

Abstract
The prognosis for children diagnosed with high-risk acute lymphoblastic leukemia (ALL) remains sub-optimal, and more potent and less toxic treatments are urgently needed. We investigated the efficacy of a novel nicotinamide phosphoribosyltransferase inhibitor, OT-82, against a panel of patient-derived xenografts (PDXs) established from high-risk and poor outcome pediatric ALL cases. OT-82 was well-tolerated and demonstrated impressive single agent in vivo efficacy, achieving significant leukemia growth delay in 95% (20/21) and disease regression in 86% (18/21) of PDXs. In addition, OT-82 enhanced the efficacy of the established drugs cytarabine and dasatinib and, as a single agent, showed similar efficacy as an induction-type regimen combining three drugs used to treat pediatric ALL. OT-82 exerted its anti-leukemic action by depleting NAD+ and ATP, inhibiting the NAD+-requiring DNA damage repair enzyme PARP-1, increasing mitochondrial ROS levels and inducing DNA damage, culminating in apoptosis induction. OT-82 sensitivity was associated with the occurrence of mutations in major DNA damage response genes, while OT-82 resistance was characterized by high expression levels of CD38. In conclusion, our study provides evidence that OT-82, as a single agent, and in combination with established drugs, is a promising new therapeutic strategy for a broad spectrum of high-risk pediatric ALL for which improved therapies are urgently needed.
INTRODUCTION
Acute lymphoblastic leukemia (ALL) is the most common childhood cancer. Long-term survival has improved dramatically over the last decades, resulting in current cure rates exceeding 80%.1, 2 However, disease prognosis remains much more guarded for patients who are classified as high-risk, including those that harbor translocations of the Mixed Lineage Leukemia (MLL/KMT2A) gene (MLLr-ALL) and BCR-ABL1 gene rearrangements (Ph+ ALL), as well as patients who relapse following standard therapy.1–8 ALL therefore remains one of the most common causes of death from disease in children.1, 7–9 In addition, existing treatment protocols are associated with significant detrimental health effects for some survivors.10 Consequently, there is an urgent need for the development of novel treatment strategies for high-risk pediatric ALL, which are more potent and less toxic than existing treatments, and which allow for dose reduction of standard chemotherapeutic agents.
Based on growing recognition of the significance of cancer cell metabolism in oncogenesis, metabolic pathways are increasingly investigated as targets for cancer therapy.11 One of the pathways of interest is the nicotinamide adenine dinucleotide (NAD+) biosynthesis pathway with nicotinamide phosphoribosyltransferase (NAMPT) as the major rate-limiting enzyme. NAD+ is a vital cellular energy source for ATP production and a substrate for several NAD+-dependent enzymes with indispensable cellular survival functions including CD38 which regulates Ca2+ signaling and the DNA damage repair enzyme polyADP ribose polymerase 1 (PARP-1) (reviewed in Garten et al.).12 Cancer cells are considered to be addicted to NAD+ due to their heightened proliferation, inefficient energy production, and dependency on PARP-mediated DNA damage repair to maintain genomic stability, making NAMPT an attractive anti-cancer target.12 NAMPT became the subject of intensive anti-cancer drug discovery research efforts (reviewed in Sampath et al.), however, the first NAMPT inhibitors failed in clinical trials due to a limited therapeutic window of these compounds.13 Despite these disappointing results, the search for safe NAMPT inhibitors has continued with novel inhibitors being reported frequently in the past few years.14–19
We recently discovered a novel NAMPT inhibitor, OT-82, which showed selective cytotoxicity against hematological malignancies without inducing any of the toxicities associated with earlier NAMPT inhibitors, as confirmed by extensive toxicity studies in mice and non-human primates (ref. Companion Manuscript 19-LEU-0662: Korotchkina L. et al.: OT-82, a novel anticancer drug candidate that targets the strong dependence of hematological malignancies on NAD biosynthesis). OT-82 was identified in a drug screen designed to discover compounds with selective cytotoxicity towards hematological cancers, which suggests that hematological malignancies may have a better therapeutic index for NAMPT inhibition than solid tumors (Korotchkina L. et al.). This hypothesis is supported by the recent discovery of the NAMPT inhibitor STF-118804 in an unbiased screen that aimed to identify selective inhibitors of MLLr-ALL.16 In addition, a recent study on the efficacy of a novel dual inhibitor of p21-activated kinase 4 and NAMPT in B-cell ALL (B-ALL), suggested that B-ALL might be more sensitive to NAMPT inhibition than solid malignancies due to reduced cellular NAD+-reserves in B-ALL cells.15
Given the preclinical safety and efficacy of OT-82 in hematological cancer models, the aim of the current study was to evaluate the therapeutic potential of OT-82 in high-risk childhood ALL using a diverse panel of molecularly annotated patient-derived xenografts (PDXs) derived from patients with high-risk and poor outcome ALL. OT-82 was tested both as a single agent and in combination with established drugs, and factors that determine responsiveness to the drug were identified.
