

Abstract
The SARS-CoV-2 spike protein presents a surface with enormous membrane binding potential to host tissues and organelles of infected cells. Its exposed trimeric head binds not only the angiotensin-converting enzyme 2 (ACE2), but also host phospholipids which are missing from all existing structures. Hence, the membrane interaction surfaces that mediate viral fusion, entry, assembly and egress remain unclear. Here the spike:membrane docking sites are identified based on membrane optimal docking area (MODA) analysis of 3D structures of spike proteins in closed and open conformations at endocytic and neutral pH levels as well as ligand complexes. This reveals multiple membrane binding sites in the closed spike head that together prefer convex membranes and are modulated by pH, fatty acids and post-translational modifications including glycosylation. The exposure of the various membrane interaction sites adjusts upon domain repositioning within the trimer, allowing formation of intermediate bilayer complexes that lead to the prefusion state while also enabling ACE2 receptor recognition. In contrast, all antibodies that target the spike head would block the membrane docking process that precedes ACE2 recognition. Together this illuminates the engagements of the spike protein with plasma, endocytic, ER or exocytic vesicle membranes that help to drive the cycle of viral infection, and offers novel sites for intervention.
Keywords: Coronavirus, Membrane docking, Lipid bilayer, Spike protein, ACE2 receptor, SARS-CoV-2
Graphical abstract
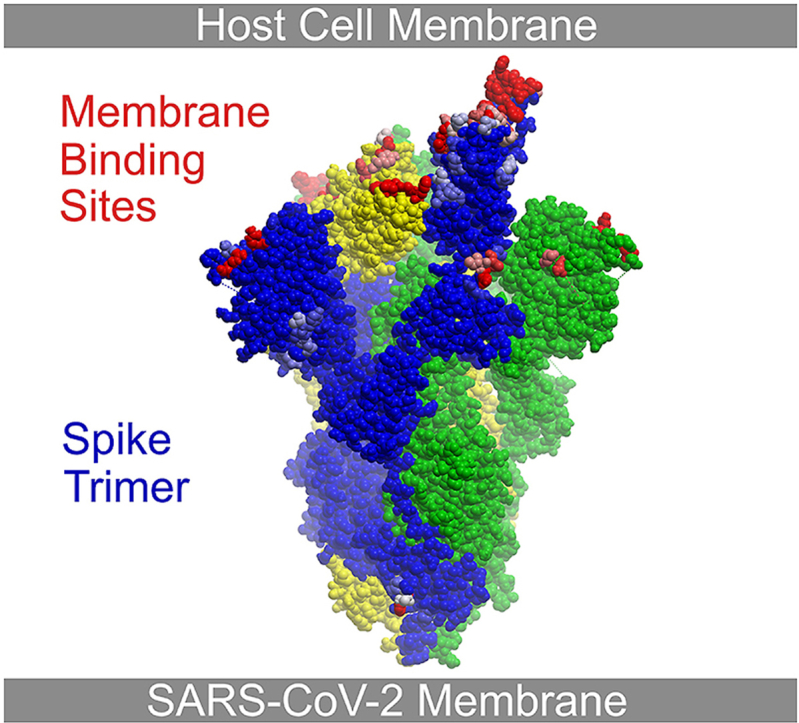
Highlights
-
•
Spike proteins are shown to have high membrane binding propensity for host cell docking.
-
•
Multiple lipid interaction sites are discovered by analysis of many trimeric spike structures.
-
•
SARS CoV-2 spike N-terminal domain is found to have a membrane recognition function.
-
•
ACE2 receptor binding is shown to be aided by membrane interactions of the spike trimer.
-
•
Therapeutic antibodies are shown to compete for binding to spike-membrane docking sites.
1. Introduction
The SARS-CoV-2 coronavirus led to a global COVID-19 pandemic for which mechanistic insights and druggable sites are needed for targeted intervention (J. Zhang et al., 2021). Following entry through the pulmonary system, the virus destroys lung tissues and invades organs including the heart, kidney, brain and liver, leading to cardiogenic shock, renal failure, neurological dysfunction and lymphopenia (Satturwar et al., 2021). Despite the profusion of structures of the viral components, how they mediate assembly on and passage through membranes while eliciting cytotoxic injury remains unclear. This is particularly true for the spike protein, which projects most prominently from the viral particle and is best positioned to mediate interactions with cell and organelle membranes. Its head is also the predominant target for vaccines and therapeutic antibodies, and hence understanding its mechanisms is critical for designing improved therapeutic agents.
The structure of the SARS-CoV-2 spike (S) protein is well defined, with several conformational states now resolved (Table 1). It comprises two subunits, namely S1 (aa 14–685) and S2 (aa 686–1273), which mediate host cell binding and fusion, respectively. The S1 subunit includes a signal peptide (SP), N terminal domain (NTD) and receptor binding domain (RBD), which recognizes the ACE2 glycoprotein through a receptor binding motif (RBM). The S2 subunit contains fusion peptide (FP), heptapeptide repeat sequences 1 and 2 (HR1 and HR2), transmembrane (TM) and cytoplasmic (CP) domains. The MODA algorithm was developed in order to identify novel phospholipid bilayer recognition sites within protein structures (Kufareva et al., 2014). This approach has since been used to resolve such binding surfaces on hundreds of eukaryotic membrane readers (Overduin and Kervin, 2021), prions (Overduin et al., 2021) and viral (Bissig et al., 2013) and bacterial proteins (Bryant et al., 2020a) but has not yet been applied to coronavirus proteins to our knowledge.
Table 1.
Structures of wild-type SARS-CoV-2 spike protein trimers with resolutions of typically under 3.5 Å. The conformations of the RBD, complexed ligands including linoleic acid (L. Acid), ACE2 or antibody proteins are indicated, as are pH values, resolution in Å and PDB entries.
| Organism | Conformation | Complex | pH | Resolution | PDB | Reference |
|---|---|---|---|---|---|---|
| SARS-CoV-2 | 3 RBD down | – | 4.0 | 2.40 | 6xlu | Zhou et al. (2020) |
| SARS-CoV-2 | 3 RBD down | – | 4.5 | 2.50 | 7jwy | Zhou et al. (2020) |
| SARS-CoV-2 | 3 RBD down | – | 5.5 | 3.10 | 6xm5 | Zhou et al. (2020) |
| SARS-CoV-2 | 3 RBD down | – | 7.3 | 2.70 | 7df3 | (Xu et al., 2021) |
| SARS-CoV-2 | 3 RBD down | – | 7.5 | 2.90 | 6xr8 | Cai et al. (2020) |
| SARS-CoV-2 | 3 RBD down | – | 7.5 | 3.00 | 7ddd | (C. Zhang, Wang, et al., 2021) |
| SARS-CoV-2 | 3 RBD down | – | 8.0 | 2.60 | 6zge | (Wrobel et al., 2020) |
| SARS-CoV-2 | 3 RBD down | – | 8.0 | 2.80 | 6vxx | (Walls et al., 2020) |
| SARS-CoV-2 | 3 RBD down | – | 8.0 | 2.80 | 7kdk | (Gobeil et al., 2021) |
| SARS-CoV-2 | 3 RBD down | – | 8.0 | 2.90 | 6zgi | (Wrobel et al., 2020) |
| SARS-CoV-2 | 3 RBD down | – | 8.0 | 2.90 | 6x79 | (McCallum et al., 2020) |
| SARS-CoV-2 | 3 RBD down | – | 8.0 | 3.00 | 6zow | (Melero et al., 2020) |
| SARS-CoV-2 | 3 RBD down | – | 8.0 | 3.01 | 7kdg | (Gobeil et al., 2021) |
| SARS-CoV-2 | 3 RBD down | – | 8.0 | 3.22 | 6x6p | (Herrera et al., 2021) |
| SARS-CoV-2 | 1 RBD up | – | 5.5 | 2.50 | 6xm3 | Zhou et al. (2020) |
| SARS-CoV-2 | 1 RBD up | – | 5.5 | 2.50 | 6xm4 | Zhou et al. (2020) |
| SARS-CoV-2 | 1 RBD up | – | 5.5 | 2.70 | 6xm0 | Zhou et al. (2020) |
| SARS-CoV-2 | 1 RBD up | – | 7.4 | 3.10 | 6zp5 | (Melero et al., 2020) |
| SARS-CoV-2 | 1 RBD up | – | 7.6 | 3.50 | 7eaz | (Yang et al., 2021) |
| SARS-CoV-2 | 1 RBD up | – | 8.0 | 3.10 | 7dx1 | Yan et al. (2021) |
| SARS-CoV-2 | 1 RBD up | – | 8.0 | 3.20 | 6vyb | (Walls et al., 2020) |
| SARS-CoV-2 | 1 RBD up | – | 8.0 | 3.30 | 6zp7 | (Melero et al., 2020) |
| SARS-CoV-2 | 1 RBD up | – | 8.0 | 3.30 | 7dwz | Yan et al. (2021) |
| SARS-CoV-2 | 1 RBD up | – | 8.0 | 3.40 | 6z97 | (Huo et al., 2020) |
| SARS-CoV-2 | 1 RBD up | – | 8.0 | 3.46 | 6vsb | (Wrapp et al., 2020) |
| SARS-CoV-2 | 3 RBD up | 1 ACE2 | 8.0 | 3.30 | 7dx5 | Yan et al. (2021) |
| SARS-CoV-2 | 3 RBD up | 1 ACE2 | 5.5 | 3.85 | 7kne | Zhou et al. (2020) |
| SARS-CoV-2 | 3 RBD up | 1 ACE2 | 8.0 | 3.00 | 7dx6 | Yan et al. (2021) |
| SARS-CoV-2 | 3 RBD up | 2 ACE2 | 5.5 | 3.74 | 7knh | Zhou et al. (2020) |
| SARS-CoV-2 | 3 RBD up | 2 ACE2 | 7.4 | 3.62 | 7kmz | Zhou et al. (2020) |
| SARS-CoV-2 | 3 RBD up | 3 ACE2 | 5.5 | 3.91 | 7kni | Zhou et al. (2020) |
| SARS-CoV-2 | 3 RBD up | 3 ACE2 | 7.4 | 3.64 | 7 kms | Zhou et al. (2020) |
| SARS-CoV-2 | 1 RBD up | 1 Ab | 7.5 | 3.10 | 7sn3 | (Nabel et al., 2021) |
| SARS-CoV-2 | 3 RBD up | 3 Ab | 8.0 | 3.50 | 7vne | (Yang et al., 2021, Wang et al., 2021) |
| SARS-CoV-2 | 3 RBD down | 3 Ab | 4.5 | 2.97 | 7l2e | Cerutti et al. (2021) |
| SARS-CoV-2 | 3 RBD down | 3 Ab | 5.5 | 3.25 | 7lqv | Cerutti et al. (2021) |
| SARS-CoV-2 | 3 RBD down | 3 Ab | 8.0 | 2.20 | 7lxy | McCallum et al. (2021) |
| SARS-CoV-2 | 3 RBD down | 3 Ab | 8.0 | 2.50 | 7ly2 | McCallum et al. (2021) |
| SARS-CoV-2 | 3 RBD down | L. acid | 7.2 | 3.60 | 7jji | Bangaru et al. (2020) |
| SARS-CoV-2 | 3 RBD down | L. acid | 7.5 | 2.85 | 6zb5 | Toelzer et al. (2020) |
| SARS-CoV-2 | 3 RBD down | L. acid | 7.5 | 3.03 | 6zb4 | Toelzer et al. (2020) |
| SARS-CoV-2 | 3 RBD down | L. acid | 8.0 | 2.70 | 7dwy | Yan et al. (2021) |
| SARS-CoV-2 | post-fusion | – | 7.5 | 3.00 | 6xra | Cai et al. (2020) |
The available S structures show various trimer conformations and their ACE2 and antibody complexeshave yet revealed little about how the virus enters cells via plasma membrane or endosomal routes (Jackson et al., 2021). The various states could also engage diverse phospholipids and glycolipids to facilitate membrane docking, budding, vesiculation and pre- or post-fusion events. There is evidence for spike interactions with membrane-associated ligands other than ACE2. Sialic acid presented on host cell surfaces has long been known to be bound by the NTD modules of coronaviruses (Künkel and Herrler, 1993; Krempl et al., 1997; W. Li et al., 2017; Park et al., 2019), and electrostatic interactions of gangliosides and membrane rafts with the SARS-CoV-2 NTD have been modelled (Fantini et al., 2021; Fantini et al., 2021). Glycolipid binding by the RBD of SARS-CoV-2 is critical for infection (Nguyen et al., 2021), and heparan sulfate proteoglycans are also bound by the RBD cooperatively with ACE2 (Clausen et al., 2020). The CoV-2 spike protein is known to bind fatty acid molecules at three sites within the trimer (Toelzer et al., 2020; Bangaru et al., 2020; Carrique et al.) that can also accommodate cholesterol (Shoemark et al., 2021), which is critical for viral entry (Sanders et al., 2021). Such interactions appear to impact not only spike protein behaviour but also COVID-19 disease risk and outcomes.
