. Am. Chem. Soc 2011, 133, 11880–11883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Warm K; Paskin A; Kuhlmann U; Bill E; Swart M; Haumann M; Dau H; Hildebrandt P; Ray K A Pseudotetrahedral Terminal Oxoiron(IV) Complex: Mechanistic Promiscuity in C–H Bond Oxidation Reactions. Angew. Chem. Int. Ed 2021, 60, 6752–6756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Rohde J-U; In J-H; Lim MH; Brennessel WW; Bukowski MR; Stubna A; Münck E; Nam W; Que L Jr. Crystallographic and Spectroscopic Characterization of a Nonheme Fe(IV)=O Complex. Science 2003, 299, 1037–1039. [DOI] [PubMed] [Google Scholar]
- (53).Jackson TA; Rohde J-U; Seo MS; Sastri CV; DeHont R; Stubna A; Ohta T; Kitagawa T; Münck E; Nam W; Que L Jr. Axial Ligand Effects on the Geometric and Electronic Structures of Nonheme Oxoiron(IV) Complexes. J. Am. Chem. Soc 2008, 130, 12394–12407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).de Visser SP; Rohde J-U; Lee Y-M; Cho J; Nam W Intrinsic properties and reactivities of mononuclear nonheme iron–oxygen complexes bearing the tetramethylcyclam ligand. Coord. Chem. Rev 2013, 257, 381–393. [Google Scholar]
- (55).Krebs C; Price JC; Baldwin J; Saleh L; Green MT; Bollinger JM Rapid Freeze-Quench 57Fe Mössbauer Spectroscopy: Monitoring Changes of an Iron-Containing Active Site during a Biochemical Reaction. Inorg. Chem 2005, 44, 742–757. [DOI] [PubMed] [Google Scholar]
- (56).Abelson CS; Aboelenen AM; Rasheed W; Que L, Synthetic Nonheme High-Valent Iron-Oxo Complexes Structures and Oxidative Function. In Comprehensive Coordination Chemistry III, Lu Y; Que L Jr., Eds. Elsevier: London, 2021; pp 412–454. [Google Scholar]
- (57).Siegbahn PEM The performance of hybrid DFT for mechanisms involving transition metal complexes in enzymes. JBIC, J. Biol. Inorg. Chem 2006, 11, 695–701. [DOI] [PubMed] [Google Scholar]
- (58).Rasheed W; Draksharapu A; Banerjee S; Young VG Jr.; Fan R; Guo Y; Ozerov M; Nehrkorn J; Krzystek J; Telser J; Que L Jr. Crystallographic Evidence for a Sterically Induced Ferryl Tilt in a Non-Heme Oxoiron(IV) Complex that Makes it a Better Oxidant. Angew. Chem. Int. Ed 2018, 57, 9387–9391. [DOI] [PubMed] [Google Scholar]
- (59).St. John PC; Guan Y; Kim Y; Kim S; Paton RS Prediction of organic homolytic bond dissociation enthalpies at near chemical accuracy with sub-second computational cost. Nat. Commun 2020, 11, 2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Laarhoven LJJ; Mulder P α-C–H Bond Strengths in Tetralin and THF: Application of Competition Experiments in Photoacoustic Calorimetry. J. Phys. Chem. B 1997, 101, 73–77. [Google Scholar]
- (61).Warren JJ; Tronic TA; Mayer JM Thermochemistry of Proton-Coupled Electron Transfer Reagents and its Implications. Chem. Rev 2010, 110, 6961–7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Hagen KS Iron(II) triflate salts as convenient substitutes for perchlorate salts: Crystal structures of [Fe(H2O)6](CF3SO3)2 and Fe(MeCN)4(CF3SO3)2. Inorg. Chem 2000, 39, 5867–5869. [DOI] [PubMed] [Google Scholar]
- (63).Seki H; Abe Y; Gunji T Stereochemistry of the reaction of cis,trans,cis-2,4,6,8-tetraisocyanato-2,4,6,8-tetramethylcyclotetrasiloxane with triphenylsilanol and 1,1,3,3-tetraphenyldisiloxane-1,3-diol. J. Organomet. Chem 2011, 696, 846–851. [Google Scholar]
- (64).Armarego WLF; Chai CLL, Purification of Laboratory Chemicals. 6th ed.; Elsevier/Butterworth-Heinemann: Amsterdam; Boston, 2009; p 760. [Google Scholar]
- (65).Fulmer GR; Miller AJM; Sherden NH; Gottlieb HE; Nudelman A; Stoltz BM; Bercaw JE; Goldberg KI NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar]
- (66).Prisecaru I WMOSS4 Mössbauer Spectral Analysis Software, Version F; 2009.