MATERIALS AND METHODS
PDXs and in vivo drug treatments
All experimental studies were conducted with approval from the Animal Care and Ethics Committee of the University of New South Wales (Sydney, Australia). Leukemia engraftment was assessed as previously described and detailed in the Supplementary Methods.20–22 Drug responses were evaluated by leukemia growth delay values T-C and T/C, with T and C representing the median event-free survival of drug-treated and vehicle control-treated cohorts, respectively, as well as by an Objective Response Measure (ORM), modeled after stringent clinical criteria as detailed in the Supplementary Methods.20 OT-82 (40 mg/kg) or vehicle (30% captisol or 2-hydroxypropyl-beta-cyclodextrin) was administered via oral gavage (p.o.) on 3 consecutive days/week for 3 weeks. For combination studies, OT-82 or vehicle was administered according to the same regimen for 2 weeks in combination with cytarabine (Clifford Hallam Healthcare, Eastern Creek, Australia) intraperitoneally (i.p.), at 25 mg/kg, 5 days/week for 2 weeks or dasatinib (Medchem Express, Monmouth Junction, USA), i.p. at 15 mg/kg, 5 days/week for 2 weeks. Doses of cytarabine and dasatinib were selected to reflect levels achievable in humans and to allow a well-tolerated combination regimen with OT-82.
Cell-based assays, cytotoxicity and synergy assays
Annexin V apoptosis assays, cell counting assays and cytotoxicity assays with leukemia cell lines and PDX cells were performed as previously described.23–25 Combination experiments were performed in a two-way matrix format using fixed ratios of drugs in a 6×6 matrix and 2-fold increasing drug concentrations. Synergy was scored based on the Bliss independence model26, 27 and visualized by Combenefit.28
Analysis of NAD+ and ATP levels
NAD+ and ATP levels were determined in frozen spleen mononuclear cells and cell line pellets with the NAD/NADH (Promega, Alexandria, Australia) and ATPlite (Perkin-Elmer, Thermo Fisher Scientific, Scoresby, Australia) kits respectively, according to the manufacturer’s recommendations.
Immunofluorescence
Cells were treated, washed in PBS and allowed to attach to glass 8-well chamber slides (LAB-TEK, Thermo Fisher Scientific, North Ryde, Australia). Subsequently cells were fixed with 4% paraformaldehyde (ProSciTech, Kirwan, Australia) for 10 minutes at room temperature and permeabilized by incubation in 0.15% Triton-X100 in 1%BSA/PBS for 10 min at room temperature. Blocking was performed by incubation in 1%BSA/PBS for 10 min at room temperature followed by overnight incubation with mouse monoclonal anti-γH2AX (#80312, Cell Signaling Technology, Danvers, Massachusetts, 1:200) and rabbit anti-53BP1 (#88439, Cell Signaling, 1:1000) in 1%BSA/PBS at 4°C in a humid environment. Secondary anti-mouse-Alexa-Fluor 488 (Life Technologies, Thermo Fisher Scientific, 1:500) and/or anti-mouse-Alexa-Fluor 647 (Life Technologies, Thermo Fisher Scientific, 1:500) in 1%BSA/PBS was added for 1h at room temperature. Slides were stained for 30 min with DAPI as a nuclear stain followed by mounting. Cells were visualized using the Leica TCS SP8 DLS confocal microscope (Leica Microsystems, Macquarie Park, Australia) and images were analysed using the Leica Application Suite Advanced Fluorescence (LAS AF) software (Leica microsystems) and ImageJ/FUJI software.
ROS measurements
Levels of mitochondrial reactive oxygen species (ROS) were determined by flow cytometry. Treated cells were stained with Mitosox (Thermo Fisher Scientific) according to the manufacturer’s instructions and the samples were analyzed on a FACSCanto (BD Biosciences, North Ryde, Australia). The mean fluorescence intensity (MFI) of OT-82 treated samples was calculated using FlowJo (BD, Australia) relative to vehicle-treated cells.
Measurement of serum visfatin
Serum visfatin concentrations were measured using the Nampt (Visfatin/PBEF) (human) ELISA and Nampt (Visfatin/PBEF) (mouse) ELISA Kits (Adipogen, San Diego, USA) as per manufacturer’s instructions.