Essentially all stages of the viral journey from entry, replication and egress involve membranes, with the protruding S heads being ideally positioned to mediate lipid interactions. Indeed, snapshots of the native states involved in the replicative cycle frequently appear to show the S head contacting ER, Golgi, vesicular and plasma membrane surfaces (Mendonça et al., 2021; Klein et al., 2020; Caldas et al., 2021; Eymieux et al., 2021). Moreover, the S1 subunit is known to interact with membranes composed of neutral lipids (Asandei et al., 2020) as well as cholesterol (Wei et al., 2020). The spike ectodomain binds to and extracts phospholipids from membranes (Luchini et al., 2021), and mediates cell-cell fusion (Meng et al., 2021). Yet the lack of structural elucidation of how lipids and membranes are recognized represents a major gap in our understanding of how the viral particles bind, assemble on and pass through cellular membranes.
The paucity of data on spike-membrane binding sites and lipid specificity leaves many mechanistic questions unresolved. After latching onto ACE2 molecules, how do the S1 subunits of the engaged trimer as well as those of many other ACE2-free spike trimers on the viral particle engage the cell membrane? This is especially pertinent given the multiple points of articulation and large freedom of movement of the dozens of hinged S proteins on each viral particle (Ke et al., 2020). Viral entry occurs via an unresolved super-complex that brings together the virus and host plasma membranes together to allow lipid bilayers to contact, fuse and form pores that dilate through a series of molecular docking and rearrangement events (J. Wang et al., 2021). An analogous process also occurs via the endosomal route of entry (Xia et al., 2020; Zhu et al., 2021). These processes are dependent on the local lipid composition, proteases, receptors, calcium and pH levels, the specific effects of which remain unclear but can act as critical triggers (White and Whittaker, 2016). For example, phosphatidyinositol 4-phosphate (PtdIns4P) and PtdIns(3,5)P2 appears to be critical for viral entry (Yang et al., 2012; Ou et al., 2020; Kang et al., 2020), and a PtdIns3P-binding sorting nexin SNX27 is involved in retromer-mediated SARS-CoV-2 entry (Zhu et al., 2021). The ACE2 receptor is poorly expressed on some infected tissues including spleen, bone marrow, brain and blood vessels (M.Y. Li et al., 2020). Could these offer distinct routes of cellular entry that rely on factors other than the presence of abundant ACE2 receptors? Once expressed inside host cells, how are the viral proteins assembled into stable particles and then transported via double membrane vesicles to the plasma membrane where fusion and release into the extracellular space occurs? Multiple functions likely reside with the spike protein (Wong and Saier, 2021), with identification of the relevant sites providing potentially valuable routes for therapeutic intervention, particularly since phospholipid treatment is known to inhibit viral infection (Perino et al., 2011; Voelker and Numata, 2019). Here the lipid interaction sites are mapped in high resolution spike structures, indicating that membrane binding is a core function of the NTD and RBD modules of the S1 subunit and is central to SARS-CoV-2 infectivity.
2. Material and methods
2.1. Structure analysis
Structures of the wild type SARS-CoV-2 spike protein solved by cryo-electron microscopy were obtained from UniProt entry P0DTC2 (Consortium, 2021) and from searches for SARS-CoV-2 structures in the RCSB Protein DataBank (PDB) (Berman et al., 2000). The 43 structures with the highest resolution, typically under 3.5 Å, were selected for atomic resolution analysis of membrane binding sites. The PDB files (Table 1) analyzed include both those of free forms in various pH-dependent conformational states as well as complexes with linoleic acid, ACE2 receptors and antibodies. Structures were analyzed with ICM Browser and PyMol (DeLano, 2014). Missing coordinates were not modelled.
2.2. Identification of high membrane binding propensity residues
The MODA program developed by Ruben Abagyan and collaborators is freely available online and is trained to predict specific residues in PDB files that bind directly to lipid bilayers (Kufareva et al., 2014). We have used this program previously to identify novel validated membrane interacting surfaces in the viral trafficking protein Alix (Bissig et al., 2013), the bacterial outer membrane binding protein DolP (Bryant et al., 2020b), matrix metalloproteinases (Koppisetti et al., 2014) and superfamilies of phox homology (PX) (Kervin and Overduin, 2021) and pleckstrin homology (PH) (Lenoir et al., 2015) domains. The output of MODA includes spreadsheets and structure files, which were compared to assess which residues in each spike assembly could recognize the membranes of diverse cellular destinations encountered by viral particles during their replication journey. Clusters of at least two residues having MODA scores above 20 in at least one structure were considered likely to participate in membrane interactions, while such clusters with residues scoring above 40 indicated a substantial likelihood. This is based on our original calibrations (Kufareva et al., 2014) and on the observation that membrane recognition generally involves multivalent phospholipid binding and bilayer insertion by several proximal basic and apolar residues (Overduin and Kervin, 2021). The confidence of such clusters can be increased by finding recurrent MODA patterns from structures of the same state. This can be seen in heatmaps of MODA values, where values of 0-40-400 are shown with a gradation of blue-yellow-red in the figures to highlight conserved patterns. The significance of changes in membrane binding propensities between structures was determined from the standard deviations of MODA signals of residues in three subunits of symmetric spike trimer structures. To reduce the possibility of false positives, MODA scores of residues adjacent to elements where electron density is missing such as exposed termini and unstructured loops were considered with caution and required validation by other similar structures with more complete density maps. Multiple PDB structures of similar conformational states of spike proteins were compared in the analysis to assess consistency.
2.3. Mapping of membrane binding sites
The SARS-CoV-2 structures were assigned to distinct conformational classes encompassing the various domain-domain rearrangements and ligand complexes. The structure of each class exhibiting the highest resolution and most complete density was selected as being representative. Its subunit with the highest MODA score was chosen to define the optimal membrane binding elements with cross validation to other high-resolution structures in the same conformational class. Pockets that could engage membrane surfaces simultaneously through multiple lipid binding motifs were identified by visual inspection of the structures. The total membrane binding propensity of each motif, pocket and domain was calculated as the sum of the MODA scores of its constituent residues.
2.4. Validation of MODA results
The MODA results were compared to analogous predictions of membrane binding residues in SARS-CoV-2 spike structures. The other fast computational programs for prediction of membrane interacting surfaces of peripheral membrane proteins include Ez-3D (Schramm et al., 2012) and PPM (M.A. Lomize et al., 2012; A.L. Lomize, Todd, and Pogozheva, 2022). Only the latter is available online to predict spatial position of a protein structure on a fluid anisotropic solvent slab that represents a membrane-like environment. Hence the PPM server was used to analyse structures of the highest resolution (resolutions of 2.8 Å or under) of spike trimers in closed conformations as these offer more complete descriptions of loop and side chain conformations for predictions of simpler, symmetric states.
2.5. Regulatory site identification
The SARS-CoV-2 spike residues that are that are glycosylated (Watanabe et al., 2020; Shajahan et al., 2020) or phosphorylated (Davidson et al., 2020; Bouhaddou et al., 2020) were identified from the literature. The proximities of modified residues to the membrane binding elements identified here in the spike protein structures was assessed by visual inspection of models of the lipid bilayer complexes as predicted by PPM 3.0 (A.L. Lomize, Todd, and Pogozheva, 2022). The regulatory impact of small molecule modifications is generally limited to distances corresponding to ∼5 residues, as these bounds were previously useful for identifying positions that, when phosphorylated or metabolite-modified, modulate specific membrane recognition by protein residues acting as PIP-stops (Kervin and Overduin, 2021) and MET-stops in membrane binding domains (Kervin et al., 2021).
