

SUMMARY
Mitogen-activated protein kinases (MAPKs) are inactivated by dual-specificity phosphatases (DUSPs), the activities of which are tightly regulated during cell differentiation. Using knockdown screening and single-cell transcriptional analysis, we demonstrate that DUSP4 is the phosphatase that specifically inactivates p38 kinase to promote megakaryocyte (Mk) differentiation. Mechanistically, PRMT1-mediated methylation of DUSP4 triggers its ubiquitinylation by an E3 ligase HUWE1. Interestingly, the mechanistic axis of the DUSP4 degradation and p38 activation is also associated with a transcriptional signature of immune activation in Mk cells. In the context of thrombocytopenia observed in myelodysplastic syndrome (MDS), we demonstrate that high levels of p38 MAPK and PRMT1 are associated with low platelet counts and adverse prognosis, while pharmacological inhibition of p38 MAPK or PRMT1 stimulates megakaryopoiesis. These findings provide mechanistic insights into the role of the PRMT1-DUSP4-p38 axis on Mk differentiation and present a strategy for treatment of thrombocytopenia associated with MDS.
In brief
Su et al. report that methylation of DUSP4 by PRMT1 triggers DUSP4 ubiquitylation by HUWE1. DUSP4 inactivates p38 kinase to promote megakaryocyte differentiation. Abnormally high expression of PRMT1 and activation of p38 kinase in blood cells of MDS patients blocks megakaryopoiesis, revealing PRMT1 as a target for MDS treatment.
Graphical Abstract
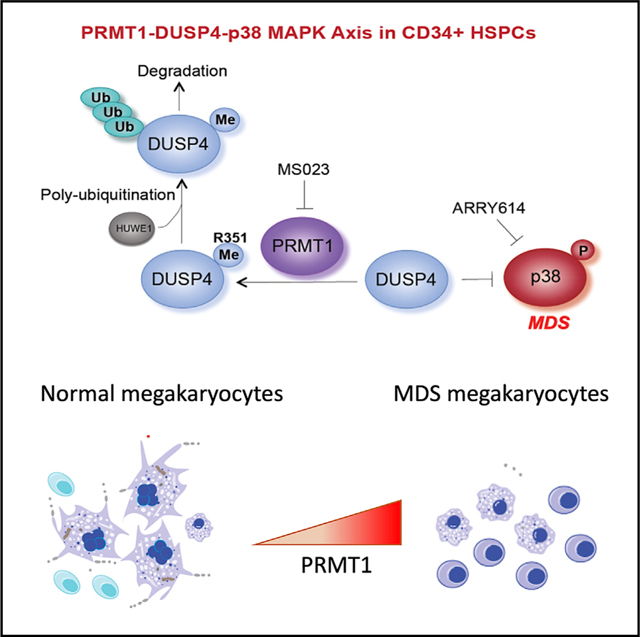
INTRODUCTION
Precisely orchestrated cellular differentiation is essential for normal development of a multicellular organism (Cheng et al., 2020). Protein posttranslational modifications such as phosphorylation, methylation, and ubiquitylation involved in the combination of signal transduction and epigenetic regulation, which broadly refers to modifications of chromatin-associated proteins, are essential for cellular differentiation (Suganuma and Workman, 2011). Conversely, erasing posttranslational modification marks attenuates or even reverses many signaling and epigenetic events. Mitogen-activated protein (MAP) kinases (MAPKs) are activated through phosphorylation of the TxY motif on their catalytic sites. MAPK signaling is in general inactivated by a collection of dual-specificity phosphatases (DUSPs), which function by dephosphorylating both threonine and tyrosine residues in the TxY motif, leading to inactivation of MAPK signaling (Caunt and Keyse, 2013; Owens and Keyse, 2007; Seternes et al., 2019; Yu and Zhang, 2018). Furthermore, the activity of DUSPs is tightly regulated by posttranslational modifications such as phosphorylation and acetylation (Chi and Flavell, 2008; Owens and Keyse, 2007).
Hematopoietic stem cells (HSCs) give rise to multiple types of lineage-specific progenitor cells with distinct gene expression patterns. A differentiation hierarchy of HSCs to erythroid (Er) and megakaryocytic (Mk) cells has been described through short-term HSCs, multipotent progenitors (MPPs), common myeloid progenitors (CMPs), and megakaryocyte-Er progenitors (MEPs) according to characteristic surface markers and transcriptional signatures (reviewed in Cheng et al., 2020). Nevertheless, recent single-cell RNA sequencing (scRNA-seq) analysis of human CD34+ cells reveals a heterogeneous nature within surface marker-defined progenitors for gradually evolved transcriptional expression topology during differentiation of hematopoietic stem/progenitor cells (HSPCs) (Pellin et al., 2019; Weinreb et al., 2020). Thus, extended data of essential signaling and epigenetic events, i.e., the so-called ‘‘hidden variables’’ (Weinreb et al., 2020), in addition to single-cell transcriptome snapshots are necessary to define potential borders between lineage-specific progenitor cells and classify differentiated hematopoietic cells into functional subtypes. Activation of distinct MAPK cascades has been implicated in differentiation regulation of Er and Mk cells. A number of studies have shown that extracellular signal-regulated kinase (ERK) promotes Mk differentiation and p38 MAPK activation promotes Er differentiation after stimulation by various cytokines (Herrera et al., 1998; Hua et al., 2013; Mazharian et al., 2009; Miyazaki et al., 2001; Nagata et al., 1998; Norman et al., 2019; Uddin et al., 2004; Whalen et al., 1997; Witt et al., 2000). However, the precise molecular mechanisms that regulate the activation of MAPKs and phosphatases during Mk differentiation into mature Mk cells have not been fully elucidated.
PRMT1, one of the nine members of the human protein arginine methyltransferase (PRMT) family, has been associated with diverse biological functions such as transcriptional regulation, RNA processing, signal transduction, and DNA repair (Guccione and Richard, 2019). In hematopoietic differentiation, PRMT1 is expressed at low levels in HSCs and is highly expressed in MEPs (Zhang et al., 2015). Constitutive expression of PRMT1 reduces the generation of CD41+CD42+ Mk cells, presumably through arginine methylation of essential substrates during Mk differentiation. Consistently, PRMT1 knockout promotes megakaryocyte differentiation (Zhu et al., 2019). One challenge to identifying PRMT1 substrates in vivo has been the lack of specific antibodies and the limitation of mass spectroscopy (MS) to detect methylarginine-containing peptides. To readily probe the activity of PRMTs, we developed the next-generation bioorthogonal profiling of protein methylation (BPPM) technology for live cells (Luo, 2018; Wang et al., 2013; Wang and Luo, 2013; Wang et al., 2011). In this study, the cofactor-binding pockets of individual PRMTs were engineered to accommodate terminal-alkyne-containing bulky SAM analogs. The bulky SAM analogs were synthesized in live cells by an engineered methionine adenosyltransferase (MAT) with cell-permeable methionine analogs and endogenous adenosine triphosphate as substrates. The engineered PRMTs can then label substrates with a terminal-alkyne moiety from the cofactor analogs inside live cells. Using the live-cell BPPM technology, we identified that dual-specificity phosphatase 4 (DUSP4 or MKP2 [MAPK phosphatase 2]) is a bona fide substrate of PRMT1 in broad cellular contexts.
In the context of Mk lineage determination, we show that PRMT1 triggers Arg351 methylation-dependent ubiquitinylation by the E3 ligase HUWE1 and thus DUSP4’s degradation, which result in activation of p38 MAPK signaling due to the inability of DUSP4 to dephosphorylate p38 MAPK. scRNA-seq analysis further confirmed the positive correlation between high levels of PRMT1 and the expression of signature genes of p38 activation, including genes associated with the pro-inflammatory response. We further show that pharmacological inhibition of p38 MAPK or PRMT1 promotes Mk differentiation both in vivo and ex vivo. These findings suggest potential pharmacological strategies for treatment of a commonly observed Mk cell differentiation defect in patients with myelodysplastic syndrome (MDS).
RESULTS
Identification of DUSP4 as a key regulator for Mk differentiation
Distinct MAPK cascades regulate differentiation and homeostasis during hematopoiesis (Baumgartner et al., 2018; Geest and Coffer, 2009). While MAPK cascades are modulated by DUSPs in general (Patterson et al., 2009), the regulatory DUSPs specific for MAPK signaling during megakaryopoiesis remain to be elucidated. To address this question, we developed a short hairpin RNA (shRNA)-based screen to identify essential DUSPs regulating Mk differentiation (Figure 1A). In this study, we targeted 10 DUSPs known to modulate the signaling of MAPKs (Owens and Keyse, 2007) and examined their specific roles in Mk differentiation. Given potential differences between factors regulating Mk cell differentiation of bone marrow (BM) cells and fetal liver cells (Liu and Sola-Visner, 2011), we tested both adult BM- and cord blood (CB)-derived human CD34+ HSPCs for their ability to differentiate into the Mk lineage when the individual DUSPs were downregulated using lentivirus-expressed shRNAs (Figure 1B). In the presence of thrombopoietin (TPO), CD34+ BM and CB cells differentiated into mature Mk cells as characterized by CD41a+ and CD42b+ expression (Figures 1C and 1D). Upon knocking down specific DUSPs, only perturbation of DUSP4 significantly inhibited differentiation to CD41a+CD42b+ megakaryocytes (∼40% reduction of the CD41a+CD42b+ population, Figures 1B and 1C). This requirement for DUSP4 during Mk differentiation was independently validated using BM and CB CD34+ cells from different donors (Figure 1D), human granulocyte colony-stimulating factor (G-CSF)-mobilized peripheral blood (PB) CD34+ cells, and two additional DUSP4 shRNAs (Figures 1E–1G). While it is challenging to target Mk differentiation in a temporal manner (early progenitor cells, Mk progenitor cells, or matured Mk cells with different degrees of polyploidies), our results collectively argue for the general requirement of DUSP4 for optimal Mk differentiation.
Figure 1. Identification of DUSP4 for optimal Mk differentiation.

(A) Schematic of shRNA-based screening assay to identify essential DUSPs for Mk-induced Mk differentiation. Human CD34+ cells infected with lentiviruses expressing shRNAs against DUSPs were cultured for Mk differentiation.
(B) Heatmap of the percentages of CD41a+CD42b+ cells upon DUSP knockdown on day 8. Fold changes were normalized to the percentage of double-positive cells with the group treated with control shRNA.
(C) Representative flow chart of FACS analysis of Mk differentiation using BM cells (top panel) and CB cells (bottom panel) cultured in TPO-containing medium.
(D) Summary of FACS analysis. Statistics are based on the data of three independent experiments (n = 3) with the bone marrow (BM) or cord blood (CB) cells from two donors. Data are shown as mean ± SD. Two-tailed paired t test, *p ≤ 0.05, **p ≤ 0.01.
(E) Schematic description of Mk differentiation of human peripheral blood-derived CD34+ cells with DUSP4 knockdown.
(F and G) Representative and complete FACS analysis of CD41a and CD42b markers for Mk differentiation with peripheral blood CD34+ cells upon DUSP4 knockdown on day 7. Representative plots (F) and statistics (G) are shown (n = 3, independent experiments). Data are shown as mean ± SD. Two-tailed paired t test, *p ≤ 0.05, **p ≤ 0.01.
DUSP4 drives the differentiation toward Mk progenitors
Megakaryocytes and erythrocytes can originate from the same progenitor subset such as MEPs (Xavier-Ferrucio and Krause, 2018) or from even earlier stem/progenitor cells (Rodriguez-Fraticelli et al., 2018; Sanjuan-Pla et al., 2013; Yamamoto et al., 2013). We knocked down DUSP4 in human CD34+ cells grown in media containing both TPO and erythropoietin (EPO), which support differentiation to Mk or Er cells, respectively (Figure 2A). The results showed that DUSP4 knockdown decreased differentiation efficiency to CD41a+ Mk cells and increased commitment to CD71+ Er cells (Figure 2B). On average, we observed up to a 2-fold reduction of CD41a+ megakaryocytes and an ∼30%–50% increase in CD71+ erythrocytes (Figure 2B). The differentiation bias observed with DUSP4 knockdown closely resembled what was observed when PRMT1 was overexpressed in CD34+ cells (Jin et al., 2017; Zhang et al., 2015).
Figure 2. DUSP4-regulated differentiation choices between Mk cells and Er cells.

(A) Schematic of differentiation experiments using CB CD34+ cells.
(B) FACS analysis of CD41a and CD71 on the cultured cells. Representative plots (top panel) and normalized statistics of three samples (bottom panel) are shown (n = 3, independent experiments). Fold changes were normalized with scramble controls.
(C) Schematic description of colony-forming unit (CFU) assays using MEP.
(D) Percentages of CFU-Mk, BFU-E, and CFU-MkE in each sorted population (n = 2, independent experiments).
To explore the potential role of DUSP4 in regulating differentiation of MEP cells into Mk or Er cells (Debili et al., 1996; Pronk et al., 2007; Sanada et al., 2016; Xavier-Ferrucio and Krause, 2018), we then isolated potential human MEPs, according to their respective cell surface marker profile (Lin−CD34+FLT3−CD45RA−MPL+ CD38midCD41a− for MEPs) (Figure 2C) (Sanada et al., 2016), and performed colony-forming unit (CFU) assays, following transduction with the lentivirus-expressed DUSP4 shRNAs. Using MEPs, DUSP4 knockdown significantly increased BFU-E (burst-forming units of erythrocytes by ∼2- to 3-fold) and decreased CFU-Mk (CFU of megakaryocytes by ∼2-fold) (Figure 2D). These results demonstrate that DUSP4 drives differentiation from biopotent MEP to Mk progenitor cells in colony formation assays.
Specific deactivation of p38 MAPK by DUSP4 during Mk terminal differentiation
The human M7 leukemia cell line MEG-01, which represents immature Mk progenitors, can be stimulated to undergo differentiation and polyploidization with PMA (phorbol 12-myristate 13-acetate) treatment. We then used this system to examine the impact of DUSP downregulation on PMA-stimulated Mk terminal differentiation in cells that were transduced with the anti-DUSP shRNA library (Figure S1A). Consistent with observations using human BM, CB, and PB CD34+ cells (Figure 1), only DUSP4 knockdown inhibited terminal Mk differentiation (∼40% reduction of efficiency, Figures S1B and S1C). During the PMA-stimulated Mk differentiation of MEG-01 cells, DUSP4 mRNA increased rapidly and reached a plateau by day 2 (Figure 3A). In contrast, DUSP4 protein levels gradually increased during the course of differentiation, indicating a positive correlation between DUSP4 protein levels and Mk differentiation (Figure 3B). We then examined whether DUSP4 overexpression would promote PMA-induced Mk differentiation of MEG-01 cells. Consistent with the necessity of DUSP4 for optimal Mk differentiation of human CD34+ HSPCs, DUSP4 overexpression directly increased the frequency of the CD41a+ population and led to a higher degree of polyploidy (Figure S1D). These data further confirm the essential role of DUSP4 in Mk maturation.
Figure 3. Crosstalk between DUSP4 and PRMT1 for MAPK signaling in Mk differentiation.

(A) DUSP4 mRNA level in PMA-treated MEG-01 cells. Cells were harvested at indicated time intervals, and the extracted mRNAs were quantified by real-time PCR. Representative statistics are shown as mean ± SD. Two-tailed unpaired t test, **p ≤ 0.01, ***p ≤ 0.001 (n = 3, independent experiments).
(B) MAPK-related proteins and PRMT1 in MEG-01 cells after PMA stimulation. Protein extracts were collected at indicated time intervals for western blotting.