Protein analysis and immunoblotting
Methods for the analysis of cellular proteins by immunoblotting have been described previously and used antibodies are listed in the Supplementary Methods.23, 25, 29
Whole transcriptome sequencing and analysis
Whole transcriptome RNA sequencing was performed using total RNA as described in the Supplementary Methods. All RNA-sequencing data are available on the pediatric cBioPortal (https://pedcbioportal.org).
Whole exome mutation analysis
Whole exome mutation analysis was performed as described in the Supplementary Methods. Mutation classification databases (ClinVar, Cosmis, Varsome, MutationTaster, MutationAssesser and Polyphen) were used to predict the pathogenicity of detected mutations in DNA damage repair genes BRCA1/2, CHEK1/2, ATM and ATR. All WES data are available on the pediatric cBioPortal (https://pedcbioportal.org).
Statistical analysis
R statistical software was used for survival curve analysis. GraphPad Prism 7 was used for the other statistical analyses. P-values <0.05 were considered significant.
RESULTS
The NAMPT inhibitor OT-82 potently decreases the viability of leukemia cells
To evaluate the efficacy of OT-82 against leukemia cells, we assessed its effect on the viability of 14 acute leukemia cell lines including cells derived from aggressive ALL subtypes such as infant MLLr-ALL and T-ALL (Supplementary Table 1).30 OT-82 reduced the viability of all cell lines in a dose-dependent manner, with IC50 values ranging from 0.2 to 4.0 nM (mean IC50 ± SD = 1.3 ± 1.0 nM) (Figure 1A). Live cell counting confirmed that OT-82 significantly inhibited the growth of leukemia cells (Figure 1B). We subsequently evaluated the effect of OT-82 on the viability of PDX cells established from patients with high-risk pediatric ALL, in short term culture ex vivo (Supplementary Table 2).21, 31 Similar to the leukemia cell lines, the PDX cells were highly sensitive to OT-82 treatment with IC50 values ranging from 0.4 to 3.6 nM (mean IC50 ± SD = 1.2 ± 0.9 nM) (Figure 1C). The leukemia cell lines and PDX cells most sensitive to OT-82 were characterized by the presence of an MLL rearrangement.
Figure 1: OT-82 potently decreases the viability of leukemia cell lines and pediatric ALL PDX cells.
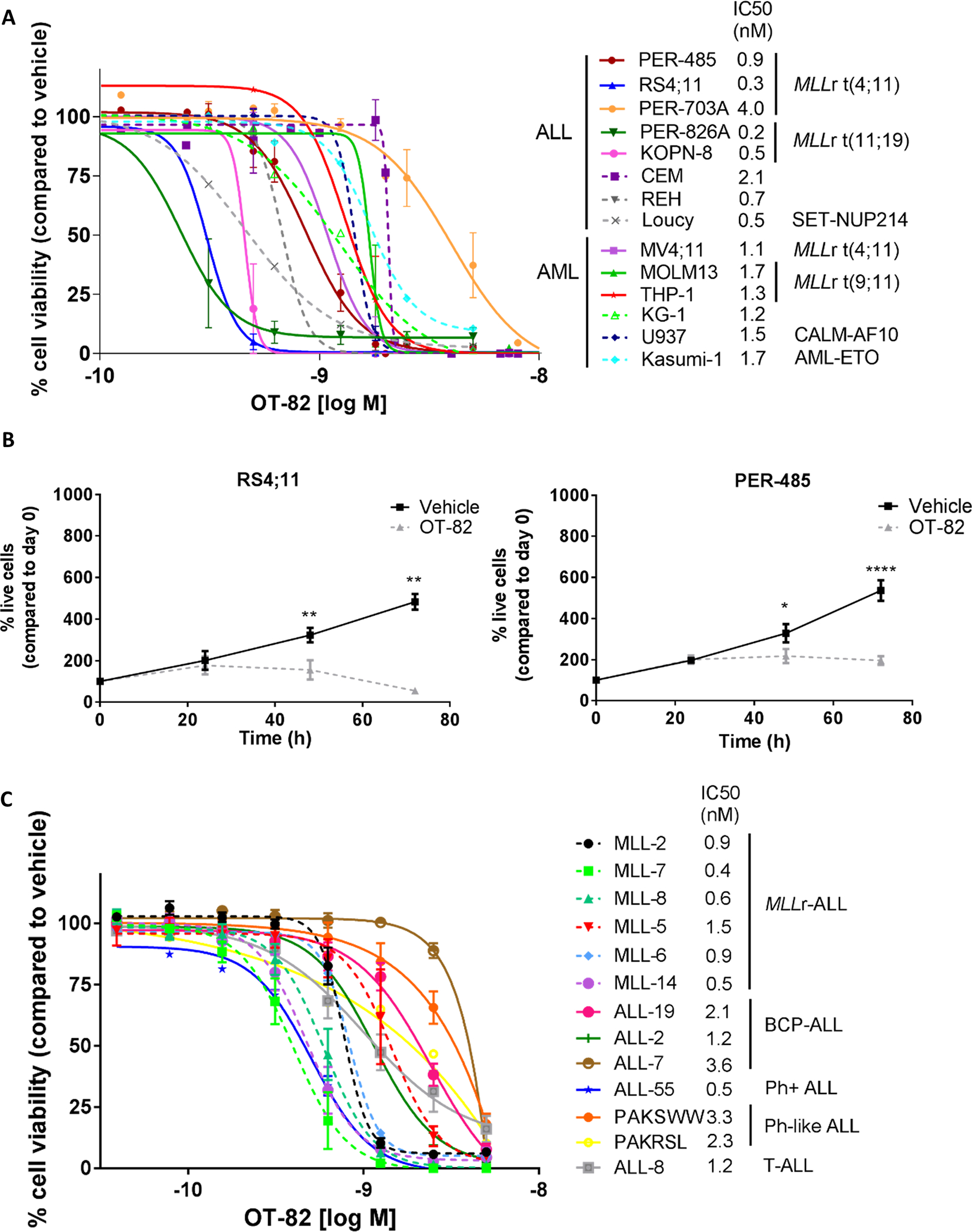