3. Results and discussion
3.1. Identification of membrane docking sites of the closed SARS-CoV-2 spike protein
The membrane docking surfaces of the highest resolution spike structures were mapped using the MODA program (Kufareva et al., 2014). This reveals motifs with high propensities for lipid interactions at the top of the prefusion head that comprises a large and discontinuous binding surface (Fig. 1a). The 2.6 Å structure of the uncleaved SARS-CoV-2 spike protein with all RBDs down (PDB: 6zge) was used to represent the most populated wild-type state, which hence is the most likely to approach membrane surfaces. The trimeric head presents three major and largely identical sites in the various closed spike structures that are predicted by MODA to bind very similarly to lipid bilayers (P = 0, R2 = 0.9842). The most central element consists of proximal KVGG motifs within the RBD, including the three V441 residues which are separated by 23 Å (Fig. 1b). Its highly exposed methyl, basic and glycine groups are among the most consistently identified membrane binding features in all subunits assessed by MODA. These protrude furthest from the viral surface such that they could simultaneously insert into a bilayer when the closed spike trimer docks to a host cell membrane at a perpendicular angle. This central protrusion is bordered by the longest predicted membrane docking element, which includes the AGSTP and VEGFNCYF motifs in the RBM and is most accessible when the RBD is positioned upwards (Fig. 1c). This element also projects from each side of the closed head, suggesting docking to a concave membrane. The circumference of the closed head projects membrane binding surfaces formed by the TQLPPA-27, HAIH-69 and VR-214 motifs in the NTD (Fig. 1d). This site offers proximal basic residues including R21, H66, H69, K77, R78, and R214, which could be expected to engage acidic lipid headgroups. This extensive docking surface of the closed head offers exposed aliphatic residues including L24, I68, V70 and V213 that could insert into the hydrophobic interior of the bilayer. The only other site on the NTD that is predicted to bind membranes is the QPFL-176 motif, which is positioned near the disordered N-terminus and offers the smallest area with significant membrane binding propensity in the closed state. Together these elements of the top and sides of the spike head offer exposed surfaces that are rich in the basic, hydrophobic and polar residues typically found in lipid binding pockets. Consistent MODA results are found in other high-resolution structures of closed spike trimers, including the furin-cleaved S1 conformation (PDB 6zgi, P = 0, R2 = 0.9923) as well as those from other studies despite their less complete electron densities (PDBs 7kdk, 6x79, 6vxx, P = 4.48 × 10−87, R2 = 0.3547), as can be seen in the heatmap (Fig. 2a). The conserved lipid binding patches and expansive membrane docking area of the spike head contrasts with the rest of the trimeric spike protein, which is essentially devoid of membrane interacting elements. Thus, we propose that the membrane docking surfaces identified here offer high avidity, multivalent lipid binding as needed to engage curved cell membranes in either perpendicular or oblique angles.
Fig. 1.

Membrane binding residues of spike protein structures. A. The residues with moderate (orange) and substantial (red) membrane binding propensities in the wildtype SARS-CoV-2 spike protein trimer’s closed state (PDB: 6zge) are shown on the surface as are the residues that are glycosylated (blue) or phosphorylated (magenta). The subunits are depicted as light purple, green and yellow surfaces. B. The peak of the spike trimer presents a central membrane binding region with KVGG motif residues shown on a silver backbone ribbon of the closed spike trimer structure (PDB 6zge). C. The membrane binding pocket surface of an RBD-up subunit of the open structure of the spike trimer (PDB 7dwz) extends furthest from the virion and closest to a host cell membrane, with residues predicted by MODA to be membrane interactive labelled and color-coded. D. The circumference of the closed spike trimer structure (PDB 6zge) presents three strips of membrane interacting residues on the subunits depicted as silver, green and yellow backbone ribbons, with residues with significant membrane binding propensities within the four sites in the NTD color-coded and labelled, as is the N-terminus. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Fig. 2.

Effect of linoleic acid on the membrane binding propensities of residues of wild-type CoV-2 S1 subunits from closed trimer structures with all RBD’s down. A) The PDB entries are indicated as is the presence of the fatty acid in the structure. Residue positions 14 to 508 of each the three subunits, which are ordered a, b, and c, are colored on a gradation of blue, yellow and red to indicate MODA scores of 0, 40 and at least 400 in the heatmap. Grey areas are missing in the structures. The predicted membrane binding motifs in the NTD and RBD are labelled on the left of the heatmap. B) The significant differences of MODA values of key residues of closed spike trimers in fatty acid-bound vs free states (7dwy vs 6zge) are shown. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.2. Validation of membrane binding residues
The Positioning of Proteins in Membranes (PPM) server can also be used to identify membrane interacting residues and was used here to confirm which spike residues are most likely to insert into the lipid bilayer. The following residues in various subunits were identified by PPM as being membrane interacting: 6zge: a483-486; 6vxx:b489-b490; 6xlu: a484-486; 7jwy:a484-486; 7df3: a445–446,b445-446,c445-446; and 7kdk:b489-490. These 20 residues exhibit significant MODA scores ranging from 48.13 to 2440.43, which is near the maximum end of the MODA range. They all are found within the major membrane binding surface of the RBM. The major difference is that PPM predicts that only 3.33 ± 1.51 residues in each spike trimer structure, typically from a single subunit, interact with membranes while MODA identifies 81.5 ± 11.7 membrane binding residues that are roughly symmetrically arrayed around each closed spike trimer structure. Hence although the two methods converge on the highest membrane binding propensity residues on one subunit, MODA provides more sensitive detection and more complete coverage of expansive sites on multi-subunit structures.
3.3. Post-translational modifications influencing membrane binding
An important feature of the spike protein, particularly regarding immune evasion, is glycosylation. The array of N-linked glycans that cover the surface of the spike protein of SARS-CoV-2 compromise antigen recognition, thus shielding the virus from the host’s immune response. However, the key functional sites including the points of proteolytic activation must remain unhindered for the virus to invade and replicate successfully. None of the identified membrane docking residues identified here in the wild type closed spike protein are glycosylated, indicating a lack of direct overlap that could otherwise compromise membrane recognition. The most prominent sites with high membrane docking propensities in the RBD are free of glycosylation and hence could be generally available to mediate host membrane interactions in the closed or open states of the free forms of the trimer. However glycosylated residue N343 displays significant membrane binding propensities in RBD-down subunit structures, particularly those prepared at low pH (Fig. 3a&4a). This suggests that bulky glycans here would occlude bilayer interactions at these locations around the periphery of the RBD’s membrane binding surface. This is consistent with the most critical interactions being mediated by the ternary membrane binding site at the central protrusion of the spike trimer. Glycosylated residues in the NTD including N17, N122, N149, N165, as well as N74 (which is unresolved in SARS-CoV-2 spike trimer structures) could also partially occlude interactions with concave membrane surfaces by the upper rim of the spike head. Although residues T29 and S31 are reported to be phosphorylated (Davidson et al., 2020; Bouhaddou et al., 2020) and are near the membrane-binding TQLPPA-27 motif and hence could theoretically regulate such interactions, the role of such modifications in trafficking and signaling pathways remains unclear. Hence we propose that bulky glycosylations (S. Zhang et al., 2022) are most likely to attenuate the dynamic interactions of the major membrane binding surface with curved bilayers as spike trimers assemble and traffic through endoplasmic reticulum, Golgi and secretory vesicle compartments that lack ACE2, as well as influencing the specificity and dynamics of host cell membrane fusion and invasion (Bagdonaite and Wandall, 2018).
Fig. 3.

Effects of pH on the membrane binding propensities of the residues of the wild-type CoV-2 S1 subunits of closed trimers with all RBD’s down collected. A) Heatmap of MODA scores of each residue of each subunit of closed spike trimers collected at the pH values indicated above the PDB codes. Colors are as in Fig. 2a). B. Structures collected at pH 4.0 and 8.0 (PDB entries 6xlu [blue] and 6zge [grey], respectively), are superimposed. Sidechains of NTD residues exhibiting membrane binding propensities in the various closed spike structures at low pH are color-coded as shown. C. The surface of the closed spike trimer structure collected at pH 5.5 (PDB 6xm5) is shown in blue, with residues having moderate and substantial MODA scores colored pink and red, respectively. The position of the C termini is labelled “C”. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Fig. 4.

Membrane binding by RBD up and down conformers of wild-type CoV-2 S1 trimers. A. The heatmap shows the membrane binding propensity of each residue of the structures from indicated PDB entries collected at various pH values. The RBD up and down positions of subunits a, b and c of each structure are shown with black up and grey down arrows below. The MODA scores of each residue of the three subunits are colored as in Fig. 2, with key positions labelled on the left. B. Top and side views of the 3D structure of the spike trimer structure with one RBD up (PDB 7dwz) is shown with a, b and c subunit surfaces colored blue, green and yellow. Residues with moderate and substantial MODA scores are in pink and red, respectively, and are labelled, as is the C-terminus. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.4. Fatty acid and detergent interactions impact membrane sites
The membrane binding sites are affected by ligand occupancy, as is evident by comparison with several spike structures containing bound linoleic acid molecules that stabilize the closed conformation (Toelzer et al., 2020; Yan et al., 2021; Bangaru et al., 2020). The similar amphipathic character of linoleic acid and phospholipid molecules prompted us to investigate whether their binding sites overlap. The fatty acid headgroup interacts with R408 and Q409, while the acyl tail inserts into a neighbouring subunit’s hydrophobic pocket lined with phenylalanine residues as seen in three structures (PDB 7dwy, 6zb4 and 6zb5). The residues involved in binding linoleic acid do not exhibit membrane binding propensities, suggesting that linoleic acid binds as a soluble rather than membrane-bound molecule. Interestingly, the linoleic acid complexes exhibit significant decreases in membrane binding propensities of the TQLPPA-27, QPFL-176 and KVGG-447 motifs (Fig. 2a), as demonstrated with residues L24, F175 and K444, respectively (Fig. 2b). These correspond to high membrane binding propensity sites of the closed state, suggesting that linoleic acid binding and release reduces and enhances the spike protein’s membrane interactivity, respectively. Apparent linoleic acid-induced increases in membrane binding propensities are seen in K147, Q183, Y248, and L249 residues that do not exhibit substantial MODA scores in the free, closed state. This is consistent with molecular dynamics simulations showing that linoleic acid removal induces allosteric changes in elements including L141-E156, C166–F186, L249-G257 (Sofia F Oliveira et al., 2021). Hence binding of free fatty acid appears to reduce the spike protein’s canonical membrane binding and could compromise its specific lipid bilayer interactions, reinforcing its value as a potentially impactful druggable site. This effect is in addition to linoleic acid’s ability to reduce ACE2 receptor affinity and hence could also contribute to its inhibition of viral replication (Toelzer et al., 2020; Yan et al., 2021; Bangaru et al., 2020).
Detergent molecules interact with shallow pockets near membrane binding sites of the spike protein (Bangaru et al., 2020). In particular, the aliphatic tail of polysorbate 80 can be accommodated by a hydrophobic pocket lined by residues including N121, V126, Y170, S172 and F175, with the latter three residues overlapping a membrane interacting site. The detergent’s polar headgroup is situated near residues R190 and H207, which lack significant membrane binding propensity. Hence the apolar portion of this bound detergent may mimic the position of biological lipids, reinforcing the notion that the QPFL-176 membrane interaction site identified here engages membrane bilayers.