(C) Regulation of MAPK signaling upon DUSP4 overexpression. MEG-01 cells were treated overnight with doxycycline to induce DUSP4 ectopically expressed from lentivirus. Cell extracts were collected for western blotting (n = 3, representative western blots).
(D) PRMT1 mRNA level in MEG-01 cells during the course of PMA-stimulated Mk differentiation. Representative statistics were shown as mean ± SD, two-tailed unpaired t test, *p ≤ 0.05 (n = 3, independent experiments).
(E and F) PRMT1-dependent regulation of DUSP4 protein. NB4 cells that conditionally express shRNA against PRMT1 were treated with doxycycline to induce PRMT1 knockdown (E). DUSP4- and PRMT1-encoding plasmids were transfected into HEK293T cells for their overexpression in the presence or absence of MG132 treatment (F).
(G) Antagonistic roles of PRMT1 and DUSP4 on Mk differentiation of human CD34+ cells. Human CD34+ cells were infected with PRMT1 lentivirus (puromycin-R) and DUSP4 lentivirus (GFP), followed by puromycin selection and Mk differentiation. Representative plots and statistics are shown (n = 3, independent experiments). Data are shown as mean ± SD. Two-tailed paired t test, *p ≤ 0.05, **p ≤ 0.01.
DUSP4 has been shown to dephosphorylate MAPKs including ERK, JNK, and p38 kinases (Keyse, 2008). To determine the specific MAPKs dephosphorylated by DUSP4, we performed western blot analysis using lysates from PMA-treated MEG-01 cells to probe phosphorylation levels of ERK2, JNK, and p38 with the former two MAPKs implicated in Mk-Er differentiation. As cells underwent Mk differentiation, levels of phosphorylated p38 kinase decreased, while levels of phosphorylated ERK2 increased (Figure 3B). Using a DUSP4-inducible cell line, we then confirmed that DUSP4 overexpression was sufficient for rapid suppression of p38 phosphorylation (Figure 3C) but not for the phosphorylation of ERK. This observation suggests that p38 signaling can be directly antagonized by high levels of DUSP4 to promote Mk differentiation. In contrast, JNK kinase phosphorylation levels were only modestly decreased during Mk differentiation (Figure S2). Therefore, the PRMT1-DUSP4 axis mainly suppresses the p38 kinase pathway for Mk differentiation. Furthermore, PRMT1-dependent activation of p38 kinase occurs in NB4 cells, which are immature granulocytes (Figure S1E). These consistent results imply that PRMT1-mediated p38 kinase activation can be achieved via direct suppression of DUSP4 in different types of cells.
PRMT1 controls DUSP4 stability by arginine methylation
We noted that the protein and mRNA expression levels of PRMT1 were inversely correlated with DUSP4 protein levels, but not with DUSP4 mRNA levels (Figures 3A, 3B, and 3D). To determine whether PRMT1 activity may directly affect DUSP4 protein, we used doxycycline-inducible shRNA to knock down PRMT1, which resulted in increased DUSP4 protein levels (Figure 3E). Conversely, overexpression of major PRMT1 isoform variants (V1 or V2) significantly decreased DUSP4 protein (Figure 3F). An increased level of phosphorylated p38 kinase was detected in NB4 cells stably overexpressing PRMT1 (Figure S1E), which was associated with the decreased level of DUSP4. Treatment of PRMT1-overexpressing cells with the proteasome inhibitor MG132 blocked PRMT1-mediated DUSP4 degradation (Figure 3F). These results collectively support that PRMT1 antagonizes DUSP4 protein levels by promoting proteasome-mediated target degradation. Of note, the PRMT1 isoform V2 exerts a stronger effect on protein degradation (compare lanes 3 and 4 in Figure 3F).
We next examined the effects of DUSP4 on PRMT1-mediated block of Mk differentiation (Figure 3G). In this study, stimulation of human CD34+ hematopoietic progenitor cells with TPO and stem cell factor (SCF) toward CD41a+CD42b+ megakaryocytes was inhibited by PRMT1 overexpression, which decreased the frequency of Mk differentiation by 3-fold (Figures 3F and S3). In contrast, expression of DUSP4 in PRMT1-overexpressing cells nearly restored the efficiency of megakaryocyte differentiation to levels comparable to CD34+ cells that do not overexpress PRMT1 (Figure 3G). These results indicate an antagonistic interplay of the PRMT1-DUSP4 axis in regulating Mk differentiation into matured CD41a+CD42b+ cells.
scRNA-seq analysis of Mk differentiation of primary human CD34+ cells
HSPCs are heterogeneous with a continuum of transcriptional expression topology (Psaila and Mead, 2019; Weinreb et al., 2020). Unknown regulatory pathways for Mk and Er differentiation have been discovered by scRNA-seq analysis (Lu et al., 2018; Tusi et al., 2018). In this context, we leveraged scRNA-seq technology in combination with fluorescence-activated cell sorting (FACS) sorting to interrogate essential modulators of Mk differentiation in human CD34+ BM cells (Figure 4A). A droplet-based scRNA-seq (Drop-seq) protocol was implemented for scRNA-seq analysis of non-stimulated (day 0) and TPO/SCF-stimulated cells (day 8 post-stimulation). The scRNA-seq data of all 4,684 cells were analyzed using the SPRING algorithm, a force-directed layout developed for visualizing continuous gene expression topologies and for assigning the cell fates of hematopoietic cells (Figure 4B) (Weinreb et al., 2018). Unstimulated human CD34+ BM consisted of a mixture of cell subpopulations with lineage-specific gene expression signatures indicative of dendritic (D), granulocytic neutrophil (GN), lymphocytic (Ly), monocytic (M), and MPP cells (Figure 4C). After 8 days of TPO/SCF stimulation, cell subsets were characterized by lineage-specific gene expression signatures of basophilic cells (or mast cells), Er, Mk, and likely their progenitors (Figure 4C), including a characteristic long ‘‘tail’’ on the SPRING plot that largely consisted of Mks. Analysis of the gene expression signature of CD41a+CD42b+ cells FACS sorted from the TPO/SCF-stimulated cell population indicated that these cells overlapped with the tail population in the day 8 TPO/SCF-stimulated cell populations. Basophilic cells (or mast cells) were embedded within the TPO/SCF-stimulated, non-CD41a+CD42b+ cells, consistent with scRNA-seq analysis of HSPCs showing that basophils can be derived from MEPs (Pellin et al., 2019; Tusi et al., 2018).
Figure 4. Negative correlation between PRMT1 and Mk differentiation revealed by single-cell RNA-seq (scRNA-seq) analysis.

(A) Experimental design for scRNA-seq analysis. Sample 1: Native CD34+ cells were cells isolated directly from BM. Sample 2: CD34+ cells were cells cultured in TPO and SCF for 8 days before sorting with CD41a and CD42b. Sample 3: The non-CD41a+CD42b+ cells were sorted from sample 2. Sample 4: The CD41a+CD42b+ cells were sorted from sample 2.
(B and C) SPRING plots of single-cell transcriptomes. Individual cells are presented according to their origins (B) or transcriptome-associated cell types (C). Ba, basophilic or mast cell; D, dendritic; Er, erythroid; GN, granulocytic neutrophil; Ly, lymphocytic; M, monocytic; Mk, megakaryocytic; MPP, multipotential progenitor.
(D) Pathway analysis of the top 400 genes with the strongest negative Spearman correlation to PRMT1 expression level in the TPO/SCF-stimulated CD41a+CD42b+ population. Red bars highlight Mk-relevant biological pathways.
(E) Spearman correlation coefficients between PRMT1 and any gene revealed by scRNA-seq in TPO/SCF-stimulated CD41a+CD42b+ cells. Representative genes with significant correlation and functional relevance are annotated.
(F) Gene set enrichment analysis (GSEA) of PRMT1-correlated genes in the TPO/SCF-stimulated CD41a+CD42b+ population. GSEA inputs are Spearman correlation coefficients of PRMT1 versus any gene with single-cell resolution.
(G) Normalized expression of representative transcripts co-plotted against that of PRMT1 transcript in TPO/SCF-stimulated CD41a+CD42b+ cells with single-cell resolution.
To further elucidate the TPO/SCF-stimulated CD41a+ CD42b+ cell population, we implemented the MAGIC algorithm, a diffusion-based imputation method (van Dijk et al., 2018), to recover potential dropouts associated with inefficient mRNA capture (Figure 4). The resultant MAGIC-normalized scRNA-seq transcripts of the TPO/SCF-stimulated, CD41a+ CD42b+ cells were analyzed using Spearman correlation between PRMT1 and other genes. The 400 transcripts with the largest and smallest Spearman correlation coefficients were then subject to pathway analysis with Reactome (Figures 4D and 4E; Table S4). With the whole set of Spearman correlation coefficients as inputs, gene set enrichment analysis (GSEA) was also conducted to identify PRMT1-associated biological pathways (Figure 4F) (Mootha et al., 2003). Remarkably, the two approaches revealed Mk differentiation and platelet production, at single-cell resolution, as the top biological processes negatively correlated with the transcript level of PRMT1 (Figures 4E and 4F). Consistently, the transcript levels of CD41 (ITGA2B), CD42B (GP1BA), PF4, MPL, CD42A (GP9), GP6, VWF, FLI1, F2R, F2RL2, and GATA1, a collection of characteristic Mk differentiation markers (Psaila and Mead, 2019; Sun et al., 2013), negatively correlated with the transcript level of PRMT1 of individual cells (Figures 4E, 4G, and S4). In contrast, IFNAR1/2, EGR1, IRF7, and IL1B— a panel of proinflammatory response genes that can trigger p38 activation (Lim et al., 1998; Wang et al., 2018) associated with blocking Mk differentiation (D’Atri et al., 2015; Verma et al., 2002)—positively correlated with the transcript level of PRMT1 (Figures 4E, 4G, and S4). We therefore conclude that there is a negative correlation between PRMT1 and Mk-platelet differentiation at a single-cell resolution, and a positive correlation between PRMT1 upregulation and the genes associated with p38 kinase activation of proinflammatory pathways.
Methylation of DUSP4 by PRMT1
Due to the antagonistic functions of PRMT1 and DUSP4 in Mk differentiation, we then examined whether DUSP4 is a substrate of PRMT1 in live cells. We developed the next-generation BPPM technology (Figures 5A and S5A) (Guo et al., 2014a). The three-step next-generation live-cell BPPM approach consisted of (1) the biosynthesis of (E)-hex-2-en-5-ynyl-SAM (Hey-SAM)—a bulky sulfonium-alkyl SAM analog that is inert to wild-type methyltransferases—from its cell-permeable methionine analog precursor Hey-Met by an engineered methionine adenosyltransferase (MAT2A I117A) within live cells; (2) in situ modification of the PRMT1 substrates by the engineered PRMT1 M48G variant with Hey-SAM as a cofactor; and (3) enrichment and quantification of the Hey-SAM-modified substrates via the azide-alkyne click reaction with an azide-containing fluorescent dye (Figures S5A and S5B) or biotin chemical reporter followed by enrichment with streptavidin-conjugated beads (Figure 5A) (Wang et al., 2011, 2013, 2014). The robust labeling of endogenous DUSP4 by PRMT1 (V1 or V2 isoforms), as well as the canonical substrate histone H4, was observed in HEK293T cells only in the presence of the complete set of the BPPM reagents, that is, the engineered PRMT1, MAT2A, and in situ produced Hey-SAM (Figure 5B), and also multiple cell lines (Figures S5C and S5D).
Figure 5. R351 methylation of DUSP4 by PRMT1.

(A) Schematic of the next-generation live-cell BPPM technology to uncover substrates of PRMT1.
(B) Immunoblotting readouts of H4 and DUSP4 as PRMT1 targets enriched via the BPPM technology using HEK293T cells (n = 3, representative western blots).
(C) Schematic description of the next-generation live-cell BPPM technology to reveal methylation sites of PRMT1. Candidates of methylated arginine (Arg) residues on a protein substrate are mutated to lysine (Lys). The resulting arginine-to-lysine mutation is expected to diminish or abolish the BPPM-associated labeling.
(D) DUSP4 sequence with functional domains and RXR motifs highlighted.
(E and F) Revealing PRMT1 methylation sites on DUSP4 with the next-generation live-cell BPPM technology. The DUSP4 variants contain dual arginine-to-lysine mutations at RXR motifs (E) and the point mutations at R351 and R353 (F), respectively (n = 3, representative western blots).
(G) Validating PRMT1-invovled R351 methylation on DUSP4 with anti-methylarginine antibodies in HEK293T cells (n = 3, representative western blots). Two anti-methyl-arginine antibodies were used to determine the presence of arginine methylation in DUSP4.
PRMT1 methylates DUSP4 at R351
We modified the BPPM procedure to map the sites of PRMT1-mediated DUSP4 methylation (Figure 5C). The potentially methylated arginine residues, such as in the GAR motif (Boisvert et al., 2005) and arginine-rich motif (Boisvert et al., 2003; Guo et al., 2014a; Larsen et al., 2016; Yamagata et al., 2008; Zhang et al., 2015; Zhao et al., 2008), were mutated into lysine residues (Figure 5D). Using the resulting R-to-K DUSP4 variants as substrate candidates of PRMT1, BPPM revealed that only the R351/352K mutations, but not other mutations, significantly abolished the DUSP4 labeling activity of PRMT1 in HEK293T cells. This indicates that PRMT1 recognizes the RER motif (amino acids 351–353) of DUSP4 in live cells (Figure 5E). With the individual R351K and R353K variants, we further defined that the R351K, but not R353K, mutation abolished the DUSP4 labeling activity of PRMT1 (Figures 5F, S5C, and S5D). Independently, PRMT1-dependent R351 methylation of DUSP4 was validated in cells expressing exogenous PRMT1 (V1 and V2 isoforms) and wild-type DUSP4, but not its R351K mutant, by two independently developed pan-anti-methyl-arginine monoclonal antibodies from Cell Signaling Technology (Figure 5G). Interestingly, the V2 isoform showed higher activity of DUSP4 methylation than did the V1 isoform (Figure 5G), consistent with the stronger effect of the former on DUSP4 degradation (Figure 3F). We then demonstrated PRMT1 to methylate the R351 residue of full-length DUSP4 as detected by mass spectrometry (Figure S5E) and in vitro methylation assays (Figure S6).
PRMT1-mediated R351 methylation of DUSP4 promotes its degradation via polyubiquitylation
Consistent with PRMT1-promoted proteasome-mediated degradation of DUSP4, overexpression of PRMT1 (V1 and V2 isoforms) in HEK293T cells led to the robust polyubiquitylation of wild-type DUSP4, which was completely suppressed on the DUSP4 R351K mutant that was inert for PRMT1 methylation (Figure 6A). Furthermore, the stability of the DUSP4 R351K mutant was independent of the level of PRMT1, which contrasted with the decreased stability of wild-type DUSP4 upon PRMT1 expression (Figure 6B). We assessed the stability of wild-type DUSP4 versus the methylation-dead R351K mutant in HEK293T cells. In these studies, wild-type DUSP4 showed a short half-life of less than 2 h, while its R351K mutant was remarkably more stable, with an increased half-life of more than 5 h (Figure 6C). These data indicate that the R351 residue of DUSP4 and likely R351 methylation by PRMT1 are essential for promoting DUSP4 degradation (Figure 6D). Consistent with these observations, the DUSP4 R351K variant was significantly more efficient than wild-type DUSP4 in promoting the Mk differentiation of human CD34+ cells (Figure 6E). Mass spectral analysis of immunoprecipitated DUSP4’s interactome revealed that HUWE1, one of the four E3 ligases associated with DUSP4, can account for the methylation-dependent degradation of DUSP4, as evidenced by HUWE1-DUSP4 interaction (Figure 6F) and the promoted polyubiquitylation and then degradation of wild-type but not R351K DUSP4 (Figures 6G–6I). Furthermore, HUWE1 knockout also promotes Mk differentiation (Figure S7G). Overall, we conclude that the antagonistic role of PRMT1 on DUSP4 acts through a methylation-dependent, polyubiquitylation- and proteome-mediated degradation of DUSP4.