- (67).Neese F Software update: the ORCA program system, version 4.0. WIREs Comput. Mol. Sci 2018, 8, e1327. [Google Scholar]
- (68).Perdew JP Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [DOI] [PubMed] [Google Scholar]
- (69).Becke AD Completely numerical calculations on diatomic molecules in the local-density approximation. Phys. Rev. A 1986, 33, 2786–2788. [DOI] [PubMed] [Google Scholar]
- (70).Becke AD Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys 1993, 98, 5648–5652. [Google Scholar]
- (71).Grimme S; Antony J; Ehrlich S; Krieg H A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys 2010, 132, 154104. [DOI] [PubMed] [Google Scholar]
- (72).Weigend F Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys 2006, 8, 1057–1065. [DOI] [PubMed] [Google Scholar]
- (73).Neese F; Wennmohs F; Hansen A; Becker U Efficient, approximate and parallel Hartree–Fock and hybrid DFT calculations. A ‘chain-of-spheres’ algorithm for the Hartree–Fock exchange. Chem. Phys 2009, 356, 98–109. [Google Scholar]
- (74).Eichkorn K; Treutler O; Öhm H; Häser M; Ahlrichs R Auxiliary basis sets to approximate Coulomb potentials. Chem. Phys. Lett 1995, 240, 283–290. [Google Scholar]
- (75).Noodleman L; Davidson ER Ligand spin polarization and antiferromagnetic coupling in transition metal dimers. Chem. Phys 1986, 109, 131–143. [Google Scholar]
- (76).Noodleman L Valence bond description of antiferromagnetic coupling in transition metal dimers. J. Chem. Phys 1981, 74, 5737–5743. [Google Scholar]
- (77).Ginsberg AP Magnetic Exchange in Transition Metal Complexes. 12. Calculation of Cluster Exchange Coupling Constants with the Xα-Scattered Wave Method. J. Am. Chem. Soc 1980, 102, 111–117. [Google Scholar]
- (78).Neese F The ORCA program system. WIREs Comput. Mol. Sci 2012, 2, 73–78. [Google Scholar]
- (79).Römelt M; Ye S; Neese F Calibration of modern density functional theory methods for the prediction of 57Fe Mossbauer isomer shifts: meta-GGA and double-hybrid functionals. Inorg. Chem 2009, 48, 784–785. [DOI] [PubMed] [Google Scholar]
- (80).Weigend F; Ahlrichs R Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys 2005, 7, 3297–3305. [DOI] [PubMed] [Google Scholar]
- (81).Schäfer A; Horn H; Ahlrichs R Fully Optimized Contracted Gaussian-Basis Sets for Atoms Li to Kr. J. Chem. Phys 1992, 97, 2571–2577. [Google Scholar]
- (82).Pápai M; Vankó G On Predicting Mössbauer Parameters of Iron-Containing Molecules with Density-Functional Theory. J. Chem. Theory Comput 2013, 9, 5004–5020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Pappas I; Chirik PJ Catalytic Proton Coupled Electron Transfer from Metal Hydrides to Titanocene Amides, Hydrazides and Imides: Determination of Thermodynamic Parameters Relevant to Nitrogen Fixation. J. Am. Chem. Soc 2016, 138, 13379–13389. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.