3.5. Influence of pH on membrane binding propensity of spike proteins
Coronaviruses such as SARS-CoV-2 can enter host cells through early and late endosomes where pH values are ∼6.3 and under 6, respectively, and exit through secretory vesicles with pH values of ∼5.5. We investigated whether the acidic environments experienced by the spike protein alter its membrane binding sites by comparing structures collected at a variety of pH values (Zhou et al., 2020). MODA analysis reveals that the HRSYL-249 element displays consistently higher membrane binding propensity in all structures collected at low pH as compared to those seen in other states of closed spikes (Fig. 3a). This element is positioned close to H69, which also exhibits higher membrane propensities in various low pH structures. This histidine-dense cluster comprises H66, H69 and H245 (Fig. 3b), and their conformation differs from that of the neutral pH form. In particular, the H66 ring is significantly closer to H69 (10.2 ± 0.4 Å vs 14.1 ± 2.0 Å between Nε1 atoms) and to H245 (14.6 ± 1.5 Å vs 18.2 ± 0.3 Å) in structures collected at pH 4.5–5.5. This is reminiscent of endosome-targeted FYVE domains which contain a conserved pair of proximal histidines that form a pH switch and move closer together upon PtdIns3P binding (Lee et al., 2005; Kutateladze and Overduin, 2001). By analogy, we propose that the triple His cluster depresses the pH at which multiple imidazole rings become positively charged, thus providing selectivity for their acidic phospholipid targets. The spike histidines form the center of an extensive membrane binding surface lined by residues that generally yield increased MODA signals at low pH, in contrast to the reduced MODA signals of their KVGG-447 motifs, suggesting a migration of binding activity from the tip to the rim of the spike head. The NTD surfaces could preferentially engage concave membrane surfaces given that they project the furthest laterally from the central axis of the spike trimer (Fig. 3c) and adjust their position at low pH (Zhou et al., 2020). This is consistent with the proposed roles of coronavirus NTDs in membrane interactions (Künkel and Herrler, 1993; Krempl et al., 1997; W. Li et al., 2017; Park et al., 2019; Fantini et al., 2021; Fantini et al., 2021), Moreover, we speculate that the H69 deletion found in Alpha and Omicron variants of concern could compromise this acidic membrane binding switch. Thus, we propose that the closed spike trimer exhibits increased acidic membrane binding via a tightened histidine-rich pocket, enhancing interactions of the rim of the spike head with phospholipids as viral particles traffic through low pH vesicles inside the cell.
3.6. Spike conformation affects its membrane docking function
We investigated whether membrane interactions could be affected by conformational rearrangements of the SARS-CoV-2 spike that enable strong ACE2 binding. Comparison of structures with one RBD up and closed states reveals significantly enhanced membrane binding propensities in the raised RBD, particularly of the L455-F456, Y473-A475 and G504-Y505 motifs (Fig. 4a) due to their greater sidechain accessibility and a reorientation of Y505 at neutral pH to a more solvent exposed position. This contributes to an extensive trimer docking surface that could bind host membranes via the uplifted RBD that projects furthest from the viral membrane (Fig. 1, Fig. 4b). The other subunit RBDs remain free to engage membranes in their down states or could flip up, leading to an encounter with more host membrane surface or an ACE2 molecule. Together this suggests that host membrane binding by spikes increases when an RBD flips up and is then mediated by RBM-proximal sites that comprise an extensive, accessible docking surface but would be partially occluded by an ACE2 protein.
We propose that the open, RBD-up state of the spike trimer is stabilized by membrane interactions, which would concentrate and transiently position viral particles on the surfaces of host membranes. This in turn could enhance the probability of productive encounters with ACE2 receptors in host cell membranes, which compete effectively with the host membrane for binding to an RBM through higher affinity interactions. This model would clearly benefit from experiments comparing the membrane and ACE2 affinities of the various domains and trimer states, and while this is beyond the scope of the present study, our identification of the residues involved could inform such studies. It is already clear that these elements are hotspots for mutations, with L455F, A475S, A475V, Y505H and Y505W (Elbe and Buckland-Merrett, 2017) spike mutations of variants of concern mapping to the enhanced membrane binding surface of the upturned RBD, along with other mutations in the NTD and down RBD sites also potentially impacting such interactions.
The conformational equilibria of NTD and RBD rearrangements in the spike trimer structures is pH-dependent. In particular dropping the pH below 5.5 disfavours single RBD-up positions and instead maintains all RBDs in down states based on computational analysis by Kwong and colleagues (Zhou et al., 2020). At pH 5.5, significantly elevated membrane binding propensities are evident in RBD-up subunit residues W64, H66, K97–I101, V213 and R214 as well as in the more ordered A243-P251 sequence due to greater solvent exposure of these NTD elements (Fig. 4a). This is reminiscent of increased membrane binding propensity of the NTD sites at low pH when all RBD’s are positioned in the down state (Section 3.5). Hence NTD:membrane binding appears to be enhanced at the lower pH values found in endosomal and secretory compartments irrespective of whether the RBD is positioned up or down, while membrane interactions of the RBD’s up conformation appear to be favoured at neutral pH values such as those found at the cell surface.
3.7. ACE2 receptor effects on spike membrane interactions
The spike trimer can bind one, two and then three ACE2 receptors molecules via its three elevated RBM surfaces, with the last, symmetric complex being best positioned to initiate membrane fusion (J. Wang et al., 2021). We investigated how this multi-step process could be mediated by comparing the membrane docking poses of the single, double and triple ACE2 protein complexes. The ACE2 receptor directly competes with membrane for binding to RBM sites with the following spike residues being close (4.5 Å) to ACE2 as well as having high membrane propensities: G446, Y449, L455, F456, Y473, A475, G476, E484, F486, N487, Y489, Q493, G496, Q498, T500, N501, G502 and Y505. Hence, there is direct competition between ACE2 receptors and membranes for attachment via the top of the spike head. The entire upper surface of the head appears to prefer binding to concave lipid bilayers with intrinsic curvature radii of 130 Å, to which it binds with a ΔGtransfer of −11.1 kcal/mol based on PPM 3.0 predictions (Fig. 6b). While this covers the top part of the NTD distal from the viral membrane, several patches around the rim of spike trimers could still potentially interact with phospholipids even if three ACE2 molecules are bound. In particular, N125, N137, F157, R214, Q218, I332, N334, L355, N360, S469, T470, I472, G482 and F490 exhibit membrane binding propensities in ACE2-complexed states (Figs. 5 and 6a). These form exposed binding surfaces on three sides of the spike head, suggesting that they could also interact with viral or host cell membranes when S is acutely tilted or after S1 is released, respectively. Moreover, asymmetric spike complexes with one or two ACE2 molecules could conceivably employ free residues of the remaining RBDs (Fig. 5a) to engage host cell membranes, allowing such assemblies to scan the host surface for additional ligands such as free ACE2 molecules to accelerate formation of a fully receptor-loaded prefusion state. Thus in addition to directly mediating the critical fusion process, we propose that the 25–130 spike trimers found on each SARS-CoV-2 virion (Bar-On et al., 2020; Ke et al., 2020; Klein et al., 2020) could also participate indirectly by using their heads for lipid bilayer tethering, apposition and disruption to support the fusion of viruses and host cells.
Fig. 6.

Comparison of the ACE2, antibody and membrane positions in the SARS-CoV-2 spike protein structures. A. The top view of the spike trimer (blue surface, PDB: 7kms) bound to 3 ACE2 molecules (ribbons colored aqua, green, grey) with the residues exhibiting moderate to substantial membrane binding propensities shown in white to red and labelled. B. A curved lipid bilayer (concave red and blue slab) is recognized by the symmetric contacts of the closed spike trimer (PDB: 7df3) based on PPM 3.0 analysis. The spike subunits are rainbow colored from the N- (blue) the C- (red) termini. Glycans are shown as sticks. C. The closed spike structure (PDB: 6zge) is shown in top and side views, with the residues exhibiting membrane binding propensities colored as in A and labelled. D. The structure of the closed spike trimer (PDB: 7lxy) bound to three RBD-specific antibodies (S2M11) and three NTD-specific antibodies (S2M28), which are drawn as colored-coded ribbons and labelled with their subunits, is shown in top and side views, along with residues exhibiting membrane binding propensities. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Fig. 5.

Membrane binding by ACE2-bound spike trimers. A. The heatmap shows the membrane binding propensity of each residue in the various ACE2 complexed SARS-CoV-2 structures for the indicated PDB entries collected at the various pH values. The ACE2 bound and free states of each subunit structure are shown with black and light grey circles below the heatmap, as are the RBD positions. B. The structure of the spike protein is shown as a surface with a, b, and c subunits in blue, green and yellow, with the three bound ACE2 proteins shown as ribbons with similar, lighter colors. The residues with moderate and substantial MODA scores are shown in pink and red, respectively, and are labelled as are the C-termini. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
The viral membrane plays a role in orienting the spike ectodomain. The only predicted membrane binding residue in S that extends toward the viral membrane is H1101. This residue exhibits membrane binding propensity in 23% of trimeric spike structures, with the aromatic sidechain oriented towards the viral membrane (Fig. 5b). Its sidechain could interact with the viral membrane given that spike ectodomains can lean over from 1 to 116° (B. Wang, Zhong, and Tieleman, 2021). Such tilted positions could allow the NTD’s membrane binding surface around the circumference of the spike head to sit on the viral membrane with or without ACE2 being bound (Figs. 3b and 7 b). Protonation of the H1101 ring at low pH could also contribute to long range electrostatic interactions with lipids in endocytic or secretory vesicles. Viral membrane interactions by the H1101 motif could be compromised by steric clashes of the glycans added to N1098, which otherwise stabilizes the exposed hairpin turn that positions their sidechains. Together this suggests that the angle and orientation of spike trimers over the virus surface could be modulated by both pH and ACE2 occupancy.