Figure 6. Polyubiquitylation and instability of DUSP4 promoted by PRMT1-involved R351 methylation.

For a Figure360 author presentation of this figure, see https://doi.org/10.1016/j.celrep.2021.109421.
(A) Polyubiquitylation of DUSP4, but not its R351K variant, is stimulated by PRMT1. 293T cells were transfected with DUSP4 (wild-type and R351K mutant), 6× His-tagged ubiquitin, and PRMT1 (V1 and V2 isoforms). Cells were treated with MG132 for 6 h prior to harvest. Fractions of pull-downs (upper panel) and inputs (bottom panel) were applied for western blotting (n = 3, representative western blots).
(B) PRMT1-dependent stability levels of wild-type and R351K DUSP4 were determined in 293T cells by western blotting (n = 3, representative western blots).
(C) Half-life time of wild-type and R351K DUSP4. Normalized protein stability curves are plotted in the right panel (n = 3, representative western blots).
(D) Mechanistic description of DUSP4 stability modulated by PRMT1-dependent R351 methylation. R351 methylation of DUSP4 by PRMT1 triggers its polyubiquitylation and thus its degradation; R351K mutation abolishes the methylation and thus suppresses polyubiquitylation and degradation (n = 3, representative western blots).
(E) Mk differentiation in the presence of wild-type and R351K DUSP4. Human CD34+ cells infected with lentiviruses expressing DUSP4s (wild-type or R351K mutant) were induced for Mk differentiation. Percentage of CD41a+CD42b+ cells was determined by FACS after 7 days. Representative plots and statistics are shown (n = 3, independent experiments). Data are shown as mean ± SD. Two-tailed paired t test, *p ≤ 0.05.
(F) Co-immunoprecipitation of HUWE1 and DUSP4. Flag-tagged DUSP4 was used to immunoprecipitate myc-tagged HUWE1 in co-transfected 293T cells (n = 3, representative western blots after immunoprecipitation).
(G) The protein ubiquitylation of DUSP4 is measured in 293T cells transfected with plasmids as shown on top of the gels (n = 3, representative western blots).
(H) Mutant and wild-type DUSP4 were expressed together with HUWE1 shRNA in 293T cells for western blotting with respective antibodies (n = 3, representative western blots).
(I) Protein ubiquitylation assays with wild-type and mutant DUSP4. DUSP4 wild-type protein and mutant protein were expressed in 293T cells transfected with or without the plasmid combination of HUWE1 and PRMT1 as indicated on the top of the gel for affinity purification with Ni-NTA beads (n = 3, representative western blots).
Pharmacological inhibition of p38 MAPK and PRMT1 stimulates Mk differentiation in MDS
MDS is characterized by low blood counts and defects in hematopoietic cell differentiation (Nimer, 2008). Mk differentiation is adversely impacted in MDS, leading to thrombocytopenia and low platelet counts that are major clinical problems associated with this malignancy and that necessitate frequent platelet transfusion (Ramsey et al., 2012). Studies have shown that p38 MAPK is activated in small cohorts of MDS patients (Navas et al., 2006), which may suggest that the PRMT1-DUSP4 axis plays a role in the defective Mk differentiation. We analyzed gene expression data of CD34+ HSPCs from MDS patients (n = 183) and age-matched controls (n = 17) (Gerstung et al., 2015). PRMT1 levels were significantly elevated in MDS HSPCs (Figure 7A). Furthermore, p38α MAPK was also significantly overexpressed in MDS HSPCs (Figure 7B), which correlated with significantly lower platelet counts (Figure 7C). We also examined gene expression profiles generated from FACS-sorted long-term (LT)-HSCs (Lin−CD34+CD38−) and short-term (ST)-HSCs from 12 acute myeloid leukemia (AML)/MDS samples with normal karyotype, del(Chr7), and complex karyotype. We observed that HUWE1 was significantly overexpressed in leukemia stem cell/MDS populations (Figure S7H). Multivariate analysis using IPSS (international prognostic scoring system; the accepted system for prognosis in MDS) showed that p38α expression was predictive of adverse overall survival even after multivariate correction using standard risk factors as variables (Figure 7D, Cox proportional hazards model, p = 0.049 for p38α expression using IPSS) (Greenberg et al., 1997). We further examined p38 phosphorylation status within megakaryocytes from 29 MDS patients using immunohistochemistry. Megakaryocytes from MDS patients had significantly higher levels of phosphorylated (activated) p38 kinase, in comparison with levels in normal control megakaryocytes (Figures 7E and 7F; Tables S5 and S6).
Figure 7. Clinical implication and pharmacological targeting of the p38-DUSP4-PRMT1 axis in MDS.

(A and B) Gene expression of PRMT1 and p38α in MDS patients and healthy donors with array-based analysis. The CD34+ HSPCs of MDS patients (N = 183) and age-matched healthy controls (N = 17) were analyzed for PRMT1 expression (A) and p38α expression (B). Data are shown as mean ± SD. Two-tailed unpaired t test, ****p ≤ 0.0001.
(C) MDS cohorts as classified by low and high expression of p38α MAPK on the basis of median expression levels. The subjects with high p38α expression showed significantly lower platelet counts. Data are shown as mean ± SD. Two-tailed unpaired t test, *p ≤ 0.05.
(D) Survival curves of MDS patients classified by low and high expression of p38α MAPK. The MDS patients with higher p38α expression in HSPCs showed significantly worse overall survival. *p ≤ 0.05.
(E and F) Immunohistochemistry (IHC) analysis for phosphorylation-activated p38α MAPK of age-matched healthy controls and MDS BM samples from a clinical trial with the p38α inhibitor pexmetinib (ARRY614, labeled as p38i). MDS BMs showed significantly higher phospho-p38α staining in megakaryocytes (E). Representative stains are shown (F). Data are shown as mean ± SD. Two-tailed unpaired t test, **p ≤ 0.01.
(G–I) Effects of the p38α inhibitor pexmetinib (labeled as p38i) on normal CD34+ cells. Normal CD34+ cells were grown in liquid culture conditions in the presence and absence of pexmetinib and analyzed for CD41 and CD42 expression by representative charts shown (G) and averaged FACS data (H) (n = 2, independent experiments). (I) Normal CD34+ cells were also grown for MegaCult assay for production of megakaryocyte colonies (n = 2, independent experiments). Data are shown as mean ± SD. Two-tailed paired t test, **p ≤ 0.05, **p ≤ 0.01.
(J and K) Analysis of BM mononuclear cells (MNCs) of MDS patients with MegaCult assay for production of megakaryocyte colonies in the presence or absence of the p38α inhibitor pexmetinib (or p38i). Six MNC samples were examined (J) with the representative images shown (K).
(L) Analysis of MNCs of MDS patients (n = 6) with MegaCult assay for production of megakaryocyte colonies in the presence or absence of a PRMT1 inhibitor MS023. Data are shown as mean ± SD. Two-tailed paired t test, **p ≤ 0.01.
(M) Mk polyploidy and platelet count analysis in C57BL6/J mice treated with MS023. BM CD41+ cells were analyzed by FACS for polyploidy. The number of platelets in peripheral blood and MPV (mean platelet volume) were analyzed by a Hemavet machine.
(N) Mechanistic description of Mk differentiation via the PRMT1-DUSP4-p38 axis. Mk progenitors undergo abnormal differentiation in MDS by upregulation of PRMT1, which leads to p38 kinase activation. The relative levels of phospho-p38 are regulated by DUSP4. DUSP4 R351 is subject to PRMT1-medidated methylation, which leads to polyubiquitylation by HUWE1 and then degradation. Collectively, the PRMT1-DUSP4-p38 axis determines generation of Mk progenitor cells and the maturation of Mk cells.
A pilot phase I clinical trial of the p38α MAPK inhibitor pexmetinib (ARRY614) did not show significant increases in hemoglobin, but it strikingly demonstrated increases in platelet counts in 12 of 16 (75%) patients with transfusion-dependent severe thrombocytopenia (7 of 16 of patients achieved partial and 5 of 16 of patients achieved complete independence from platelet transfusion (ClinicalTrials.gov: NCT01496495) (Garcia-Manero et al., 2015). For ex vivo validation of this striking observation, we examined terminal differentiation of human primary CD34+ cells in a liquid culture after p38 kinase inhibition (Bachegowda et al., 2016). Pexmetinib treatment led to a modest increase in normal Mk terminal differentiation (Figures 7G and 7H). Furthermore, in MegaCult assays that quantify MEP progenitor cells with the potential to differentiate to the Mk lineage, p38 kinase inhibition significantly promoted Mk colony formation (Figure 7I). Importantly, when primary mononuclear cells from MDS patients were plated in the MegaCult assays after being treated ex vivo with pexmetinib, we observed significant increases in Mk colonies for all six cases (Figures 7J and 7K). The enhancement of Mk differentiation with pexmetinib treatment was also recapitulated using MS023 (Eram et al., 2016; Fong et al., 2019), an inhibitor of type I PRMTs, including PRMT1 (Figure 7l). MS023 of 200 nM inhibited more than half of the global protein arginine methylation as well as the methylation of two representative substrates RBM15 and DUSP4 (Figures S7A and S7B). Furthermore, MS023 promoted Mk differentiation (Figures S7C, S7D and S7F) and increased polyploidy (Figure S7E). We then treated wild-type C57BL6/J mice with MS023 inhibitor, which led to increased levels of high polyploid Mk cells and platelets (Figure 7M), which is consistent with data from PRMT1 conditional knockout mice (Zhu et al., 2019). These data indicate that pharmacological inhibition of p38 MAPK or PRMT1 may be a promising therapeutic strategy for treatment of thrombocytopenia in MDS patients.
DISCUSSION
The next-generation live-cell BPPM technology reveals DUSP4 as a substrate of PRMT1
While the BPPM technology was initially developed to interrogate methylome (a collection of methylation substrates) of designated protein methyltransferases, our work has adapted the technology to allow identification of individual substrate candidates and to reveal specific modification sites of PRMT1 in relevant cellular contexts. Given that the M48 residue of PRMT1 is conserved across the PRMT family and the homologous M233G variant of PRMT3 also utilizes bulky SAM analog cofactors (Guo et al., 2014b), we envision that the next-generation live-cell BPPM approach will be generally applicable for profiling and validating PRMT targets.
To characterize proteome-wide arginine methylation, conventional approaches rely on MS or antibodies to trace characteristic methylarginine-containing peptides (Guo et al., 2014a). However, because of the low abundance of many arginine methylation events, MS-based detection approaches often need to be coupled with antibody-based enrichment strategies. Furthermore, methylation has minimal effects on the overall size and electrostatic property of the targeted arginine residues. The small difference in the biophysical properties between methylated and unmodified basic residues such as lysine and arginine thus makes it challenging to develop high-quality antibodies to probe methylation events in an unambiguous manner. This is particularly the case for arginine methylation of low abundance (Luo, 2012, 2018). This situation is further complicated when arginine methylation is embedded in regions of the protein that are rich in other posttranslational modifications. Consequently, with conventional assays, many arginine methylation events and the associated PRMT activities are ‘‘invisible’’ in a cellular context. In contrast, BPPM technology allows effective enrichment of the targets of designated (engineered) PRMTs via distinct chemical labeling, whose detection is independent of neighboring sequences and posttranslational modifications. Additionally, the BPPM approach can unambiguously assign the peptides containing the distinctly labeled arginine residues as the direct targets of a specific (engineered) PRMT activity. BPPM technology can also be coupled with conventional antibody- or MS-based assays to show that the BPPM-revealed arginine sites can be subject to native methylation (Luo, 2012, 2018).
Methylation of DUSP4 by PRMT1 promotes ubiquitylation
DUSP4 (R351me) is in a SGPLR(351)ERGKTPAYP sequence, which also contains serine and threonine residues that have been shown to be phosphorylated (PhosphoSite.org). Other RXRXXS/T-containing substrates of PRMT1 such as FOXO1 and BAD are methylated by PRMT1 on both arginine sites (Sakamaki et al., 2011; Yamagata et al., 2008), which blocks AKT-mediated phosphorylation on the nearby serine/threonine residues. The potential regulatory crosstalk between AKT phosphorylation and PRMT1 methylation of DUSP4 within this sequence remains to be studied. Notably, R351 is not conserved between DUSP1 and DUSP4. Although DUSP1 and DUSP4 share many biological pathways (Auger-Messier et al., 2013), upregulation of DUSP1 renders cancer cells resistant to chemotherapy and BCR-ABL kinase inhibitors (Fang et al., 2018; Kesarwani et al., 2017), while downregulation of DUSP4 enhances chemoresistance (Balko et al., 2012). Differential regulation via arginine methylation between DUSP1 and DUSP4 may distinguish their unique roles in hematopoiesis and in other developmental contexts.
Interestingly, methylarginine-promoted protein degradation has also been observed for other PRMT substrates such as SRC3 (Naeem et al., 2007) and RBM15 (Zhang et al., 2015). Intriguingly, we found that the PRMT1 isoform V2 more efficiently methylates DUSP4 than does the isoform V1 in cell lines, an effect that may be mediated by the extra amino acids on the N-terminal region of V2. Since V2 may play a critical role in breast cancer cell proliferation (Baldwin et al., 2012; Mathioudaki et al., 2011), understanding the structure and function of V2 warrants future study. One possible mechanism underlying methylarginine-promoted degradation is that an E3 ligase directly recognizes the methylarginine for binding. Alternatively, arginine methylation may directly trigger a change of protein conformation that enhances recognition by E3 ligases. Protein arginine methylation has been demonstrated to change protein conformation for liquid phase transition (Hofweber et al., 2018; Qamar et al., 2018). Previously, we have shown that methylation of RBM15 by PRMT1 can trigger RBM15 ubiquitylation at a lysine site far from the methylation site (Zhang et al., 2015). In addition, the E3 ligase responsible for RBM15 ubiquitylation is a RING domain E3 ligase CNOT4, while the E3 ligase responsible for DUSP4 is a HECT domain E3 ligase HUWE1. The two E3 ligases share no similar domains that can be predicted to recognize methylated arginines on their substrates. It will therefore be interesting to investigate whether arginine methylation is a commonly utilized signal for protein degradation via unconventional ways such as arginine methylation-triggered liquid phase separation. A previous report demonstrated that HUWE1 recognizes phosphorylated protein for degradation (Wan et al., 2018), although the HUWE1 domains that recognize protein phosphorylation or methylation have not yet been identified.
PRMT1 participates MAPK signaling in megakaryopoiesis via methylation of DUSP4
While DUSP4 can dephosphorylate ERK, p38, and JNK MAPKs in vitro, DUSP4 preferentially dephosphorylates ERK, JNK, or p38 in a highly cell type-specific manner (Al-Mutairi et al., 2010; Chen et al., 2001; Guan and Butch, 1995; Robinson et al., 2001). In this study, we identified a specific developmental context wherein DUSP4 fine-tunes MAPK signaling for determination of Mk cell fate at multiple differentiation steps (Figure 7N). Given that the FACS-sorted progenitor cells are still heterogeneous, we cannot examine whether DUSP4 determines the binary cell fate choice at the level of a single MEP cell. Moreover, DUSP4 upregulation could happen at the stem cell stage (Reya et al., 2003), and p38 activation mobilizes hematopoietic stem cells for differentiation (Ito et al., 2006; Karigane et al., 2016). Specifically, we showed that DUSP4 selectively dephosphorylates p38 (Figure 3), while upregulation of ERK2 phosphorylation was not affected by DUSP4 expression during Mk maturation. Signal transduction regulators such as p38 and ERK2 can be potential epigenetic regulators given their association with chromatin (Klein et al., 2013), and PRMT1 is a known epigenetic regulator that binds to chromatin and methylates histones as well as transcription factors. Thus, the intersection between the p38-DUSP4 and DUSP4-PRMT1 axis highlights the intrinsic connection between signaling events and epigenetic regulation (Figure 7N).