(A) Cytotoxicity of OT-82 against a panel of leukemia cell lines (n=14) as evaluated by resazurin reduction assays 72h after compound administration. Each data point represents the mean % viability (relative to vehicle-treated cells) ± SEM of at least 3 independent experiments. IC50 values were estimated by non-linear regression of transformed data. (B) Number of live RS4;11 and PER-485 cells after treatment with 1 nM OT-82 or vehicle compared to the number of living cells on the day of seeding as determined by trypan blue exclusion assay. Each data point represents the mean ± SEM of at least 3 independent experiments. Mean % of live cells after OT-82 and vehicle treatment for each time point were compared by paired t-test. (C) Cytotoxicity of OT-82 against a panel of PDX cells derived from pediatric patients with high-risk or poor outcome ALL as evaluated by resazurin reduction assays 72h after compound administration. Each data point represents the mean % viability (relative to vehicle-treated cells) ± SEM of at least 2 independent experiments, except for PAKRSL (n=1). IC50 values were estimated by point-by-point regression of transformed data. **, P<0.01; ****, P<0.0001.
OT-82 exerts its anti-leukemic action by reducing cellular NAD+ and inducing apoptosis
To confirm that OT-82 reduced the viability of leukemia cells by inhibition of NAMPT, we firstly assessed the effects of OT-82 on intracellular levels of NAD+ and ATP. OT-82 rapidly induced a complete depletion of NAD+ in the highly sensitive RS4;11 cells (IC50 = 0.3 nM) within 24h, which was followed by a near complete reduction in intracellular ATP levels within 48h of treatment (Figure 2A). Similar reductions in NAD+ and ATP levels were observed in the sensitive PER-485 cells (IC50 = 0.9 nM) when exposed to the same concentration of OT-82 (1 nM), albeit the decreases were slightly delayed and the achieved ATP depletion up to 72h was less pronounced compared to that observed in the RS4;11 cells (Figure 2A). However, in line with our previous demonstration that OT-82 induces concentration and time-dependent decreases in NAD+ and ATP levels (Korotchkina L et al.), higher OT-82 concentrations and longer exposure times caused near complete ATP depletion in PER-485 cells (Supplementary Figure 1A). Furthermore, for the less sensitive CEM and PER-703A cells (2.1 and 4.0 nM IC50, respectively), the NAD+ depletion induced by 1 nM OT-82 was further delayed compared to the RS4;11 and PER-485 cells and remained incomplete up to 72h (Supplementary Figure 1B). No significant downstream decreases in ATP levels were observed in these cell lines indicating that a near complete depletion of NAD+ is needed before ATP levels start to decrease (Supplementary Figure 1B) which is in line with previous studies on other NAMPT inhibitors.32–34 Thus, the in vitro sensitivity of the leukemia cells to OT-82 in viability assays aligned with the extent of induced NAD+ and ATP decreases and the rapidity of these effects.
Figure 2: OT-82 decreases intracellular NAD+ levels culminating in apoptosis induction in leukemia cells.