Following ACE2 binding and proteolytic cleavage of the spike protein into S1 and S2 fragments, the latter rearranges into a thin stalk (Cai et al., 2020). The S1 subunit could conceivably interact with the host membrane via the membrane interaction sites identified here (Fig. 5b), allowing it to remain localized and contribute to the fusion process. The available post-fusion S2 structure (PDB 6xra) displays only a few residues with significant membrane binding propensities, i.e. T1100, H1101, I1132 and N1134. Membrane binding by S2 subunit at these positions would be diminished by the addition of glycans to residues N1098 and N1134, which would sterically occlude docking to lipid bilayers via these motifs. Thus, the majority of membrane binding by S2 is likely mediated by other elements such as the FP, HR1, and HR2 elements. A 3-helix bundle is formed by the FP at the N-terminal end of S2, forming a wedge that inserts into the host membrane (Koppisetti et al., 2021). The heptad repeats form α helices that interact with phospholipid bilayers and oligomerize, thus pulling the viral and host membranes closer together (Chiliveri et al., 2021). The transmembrane region forms an α helical trimer that anchors into the viral membrane (Fu and Chou, 2021), while palmitoylation of ten C-terminal cysteines promotes interactions with the inner membrane leaflet by the cytoplasmic domain and enhances fusion capacity (Mesquita et al., 2021). The membrane interactions of the downstream intermediates such as the elongated stalk and hemi-fusion and fusion pore states (J. Wang et al., 2021) remain to be elucidated. While characterization of the lipid bilayer interactions of such spike components would also benefit from further experimental studies, MODA could pinpoint the residues involved, providing opportunities for defining their conformationally-specific contributions to membrane docking.
3.8. Antibody targeting of membrane docking sites
Monoclonal antibodies that specifically target the spike protein are widely used to treat high-risk cases of COVID-19 (Ledford, 2021). While those that target the RBD are known to block ACE2 binding directly, the mechanisms whereby those that target the NTD remain unclear. The latter are known to bind to an antigenic site that involves Y144, K147, K150, E156, R158, R246, S247 and Y248 (Cerutti et al., 2021; McCallum et al., 2021). These residues are located near a membrane binding surface identified here within the NTD. Hence this antigenic NTD supersite overlaps a membrane docking site, and antibody binding here would interfere directly with membrane interactions. The same holds true for the RBD’s antigenic site, which overlaps the surface that is normally able to dock to membranes based on our analysis. When antibodies occupy the closed spike trimer’s RBD and NTD domains, significant membrane binding propensity is only retained by the exposed NKSW-152 and NLVRD-215 motifs (Fig. 6D). However, these are largely inaccessible to host cells, being tucked under the head and overshadowed by the Fabs. Hence our results suggest a unifying hypothesis whereby the neutralizing activity of therapeutic antibodies is due not only to competition with RBM-mediated ACE2 binding but also through competition of host cell membrane interactions by key sites on both the spike NTD and RBD domains. This explains the otherwise unclear effects of NTD-specific antibodies (Liu et al., 2020; Chi et al., 2020) and infers that multivalent membrane binding is essential to viral infection.
Despite the impressive progress in the structural elucidation of spike complexes, the exact mechanisms occurring throughout the process of viral infection and replication remain challenging to discern. Whether specific phospholipids and glycolipids are recognized by the various pockets identified is unclear. The precise effects of mutations, post-translational modifications, membrane order and bilayer curvature on binding and fusion events warrant further experimental investigation. The dynamics between domains, subunits and receptors of the spike protein require computational approaches to simulate the journey of viral entry, assembly and egress. Although lipids are thought to play central roles in viral replication, how the protein-lipid interactions involved could be exploited to effectively alter COVID-19 disease risk and outcomes remains to be seen. Nonetheless, the advances by many research groups indicate that these interactions are key to the infection cycle, with the visualization provided here unlocking the potential of such sites.
Funding
This work was supported by NSERC Discovery Grant (RGPIN- 2018–04994) and Campus Alberta Innovates Program (#RCP-12-002C) grants to MO.
CRediT authorship contribution statement
Anh Tran: Formal analysis. Troy A. Kervin: Formal analysis. Michael Overduin: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
We thank Molsoft LLC for making the ICM program (MODA’s engine) and MODA available.
Handling editor: Natalie Strynadka
References
- Asandei A., Mereuta L., Schiopu I., Park J., Seo C.H., Park Y., Luchian T. Non-receptor-mediated lipid membrane permeabilization by the SARS-CoV-2 spike protein S1 subunit. ACS Appl. Mater. Interfaces. 2020;12(50):55649–55658. doi: 10.1021/acsami.0c17044. https://www.ncbi.nlm.nih.gov/pubmed/33270413 [DOI] [PubMed] [Google Scholar]
- Bagdonaite I., Wandall H.H. Global aspects of viral glycosylation. Glycobiology. 2018;28(7):443–467. doi: 10.1093/glycob/cwy021. https://www.ncbi.nlm.nih.gov/pubmed/29579213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangaru S., Ozorowski G., Turner H.L., Antanasijevic A., Huang D., Wang X., Torres J.L., Diedrich J.K., Tian J.H., Portnoff A.D., Patel N., Massare M.J., Yates J.R., Nemazee D., Paulson J.C., Glenn G., Smith G., Ward A.B. Structural analysis of full-length SARS-CoV-2 spike protein from an advanced vaccine candidate. Science. 2020;370(6520):1089–1094. doi: 10.1126/science.abe1502. https://www.ncbi.nlm.nih.gov/pubmed/33082295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-On Y.M., Flamholz A., Phillips R., Milo R. SARS-CoV-2 (COVID-19) by the numbers. Elife. 2020;9 doi: 10.7554/eLife.57309. https://www.ncbi.nlm.nih.gov/pubmed/32228860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. The protein data bank. Nucleic Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. https://www.ncbi.nlm.nih.gov/pubmed/10592235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissig C., Lenoir M., Velluz M.C., Kufareva I., Abagyan R., Overduin M., Gruenberg J. Viral infection controlled by a calcium-dependent lipid-binding module in ALIX. Dev. Cell. 2013;25(4):364–373. doi: 10.1016/j.devcel.2013.04.003. https://www.ncbi.nlm.nih.gov/pubmed/23664863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouhaddou M., Memon D., Meyer B., White K.M., Rezelj V.V., Correa Marrero M., Polacco B.J., Melnyk J.E., Ulferts S., Kaake R.M., Batra J., Richards A.L., Stevenson E., Gordon D.E., Rojc A., Obernier K., Fabius J.M., Soucheray M., Miorin L., Moreno E., Koh C., Tran Q.D., Hardy A., Robinot R., Vallet T., Nilsson-Payant B.E., Hernandez-Armenta C., Dunham A., Weigang S., Knerr J., Modak M., Quintero D., Zhou Y., Dugourd A., Valdeolivas A., Patil T., Li Q., Hüttenhain R., Cakir M., Muralidharan M., Kim M., Jang G., Tutuncuoglu B., Hiatt J., Guo J.Z., Xu J., Bouhaddou S., Mathy C.J.P., Gaulton A., Manners E.J., Félix E., Shi Y., Goff M., Lim J.K., McBride T., O'Neal M.C., Cai Y., Chang J.C.J., Broadhurst D.J., Klippsten S., De Wit E., Leach A.R., Kortemme T., Shoichet B., Ott M., Saez-Rodriguez J., tenOever B.R., Mullins R.D., Fischer E.R., Kochs G., Grosse R., García-Sastre A., Vignuzzi M., Johnson J.R., Shokat K.M., Swaney D.L., Beltrao P., Krogan N.J. The global phosphorylation landscape of SARS-CoV-2 infection. Cell. 2020;182(3):685–712. doi: 10.1016/j.cell.2020.06.034. https://www.ncbi.nlm.nih.gov/pubmed/32645325 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant J.A., Morris F.C., Knowles T.J., Maderbocus R., Heinz E., Boelter G., Alodaini D., Colyer A., Wotherspoon P.J., Staunton K.A., Jeeves M., Browning D.F., Sevastsyanovich Y.R., Wells T.J., Rossiter A.E., Bavro V.N., Sridhar P., Ward D.G., Chong Z.S., Goodall E.C.A., Icke C., Teo A., Chng S.S., Roper D.I., Lithgow T., Cunningham A.F., Banzhaf M., Overduin M., Henderson I.R. Structure of dual-BON domain protein DolP identifies phospholipid binding as a new mechanism for protein localization. Elife. 2020;9 doi: 10.7554/eLife.62614. https://www.ncbi.nlm.nih.gov/pubmed/33315009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant J.A., Morris F.C., Knowles T.J., Maderbocus R., Heinz E., Boelter G., Alodaini D., Colyer A., Wotherspoon P.J., Staunton K.A., Jeeves M., Browning D.F., Sevastsyanovich Y.R., Wells T.J., Rossiter A.E., Bavro V.N., Sridhar P., Ward D.G., Chong Z.S., Goodall E.C., Icke C., Teo A.C., Chng S.S., Roper D.I., Lithgow T., Cunningham A.F., Banzhaf M., Overduin M., Henderson I.R. Structure of dual BON-domain protein DolP identifies phospholipid binding as a new mechanism for protein localisation. Elife. 2020;9 doi: 10.7554/eLife.62614. https://www.ncbi.nlm.nih.gov/pubmed/33315009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y., Zhang J., Xiao T., Peng H., Sterling S.M., Walsh R.M., Rawson S., Rits-Volloch S., Chen B. Distinct conformational states of SARS-CoV-2 spike protein. Science. 2020;369(6511):1586–1592. doi: 10.1126/science.abd4251. https://www.ncbi.nlm.nih.gov/pubmed/32694201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldas L.A., Carneiro F.A., Monteiro F.L., Augusto I., Higa L.M., Miranda K., Tanuri A., de Souza W. Intracellular host cell membrane remodelling induced by SARS-CoV-2 infection in vitro. Biol. Cell. 2021;113(6):281–293. doi: 10.1111/boc.202000146. https://www.ncbi.nlm.nih.gov/pubmed/33600624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrique, Loic, Helen ME Duyvesteyn, Tomas Malinauskas, Yuguang Zhao, Jingshan Ren, Daming Zhou, Thomas S. Walter, Julika Radecke, Jiandong Huo, Reinis R. Ruza, Pranav NM Shah, Elizabeth Fry, and David I Stuart. The SARS-CoV-2 Spike Harbours a Lipid Binding Pocket Which Modulates Stability of the Prefusion Trimer. 10.2139/ssrn.3656643%20SSRN: https://ssrn.com/abstract=3656643. [DOI]