DUSP4 knockdown in MEP cells specifically suppressed Mk colony-forming ability and promoted the generation of Er colonies (Figure 2). As many stress signals can promote Er differentiation, the effects of DUSP4 on erythropoiesis could be indirect. Relatedly, at the erythrocyte maturation stage, excessive p38 kinase activity has been demonstrated to restrain excessive erythropoiesis by inducing apoptosis (Hu et al., 2018), suggesting that the role of p38 at different stages of Er differentiation could be different.
Our data suggest that the later stage of Mk development and p38 MAPK activity are regulated by DUSP4, which also promotes Mk polyploidy (Figure S1D). Consistent with our scRNA-seq data, it is reported that Mks with proinflammatory gene expression profiles are low polyploidy and reside in lung (Lefrançais et al., 2017). Data using the MDS patient samples further strengthen the model that inhibition of PRMT1 is sufficient for the promotion of Mk differentiation (Figures 7 and S7). Our results and other studies have shown that PRMT1 expression and p38 MAPK activity are positively correlated with the interferon a response (Figure 4E) (Verma et al., 2002), which is known to repress differentiation of Mks (Wang et al., 2000). These findings suggest that an additional role of DUSP4 in Mk differentiation could be to suppress expression of interferon response genes. Primary myelofibrosis (PMF) is a type of myeloid proliferative disease that is also characterized by abnormal megakaryopoiesis. The p38 kinase is activated by FLT3 kinase in megakaryocytes of PMF (Desterke et al., 2011). In PMF, inhibition of p38 kinase promotes Mk differentiation, and FLT3 kinase is directly methylated by PRMT1 for activation (He et al., 2019). Therefore, it would be interesting to further test PRMT1 inhibitors for PMF treatment.
Anticipated benefit of inhibition of PRMT1 and p38 MAPK signaling for stimulating platelet production in MDS
Constitutive activation of p38 kinase in a subset of MDS cases (Navas et al., 2006) and its high expression can exhaust normal HSCs (Ito et al., 2006). In light of the PRMT1-DUSP4-p38 axis, we anticipated that suppression of overall p38 kinase activity in MDS cells using inhibitors such as pexmetinib, or MS023, could rescue Mk development and enhance platelet production. In a pilot phase I clinical trial, a large proportion of MDS patients with severe transfusion-dependent thrombocytopenia benefited from the p38α MAPK inhibitor pexmetinib, thus validating our preclinical findings. Our discovery of the inhibitory role of the PRMT1-DUSP4-p38 axis in megakaryopoiesis (Figure 7N) provides a preclinical rationale for therapeutic targeting of components of this pathway in future clinical trials in patients with myeloid malignancies and thrombocytopenia.
Limitations of the study
The biological importance of the PRMT1-DUSP4-HUWE1 axis for megakaryocyte differentiation needs to be further tested in animal models and with more MDS samples. While the feasibility of using PRMT1 inhibitors to boost platelet production in mice (Figure 7M) was demonstrated, optimizing the in vitro cell culture conditions to produce platelets from MDS samples with diverse genetic mutation backgrounds are still ongoing. These pre-clinical studies of MDS will further address the therapeutic potential of targeting PRMT1.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Xinyang Zhao (zhaox88@uab.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The original/source data for Figure 4 is available at GEO: GSE174261.
No original code was written. The bioinformatic analysis pipeline is in method details.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human primary CD34+ cells
The three sources of human primary CD34+ cells are peripheral blood, bone marrow and cord blood. The G-CSF-mobilized peripheral blood CD34+ cells were obtained in two steps: Ficoll-Paque Plus (GE Healthcare) to separate mononuclear cells and then CD34 MultiSort Kit (Miltenyi) for CD34 enrichment according to the description of the manufacturers. Human bone marrow and cord blood CD34+ cells were purchased from Lonza Inc. Human bone marrow samples were originated from a 35-year-old female and a 32-year-old male. The information of the donor(s) of cord blood is not available. These CD34+ cells were cultured in IMDM medium supplemented with 20% BIT (Stem Cell Technology) and a cytokine mixture consisting of 100 ng/mL SCF, 100 ng/mL FLT3 ligand, 50 ng/mL IL-6 and 20 ng/mL of TPO in 5% CO2 incubators at 37°C before Mk stimulation. For viral transduction, these primary cells were spin-infected twice with corresponding lentiviruses (2 × 805 g for 2 hours with 8-hour interval).
Cell lines
All of the used human cell lines were cultured in 5% CO2 incubators at 37°C. HEK293T cells (RRID: CVCL_0063) were cultured in DMEM medium supplemented with 10% FBS. MEG-01 (human chronic myeloid leukemia cell line derived from a 55-year-old male, RRID: CVCL_0425), CMK (human megakaryocytic leukemia cell line derived from 10-year-old male with down syndrome, RRID: CVCL_0216), and NB4 (human acute promyelocytic cell line derived from a 23-year-old female, RRID: CVCL_0005) were cultured in RPMI1640 medium supplemented with 10% FBS. K562 (human chronic myeloid leukemia cell line derived from a 53-year-old female, RRID: CVCL_0004) cells were cultured in IMDM medium supplemented with 10% FBS.
Bacteria
BL21 (DE3) and Rosetta (DE3) cells were cultured for recombinant protein production in LB medium at 37°C.
C57BL6/J mice
Wild-type C57/Bl6J mice of 6 to 16 weeks (n = 8 with equal numbers of male and female mice) were injected intraperitoneally with 80 mg/kg of MS023 every other day for one month under the animal protocol IACUC-21569 approved by animal core facility at the University of Alabama at Birmingham. Bone marrow cells from femurs were stained with antibodies and pyronin Y for FACS analysis of polyploidy of Lin−CD41+ cells. The number of platelets and mean platelet volume (MPV) were measured by Hemavet machine (Drew Scientific).
METHOD DETAILS
Synthesis of Hey-Met
Hey-methionine analog (Hey-Met) was synthesized as described previously (Wang et al., 2013). Briefly, into 10 mL of diethyl ether, add 165 mg (2.0 mmol) of (E)-pent-2-en-4-yn-1-ol (ASW MedChem, cat. no. DH-33159) and 480 mg (2.4 mmol) of p-toluenesulfonyl chloride (Alfa Aesar, cat. no. A14547–30) at ‒5°C with a saline ice water bath. Into the solution, add 560 mg (10 mmol) of potassium hydroxide. This mixture was stirred at ‒5°C for 1 hour and then at ambient temperature (22 oC) overnight. The reaction was quenched with 20 mL of ice-cold ddH2O water. The resulting organic phase was collected. The residual aqueous phase was extracted with 10 mL of diethyl ether twice. All organic phase components were combined, washed stepwise with 15 mL of ddH2O, 1 N HCl, and brine, and then dried with anhydrous MgSO4 for 3 hours. The volatile component was then removed with a rotary evaporator. The intermediate (E)-pent-2-en-4-ynyl tosylate was obtained with 85% yield as a light brown solid. A round-bottom flask containing S-Benzyl-L-homocysteine (255 mg, 1 mmol) was then placed in a dry ice-ethanol bath. Liquid ammonia (20 mL) and then sodium metal (50 mg, 2.2 mmol) were added the solution, When the mixture became colorless, the dry ice-ethanol bath was removed to allow the evaporation of ammonia, followed by a flow of argon and then vacuum of an oil pump to remove the residual ammonia. The resulting white solid was dissolved in dry ethanol (10 mL) and cooled with an ice-water bath to 0°C. A solution of the intermediate (E)-pent-2-en-4-ynyl tosylate in ethanol (3 mL) was added. This reaction mixture was stirred at 0°C for 1 h, followed by overnight at ambient temperature (22°C). After removing the ethanol, the resultant mixture was re-dissolved in distilled de-ionized (DD) water (5 mL) and passed through a Dowex 50® (H+) ion-exchange column. After washing the column with DD water until neutral, the desired product was then eluted with diluted ammonia hydroxide (5% DD water solution) and concentrated on a rotary evaporator to yield a white solid. This crude product was then dissolved in 0.1 N HCl and further purified with a preparative reversed-phase HPLC (XBridge Prep C18 5μm OBD 19 × 150mm). The desired fractions were combined and lyophilized to yield Hey-Met a white powder with 35% yield. 1H-NMR of Hey-Met (500MHz, D2O): 2.13–2.19(m, 1H), 2.22–2.28(m, 1H), 2.52(t, 1H, J = 2.6Hz), 2.66(t, 2H, J = 7.5Hz), 3.00–3.02(m, 2H), 3.23(dd, 2H, J = 7.2, 0.7Hz), 4.14(t, 1H, J = 6.3Hz), 5.64–5.70(m, 1H), 5.75–5.81(m, 1H).
Protein expression
Native PRMT1 with N-terminal 6xHis tag was expressed in E. Coli Rosetta (DE3) Strain and purified by Ni-NTA agarose (QIAGEN) and then HiTrap Q Sepharose Fast Flow Column (GE HealthCare) as described previously (Wang et al., 2011). For DUSP4 expression, the plasmid harboring human DUSP4 with an N-terminal GST tag was transformed into BL21 (DE3) cells. The selected colony was cultured at 37°C to OD600 of 0.6∼0.8. IPTG (0.1 mM) was added to induce protein expression for 4 hours. The cells were collected by centrifuging at 7,000 g at 4°C for 30 minutes. The resultant cell pellet was dissolved in a lysis buffer (50 mM Tris-Cl pH 8.0, 50 mM NaCl, 10% Glycerol and 0.1% Tween-20) supplemented with DNase I, lysozyme, and protease inhibitor cocktail (Roche), followed by French press for 4 times. This lysate was centrifuged at 30,000 g at 4°C for 1 hour. The supernatant was collected and applied to Glutathione Sepharose 4 Fast Flow GST-tagged protein purification resin (GE Healthcare) for DUSP4 purification. An elution buffer (50 mM Tris-Cl, pH 9.0, 50 mM NaCl, 10% Glycerol and 40 mM Glutathione) of 10 mL was added onto column, followed by ten collections of 1 mL fraction of eluates. To visualize and quantify DUSP4, Laemmli sample buffer was used to dissolve the samples. The resultant mixture was boiled and then resolved by SDS-PAGE. The gels were stained with Coomassie Brilliant Blue to determine the amount of DUSP4. Eluted fractions containing DUSP4 were combined and concentrated by centrifugation at 1,800 g at 4°C with 30 kDa MWCO (Millipore). The concentrated DUSP4 were further purified by overnight dialysis in a dialysis buffer (50 mM Tris-HCl, pH 7.5, 100 mM NaCl and 15% Glycerol) at 4°C with 20 kDa MWCO (Millipore).
Mass spectrometry
To prepare the Arg-methylated DUSP4 sample, 10 μM of GST-DUSP4 was incubated with 50 μM of CH3-SAM and 2 μM PRMT1 in a reaction buffer (50 mM HEPES pH 8.0, 0.005% Tween-20, 0.0005% BSA and 1 mM TCEP) at ambient temperature (22°C) overnight. The resulting reaction mixture was resolved by 4%–12% Bis-Tris gel (Bio-Rad) and stained with Coomassie Brilliant Blue. The gel band corresponding to the methylated GST-DUSP4 was extracted and digested with trypsin (Promega). Liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis was performed to map potential methylation sites as described previously (Zhang et al., 2015). Briefly, the digested product was resolved by a 60 min gradient elution at a flow rate 0.30 mL/min using an UltiMate 3000 RSLCnano System (Thermo Scientific, USA) interfaced with a Thermo Q Exactive benchtop mass spectrometer. The analytical column was a home-made fused silica capillary column (75 mm ID, 150 mm length; Upchurch, Oak Harbor, WA) packed with C-18 resin (300 A°, 5 mm, Varian, Lexington, MA). Mobile phase A consisted of 0.1% formic acid and mobile phase B consisted of 100% acetonitrile and 0.1% formic acid. The mass spectrometer was operated in the data-dependent acquisition mode using the Xcalibur 2.2.0 software and there is a single full-scan mass spectrum in the Orbitrap (400–1800 m/z, 70,000 resolution) followed by 8 MS/MS scans under the higher energy collision dissociation (HCD). The MS/MS spectra from each LC-MS/MS run were searched for potential methylation sites against the selected database using an in-house Mascot or Proteome Discovery searching algorithm.
To reveal the DUSP4 interactome with mass spectroscopy, endogenous DUSP4 in 293T cells was immunoprecipitated with the Flag antibody. DUSP4 and its binding partners were eluted, resolved by 4%–12% Bis-Tris gel (Bio-Rad), and stained with Coomassie Brilliant Blue. The stained gel was cut into seven equal pieces. The protein identities in the gel pieces were analyzed in a similar manner as described above to reveal the identifies of the DUSP4-interacting proteins. The raw data is in Table S3.
Single-cell RNA sequencing (scRNA-seq) analysis
Sample preparation
Human bone marrow-derived CD34+ cells were cultured in a prestimulation medium (IMDM + 20% BIT + Basic Cytokine Mix: 100 ng/mL SCF, 20 ng/mL TPO, 100 ng/mL FLT3 Ligand and 50 ng/mL IL-6) for four days and examined for the surface markers CD41a/CD42b before processing for scRNA-seq. For un-stimulated cells (Sample 1 in Figure 4), approximately 100,000 cells were harvested. The rest of CD34+ cells were cultured in a Mkstimulating medium (IMDM + 20% BIT + MK Cytokine Mix: 25 ng/mL SCF, 50 ng/mL TPO), which was replaced with fresh Mk-stimulating medium on Day 4 and Day 7. On Day 8 poststimulation, approximately 100,000 cells were collected as TPO/SCF-stimulated cells (Sample 2 in Figure 4). FACS sorting of the TPO/SCF-stimulated cells allowed the collection of approximately 88,000 TPO/SCF-stimulated, CD41a+CD42b+ cells (Sample 4 in Figure 4) and 100,000 TPO/SCF-stimulated, non-CD41a+CD42b+ cells (Sample 3 in Figure 4). The four sets of cell samples were immediately subject to Drop-Seq-based single cell RNA sequencing as detailed below.
Library preparation
The procedure of scRNA-seq followed the C version 3.1 from Steve McCarroll’s lab (http://mccarrolllab.org/wp-content/uploads/2015/05/Online-Dropseq-Protocol-v.−3.1-Dec-2015.pdf). Briefly, 1 mL of the samples containing approximately 100,000 cells with 90% viability were processed using microfluidic devices (PMDS Microfluidic Devices, FlowJEM). 8,000∼10,000 cells were encapsulated in microfluidic droplets with 3000∼4000 single cells recovered after washing and enzymatic steps. The resultant emulsion droplets were broken and subject to reverse transcription, followed by 14 cycles of PCR amplification−95°C for 3 min, 4 x (98°C for 20 s, 65°C for 45 s, 72°C for 3 min), 10 x (98°C for 20 s, 67°C for 20 s, 72°C for 3 min), and 72°C for 5 min. After purification with AMPure XP beads (Beckman Coulter), 600 pg barcoded cDNA was fragmented by Amplicon Tagment enzyme using Nextera XT Kit (Illumina), followed by indexing PCR–95°C for 30 s, 12 x (95°C for 10 s, 55°C for 30 s, 72°C for 30 s), and 72°C for 5 min. The resulting DNA library was further purified with 0.6 x AMPure XP beads before sequencing.