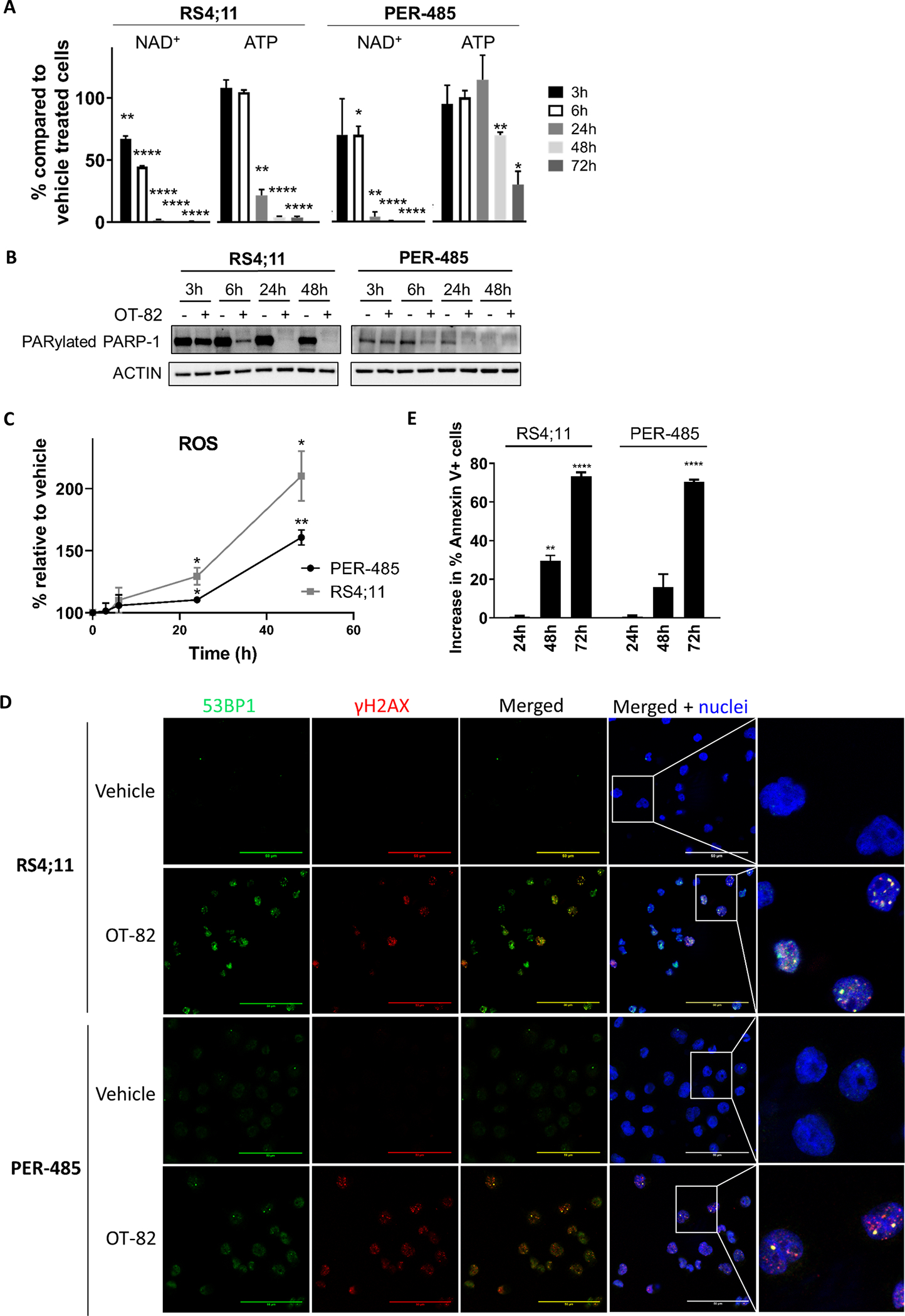
(A) % NAD+ and ATP in cells treated with 1 nM OT-82 compared to vehicle-treated cells (equal number of cells used). Bar graphs depict the mean ± SEM of at least 3 independent experiments. The significance of the decreases in % NAD+ and ATP in response to OT-82 treatment for each time point was evaluated by one sample t-test. (B) Immunoblotting of lysates from leukemia cells treated with 1 nM OT-82 up to 48h representative of two independent experiments. (C) Relative increase in mitochondrial ROS levels in cells treated with 1 nM OT-82 compared to cells treated with vehicle. The significance of the increase in ROS levels relative to vehicle-treated cells was assessed for each time point by one sample t-test. (D) Immunofluorescent staining for γH2AX (red) and 53BP1 (green) of RS4;11 and PER-485 cells treated with vehicle or OT-82 (0.5 nM and 1 nM respectively) for 24h. Pictures are representative for results obtained in 2 independent experiments. Scale bars represent 50 μM (E) Increase in % of Annexin V-positive cells after treatment with 1 nM OT-82 compared to vehicle-treated cells. Mean ± SEM of at least 3 independent experiments is shown. The mean percentages of Annexin V-positive cells after OT-82 treatment were compared to the percentage of Annexin V-positive cells in vehicle-treated cells by t-tests. *, P<0.05; **, P<0.01; ****, P<0.0001.