- Cerutti G., Guo Y., Zhou T., Gorman J., Lee M., Rapp M., Reddem E.R., Yu J., Bahna F., Bimela J., Huang Y., Katsamba P.S., Liu L., Nair M.S., Rawi R., Olia A.S., Wang P., Zhang B., Chuang G.Y., Ho D.D., Sheng Z., Kwong P.D., Shapiro L. Potent SARS-CoV-2 neutralizing antibodies directed against spike N-terminal domain target a single supersite. Cell Host Microbe. 2021;29(5):819–833. doi: 10.1016/j.chom.2021.03.005. https://www.ncbi.nlm.nih.gov/pubmed/33789084 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi X., Yan R., Zhang J., Zhang G., Zhang Y., Hao M., Zhang Z., Fan P., Dong Y., Yang Y., Chen Z., Guo Y., Li Y., Song X., Chen Y., Xia L., Fu L., Hou L., Xu J., Yu C., Li J., Zhou Q., Chen W. A neutralizing human antibody binds to the N-terminal domain of the Spike protein of SARS-CoV-2. Science. 2020;369(6504):650–655. doi: 10.1126/science.abc6952. https://www.ncbi.nlm.nih.gov/pubmed/32571838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiliveri S.C., Louis J.M., Ghirlando R., Bax A. Transient lipid-bound states of spike protein heptad repeats provide insights into SARS-CoV-2 membrane fusion. Sci. Adv. 2021;7(41):eabk2226. doi: 10.1126/sciadv.abk2226. https://www.ncbi.nlm.nih.gov/pubmed/34623907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen T.M., Sandoval D.R., Spliid C.B., Pihl J., Perrett H.R., Painter C.D., Narayanan A., Majowicz S.A., Kwong E.M., McVicar R.N., Thacker B.E., Glass C.A., Yang Z., Torres J.L., Golden G.J., Bartels P.L., Porell R.N., Garretson A.F., Laubach L., Feldman J., Yin X., Pu Y., Hauser B.M., Caradonna T.M., Kellman B.P., Martino C., M Gordts P.L.S., Chanda S.K., Schmidt A.G., Godula K., Leibel S.L., Jose J., Corbett K.D., Ward A.B., Carlin A.F., Esko J.D. SARS-CoV-2 infection depends on cellular heparan sulfate and ACE2. Cell. 2020;183(4):1043–1057. doi: 10.1016/j.cell.2020.09.033. https://www.ncbi.nlm.nih.gov/pubmed/32970989 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium UniProt. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 2021;49(D1):D480–D489. doi: 10.1093/nar/gkaa1100. https://www.ncbi.nlm.nih.gov/pubmed/33237286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson A.D., Williamson M.K., Lewis S., Shoemark D., Carroll M.W., Heesom K.J., Zambon M., Ellis J., Lewis P.A., Hiscox J.A., Matthews D.A. Characterisation of the transcriptome and proteome of SARS-CoV-2 reveals a cell passage induced in-frame deletion of the furin-like cleavage site from the spike glycoprotein. Genome Med. 2020;12(1):68. doi: 10.1186/s13073-020-00763-0. https://www.ncbi.nlm.nih.gov/pubmed/32723359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano W.L. Schrödinger LLC; 2014. The PyMOL Molecular Graphics System, Version 1.8.http://www.pymol.org [DOI] [Google Scholar]
- Elbe S., Buckland-Merrett G. Data, disease and diplomacy: GISAID's innovative contribution to global health. Glob Chall. 2017;1(1):33–46. doi: 10.1002/gch2.1018. https://www.ncbi.nlm.nih.gov/pubmed/31565258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eymieux S., Rouillé Y., Terrier O., Seron K., Blanchard E., Rosa-Calatrava M., Dubuisson J., Belouzard S., Roingeard P. Ultrastructural modifications induced by SARS-CoV-2 in Vero cells: a kinetic analysis of viral factory formation, viral particle morphogenesis and virion release. Cell. Mol. Life Sci. 2021;78(7):3565–3576. doi: 10.1007/s00018-020-03745-y. https://www.ncbi.nlm.nih.gov/pubmed/33449149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantini J., Chahinian H., Yahi N. Leveraging coronavirus binding to gangliosides for innovative vaccine and therapeutic strategies against COVID-19. Biochem. Biophys. Res. Commun. 2021;538:132–136. doi: 10.1016/j.bbrc.2020.10.015. https://www.ncbi.nlm.nih.gov/pubmed/33097184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantini J., Yahi N., Azzaz F., Chahinian H. Structural dynamics of SARS-CoV-2 variants: a health monitoring strategy for anticipating Covid-19 outbreaks. J. Infect. 2021;83(2):197–206. doi: 10.1016/j.jinf.2021.06.001. https://www.ncbi.nlm.nih.gov/pubmed/34089757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Q., Chou J.J. A trimeric hydrophobic zipper mediates the intramembrane assembly of SARS-CoV-2 spike. J. Am. Chem. Soc. 2021;143(23):8543–8546. doi: 10.1021/jacs.1c02394. https://www.ncbi.nlm.nih.gov/pubmed/34086443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobeil S.M., Janowska K., McDowell S., Mansouri K., Parks R., Manne K., Stalls V., Kopp M.F., Henderson R., Edwards R.J., Haynes B.F., Acharya P. D614G mutation alters SARS-CoV-2 spike conformation and enhances protease cleavage at the S1/S2 junction. Cell Rep. 2021;34(2):108630. doi: 10.1016/j.celrep.2020.108630. https://www.ncbi.nlm.nih.gov/pubmed/33417835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo J., Zhao Y., Ren J., Zhou D., Duyvesteyn H.M.E., Ginn H.M., Carrique L., Malinauskas T., Ruza R.R., Shah P.N.M., Tan T.K., Rijal P., Coombes N., Bewley K.R., Tree J.A., Radecke J., Paterson N.G., Supasa P., Mongkolsapaya J., Screaton G.R., Carroll M., Townsend A., Fry E.E., Owens R.J., Stuart D.I. Neutralization of SARS-CoV-2 by Destruction of the Prefusion Spike. Cell Host Microbe. 2020;28(3):445–454. doi: 10.1016/j.chom.2020.06.010. https://www.ncbi.nlm.nih.gov/pubmed/32585135 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson C.B., Farzan M., Chen B., Choe H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2021;23(1):3–20. doi: 10.1038/s41580-021-00418-x. https://www.ncbi.nlm.nih.gov/pubmed/34611326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y.L., Chou Y.Y., Rothlauf P.W., Liu Z., Soh T.K., Cureton D., Case J.B., Chen R.E., Diamond M.S., Whelan S.P.J., Kirchhausen T. Inhibition of PIKfyve kinase prevents infection by Zaire ebolavirus and SARS-CoV-2. Proc. Natl. Acad. Sci. U. S. A. 2020;117(34):20803–20813. doi: 10.1073/pnas.2007837117. https://www.ncbi.nlm.nih.gov/pubmed/32764148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke Z., Oton J., Qu K., Cortese M., Zila V., McKeane L., Nakane T., Zivanov J., Neufeldt C.J., Cerikan B., Lu J.M., Peukes J., Xiong X., Kräusslich H.G., Scheres S.H.W., Bartenschlager R., Briggs J.A.G. Structures and distributions of SARS-CoV-2 spike proteins on intact virions. Nature. 2020;588(7838):498–502. doi: 10.1038/s41586-020-2665-2. https://www.ncbi.nlm.nih.gov/pubmed/32805734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kervin T.A., Overduin M. Regulation of the phosphoinositide code by phosphorylation of membrane readers. Cells. 2021;10(5) doi: 10.3390/cells10051205. https://www.ncbi.nlm.nih.gov/pubmed/34069055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kervin T.A., Wiseman B.C., Overduin M. Phosphoinositide recognition sites are blocked by metabolite attachment. Front. Cell Dev. Biol. 2021;9:690461. doi: 10.3389/fcell.2021.690461. https://www.ncbi.nlm.nih.gov/pubmed/34368138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein S., Cortese M., Winter S.L., Wachsmuth-Melm M., Neufeldt C.J., Cerikan B., Stanifer M.L., Boulant S., Bartenschlager R., Chlanda P. SARS-CoV-2 structure and replication characterized by in situ cryo-electron tomography. Nat. Commun. 2020;11(1):5885. doi: 10.1038/s41467-020-19619-7. https://www.ncbi.nlm.nih.gov/pubmed/33208793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppisetti R.K., Fulcher Y.G., Jurkevich A., Prior S.H., Xu J., Lenoir M., Overduin M., Van Doren S.R. Ambidextrous binding of cell and membrane bilayers by soluble matrix metalloproteinase-12. Nat. Commun. 2014;5:5552. doi: 10.1038/ncomms6552. https://www.ncbi.nlm.nih.gov/pubmed/25412686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppisetti R.K., Fulcher Y.G., Van Doren S.R. Fusion peptide of SARS-CoV-2 spike rearranges into a wedge inserted in bilayered micelles. J. Am. Chem. Soc. 2021;143(33):13205–13211. doi: 10.1021/jacs.1c05435. https://www.ncbi.nlm.nih.gov/pubmed/34375093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krempl C., Schultze B., Laude H., Herrler G. Point mutations in the S protein connect the sialic acid binding activity with the enteropathogenicity of transmissible gastroenteritis coronavirus. J. Virol. 1997;71(4):3285–3287. doi: 10.1128/JVI.71.4.3285-3287.1997. https://www.ncbi.nlm.nih.gov/pubmed/9060696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kufareva I., Lenoir M., Dancea F., Sridhar P., Raush E., Bissig C., Gruenberg J., Abagyan R., Overduin M. Discovery of novel membrane binding structures and functions. Biochem. Cell. Biol. 2014;92(6):555–563. doi: 10.1139/bcb-2014-0074. https://www.ncbi.nlm.nih.gov/pubmed/25394204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutateladze T., Overduin M. Structural mechanism of endosome docking by the FYVE domain. Science. 2001;291(5509):1793–1796. doi: 10.1126/science.291.5509.1793. https://www.ncbi.nlm.nih.gov/pubmed/11230696 [DOI] [PubMed] [Google Scholar]