Sequencing and fastq generation
Libraries were sequenced on Illumina NextSeq 500 platform (R1–20 cycles, index1–8 cycles, R2–64 cycles). Samples were demultiplexed and raw fastq data were generated using bcl2fastq2 v2.19.
Single-cell expression matrix generation
Raw fastq data were processed and converted to single-cell expression counts matrix following the standard Drop-seq computational pipeline v1.12 developed by McCarrol lab (http://mccarrolllab.org/dropseq/). Briefly, barcodes and unique molecular identifiers (UMIs) were extracted and corrected for bead synthesis errors. Raw reads were trimmed for adaptor and ploy-A sequences, followed by alignment to human GRCh37 reference genome using STAR v2.5.2b (Dobin et al., 2013). The number of captured cells was determined by selecting the ‘‘knee’’ point in the cumulative distribution of reads. The matrices of single-cell expression count were generated by counting UMIs per gene in captured cells.
Single-cell data visualization with SPRING
The cells detected with less than 100 genes or more than 4,000 genes or with > 20% mitochondrial RNA contents were excluded. After the sample filtering step, the scRNA-seq data of 1,813 cells, 1,077 cells, 864 cells, and 924 cells were obtained for Samples 1–4, respectively. The expression counts matrices on these filtered cells were uploaded to SPRING webserver (https://kleintools.hms.harvard.edu/tools/spring.html) for data visualization (Weinreb et al., 2018).
Annotating cell types with signature genes
Eight cell types were annotated with the current scRNA-seq dataset using the algorithm code developed previously for mouse samples with minor modification (https://github.com/AllonKleinLab/klunctions/tree/master/sam/Paper_Code/Tusi_et_al_2018) (Tusi et al., 2018). Briefly, the lineage-specific mouse genes used in the original code were converted into human homologs: for E (Erythrocyte) with the mouse gene set of Hba-a2, Hbaa1, Alas2 and Bpgm replaced by the human gene set of HBA2, HBA1, ALAS2 and BPGM; for GN (granulocytes) with the mouse gene set of Lcn2, S100a8, Ltf, Lyz2 and S100a9 replaced by the human gene set of LCN2, S100A8, LTF, LYZ and S100A9; for Ly (Lymphocytic) with the mouse gene set of Cd79a, Vpreb3, Vpreb1 and Lef1 replaced by the human gene set of CD79A, VPREB3, VPREB1 and LEF1; for D (Dendritic) with the mouse gene set of Cd74, H2-Ab1 and Cst3 replaced by human gene set of CD74, HLA-DQB1 and CST3; for Meg (Megakaryocyte) with the mouse gene set of Pf4, Itga2b, Vwf, Pbx1 and Mef2c replaced by the human gene set of PF4, ITGA2B, VWF, PBX1 and MEF2C; for M (monocyte) with the mouse gene set of Csf1r and Ccr2 replaced by the human gene set of CSF1R and CCR2; for Ba (Basophilic) with the mouse gene set of Lmo4, Ifitm1, Ly6e and Srgn replaced by the human gene set of LMO4, IFITM1, LY6E and SRGN; for MPP (multipotent progenitor) with the mouse gene set of Hlf and Gcnt2 replaced by the human gene set of HLF and GCNT2.
Recovering gene-gene expression relationship with MAGIC
Certain gene-gene relationships in the current set of scRNA-seq data can be subject to random loss because of their low expressing and thus poor recovery. In order to overcome this technological limitation, MAGIC with default parameters (van Dijk et al., 2018) was applied to process the scRNA-seq data of the TPO/SCF-stimulated, CD41a+CD42b+ cells. Spearman correlations of PRMT1 transcript with other genes of individual TPO/SCF-stimulated, CD41a+CD42b+ cells were calculated with the following equation:
Here, ρrPRMT 1 and ρrx are the standard deviations of the rank variables. COV(rPRMT1, rx) is the covariance of the rank variables between PRMT1 and individual target genes.
Pathway enrichment analysis.
Genes were ranked by spearman correlation coefficients. Reactome pathway enrichment analysis (https://reactome.org/) was performed using top 400 genes with positive and negative spearman correlation, respectively. Gene Set Enrichment Analysis (GSEA) analysis was performed using the full ranked list (10,656 genes) on curated gene sets (MSigDB, https://www.broadinstitute.org/msigdb).
Virus production and stable cell line establishment
Viral constructs were co-transfected with envelope vectors and packaging vectors in HEK293T cells by the methods of calcium phosphate DNA transfection or lipofectamine 3000 (Invitrogen). psPAX2 and pMD2.G were used for lentiviral vectors; pCMV-VSVG and pCMV-dR8.2 were used for retroviral vectors. Harvested viral supernatants were used to spin-infect target cells. Stable cell lines were selected by either addition of 400 μg/mL G148, 2 μg/mL puromycin or by GFP-based flow cytometry sorting using BD FACSAria II system (BD).
Bioorthogonal profiling of protein methylation (BPPM)
Intracellular labeling of PRMT1 substrates
The living-cell BPPM procedure described previously was followed with minor modification (Wang et al., 2013). Briefly, constructs of PRMT1-M48G, His-tag MAT2A-I117A and Flag-tag DUSP4 (WT or R351K) were transfected into HEK293T cells. At 24 hours post transfection, cells were washed with PBS and replenished with fresh DMEM medium. At 48 hours post transfection, the old medium was removed and cells were incubated with a methionine-deficient medium (DMEM without methionine, glutamine and cysteine, supplemented with 10% dialyzed FBS, L-glutamine at 0.584 g/L and Lcystine.2HCl 0.0626 g/L) for 30 minutes. 0.5 mM Hey-Met was then added into the medium for 8-hour incubation before harvest. Alternatively, the cells that stably expressed PRMT1-M48G, MAT2A or DUSP4 were directly incubated in the methionine-deficient medium for 30 minutes before Hey-Met addition.
Cell harvest and lysate preparation
After intracellular labeling with Hey-SAM processed from Hey-Met, cells were harvested, washed with PBS and then lysed in HEPES buffer (50 mM HEPES pH 8.0, 0.05% Tween-20, 1 mM TCEP, and Roche protease inhibitor cocktail) for 20 minutes on ice, followed by 20 minutes of sonication. Sample was centrifuged at 13,400 g at 4°C for 30 minutes and the resulting supernatant was saved as lysates. Protein concentrations of the lysates were determined by Bradford assay and the expression of PRMT1, MAT2 or DUSP4 was verified by western blotting. Equal amounts of proteins (10∼15 mg) were applied to methanol precipitation at −80°C overnight as described previously (Wang et al., 2013).
Click reaction of Hey-SAM-labeled proteins with biotin-containing azide probes
The click reaction procedure described previously was followed with minor modification (Blum et al., 2013). Briefly, after the step of methanol precipitation, the protein pellets (usually 5∼10 mg) containing Hey-SAM-labeled PRMT1 substrates were resuspended in 1 mL of the resuspension buffer consisting of 50 mM Triethanolamine pH 7.4, 150 mM NaCl, 4% SDS and the Roche protease inhibitor cocktail. The sample was diluted up to 3.45 mL with 50 mM Triethanolamine (pH 7.4). Click reaction cocktail was prepared by mixing 200 μL of fresh 20 mM CuSO4 and 200 μL of 40 mM BTTP (Click Chemistry Tools) for 30–60 minutes. 50 μL of freshly prepared 200 mM sodium ascorbate was added to the pre-mixed CuSO4/BTTP, as the blue color disappears, followed by adding 100 μL of 10 mM diazo biotin-azide (Click chemistry Tools) DMSO stock solution. 550 μL of the click reaction cocktail was then added to the resuspension of protein lysates to reach the final condition: 1 mM CuSO4, 2 mM BTTP ligand, 2.5 mM sodium ascorbate and 250 μM diazo biotin-azide. The sample was incubated at ambient temperature (22°C) for 2 hours with shaking. Pre-chilled methanol was added to the click reaction mixture for overnight protein precipitation at −80°C, followed by centrifugation at 3,200 g at 4°C for 30 minutes. Protein pellets were washed twice with pre-chilled methanol then air-dried for further analysis.
Biotin-streptavidin pull-down
Biotin-labeled proteins were then subject for Streptavindin pulldown as described previously with minor modification (Blum et al., 2013). Protein pellets obtained from the click reaction of the diazo biotin-azide probe were resuspended in 1 mL of resuspension buffer containing 50 mM Triethanolamine pH 7.4, 150 mM NaCl, and 4% SDS with Roche EDTA-free protease inhibitors. The samples were resuspended to the equal final concentrations of the total proteins (1∼5 mg per mL) determined by Bradford assay. Equal amounts of proteins (1∼5 mg per mL) were then transferred into new tubes containing 2 mL of the dilution buffer consisting 50 mM Triethanolamine pH 7.4, 150 mM NaCl and 1% Brij97. Pierce High Capacity Streptavidin Agarose beads (Thermo) were pre-equilibrated with PBS for three times and then resuspended with the dilution buffer. The diluted protein samples were mixed with 200 μL of streptavidin beads and incubated for 1 hour at ambient temperature (22°C) with end-over rotation. The beads were thoroughly washed with PBS containing 0.2% SDS (volume%) once, with PBS twice and 250 mM ammonium bicarbonate twice. During each wash, the samples were centrifuged at 3, 200 g for 2 minutes at 4°C. The washed beads were transferred to spin columns (Amicon Ultra-0.5 mL Centrifugal Filter). Labeled proteins were cleaved from the beads by the treatment of 250 μL of the elution buffer consisting of 100 mM freshly prepared Na2S2O4, in a buffer containing 250 mM Ammonium bicarbonate and 1% SDS for 30 minutes at ambient temperature (22°C) with shaking. Eluted proteins were collected by centrifuging at 1,000 g at 4°C for 2 minutes. The elution step was repeated once. The combined eluates were then transferred to 3 NMWL Centricon filter device (Millipore) to remove recessive Na2S2O4. The protein samples were concentrated through lyophilization and then dissolved in laemmli loading buffer for further analysis.
Click reaction of Hey-SAM-labeled proteins with fluorescent azide probes for in-gel analysis
Hey-SAM-labeled proteins were subject to click reaction with an azide dye for in-gel fluorescence visualization as described previously with minor modification (Blum et al., 2013). After the step of methanol precipitation of Hey-SAM-labeled PRMT1 substrates as describe above, the protein pellets (5∼10 mg) were resuspended in 1 mL of the resuspension buffer consisting of 50 mM Triethanolamine pH 7.4, 150 mM NaCl, 4% SDS and the Roche protease inhibitor cocktail. For in-gel fluorescence analysis, around 400 μg aliquant of individual protein samples was diluted up to 4–5 mg/mL with the resuspension buffer mentioned above. The 25 μL aliquant of this protein mixture was diluted up to 85 μL with 50 mM Triethanolamine (pH 7.4). Click reaction cocktail was prepared by mixing 5 μL of 20 mM freshly prepared CuSO4 and 5 μL of 40 mM BTTP for 30–60 minutes. 2.5 μL of freshly prepared 200 mM sodium ascorbate was added to the pre-mixed CuSO4/BTTP, as the blue color disappears, followed by adding 2.5 μL of 10 mM TAMRA-Azide (Thermo Fisher Scientific) DMSO stock solution. 15 μL of the click reaction cocktail was then added to the resuspension of protein lysates. The sample was incubated at ambient temperature (22°C) for 2 hours with shaking. Pre-chilled methanol was added to the click reaction mixture for overnight protein precipitation at −80°C, followed by centrifugation at 3,200 g at 4°C for 30 minutes. Airdried protein pellets were resuspended in a loading buffer consisting of 40 mM Tris pH = 6.8, 70 mM SDS, 10 mM EDTA, 10% glycerol and 10% β-ME (without dye to avoid interference with fluorescence signal) and resolved by SDSPAGE. The gels were then fixed in mixture of 40% methanol and 10% acetic acid (volume%) overnight to remove unreacted TAMRA dye. The gels were then rinsed in H2O for rehydration and scanned for fluorescence signal using the TAMRA channel on a Typhoon TRIO variable mode imager (Amersham Bioscience). The scanned gel was then stained with Coomassie Blue to examine the protein loading.
Western blotting
Laemmli sample buffer was added to cell lysates and enriched proteins. Samples were boiled, sonicated (WCL), resolved by SDS-PAGE, and then transferred to PVDF membranes (Millipore) or nitrocellulose membranes (Bio-Rad). Membranes were blocked in 5% non-fat milk, followed by blotting of indicated primary and secondary antibodies. Luminata Western Chemiluminescent reagent (Millipore) and Bio-Rad ChemiDoc MP system (Bio-Rad) were used for final visualization and quantification.
FLAG immunoprecipitation
Calcium phosphate transfected HEK293T cells (107cells) were lysed in 1 mL H-Lysis buffer (20 mM HEPES pH 7.9, 150 mM NaCl, 1 mM MgCl2, 0.5% NP40, 10 mM NaF, 0.2 mM NaVO4, 10 mM β-glycerol phosphate and 5% glycerol) containing freshly prepared dithiothreitol/DTT (1 mM), PMSF (200 μM) and the Roche protease inhibitor cocktail. Cells were incubated on ice for 15∼30 minutes and then sonicated by Bioruptor Ultra-sonication system (Diagenode). The sonicated cell extracts were cleared by centrifugation at 12,000 g at 4°C for 10 minutes and then applied for immunoprecipitations. 40 μL of pre-equilibrated 50% antiFLAG M2 agarose (SIGMA) was added to samples, followed by overnight rotation at 4°C. Precipitates were collected by centrifugation at 135 g at 4°C for 2 minutes and sequentially washed twice by each of three buffers: (1) 25 mM HEPES pH 7.9 and 150 mM NaCl; (2) PBS + 0.2% Triton X-100; and (3) PBS. Hot 1x Laemmli sample buffer was added to washed beads, followed by 5-minute boiling to release bound proteins.
Ubiquitylation assay
HEK293T cells were transfected with the DUSP4 (WT and mutant) construct and the construct of His-ubiquitin as previously described with minor modification (Liu et al., 2012). Briefly, MG132 was added to the culture at 40 hours post transfection and kept in the culture for 6 hours before harvest. Approximately 107 cells were lysed in 1 mL of buffer A (6 M guanidine-HCl, 0.1 M Na2HPO4/NaH2PO4 pH 7.5, 10 mM imidazole) on ice for 10 minutes. Lysate was briefly sonicated (< 5 cycles) using Bioruptor (Diagenode). Lysates were centrifuged at 12,000 g for 10 minutes at 4°C. Supernatants were collected and incubated with 100 μL of pre-balanced Ni2+-NTA beads (QIAGEN) for 3 hours at ambient temperature with occasional mixing. The NTA beads were collected by centrifugation and washed with buffer A, buffer B (1.5 M guanidine-HCl, 25 mM Na2HPO4/NaH2PO4, 20 m M Tris-Cl pH 6.8, 17.5 mM imidazole) and buffer TI (25 mM Tris-Cl pH 6.8, 10 mM imidazole). After the final washing, the beads were resuspended in hot 1x Laemmli sample buffer supplemented with 200mM imidazole. Samples were boiled and then used for western blotting.
Half-life assay
HEK293T cells were used for transfection of DUSP4 constructs (WT or R351K). At 36 hours post transfection, cells were supplemented with fresh medium containing 25 μg/mL cycloheximide (CHX). Extracts were collected at 0, 0.5, 1, 2, 3 and 5 hours post CHX treatment. Laemmli sample buffer was added to extracts. Samples were boiled and used for western blotting.