In addition to ATP and energy depletion, OT-82 impacted several other pathways downstream of NAD+ depletion. OT-82 inhibited the activity of the NAD+-dependent DNA damage repair enzyme PARP-1 in treated leukemia cell lines, evidenced by a decrease in PARylated PARP-1 levels within a few hours of NAD+ reduction (Figure 2B).
Given the importance of NAD+ in mitochondrial respiration and ROS level regulation, NAMPT inhibitors have also been reported to increase levels of intracellular ROS.35–37 In agreement with other NAMPT inhibitors, OT-82 induced small but significant increases in mitochondrial ROS in leukemia cells within 24h of treatment (Figure 2C).
As increased ROS levels can induce DNA damage, while an inhibition of PARP-1 will limit DNA repair, we subsequently investigated whether OT-82 induced DNA damage in treated cells. Indeed, OT-82 induced an increase in γH2AX and 53BP1 double positive foci in leukemia cells from different lineages within 24h of treatment (Figure 2D, Supplementary Figure 1C), indicating a rapid accumulation of DNA damage.
All these cellular effects were followed by a significant increase in the percentage of Annexin V-positive cells within 48h of treatment (Figure 2E), indicating that OT-82 reduced leukemia cell viability by inducing apoptosis.
OT-82 induces significant leukemia regression in vivo in a large panel of ALL PDXs derived from high-risk and poor outcome pediatric patients
We previously reported that OT-82 was highly efficacious against two PDXs established from high-risk pediatric ALL patients (Korotchkina L et al.). To thoroughly investigate the in vivo efficacy of the compound against pediatric high-risk ALL and to determine how broadly applicable OT-82 might be to different high-risk/poor outcome ALL subtypes, we analyzed 19 additional, molecularly characterized and validated PDXs derived from patients with either infant MLLr-ALL (n=5), B-cell precursor (BCP)-ALL (n=8) or T-ALL (n=6) (Supplementary Table 2). The PDXs accurately recapitulate the cellular and molecular features of the original disease, with their in vivo responses to commonly used chemotherapeutic drugs correlating significantly with the clinical outcome of donor patients.21, 31, 38–40 Of the BCP-ALL PDXs, two were derived from patients with Ph+ ALL, three from children with Ph-like ALL, while 3/6 T-ALL PDXs were established from patients with early T-cell precursor (ETP) ALL, all classified as high-risk ALL.4, 21, 31, 41, 42 The remaining BCP-ALL and non-ETP T-ALL PDXs were derived from patients who died from their disease.21, 31
Oral treatment of engrafted mice with a previously optimized administration scheme of OT-82(Korotchkina L. et al.) was well tolerated as indicated by low percentages of weight loss (Supplementary Table 3). When combining the efficacy data of OT-82 on these 19 additional PDXs with the previously reported data on two PDXs, OT-82 significantly extended the survival of all but one PDX (20/21, 95%). The extension of event-free survival (EFS) by OT-82 treatment, T-C (EFS OT-82-treated (T) - EFS control-treated cohort (C)), ranged from 10.9 to 73.6 days and T/C values varied from 2.4 to 10.7 (Figure 3A–C, Table 1, Supplementary Table 3). When analyzed using stringent objective response criteria that are modeled after the clinical setting,20 OT-82 treatment elicited objective responses in 18/21 (86%) PDXs with 8 PDXs achieving Maintained Complete Responses (MCRs), 7 Complete Responses (CRs) and 3 Partial Responses (PRs) (Table 1, Figure 3 A–C, Supplementary Figure 2A). Consistent with our in vitro findings, there was a trend for the MLLr-ALL PDXs to be the most responsive to OT-82 treatment, however this was not statistically significant (Table 1; Supplementary Figure 2B–C). OT-82 was effective against all tested ALL PDX subgroups indicating the broad applicability of the compound in high-risk ALL. Responsiveness of the PDXs to OT-82 in vivo was significantly correlated with in vitro sensitivity (Supplementary Figure 3).
Figure 3: OT-82 induces regressions in vivo in a broad panel of pediatric ALL PDX models.