- Künkel F., Herrler G. Structural and functional analysis of the surface protein of human coronavirus OC43. Virology. 1993;195(1):195–202. doi: 10.1006/viro.1993.1360. https://www.ncbi.nlm.nih.gov/pubmed/8317096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledford H. COVID antibody treatments show promise for preventing severe disease. Nature. 2021;591(7851):513–514. doi: 10.1038/d41586-021-00650-7. https://www.ncbi.nlm.nih.gov/pubmed/33712752 [DOI] [PubMed] [Google Scholar]
- Lee S.A., Eyeson R., Cheever M.L., Geng J., Verkhusha V.V., Burd C., Overduin M., Kutateladze T.G. Targeting of the FYVE domain to endosomal membranes is regulated by a histidine switch. Proc. Natl. Acad. Sci. U. S. A. 2005;102(37):13052–13057. doi: 10.1073/pnas.0503900102. https://www.ncbi.nlm.nih.gov/pubmed/16141328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenoir M., Kufareva I., Abagyan R., Overduin M. Membrane and protein interactions of the pleckstrin homology domain superfamily. Membranes. 2015;5(4):646–663. doi: 10.3390/membranes5040646. https://www.ncbi.nlm.nih.gov/pubmed/26512702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M.Y., Li L., Zhang Y., Wang X.S. Expression of the SARS-CoV-2 cell receptor gene ACE2 in a wide variety of human tissues. Infect. Dis. Poverty. 2020;9(1):45. doi: 10.1186/s40249-020-00662-x. https://www.ncbi.nlm.nih.gov/pubmed/32345362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Hulswit R.J.G., Widjaja I., Raj V.S., McBride R., Peng W., Widagdo W., Tortorici M.A., van Dieren B., Lang Y., van Lent J.W.M., Paulson J.C., de Haan C.A.M., de Groot R.J., van Kuppeveld F.J.M., Haagmans B.L., Bosch B.J. Identification of sialic acid-binding function for the Middle East respiratory syndrome coronavirus spike glycoprotein. Proc. Natl. Acad. Sci. U. S. A. 2017;114(40):E8508–E8517. doi: 10.1073/pnas.1712592114. https://www.ncbi.nlm.nih.gov/pubmed/28923942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L., Wang P., Nair M.S., Yu J., Rapp M., Wang Q., Luo Y., Chan J.F., Sahi V., Figueroa A., Guo X.V., Cerutti G., Bimela J., Gorman J., Zhou T., Chen Z., Yuen K.Y., Kwong P.D., Sodroski J.G., Yin M.T., Sheng Z., Huang Y., Shapiro L., Ho D.D. Potent neutralizing antibodies against multiple epitopes on SARS-CoV-2 spike. Nature. 2020;584(7821):450–456. doi: 10.1038/s41586-020-2571-7. https://www.ncbi.nlm.nih.gov/pubmed/32698192 [DOI] [PubMed] [Google Scholar]
- Lomize A.L., Todd S.C., Pogozheva I.D. Spatial arrangement of proteins in planar and curved membranes by PPM 3.0. Protein Sci. 2022;31(1):209–220. doi: 10.1002/pro.4219. https://www.ncbi.nlm.nih.gov/pubmed/34716622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomize M.A., Pogozheva I.D., Joo H., Mosberg H.I., Lomize A.L. OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res. 2012;40:D370–D376. doi: 10.1093/nar/gkr703. https://www.ncbi.nlm.nih.gov/pubmed/21890895 Database issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchini A., Micciulla S., Corucci G., Batchu K.C., Santamaria A., Laux V., Darwish T., Russell R.A., Thepaut M., Bally I., Fieschi F., Fragneto G. Lipid bilayer degradation induced by SARS-CoV-2 spike protein as revealed by neutron reflectometry. Sci. Rep. 2021;11(1):14867. doi: 10.1038/s41598-021-93996-x. https://www.ncbi.nlm.nih.gov/pubmed/34290262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCallum M., Walls A.C., Bowen J.E., Corti D., Veesler D. Structure-guided covalent stabilization of coronavirus spike glycoprotein trimers in the closed conformation. Nat. Struct. Mol. Biol. 2020;27(10):942–949. doi: 10.1038/s41594-020-0483-8. https://www.ncbi.nlm.nih.gov/pubmed/32753755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCallum M., De Marco A., Lempp F.A., Tortorici M.A., Pinto D., Walls A.C., Beltramello M., Chen A., Liu Z., Zatta F., Zepeda S., di Iulio J., Bowen J.E., Montiel-Ruiz M., Zhou J., Rosen L.E., Bianchi S., Guarino B., Fregni C.S., Abdelnabi R., Foo S.C., Rothlauf P.W., Bloyet L.M., Benigni F., Cameroni E., Neyts J., Riva A., Snell G., Telenti A., Whelan S.P.J., Virgin H.W., Corti D., Pizzuto M.S., Veesler D. N-terminal domain antigenic mapping reveals a site of vulnerability for SARS-CoV-2. Cell. 2021;184(9):2332–2347. doi: 10.1016/j.cell.2021.03.028. https://www.ncbi.nlm.nih.gov/pubmed/33761326 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melero R., Sorzano C.O.S., Foster B., Vilas J.L., Martínez M., Marabini R., Ramírez-Aportela E., Sanchez-Garcia R., Herreros D., Del Caño L., Losana P., Fonseca-Reyna Y.C., Conesa P., Wrapp D., Chacon P., McLellan J.S., Tagare H.D. Continuous flexibility analysis of SARS-CoV-2 spike prefusion structures. IUCrJ. 2020;7(Pt 6) doi: 10.1107/S2052252520012725. https://www.ncbi.nlm.nih.gov/pubmed/33063791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendonça L., Howe A., Gilchrist J.B., Sheng Y., Sun D., Knight M.L., Zanetti-Domingues L.C., Bateman B., Krebs A.S., Chen L., Radecke J., Li V.D., Ni T., Kounatidis I., Koronfel M.A., Szynkiewicz M., Harkiolaki M., Martin-Fernandez M.L., James W., Zhang P. Correlative multi-scale cryo-imaging unveils SARS-CoV-2 assembly and egress. Nat. Commun. 2021;12(1):4629. doi: 10.1038/s41467-021-24887-y. https://www.ncbi.nlm.nih.gov/pubmed/34330917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng B., Kemp S.A., Papa G., Datir R., M Ferreira I.A.T., Marelli S., Harvey W.T., Lytras S., Mohamed A., Gallo G., Thakur N., Collier D.A., Mlcochova P., Duncan L.M., Carabelli A.M., Kenyon J.C., Lever A.M., De Marco A., Saliba C., Culap K., Cameroni E., Matheson N.J., Piccoli L., Corti D., James L.C., Robertson D.L., Bailey D., Gupta R.K., COVID-19 Genomics UK (COG-UK) Consortium Recurrent emergence of SARS-CoV-2 spike deletion H69/V70 and its role in the Alpha variant B.1.1.7. Cell Rep. 2021;35(13) doi: 10.1016/j.celrep.2021.109292. https://www.ncbi.nlm.nih.gov/pubmed/34166617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesquita F.S., Abrami L., Sergeeva O., Turelli P., Qing E., Kunz B., Raclot C., Paz Montoya J., Abriata L.A., Gallagher T., Dal Peraro M., Trono D., D'Angelo G., van der Goot F.G. S-acylation controls SARS-CoV-2 membrane lipid organization and enhances infectivity. Dev. Cell. 2021;56(20):2790–2807. doi: 10.1016/j.devcel.2021.09.016. https://www.ncbi.nlm.nih.gov/pubmed/34599882 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabel K.G., Clark S.A., Shankar S., Pan J., Clark L.E., Yang P., Coscia A., McKay L.G.A., Varnum H.H., Brusic V., Tolan N.V., Zhou G., Desjardins M., Turbett S.E., Kanjilal S., Sherman A.C., Dighe A., LaRocque R.C., Ryan E.T., Tylek C., Cohen-Solal J.F., Darcy A.T., Tavella D., Clabbers A., Fan Y., Griffiths A., Correia I.R., Seagal J., Baden L.R., Charles R.C. Structural basis for continued antibody evasion by the SARS-CoV-2 receptor binding domain. Science. 2021;6251 doi: 10.1126/science.abl6251. https://www.ncbi.nlm.nih.gov/pubmed/34855508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L., McCord K.A., Bui D.T., Bouwman K.M., Kitova E.N., Elaish M., Kumawat D., Daskhan G.C., Tomris I., Han L., Chopra P., Yang T.J., Willows S.D., Mason A.L., Mahal L.K., Lowary T.L., West L.J., Hsu S.D., Hobman T., Tompkins S.M., Boons G.J., de Vries R.P., Macauley M.S., Klassen J.S. Sialic acid-containing glycolipids mediate binding and viral entry of SARS-CoV-2. Nat. Chem. Biol. 2021;18(1):81–90. doi: 10.1038/s41589-021-00924-1. https://www.ncbi.nlm.nih.gov/pubmed/34754101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou X., Liu Y., Lei X., Li P., Mi D., Ren L., Guo L., Guo R., Chen T., Hu J., Xiang Z., Mu Z., Chen X., Chen J., Hu K., Jin Q., Wang J., Qian Z. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020;11(1):1620. doi: 10.1038/s41467-020-15562-9. https://www.ncbi.nlm.nih.gov/pubmed/32221306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overduin M., Kervin T.A. The phosphoinositide code is read by a plethora of protein domains. Expert Rev. Proteomics. 2021;18(7):483–502. doi: 10.1080/14789450.2021.1962302. https://www.ncbi.nlm.nih.gov/pubmed/34351250 [DOI] [PubMed] [Google Scholar]
- Overduin M., Wille H., Westaway D. Multisite interactions of prions with membranes and native nanodiscs. Chem. Phys. Lipids. 2021;236:105063. doi: 10.1016/j.chemphyslip.2021.105063. https://www.ncbi.nlm.nih.gov/pubmed/33600804 [DOI] [PubMed] [Google Scholar]
- Park Y.J., Walls A.C., Wang Z., Sauer M.M., Li W., Tortorici M.A., Bosch B.J., DiMaio F., Veesler D. Structures of MERS-CoV spike glycoprotein in complex with sialoside attachment receptors. Nat. Struct. Mol. Biol. 2019;26(12):1151–1157. doi: 10.1038/s41594-019-0334-7. https://www.ncbi.nlm.nih.gov/pubmed/31792450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perino J., Crouzier D., Spehner D., Debouzy J.C., Garin D., Crance J.M., Favier A.L. Lung surfactant DPPG phospholipid inhibits vaccinia virus infection. Antivir. Res. 2011;89(1):89–97. doi: 10.1016/j.antiviral.2010.11.009. https://www.ncbi.nlm.nih.gov/pubmed/21095206 [DOI] [PubMed] [Google Scholar]