MDS cells in vitro assays
For characterization of Mk colonies (CFU-Mk), MegaCult-C CFU-Mk was used. Patient bone marrow- or peripheral blood-derived mononuclear cells (10,000 cells/mL) were used according to IRB (project number: 2005–536) approved protocol approved by Albert Einstein College of Medicine for each assay and incubated for 14 days before colony quantification. MS023 (500 nM) and ARRY-614 (200 nM) were added into culture in respective assays. For MDS cells in liquid culture assays, the MDS cells were culture in IMDM medium with 20% BIT 50 ng/mL TPO and 2 ng/mL SCF with TPO and SCF for day 6, then switched to TPO alone until day 14.
Immunohistochemistry
Paraffin-mounted bone marrow core biopsy sections were collected from MDS patients or control subjects with consent. Bone marrow section staining was performed with anti-p-p38 antibody (Cell signaling, Cat #9211) as previously described (Navas et al., 2006).
In vitro arginine methylation assay
The in vitro methylation of DUSP4 was measured by a previously reported filter plate assay (Blum et al., 2013; Ibanez et al., 2012) with some modifications. The reaction mixture (a total volume of 20 μL) contains 100 nM SETD8 protein (N-terminal 6 3 His tagged), 1 μg DUSP4 protein (N-terminal GST-tagged), 0.75 μM [3H-Me]SAM (PerkinElmer Life Sciences), and reaction buffer (50 mM HEPES, pH = 8.0, 0.005% BSA, 1 mM TCEP). The resulting mixture was allowed to react at ambient temperature (22°C) overnight. Each reaction mixture was split into three aliquots and quenched by spotting on phosphor cellulose (P-81) filter paper, followed by 2 hour air-dry. The dried filter paper was then washed 5 times with 50 mM Na2CO3/NaHCO3 solution (pH = 9.2). The washed filter paper was then transferred into a scintillation vial, well mixed with 0.5 mL ddH2O and 5 mL Ultima Gold, and analyzed by a Liquid Scintillation Analyzer (Perkin Elmer Tri-Carb 2910 TR).
MDS patient sample gene expression dataset and survival analysis
Gene expression data from 183 MDS CD34+ samples and 17 controls were obtained from Gene Expression Omnibus (GEO) (GSE19429) (Pellagatti et al., 2010). Survival analysis and blood counts were correlated with expression of p38 MAPK and PRMT1 as done previously (Schinke et al., 2015) with R program.
Mk differentiation of primary CD34+ cells
For screening DUSPs relevant to Mk differentiation, lentiviruses encoding shRNAs against individual DUSP transcripts were used to infect CD34+ cells. The transfected cells were stimulated with 50 ng/mL TPO and 25 ng/mL SCF for eight days before FACS analysis of GFP/CD41a+/CD42b+ population on BD Accuri C6 (BD Biosciences). To further validate Mk differentiation perturbed by DUSP4 shRNAs, lentivirusinfected CD34+ cells were cultured in IMDM supplemented with 20% BIT, 50 ng/mL TPO and 2 ng/mL SCF for seven days. Expression of CD41a and CD42b were determined by flow cytometry on BD LSRFortessa (BD Biosciences). To examine binary Mk/Er differentiation, CD34+ cells were culture in IMDM with supplement of 20% BIT, 50 ng/mL TPO, 2 ng/mL of SCF and 5 U/mL EPO for ten days. Expression of CD41a and CD71 were determined by flow cytometry on BD LSRFortessa (BD Biosciences).
Mk differentiation of MEG-01 cells
The protocol is reported previously (Zhang et al., 2015). Briefly, MEG-01 cells or lentivirus-infected MEG-01 cells after sorted with GFP coexpressed from lentivirus vector were cultured in RPMI1640 with 10% fetal bovine serum (FBS). The cells at 1X105 cells per mililiter were split into non-tissue culture treated 6 well plates the day before differentiation assay. 20 nM PMA (phorbol myristate acetate) were added to the medium to promote Mk differentiation. At different time points, the cells were collected by low speed centrifuge (200Xg) for 5 minutes. After wash with ice cold PBS, the cells were incubated with FITC conjugated anti-CD41a antibody at a hundred fold dilution in PBS. Expression of CD41a was determined by flow cytometry at different time points of differentiation. Propidium iodide (PI) staining was used to assess the total DNA content. The cell population with > 4N DNA was characterized as polyploidy.
Dual CFU-Mk/Er assay
The populations of MEP were purified from parental or lentivirus-infected CD34+ cells via FACS-sorting as previously described (Sanada et al., 2016). MEPs were sorted with Lin-CD34+FLT3-CD45RA−MPL+CD38midCD41a. For each assay, 500 sorted cells were plated on MegaCult™-C Medium with lipids and the supplement of TPO (50ng/mL), EPO (3U/mL), IL-3 (10ng/mL), IL-6 (10ng/mL) and SCF (25ng/mL). At 14 days of culturing, colonies were fixed and stained with anti-GlyA and anti-CD41a. Colonies were counted and categorized based on the stain of GlyA and CD41a: CFU-Mk (GlyA-CD41a+), BFU-E (GlyA+CD41a-) and CFU-Mk/E (GlyA+CD41a+).
Real-time PCR
Total RNA was prepared using Direct-Zol RNAprep Kit (Zymo research). cDNA was generated by the Verso cDNA Synthesis Kit (Thermo Scientific) with random hexamer primers. Real-time PCR assays were performed with Absolute Blue qPCR SYBR green Mix (Thermo Scientific) on a ViiA 7 system (Applied Biosystems). The primers are listed in Table S1.
Staining intracellular phospho-p38 for FACS analysis
Approximately 1×106 cells of NB4 cells were collected, washed twice with cold PBS, and fixed in 0.4% PFA for 7 minutes on ice. The cells were then washed with cold PBS and fully fixed in 4% PFA for 15 minutes at ambient temperature (22°C). The fixed cells were washed twice with FACS Buffer (PBS + 2% BSA + 0.1% NaN3) and then permeabilized with methanol for 30 minutes at −20°C. The resultant cells were washed with the FACS buffer two more times before staining with antibodies for 45 minutes at ambient temperature (22°C). The antibody-stained cells were then washed with the FACS buffer twice before FACS analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
The statistical details of all experiments can be found in figure legends. The 2-tailed Student’s t test was used for significance testing in the bar graphs. P values less than 0.05 were considered significant. Quantification data are presented as mean ± SEM. R program package and Prism 6 GraphPad were used for statistical analysis.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-6x His tag | Abcam | Cat# 18184; RRID: AB_444306 |
| anti-actin | Abcam | Cat# 3280; RRID: AB_303668 |
| anti-DiMethyl-R #1 | Guo et. al. 2014a | Not commercially available |
| anti-DiMethyl-R #2 | Guo et. al. 2014a | Not commercially available |
| anti-DUSP4 | Cell Signaling | Cat# 5149; RRID:AB_2750867 |
| anti-Erk | Cell Signaling | Cat# 9102, RRID:AB_330744 |
| anti-FLAG | Sigma-Aldrich | Cat# F3165; RRID:AB_259529 |
| anti-GAPDH | Thermo | Cat# MA5-15738; RRID:AB_10977387 |
| anti-CD41a | BD Biosciences | Cat# 555465; RRID:AB_395857 |
| anti-CD235a | Bio-Rad | Cat# MCA506; RRID:AB_323506 |
| anti-H4 | Cell Signaling | Cat# 13919; RRID: AB_2798345 |
| anti-HA Tag | Millipore | Cat# 05-904; RRID: AB_11213751 |
| anti-6x His tag | Abcam | Cat# 18184; RRID: AB_444306 |
| anti-actin | Abcam | Cat# 3280; RRID: AB_303668 |
| anti-DiMethyl-R #1 | Guo et. al. 2014a | Not commercially available |
| anti-DiMethyl-R #2 | Guo et. al. 2014a | Not commercially available |
| anti-DUSP4 | Cell Signaling | Cat# 5149; RRID:AB_2750867 |
| anti-Erk | Cell Signaling | Cat# 9102, RRID:AB_330744 |
| anti-FLAG | Sigma-Aldrich | Cat# F3165; RRID:AB_259529 |
| anti-GAPDH | Thermo | Cat# MA5-15738; RRID:AB_10977387 |
| anti-CD41a | BD Biosciences | Cat# 555465; RRID:AB_395857 |
| anti-CD235a | Bio-Rad | Cat# MCA506; RRID:AB_323506 |
| anti-H4 | Cell Signaling | Cat# 13919; RRID: AB_2798345 |
| anti-HA Tag | Millipore | Cat# 05-904; RRID: AB_11213751 |
| anti-Mouse HRP | Santa Cruz | Cat# SC-2314; RRID: AB_641170 |
| anti-MKP2 | Santa Cruz | Cat# SC-1200; RRID: AB_2095314 |
| anti-p38 | Santa Cruz | Cat# SC-81621, RRID:AB_1127392 |
| anti-Phospho-Erk | Santa Cruz | Cat# SC-7976, RRID:AB_2297323 |
| anti-Phospho-p38 | Santa Cruz | Cat# SC-17852-R, RRID:AB_2139810 |
| anti-PRMT1 | Millipore | Cat # 07-404; RRID: AB_310588 |
| anti-Rabbit HRP | Santa Cruz | Cat# SC-2004; RRID: AB_631746 |
| anti-Erk | Cell Signaling | Cat# 9102, RRID:AB_330744 |
| anti-FLAG | Sigma-Aldrich | Cat# F3165; RRID:AB_259529 |
| anti-GAPDH | Thermo | Cat# MA5-15738; RRID:AB_10977387 |
| anti-CD41a | BD Biosciences | Cat# 555465; RRID:AB_395857 |
| anti-CD235a | Bio-Rad | Cat# MCA506; RRID:AB_323506 |
| anti-H4 | Cell Signaling | Cat# 13919; RRID: AB_2798345 |
| anti-HA Tag | Millipore | Cat# 05-904; RRID: AB_11213751 |
| anti-Mouse HRP | Santa Cruz | Cat# SC-2314; RRID: AB_641170 |
| anti-MKP2 | Santa Cruz | Cat# SC-1200; RRID: AB_2095314 |
| anti-p38 | Santa Cruz | Cat# SC-81621, RRID:AB_1127392 |
| anti-Phospho-Erk | Santa Cruz | Cat# SC-7976, RRID:AB_2297323 |
| anti-Phospho-p38 | Santa Cruz | Cat# SC-17852-R, RRID:AB_2139810 |
| anti-PRMT1 | Millipore | Cat # 07-404; RRID: AB_310588 |
| anti-Rabbit HRP | Santa Cruz | Cat# SC-2004; RRID: AB_631746 |
| anti-Tubulin | Proteintech | Cat# 66031-1; RRID: AB_11042766 |
| anti-Ubiquitin | Sigma-Aldrich | Cat# 07-2130; RRID:AB_11205591 |
| APC Mouse anti-human CD42b | eBioscience | Cat# 17-0429-42, RRID: AB_2573146 |
| APC Mouse anti-human CD42b | BD Biosciences | Cat# 551061; RRID:AB_398486 |
| APC Mouse anti-human CD71 | BD Biosciences | Cat# 551374; RRID:AB_3985004 |
| FITC Rat anti-Mouse CD41 | BD Biosciences | Cat# 553848 RRID:AB_395085 |
| ImmPRESS Anti-Rabbit Ig Reagent antibody | Vector Laboratories | Cat# MP-7401; RRID:AB_2336529 |
| PE Mouse anti-human CD41a | BD Biosciences | Cat# 557297; RRID:AB_396624 |
| PE Mouse anti-human CD41a | BD Biosciences | Cat# 555467; RRID:AB_395859 |
| PE Phospho-p38 MAPK | Invitrogen | Cat# 12-9078-42; RRID:AB_2572691 |
| PE Phospho-pS6 Ribosome Protein | Cell Signaling | Cat# 5316S; RRID:AB_10694989 |
| PE Rat anti-Mouse CD41 | BD Biosciences | Cat# 561850 RRID:AB_10896980 |
| PerCP/Cy5.5 anti-human CD41 | Biolegend | Cat# 303720; RRID: AB_2561732 |
| Bacterial and virus strains | ||
| One Shot BL21(DE3) | Invitrogen | Cat# C6000033 |
| Biological samples | ||
| Human bone marrow CD34+ progenitor cells | Lonza | 2M-101B |
| Human cord blood CD34+ progenitor cells | Lonza | 2C-101A |
| Human bone marrow CD34+ progenitor cells | Lonza | 2M-101B |
| Human cord blood CD34+ progenitor cells | Lonza | 2C-101A |
| Chemicals, peptides, and recombinant proteins | ||
| [3H-Me]-SAM | PerkinElmer Life Sciences | Cat# NET155V |
| 4-12% Bis-Tris gel | Bio-Rad | Cat#3450124 |
| Ammonium bicarbonate | Sigma-Aldrich | Cat# 9830 |
| anti-FLAG M2 agarose | Sigma-Aldrich | Cat# A2220 |
| BIT | Stem Cell Technology | Cat3 09500 |
| BTTP ligand | Chemical Synthesis Core, Albert Einstein College of Medicine |
N/A |
| complete Protease Inhibitor Cocktail | Roche | Cat# 11697498001 |
| Copper(II) sulfate pentahydrate | Sigma-Aldrich | Cat# 203165 |
| Cycloheximide | Cayman | Cat# 14126 |
| Dialyzed Fetal Bovine Serum | GIBCO | Cat# 26400-044 |
| Diazo Biotin-Azide | Click Chemistry Tools | Cat# 1041-25 |
| DMEM without glutamine, methionine, and cystine | GIBCO | Cat# 21013024 |
| DMSO | Sigma-Aldrich | Cat# D8418 |
| Dnase I | Roche | Cat# 10104159001 |
| DPBS | Corning | Cat# 21-030-CV |
| Dulbecco’s Modified Eagles Medium (DMEM) |
GE Healthcare | Cat# SH30243.01 |
| Fetal Bovine Serum | GE Healthcare | Cat# SH30910.03 |
| Glutathione Sepharose 4 Fast Flow GST-tagged protein purification resin | GE Healthcare | Cat# 17513202 |
| Human EPO | Amgen | N/A |
| Human EPO | Peprotech | Cat# 100-64 |
| Human FLT3L | Peprotech | Cat# 300-19 |
| Human IL3 | ConnStem | Cat# I 1003-A |
| Human IL6 | ConnStem | Cat# I 1006 |
| Human IL6 | Peprotech | Cat# 200-06 |
| Human Stem Cell Factor | ConnStem | Cat# S 1000 |
| Human Stem Cell Factor | Peprotech | Cat# 300-07 |
| Human TPO | ConnStem | Cat# T 1002 |
| Human TPO | Peprotech | Cat# 300-18 |
| Iscove’s Modified Dulbecco’s Medium (IMDM) |
GE Healthcare | Cat# SH30228.01 |
| Lipofectamine 3000 | Invitrogen | Cat# L3000008 |
| Luminata Western Chemiluminescent reagent | Millipore | Cat# WBLUC0500 |
| Lysozyme | MP Biochemicals | Cat# 100834 |
| MG132 | Cayman | Cat# 10012628 |
| Ni2+-NTA beads | QIAGEN | Cat# 1018244 |
| Nitrocellulose Membrane | Bio-Rad | Cat# 1620167 |
| Pierce High Capacity Streptavidin Agarose beads | Thermo | Cat# 20359 |
| PMA | Cayman | Cat# 10008014 |
| PVDF Membrane | Millipore | Cat# PFL00010 |
| RPMI 1640 | Corning | Cat# 10-040-CV |
| RPMI 1640 without methionine | GIBCO | Cat# A1451701 |
| SAM (S-Adenosyl-Methionine) | Sigma-Aldrich | Cat# A7007 |
| Sodium hydrosulfite | Sigma-Aldrich | Cat# 157953 |
| Triethanolamine | Sigma-Aldrich | Cat# 90279 |
| Trypsin | Promega | Cat# V5280 |
| Critical commercial assays | ||
| CellTiter-Glo Viability Assay Kit | Promega | Cat# G7572 |
| Direct-Zol RNAprep Kit | ZYMO | Cat# R2062 |
| ImmPACT Vector Red Substrate Kit | Vector Laboratories | Cat# SK-5105; RRID:AB_2336524 |
| ImmPRESS HRP Anti-Mouse IgG Polymer Detection Kit |
Vector Laboratories | Cat# MP-7452; RRID:AB_2744550 |
| MegaCult-C Medium with Lipids | STEMCELL | Cat# 04850 |
| MegaCult-C Complete Kit with Cytokines; Collagen |
STEMCELL | Cat# 04901, 04902 |
| Methylcellulose Base Media kit | R&D | Cat# HSC002 |
| Verso cDNA synthesis Kit | Thermo | Cat# AB1453 |
| Deposited data | ||
| sc-RNA sequencing analysis | NCBI | GSE174261 |
| Experimental models: Cell lines | ||
| CMK | DSMZ | Cat# ACC-392; RRID: CVCL_0216 |
| HEK293T | ATCC | Cat# CRL-3216; RRID: CVCL_0063 |
| K562 | ATCC | Cat# CCL-243; RRID: CVCL_0004 |
| MEG-01 | ATCC | Cat# CRL-2021; RRID: CVCL_0005 |
| NB4 | Dr. Stephen Nimer | RRID: CVCL_0005 |
| Experimental models: Organisms/strains | ||
| C57BL/6j mouse | Jackson Laboratory | |
| Oligonucleotides | ||
| PRMT1-Fwd real-time PCR: 5′ CCA GTG GAG AAG GTG GAC AT |
This Study | N/A |
| PRMT1-Rev real-time PCR: 5′ CTC CCA GTG GAT CTT GT |
This Study | N/A |
| DUSP4-Fwd real-time PCR: 5′ AGG CGG CTA TGA GAG GTT TT |
This Study | N/A |
| DUSP4-Rev real-time PCR: 5′ CAC TGC CGA GGT AGA GGA AG |
This Study | N/A |
| HPRT1-Fwd real-time PCR: 5′ CAC CCT TTC CAA ATC CTC AG |
This Study | N/A |
| HPRT1-Rev real-time PCR: 5′ CTC CGT TAT GGC GAC CCG CA |
This Study | N/A |
| Recombinant DNA | ||
| Plasmid: pcDNA3.1 | Thermo Fisher | Cat# V79020; Addgene: #2093 |
| Plasmid: pGEX-6p-1 | GE HealthCare | Cat# 28-9546-48; Addgene: #2887 |
| Plasmid: pLKO.1 | David Root | Addgene: #10878 |
| Plasmid: pLEX_307 | David Root | Addgene: #41392 |
| Plasmid: pBGJR-GFP | Dr. Vladimir Jankovic | Not commercially available |
| Plasmid: pLV-EF1a-IRES-Neo | Hayer et al., 2016 | Addgene: #85139 |
| Plasmid: pTripZ | Thermo Fisher | Addgene: #5561 |
| pLKO.1-shDUSP4#1 | Sigma-Aldrich | target sequence: GCCTACCTGATGATGAAGAAA |
| pLKO.1-shDUSP4#2 | Sigma-Aldrich | target sequence: CCCAGTGGAAGATAACCACAA |
| pLKO.1-shHUWE1 | Sigma-Aldrich | target sequence: AAACCCAGGGCTGCCTTGGAAAAG |
| Software and algorithms | ||
| STAR | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| SPRING | Weinreb et al., 2018 | https://kleintools.hms.harvard.edu/tools/spring.html |
| MAGIC | van Dijk et al., 2018 | https://www.krishnaswamylab.org/projects/magic |
| Other | ||
| BD Accuri C6 | BD Biosciences | N/A |
| BD FACSAria II | BD Biosciences | N/A |
| BD LSRFortessa Special Order System | BD Biosciences | N/A |
| Bio-Rad ChemiDoc MP system | Bio-Rad | Cat# 170-8280 |
| Bioruptor Ultra-sonication system | Diagenode | Cat# UCD-300 |
| PDMS Microfluidic Devices | FlowJEM | N/A |
| Synergy H1 plate reader | BioTek | Cat# 8041000 |
| TRI-CARB 4910TR 110 V Liquid Scintillation Counter |
PerkinElmer Life Sciences | Cat# A491000 |
Highlights.