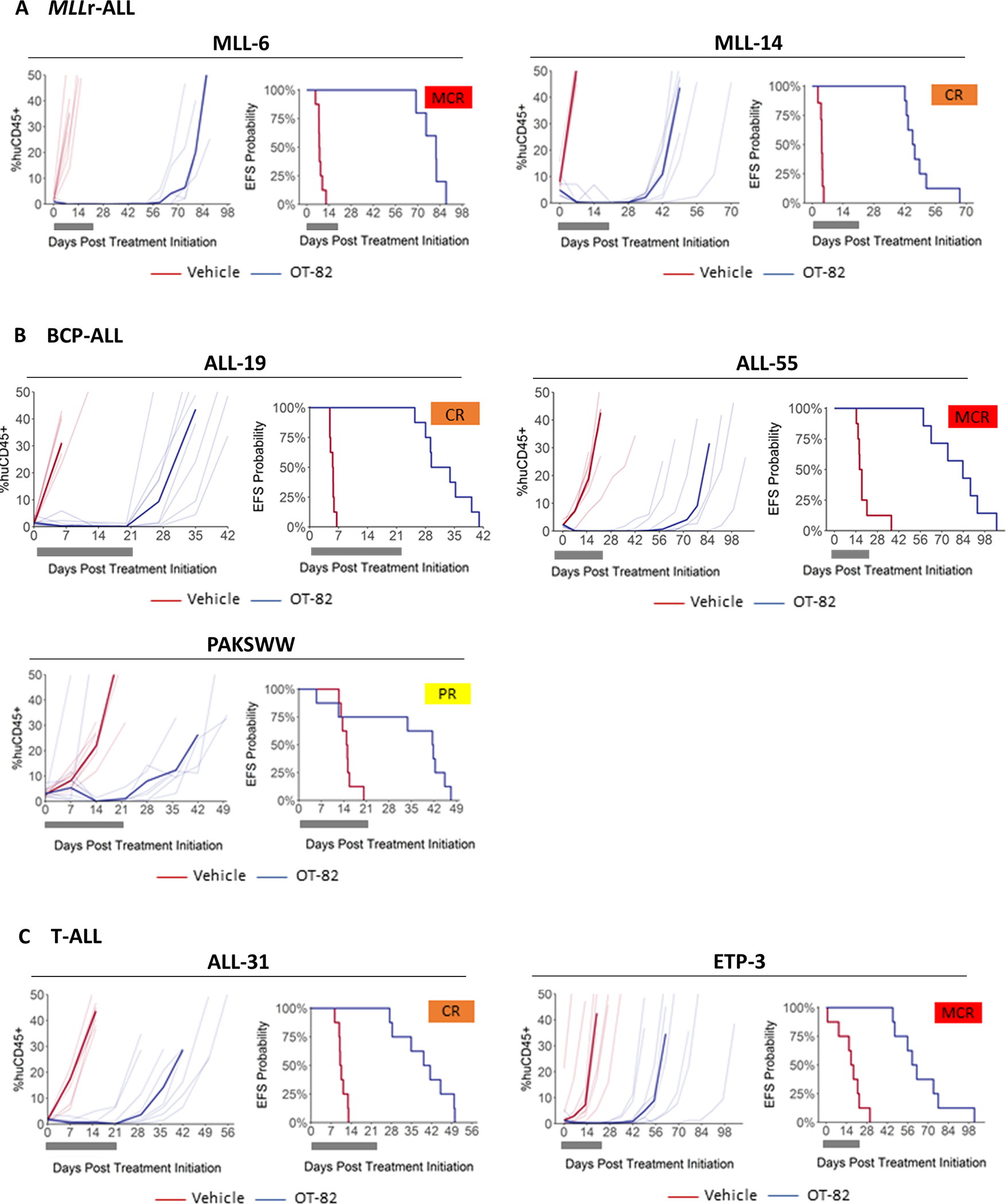
Responses of representative (A) MLLr-ALL, (B) BCP-ALL and (C) T-ALL PDXs treated with OT-82 (40 mg/kg p.o., 3 days on/4 days off, 3 weeks) (blue lines) or vehicle (red lines). For each PDX the left panels represent the % huCD45+ cells of individual mice (thin lines) and group median % huCD45+ (thick lines) over time. The right panels show the proportion of mice that remain event-free in a Kaplan-Meier plot. Gray blocks represent the treatment period (21 days). MCR, Maintained Complete Response; CR, Complete Response; PR, Partial Response.