- Sanders D.W., Jumper C.C., Ackerman P.J., Bracha D., Donlic A., Kim H., Kenney D., Castello-Serrano I., Suzuki S., Tamura T., Tavares A.H., Saeed M., Holehouse A.S., Ploss A., Levental I., Douam F., Padera R.F., Levy B.D., Brangwynne C.P. SARS-CoV-2 requires cholesterol for viral entry and pathological syncytia formation. Elife. 2021;10 doi: 10.7554/eLife.65962. https://www.ncbi.nlm.nih.gov/pubmed/33890572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satturwar S., Fowkes M., Farver C., Wilson A.M., Eccher A., Girolami I., Pujadas E., Bryce C., Salem F., El Jamal S.M., Paniz-Mondolfi A., Petersen B., Gordon R.E., Reidy J., Fraggetta F., Marshall D.A., Pantanowitz L. Postmortem findings associated with SARS-CoV-2: systematic review and meta-analysis. Am. J. Surg. Pathol. 2021;45(5):587–603. doi: 10.1097/PAS.0000000000001650. https://www.ncbi.nlm.nih.gov/pubmed/33481385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm C.A., Hannigan B.T., Donald J.E., Keasar C., Saven J.G., Degrado W.F., Samish I. Knowledge-based potential for positioning membrane-associated structures and assessing residue-specific energetic contributions. Structure. 2012;20(5):924–935. doi: 10.1016/j.str.2012.03.016. https://www.ncbi.nlm.nih.gov/pubmed/22579257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shajahan A., Supekar N.T., Gleinich A.S., Azadi P. Deducing the N- and O-glycosylation profile of the spike protein of novel coronavirus SARS-CoV-2. Glycobiology. 2020;30(12):981–988. doi: 10.1093/glycob/cwaa042. https://www.ncbi.nlm.nih.gov/pubmed/32363391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemark D.K., Colenso C.K., Toelzer C., Gupta K., Sessions R.B., Davidson A.D., Berger I., Schaffitzel C., Spencer J., Mulholland A.J. Molecular simulations suggest vitamins, retinoids and steroids as ligands of the free fatty acid pocket of the SARS-CoV-2 spike protein. Angew Chem. Int. Ed. Engl. 2021;60(13):7098–7110. doi: 10.1002/anie.202015639. https://www.ncbi.nlm.nih.gov/pubmed/33469977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofia F Oliveira A., Shoemark D.K., Avila Ibarra A., Davidson A.D., Berger I., Schaffitzel C., Mulholland A.J. The fatty acid site is coupled to functional motifs in the SARS-CoV-2 spike protein and modulates spike allosteric behaviour. Comput. Struct. Biotechnol. J. 2021;20:139–147. doi: 10.1016/j.csbj.2021.12.011. https://www.ncbi.nlm.nih.gov/pubmed/34934478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toelzer C., Gupta K., Yadav S.K.N., Borucu U., Davidson A.D., Kavanagh Williamson M., Shoemark D.K., Garzoni F., Staufer O., Milligan R., Capin J., Mulholland A.J., Spatz J., Fitzgerald D., Berger I., Schaffitzel C. Free fatty acid binding pocket in the locked structure of SARS-CoV-2 spike protein. Science. 2020;370(6517):725–730. doi: 10.1126/science.abd3255. https://www.ncbi.nlm.nih.gov/pubmed/32958580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voelker D.R., Numata M. Phospholipid regulation of innate immunity and respiratory viral infection. J. Biol. Chem. 2019;294(12):4282–4289. doi: 10.1074/jbc.AW118.003229. https://www.ncbi.nlm.nih.gov/pubmed/30733339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls A.C., Park Y.J., Tortorici M.A., Wall A., McGuire A.T., Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;181(2):281–292. doi: 10.1016/j.cell.2020.02.058. https://www.ncbi.nlm.nih.gov/pubmed/32155444 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B., Zhong C., Tieleman D.P. Supramolecular organization of SARS-CoV and SARS-CoV-2 virions revealed by coarse-grained models of intact virus envelopes. J. Chem. Inf. Model. 2021;62(1):176–186. doi: 10.1021/acs.jcim.1c01240. https://www.ncbi.nlm.nih.gov/pubmed/34911299 [DOI] [PubMed] [Google Scholar]
- Wang J., Maschietto F., Guberman-Pfeffer M.J., Reiss K., Allen B., Xiong Y., Lolis E., Batista V.S. Computational insights into the membrane fusion mechanism of SARS-CoV-2 at the cellular level. Comput. Struct. Biotechnol. J. 2021;19:5019–5028. doi: 10.1016/j.csbj.2021.08.053. https://www.ncbi.nlm.nih.gov/pubmed/34540146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y., Allen J.D., Wrapp D., McLellan J.S., Crispin M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science. 2020;369(6501):330–333. doi: 10.1126/science.abb9983. https://www.ncbi.nlm.nih.gov/pubmed/32366695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei C., Wan L., Yan Q., Wang X., Zhang J., Yang X., Zhang Y., Fan C., Li D., Deng Y., Sun J., Gong J., Wang Y., Li J., Yang H., Li H., Zhang Z., Wang R., Du P., Zong Y., Yin F., Zhang W., Wang N., Peng Y., Lin H., Feng J., Qin C., Chen W., Gao Q., Zhang R., Cao Y., Zhong H. HDL-scavenger receptor B type 1 facilitates SARS-CoV-2 entry. Nat. Metab. 2020;2(12):1391–1400. doi: 10.1038/s42255-020-00324-0. https://www.ncbi.nlm.nih.gov/pubmed/33244168 [DOI] [PubMed] [Google Scholar]
- White J.M., Whittaker G.R. Fusion of enveloped viruses in endosomes. Traffic. 2016;17(6):593–614. doi: 10.1111/tra.12389. https://www.ncbi.nlm.nih.gov/pubmed/26935856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong N.A., Saier M.H. The SARS-coronavirus infection cycle: a survey of viral membrane proteins, their functional interactions and pathogenesis. Int. J. Mol. Sci. 2021;22(3) doi: 10.3390/ijms22031308. https://www.ncbi.nlm.nih.gov/pubmed/33525632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrapp D., Wang N., Corbett K.S., Goldsmith J.A., Hsieh C.L., Abiona O., Graham B.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367(6483):1260–1263. doi: 10.1126/science.abb2507. https://www.ncbi.nlm.nih.gov/pubmed/32075877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrobel A.G., Benton D.J., Xu P., Roustan C., Martin S.R., Rosenthal P.B., Skehel J.J., Gamblin S.J. SARS-CoV-2 and bat RaTG13 spike glycoprotein structures inform on virus evolution and furin-cleavage effects. Nat. Struct. Mol. Biol . 2020;27(8):763–767. doi: 10.1038/s41594-020-0468-7. https://www.ncbi.nlm.nih.gov/pubmed/32647346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia S., Lan Q., Su S., Wang X., Xu W., Liu Z., Zhu Y., Wang Q., Lu L., Jiang S. The role of furin cleavage site in SARS-CoV-2 spike protein-mediated membrane fusion in the presence or absence of trypsin. Signal Transduct. Targeted Ther. 2020;5(1):92. doi: 10.1038/s41392-020-0184-0. https://www.ncbi.nlm.nih.gov/pubmed/32532959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C., Wang Y., Liu C., Zhang C., Han W., Hong X., Hong Q., Wang S., Zhao Q., Yang Y., Chen K., Zheng W., Kong L., Wang F., Zuo Q., Huang Z. Conformational dynamics of SARS-CoV-2 trimeric spike glycoprotein in complex with receptor ACE2 revealed by cryo-EM. Sci. Adv. 2021;7(1) doi: 10.1126/sciadv.abe5575. https://www.ncbi.nlm.nih.gov/pubmed/33277323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan R., Zhang Y., Li Y., Ye F., Guo Y., Xia L., Zhong X., Chi X., Zhou Q. Structural basis for the different states of the spike protein of SARS-CoV-2 in complex with ACE2. Cell Res. 2021;31(6):717–719. doi: 10.1038/s41422-021-00490-0. https://www.ncbi.nlm.nih.gov/pubmed/33737693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N., Ma P., Lang J., Zhang Y., Deng J., Ju X., Zhang G., Jiang C. Phosphatidylinositol 4-kinase IIIβ is required for severe acute respiratory syndrome coronavirus spike-mediated cell entry. J. Biol. Chem. 2012;287(11):8457–8467. doi: 10.1074/jbc.M111.312561. https://www.ncbi.nlm.nih.gov/pubmed/22253445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T.J., Yu P.Y., Chang Y.C., Hsu S.D. D614G mutation in the SARS-CoV-2 spike protein enhances viral fitness by desensitizing it to temperature-dependent denaturation. J. Biol. Chem. 2021;297(4):101238. doi: 10.1016/j.jbc.2021.101238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z., Wang Y., Jin Y., Zhu Y., Wu Y., Li C., Kong Y., Song W., Tian X., Zhan W., Huang A., Zhou S., Xia S., Peng C., Chen C., Shi Y., Hu G., Du S., Xie Y., Jiang S., Lu L., Sun L., Song Y., Ying T. A non-ACE2 competing human single-domain antibody confers broad neutralization against SARS-CoV-2 and circulating variants. Signal Transduct. Target Ther. 2021;6(1):378. doi: 10.1038/s41392-021-00810-1. https://www.ncbi.nlm.nih.gov/pubmed/34732694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Xiao T., Cai Y., Chen B. Structure of SARS-CoV-2 spike protein. Curr. Opin. Virol. 2021;50:173–182. doi: 10.1016/j.coviro.2021.08.010. https://www.ncbi.nlm.nih.gov/pubmed/34534731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S., Go E.P., Ding H., Anang S., Kappes J.C., Desaire H., Sodroski J.G. Analysis of glycosylation and disulfide bonding of wild-type SARS-CoV-2 spike glycoprotein. J. Virol. 2022;96(3) doi: 10.1128/JVI.01626-21. https://www.ncbi.nlm.nih.gov/pubmed/34817202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T., Tsybovsky Y., Gorman J., Rapp M., Cerutti G., Chuang G.Y., Katsamba P.S., Sampson J.M., Schön A., Bimela J., Boyington J.C., Nazzari A., Olia A.S., Shi W., Sastry M., Stephens T., Stuckey J., Teng I.T., Wang P., Wang S., Zhang B., Friesner R.A., Ho D.D., Mascola J.R., Shapiro L., Kwong P.D. Cryo-EM structures of SARS-CoV-2 spike without and with ACE2 reveal a pH-dependent switch to mediate endosomal positioning of receptor-binding domains. Cell Host Microbe. 2020;28(6):867–879. doi: 10.1016/j.chom.2020.11.004. https://www.ncbi.nlm.nih.gov/pubmed/33271067 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y., Feng F., Hu G., Wang Y., Yu Y., Xu W., Cai X., Sun Z., Han W., Ye R., Qu D., Ding Q., Huang X., Chen H., Xie Y., Cai Q., Yuan Z., Zhang R. A genome-wide CRISPR screen identifies host factors that regulate SARS-CoV-2 entry. Nat. Commun. 2021;12(1):961. doi: 10.1038/s41467-021-21213-4. https://www.ncbi.nlm.nih.gov/pubmed/33574281 [DOI] [PMC free article] [PubMed] [Google Scholar]