Arginine methylation of DUSP4 by PRMT1 triggers HUWE1-mediated ubiquitylation
Megakaryocytes have heterogeneous PRMT1 expression levels
DUSP4 promotes megakaryocyte differentiation and polyploidization
Inhibition of PRMT1 or p38 kinase activities restores differentiation of MDS cells
ACKNOWLEDGMENTS
This work was supported by grants from the NIH (1R21CA202390 and 1R01DK110574 and the Pardee Foundation to X.Z., R35GM131858 to M.L., R01GM122749 to J.J., and R01GM126154 to Y.G.Z.), the NCI (5P30 CA008748 to M.L. via MSKCC), the Tri-Institutional PhD Program in Chemical Biology (to H.G.), and the US Department of Veterans Affairs (BX0003617 and BX004426 to Y.C.). We would like to thank Dr. Iannis Aifantis at NYU for HUWE1 reagents.
DECLARATION OF INTERESTS
M.L. has served on the Scientific Advisory Board for Epi One. A.V. has received research funding from GlaxoSmithKline, Incyte, MedPacto, Novartis, Curis, and Eli Lilly and Company; has received compensation as a scientific advisor to Novartis, Stelexis Therapeutics, Acceleron Pharma, and Celgene; and has equity ownership in Stelexis Therapeutics. The remaining authors declare no competing interests. Array BioPharma provided the p38 inhibitor pexmetinib (ARRY614) and participated in its phase I study (ClinicalTrials.gov: NCT01496495).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109421.
INCLUSION AND DIVERSITY
We worked to ensure sex balance in the selection of non-human subjects. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community.
REFERENCES
- Al-Mutairi MS, Cadalbert LC, McGachy HA, Shweash M, Schroeder J, Kurnik M, Sloss CM, Bryant CE, Alexander J, and Plevin R (2010). MAP kinase phosphatase-2 plays a critical role in response to infection by Leishmania mexicana. PLoS Pathog. 6, e1001192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auger-Messier M, Accornero F, Goonasekera SA, Bueno OF, Lorenz JN, van Berlo JH, Willette RN, and Molkentin JD (2013). Unrestrained p38 MAPK activation in Dusp1/4 double-null mice induces cardiomyopathy. Circ. Res. 112, 48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachegowda L, Morrone K, Winski SL, Mantzaris I, Bartenstein M, Ramachandra N, Giricz O, Sukrithan V, Nwankwo G, Shahnaz S, et al. (2016). Pexmetinib: A novel dual inhibitor of Tie2 and p38 MAPK with efficacy in preclinical models of myelodysplastic syndromes and acute myeloid leuke mia. Cancer Res. 76, 4841–4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin RM, Morettin A, Paris G, Goulet I, and Côté J (2012). Alternatively spliced protein arginine methyltransferase 1 isoform PRMT1v2 promotes the survival and invasiveness of breast cancer cells. Cell Cycle 11, 4597–4612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balko JM, Cook RS, Vaught DB, Kuba MG, Miller TW, Bhola NE, Sanders ME, Granja-Ingram NM, Smith JJ, Meszoely IM, et al. (2012). Profiling of residual breast cancers after neoadjuvant chemotherapy identifies DUSP4 deficiency as a mechanism of drug resistance. Nat. Med. 18, 1052–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgartner C, Toifl S, Farlik M, Halbritter F, Scheicher R, Fischer I, Sexl V, Bock C, and Baccarini M (2018). An ERK-dependent feedback mechanism prevents hematopoietic stem cell exhaustion. Cell Stem Cell 22, 879–892.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum G, Bothwell I, Islam K, and Luo M (2013). Profiling protein methylation with cofactor analog containing terminal alkyne functionality. Curr. Protoc. Chem. Biol. 5, 67–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum G, Bothwell IR, Islam K, and Luo M (2013). Profiling protein methylation with cofactor analog containing terminal alkyne functionality. Curr. Protoc. Chem. Biol. 5, 67–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisvert FM, Côté J, Boulanger MC, and Richard S (2003). A proteomic analysis of arginine-methylated protein complexes. Mol. Cell. Proteomics 2, 1319–1330. [DOI] [PubMed] [Google Scholar]
- Boisvert FM, Hendzel MJ, Masson JY, and Richard S (2005). Methylation of MRE11 regulates its nuclear compartmentalization. Cell Cycle 4, 981–989. [DOI] [PubMed] [Google Scholar]
- Caunt CJ, and Keyse SM (2013). Dual-specificity MAP kinase phosphatases (MKPs): Shaping the outcome of MAP kinase signalling. FEBS J. 280, 489–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Hutter D, Yang X, Gorospe M, Davis RJ, and Liu Y (2001). Discordance between the binding affinity of mitogen-activated protein kinase subfamily members for MAP kinase phosphatase-2 and their ability to activate the phosphatase catalytically. J. Biol. Chem. 276, 29440–29449. [DOI] [PubMed] [Google Scholar]
- Cheng H, Zheng Z, and Cheng T (2020). New paradigms on hematopoietic stem cell differentiation. Protein Cell 11, 34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi H, and Flavell RA (2008). Acetylation of MKP-1 and the control of inflammation. Sci. Signal. 1, pe44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Atri LP, Etulain J, Rivadeneyra L, Lapponi MJ, Centurion M, Cheng K, Yin H, and Schattner M (2015). Expression and functionality of Toll-like receptor 3 in the megakaryocytic lineage. J. Thromb. Haemost. 13, 839–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debili N, Coulombel L, Croisille L, Katz A, Guichard J, Breton-Gorius J, and Vainchenker W (1996). Characterization of a bipotent erythro-megakaryocytic progenitor in human bone marrow. Blood 88, 1284–1296. [PubMed] [Google Scholar]
- Desterke C, Bilhou-Nabéra C, Guerton B, Martinaud C, Tonetti C, Clay D, Guglielmelli P, Vannucchi A, Bordessoule D, Hasselbalch H, et al. ; French Intergroup of Myeloproliferative Disorders; French INSERM; European EUMNET Networks on Myelofibrosis (2011). FLT3-mediated p38-MAPK activation participates in the control of megakaryopoiesis in primary myelofibrosis. Cancer Res. 71, 2901–2915. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eram MS, Shen Y, Szewczyk M, Wu H, Senisterra G, Li F, Butler KV, Kaniskan HU, Speed BA, Dela Seña C, et al. (2016). A potent, selective, and cell-active inhibitor of human type I protein arginine methyltransferases. ACS Chem. Biol. 11, 772–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang J, Ye Z, Gu F, Yan M, Lin Q, Lin J, Wang Z, Xu Y, and Wang Y (2018). DUSP1 enhances the chemoresistance of gallbladder cancer via the modulation of the p38 pathway and DNA damage/repair system. Oncol. Lett. 16, 1869–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong JY, Pignata L, Goy PA, Kawabata KC, Lee SC, Koh CM, Musiani D, Massignani E, Kotini AG, Penson A, et al. (2019). Therapeutic targeting of RNA splicing catalysis through inhibition of protein arginine methylation. Cancer Cell 36, 194–209.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Manero G, Khoury HJ, Jabbour E, Lancet J, Winski SL, Cable L, Rush S, Maloney L, Hogeland G, Ptaszynski M, et al. (2015). A phase I study of oral ARRY-614, a p38 MAPK/Tie2 dual inhibitor, in patients with low or intermediate-1 risk myelodysplastic syndromes. Clin. Cancer Res. 21, 985–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geest CR, and Coffer PJ (2009). MAPK signaling pathways in the regulation of hematopoiesis. J. Leukoc. Biol. 86, 237–250. [DOI] [PubMed] [Google Scholar]
- Gerstung M, Pellagatti A, Malcovati L, Giagounidis A, Porta MG, Jädersten M, Dolatshad H, Verma A, Cross NC, Vyas P, et al. (2015). Combining gene mutation with gene expression data improves outcome prediction in myelodysplastic syndromes. Nat. Commun. 6, 5901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg P, Cox C, LeBeau MM, Fenaux P, Morel P, Sanz G, Sanz M, Vallespi T, Hamblin T, Oscier D, et al. (1997). International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 89, 2079– 2088. [PubMed] [Google Scholar]
- Guan KL, and Butch E (1995). Isolation and characterization of a novel dual specific phosphatase, HVH2, which selectively dephosphorylates the mitogen-activated protein kinase. J. Biol. Chem. 270, 7197–7203. [DOI] [PubMed] [Google Scholar]
- Guccione E, and Richard S (2019). The regulation, functions and clinical relevance of arginine methylation. Nat. Rev. Mol. Cell Biol. 20, 642–657. [DOI] [PubMed] [Google Scholar]
- Guo A, Gu H, Zhou J, Mulhern D, Wang Y, Lee KA, Yang V, Aguiar M, Kornhauser J, Jia X, et al. (2014a). Immunoaffinity enrichment and mass spectrometry analysis of protein methylation. Mol. Cell. Proteomics 13, 372–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Wang R, Zheng W, Chen Y, Blum G, Deng H, and Luo M (2014b). Profiling substrates of protein arginine N-methyltransferase 3 with S-adenosyl-L-methionine analogues. ACS Chem. Biol. 9, 476–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayer A, Shao L, Chung M, Joubert LM, Yang HW, Tsai FC, Bisaria A, Betzig E, and Meyer T (2016). Engulfed cadherin fingers are polarized junctional structures between collectively migrating endothelial cells. Nat. Cell Biol. 18, 1311–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Zhu Y, Lin YC, Li M, Du J, Dong H, Sun J, Zhu L, Wang H,Ding Z, et al. (2019). PRMT1-mediated FLT3 arginine methylation promotes maintenance of FLT3-ITD+ acute myeloid leukemia. Blood 134, 548–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera R, Hubbell S, Decker S, and Petruzzelli L (1998). A role for the MEK/MAPK pathway in PMA-induced cell cycle arrest: Modulation of megakaryocytic differentiation of K562 cells. Exp. Cell Res. 238, 407–414. [DOI] [PubMed] [Google Scholar]
- Hofweber M, Hutten S, Bourgeois B, Spreitzer E, Niedner-Boblenz A, Schifferer M, Ruepp MD, Simons M, Niessing D, Madl T, and Dormann D (2018). Phase separation of FUS is suppressed by its nuclear import receptor and arginine methylation. Cell 173, 706–719.e13. [DOI] [PubMed] [Google Scholar]
- Hu P, Nebreda AR, Hanenberg H, Kinnebrew GH, Ivan M, Yoder MC, Filippi MD, Broxmeyer HE, and Kapur R (2018). P38α/JNK signaling restrains erythropoiesis by suppressing Ezh2-mediated epigenetic silencing of Bim. Nat. Commun. 9, 3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua WK, Chang YI, Yao CL, Hwang SM, Chang CY, and Lin WJ (2013). Protein arginine methyltransferase 1 interacts with and activates p38α to facilitate erythroid differentiation. PLoS ONE 8, e56715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibanez G, Shum D, Blum G, Bhinder B, Radu C, Antczak C, LUo M, and Djaballah H (2012). A high throughput scintillation proximity imaging assay for protein methyltransferases. Combinatorial chemistry & high throughput screening 15, 359–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Hirao A, Arai F, Takubo K, Matsuoka S, Miyamoto K, Ohmura M, Naka K, Hosokawa K, Ikeda Y, and Suda T (2006). Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat. Med. 12, 446–451. [DOI] [PubMed] [Google Scholar]
- Jin S, Su H, Tran NT, Song J, Lu SS, Li Y, Huang S, Abdel-Wahab O, Liu Y, and Zhao X (2017). Splicing factor SF3B1K700E mutant dysregulates erythroid differentiation via aberrant alternative splicing of transcription factor TAL1. PLoS ONE 12, e0175523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karigane D, Kobayashi H, Morikawa T, Ootomo Y, Sakai M, Nagamatsu G, Kubota Y, Goda N, Matsumoto M, Nishimura EK, et al. (2016). p38α activates purine metabolism to initiate hematopoietic stem/progenitor cell cycling in response to stress. Cell Stem Cell 19, 192–204. [DOI] [PubMed] [Google Scholar]
- Kesarwani M, Kincaid Z, Gomaa A, Huber E, Rohrabaugh S, Siddiqui Z, Bouso MF, Latif T, Xu M, Komurov K, et al. (2017). Targeting c-FOS and DUSP1 abrogates intrinsic resistance to tyrosine-kinase inhibitor therapy in BCR-ABL-induced leukemia. Nat. Med. 23, 472–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyse SM (2008). Dual-specificity MAP kinase phosphatases (MKPs) and cancer. Cancer Metastasis Rev. 27, 253–261. [DOI] [PubMed] [Google Scholar]
- Klein AM, Zaganjor E, and Cobb MH (2013). Chromatin-tethered MAPKs. Curr. Opin. Cell Biol. 25, 272–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen SC, Sylvestersen KB, Mund A, Lyon D, Mullari M, Madsen MV, Daniel JA, Jensen LJ, and Nielsen ML (2016). Proteome-wide analysis of arginine monomethylation reveals widespread occurrence in human cells. Sci. Signal. 9, rs9. [DOI] [PubMed] [Google Scholar]
- Lefrançais E, Ortiz-Muñoz G, Caudrillier A, Mallavia B, Liu F, Sayah DM, Thornton EE, Headley MB, David T, Coughlin SR, et al. (2017). The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature 544, 105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim CP, Jain N, and Cao X (1998). Stress-induced immediate-early gene, egr-1, involves activation of p38/JNK1. Oncogene 16, 2915–2926. [DOI] [PubMed] [Google Scholar]
- Liu ZJ, and Sola-Visner M (2011). Neonatal and adult megakaryopoiesis. Curr. Opin. Hematol. 18, 330–337. [DOI] [PubMed] [Google Scholar]
- Liu M, Hsu J, Chan C, Li Z, and Zhou Q (2012). The ubiquitin ligase Siah1 controls ELL2 stability and formation of super elongation complexes to modulate gene transcription. Mol. Cell 46, 325–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YC, Sanada C, Xavier-Ferrucio J, Wang L, Zhang PX, Grimes HL, Venkatasubramanian M, Chetal K, Aronow B, Salomonis N, and Krause DS. (2018). The molecular signature of megakaryocyte-erythroid progenitors reveals a role for the cell cycle in fate specification. Cell Rep. 25, 3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo M (2012). Current chemical biology approaches to interrogate protein methyltransferases. ACS Chem. Biol. 7, 443–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo M (2018). Chemical and biochemical perspectives of protein lysine methylation. Chem. Rev. 118, 6656–6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathioudaki K, Scorilas A, Ardavanis A, Lymberi P, Tsiambas E, Devetzi M, Apostolaki A, and Talieri M (2011). Clinical evaluation of PRMT1 gene expression in breast cancer. Tumour Biol. 32, 575–582. [DOI] [PubMed] [Google Scholar]
- Mazharian A, Watson SP, and Séverin S (2009). Critical role for ERK1/2 in bone marrow and fetal liver-derived primary megakaryocyte differentiation, motility, and proplatelet formation. Exp. Hematol. 37, 1238–1249.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki R, Ogata H, and Kobayashi Y (2001). Requirement of thrombopoietin-induced activation of ERK for megakaryocyte differentiation and of p38 for erythroid differentiation. Ann. Hematol. 80, 284–291. [DOI] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, et al. (2003). PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 34, 267–273. [DOI] [PubMed] [Google Scholar]
- Naeem H, Cheng D, Zhao Q, Underhill C, Tini M, Bedford MT, and Torchia J (2007). The activity and stability of the transcriptional coactivator p/CIP/SRC-3 are regulated by CARM1-dependent methylation. Mol. Cell. Biol. 27, 120–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata Y, Takahashi N, Davis RJ, and Todokoro K (1998). Activation of p38 MAP kinase and JNK but not ERK is required for erythropoietin-induced erythroid differentiation. Blood 92, 1859–1869. [PubMed] [Google Scholar]
- Navas TA, Mohindru M, Estes M, Ma JY, Sokol L, Pahanish P, Parmar S, Haghnazari E, Zhou L, Collins R, et al. (2006). Inhibition of overactivated p38 MAPK can restore hematopoiesis in myelodysplastic syndrome progenitors. Blood 108, 4170–4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimer SD (2008). Myelodysplastic syndromes. Blood 111, 4841–4851. [DOI] [PubMed] [Google Scholar]
- Norman TM, Horlbeck MA, Replogle JM, Ge AY, Xu A, Jost M, Gilbert LA, and Weissman JS (2019). Exploring genetic interaction manifolds constructed from rich single-cell phenotypes. Science 365, 786–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens DM, and Keyse SM (2007). Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene 26, 3203–3213. [DOI] [PubMed] [Google Scholar]
- Patterson KI, Brummer T, O’Brien PM, and Daly RJ (2009). Dual-specificity phosphatases: Critical regulators with diverse cellular targets. Biochem. J. 418, 475–489. [DOI] [PubMed] [Google Scholar]
- Pellagatti A, Cazzola M, Giagounidis A, Perry J, Malcovati L, Della Porta MG, Jädersten M, Killick S, Verma A, Norbury CJ, et al. (2010). Deregulated gene expression pathways in myelodysplastic syndrome hematopoietic stem cells. Leukemia 24, 756–764. [DOI] [PubMed] [Google Scholar]
- Pellin D, Loperfido M, Baricordi C, Wolock SL, Montepeloso A, Weinberg OK, Biffi A, Klein AM, and Biasco L (2019). A comprehensive single cell transcriptional landscape of human hematopoietic progenitors. Nat. Commun. 10, 2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pronk CJ, Rossi DJ, Månsson R, Attema JL, Norddahl GL, Chan CK, Sigvardsson M, Weissman IL, and Bryder D (2007). Elucidation of the phenotypic, functional, and molecular topography of a myeloerythroid progenitor cell hierarchy. Cell Stem Cell 1, 428–442. [DOI] [PubMed] [Google Scholar]
- Psaila B, and Mead AJ (2019). Single-cell approaches reveal novel cellular pathways for megakaryocyte and erythroid differentiation. Blood 133, 1427–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qamar S, Wang G, Randle SJ, Ruggeri FS, Varela JA, Lin JQ, Phillips EC, Miyashita A, Williams D, Ströhl F, et al. (2018). FUS phase separation is modulated by a molecular chaperone and methylation of arginine cation-π interactions. Cell 173, 720–734.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey SD, McCune JS, Blough DK, McDermott CL, Beck SJ, López JA, and Deeg HJ (2012). Patterns of blood product use among patients with myelodysplastic syndrome. Vox Sang. 102, 331–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reya T, Duncan AW, Ailles L, Domen J, Scherer DC, Willert K, Hintz L, Nusse R, and Weissman IL (2003). A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature 423, 409–414. [DOI] [PubMed] [Google Scholar]
- Robinson CJ, Sloss CM, and Plevin R (2001). Inactivation of JNK activity by mitogen-activated protein kinase phosphatase-2 in EAhy926 endothelial cells is dependent upon agonist-specific JNK translocation to the nucleus. Cell. Signal. 13, 29–41. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Fraticelli AE, Wolock SL, Weinreb CS, Panero R, Patel SH, Jankovic M, Sun J, Calogero RA, Klein AM, and Camargo FD (2018). Clonal analysis of lineage fate in native haematopoiesis. Nature 553, 212–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamaki J, Daitoku H, Ueno K, Hagiwara A, Yamagata K, and Fukamizu A (2011). Arginine methylation of BCL-2 antagonist of cell death (BAD) counteracts its phosphorylation and inactivation by Akt. Proc. Natl. Acad. Sci. USA 108, 6085–6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanada C, Xavier-Ferrucio J, Lu YC, Min E, Zhang PX, Zou S, Kang E, Zhang M, Zerafati G, Gallagher PG, and Krause DS (2016). Adult human megakaryocyte-erythroid progenitors are in the CD34+CD38mid fraction. Blood 128, 923–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuan-Pla A, Macaulay IC, Jensen CT, Woll PS, Luis TC, Mead A, Moore S, Carella C, Matsuoka S, Bouriez Jones T, et al. (2013). Platelet-biased stem cells reside at the apex of the haematopoietic stem-cell hierarchy. Nature 502, 232–236. [DOI] [PubMed] [Google Scholar]
- Schinke C, Giricz O, Li W, Shastri A, Gordon S, Barreyro L, Bhagat T, Bhattacharyya S, Ramachandra N, Bartenstein M, et al. (2015). IL8-CXCR2 pathway inhibition as a therapeutic strategy against MDS and AML stem cells. Blood 125, 3144–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seternes OM, Kidger AM, and Keyse SM (2019). Dual-specificity MAP kinase phosphatases in health and disease. Biochim. Biophys. Acta Mol. Cell Res. 1866, 124–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suganuma T, and Workman JL (2011). Signals and combinatorial functions of histone modifications. Annu. Rev. Biochem. 80, 473–499. [DOI] [PubMed] [Google Scholar]
- Sun S, Wang W, Latchman Y, Gao D, Aronow B, and Reems JA (2013). Expression of plasma membrane receptor genes during megakaryocyte development. Physiol. Genomics 45, 217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusi BK, Wolock SL, Weinreb C, Hwang Y, Hidalgo D, Zilionis R, Waisman A, Huh JR, Klein AM, and Socolovsky M (2018). Population snapshots predict early haematopoietic and erythroid hierarchies. Nature 555, 54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uddin S, Ah-Kang J, Ulaszek J, Mahmud D, and Wickrema A (2004). Differentiation stage-specific activation of p38 mitogen-activated protein kinase isoforms in primary human erythroid cells. Proc. Natl. Acad. Sci. USA 101, 147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk D, Sharma R, Nainys J, Yim K, Kathail P, Carr AJ, Burdziak C, Moon KR, Chaffer CL, Pattabiraman D, et al. (2018). Recovering gene interactions from single-cell data using data diffusion. Cell 174, 716–729.e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma A, Deb DK, Sassano A, Uddin S, Varga J, Wickrema A, and Platanias LC (2002). Activation of the p38 mitogen-activated protein kinase mediates the suppressive effects of type I interferons and transforming growth factor-b on normal hematopoiesis. J. Biol. Chem. 277, 7726–7735. [DOI] [PubMed] [Google Scholar]
- Wan W, You Z, Zhou L, Xu Y, Peng C, Zhou T, Yi C, Shi Y, and Liu W (2018). mTORC1-regulated and HUWE1-mediated WIPI2 degradation controls autophagy flux. Mol. Cell 72, 303–315.e6. [DOI] [PubMed] [Google Scholar]
- Wang R, and Luo M (2013). A journey toward bioorthogonal profiling of protein methylation inside living cells. Curr. Opin. Chem. Biol. 17, 729–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Miyakawa Y, Fox N, and Kaushansky K (2000). Interferon-α directly represses megakaryopoiesis by inhibiting thrombopoietin-induced signaling through induction of SOCS-1. Blood 96, 2093–2099. [PubMed] [Google Scholar]
- Wang R, Zheng W, Yu H, Deng H, and Luo M (2011). Labeling substrates of protein arginine methyltransferase with engineered enzymes and matched S-adenosyl-L-methionine analogues. J. Am. Chem. Soc. 133, 7648–7651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Islam K, Liu Y, Zheng W, Tang H, Lailler N, Blum G, Deng H, and Luo M (2013). Profiling genome-wide chromatin methylation with engineered posttranslation apparatus within living cells. J. Am. Chem. Soc. 135, 1048–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Zheng W, and Luo M (2014). A sensitive mass spectrum assay to characterize engineered methionine adenosyltransferases with S-alkyl methionine analogues as substrates. Anal. Biochem. 450, 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Reszka-Blanco N, Cheng L, Li G, Zhang L, and Su L (2018). p38 MAPK is critical for nuclear translocation of IRF-7 during CpG-induced type I IFN expression in human plasmacytoid dendritic cells. J. Immunol. 200 (Suppl), 109.106. [Google Scholar]
- Weinreb C, Wolock S, and Klein AM (2018). SPRING: A kinetic interface for visualizing high dimensional single-cell expression data. Bioinformatics 34, 1246–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreb C, Rodriguez-Fraticelli A, Camargo FD, and Klein AM (2020). Lineage tracing on transcriptional landscapes links state to fate during differentiation. Science 367, eaaw3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalen AM, Galasinski SC, Shapiro PS, Nahreini TS, and Ahn NG (1997). Megakaryocytic differentiation induced by constitutive activation of mitogen-activated protein kinase kinase. Mol. Cell. Biol. 17, 1947–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witt O, Sand K, and Pekrun A (2000). Butyrate-induced erythroid differentiation of human K562 leukemia cells involves inhibition of ERK and activation of p38 MAP kinase pathways. Blood 95, 2391–2396. [PubMed] [Google Scholar]
- Xavier-Ferrucio J, and Krause DS (2018). Concise review: Bipotent megakaryocytic-erythroid progenitors: Concepts and controversies. Stem Cells 36, 1138–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata K, Daitoku H, Takahashi Y, Namiki K, Hisatake K, Kako K, Mukai H, Kasuya Y, and Fukamizu A (2008). Arginine methylation of FOXO transcription factors inhibits their phosphorylation by Akt. Mol. Cell 32, 221–231. [DOI] [PubMed] [Google Scholar]
- Yamamoto R, Morita Y, Ooehara J, Hamanaka S, Onodera M, Rudolph KL, Ema H, and Nakauchi H (2013). Clonal analysis unveils self-renewing lineage-restricted progenitors generated directly from hematopoietic stem cells. Cell 154, 1112–1126. [DOI] [PubMed] [Google Scholar]
- Yu ZH, and Zhang ZY (2018). Regulatory mechanisms and novel therapeutic targeting strategies for protein tyrosine phosphatases. Chem. Rev. 118, 1069–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Tran NT, Su H, Wang R, Lu Y, Tang H, Aoyagi S, Guo A, Khodadadi-Jamayran A, Zhou D, et al. (2015). Cross-talk between PRMT1-mediated methylation and ubiquitylation on RBM15 controls RNA splicing. eLife 4, e07938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Jankovic V, Gural A, Huang G, Pardanani A, Menendez S, Zhang J, Dunne R, Xiao A, Erdjument-Bromage H, et al. (2008). Methylation of RUNX1 by PRMT1 abrogates SIN3A binding and potentiates its transcriptional activity. Genes Dev. 22, 640–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, He X, Dong H, Sun J, Wang H, Zhu Y, Huang F, Zou J, Chen Z, Zhao X, and Li L (2019). Protein arginine methyltransferase 1 is required for maintenance of normal adult hematopoiesis. Int. J. Biol. Sci. 15, 2763–2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The original/source data for Figure 4 is available at GEO: GSE174261.
No original code was written. The bioinformatic analysis pipeline is in method details.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.