Table 1:
In vivo responses of PDXs of high-risk pediatric ALL to OT-82
| EFS (days) |
Objective Response Measure (ORM)4 |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| ALL lineage | PDX ID | Vehicle control | OT-82 | T-C (Days)1 | T/C2 | P-value3 | Median ORM | ORM Heatmap | |
|
| |||||||||
| MLLr-ALL | MLL-25,6 | 10.8 | 68.6 | 57.8 | 6.4 | <0.001 | 8 | CR | |
| MLL-5 | 4.9 | 23.9 | 19.0 | 4.9 | <0.001 | 2 | PD2 | ||
| MLL-6 | 8.0 | 81.6 | 73.6 | 10.2 | 0.003 | 10 | MCR | ||
| MLL-7 | 7.4 | 62.8 | 55.5 | 8.5 | <0.001 | 10 | MCR | ||
| MLL-8 | 9.9 | 58.3 | 48.4 | 5.9 | <0.001 | 10 | MCR | ||
| MLL-14 | 4.3 | 46.1 | 41.8 | 10.7 | <0.001 | 8 | CR | ||
|
| |||||||||
| BCP-ALL | ALL-2 | 11.9 | 76.4 | 64.5 | 6.4 | <0.001 | 10 | MCR | |
| ALL-7 | 6.4 | 27.5 | 21.1 | 4.3 | <0.001 | 6 | PR | ||
| ALL-19 | 5.6 | 31.7 | 26 | 5.7 | <0.001 | 8 | CR | ||
|
| |||||||||
| Ph+ ALL | ALL-45,7 | 5.5 | 45.9 | 40.4 | 8.3 | 0.003 | 10 | MCR | |
| ALL-55 | 17.1 | 83.7 | 66.6 | 4.9 | <0.001 | 10 | MCR | ||
| ALL-56 | 5.5 | 34 | 28.5 | 6.2 | <0.001 | 8 | CR | ||
|
| |||||||||
| Ph-like ALL | PAKRSL | 5.6 | 24.9 | 19.3 | 4.4 | <0.001 | 6 | PR | |
| PAKSWW | 15 | 41.6 | 26.6 | 2.8 | 0.081 | 6 | PR | ||
| PAKYEP | 10.9 | 55.1 | 44.2 | 5.1 | <0.001 | 10 | MCR | ||
|
| |||||||||
| T-ALL | ALL-8 | 7.6 | 18.5 | 10.9 | 2.4 | 0.007 | 2 | PD2 | |
| ALL-31 | 10.3 | 40.3 | 30.0 | 3.9 | <0.001 | 8 | CR | ||
| ALL-39 | 5.5 | 34 | 28.5 | 6.2 | <0.001 | 8 | CR | ||
|
| |||||||||
| ETP | ETP-3 | 17.9 | 60.7 | 42.7 | 3.4 | <0.001 | 10 | MCR | |
| ETP-4 | 6.5 | 60.3 | 53.8 | 9.3 | <0.001 | 10 | MCR | ||
| ETP-6 | 12.8 | 31.1 | 18.3 | 2.4 | <0.001 | 2 | PD2 | ||
T-C = median EFS (OT-82) - median EFS (Vehicle)
T/C = median EFS (OT-82) / median EFS (Vehicle)
P-value as determined by Gehan-Wilcoxon survival analysis
Objective Response Measure (ORM): MCR, Maintained Complete Response; CR, Complete Response; PR, Partial Response; PD2, Progressive Disease 2
Data previously reported (Korotchkina L. et al.)
OT-82 administered orally at 40 mg/kg for 6 weeks, 3 days/week
Non-leukemia related toxicity in 3/8 mice treated with OT-82
OT-82 has similar efficacy against ALL PDXs as an induction-type chemotherapeutic protocol used in the treatment of high-risk pediatric ALL
To further evaluate the significance of the observed anti-leukemic potential of OT-82, we compared the responsiveness of the PDXs to OT-82, with their previously established sensitivity to an induction-type regimen used to treat pediatric ALL.43, 44 Treatment with OT-82 as a single agent for 3 weeks was almost as effective as a 4-week combination protocol of three chemotherapeutics: vincristine, dexamethasone and L-asparaginase (VXL) (Supplementary Figure 4A).43 There was no correlation between in vivo responsiveness of the PDXs to OT-82 and their sensitivity to VXL with ALL PDXs resistant to VXL being sensitive to OT-82 (Supplementary Figure 4B).
OT-82 reduces NAD+ and ATP levels and induces apoptosis in ALL PDXs in vivo
To confirm the mechanism of action of OT-82 in vivo, the levels of NAD+, ATP and the activity of PARP-1 were measured in purified spleen mononuclear cells (containing >95% of huCD45+ cells, Supplementary Table 4) isolated from a representative subset of PDXs treated with OT-82 or vehicle for 3 days. Consistent with the effects observed in vitro, a 3-day treatment with OT-82 significantly reduced mean NAD+ and ATP levels (Figure 4A, Supplementary Figure 5). The magnitude of the OT-82 induced ATP reduction correlated with the observed NAD+ decrease measured in the PDXs at this time point (Supplementary Figure 5E), indicating that the extent of the NAD+ decrease dictates the magnitude of ATP reduction, in line with our observations in vitro. In contrast to our in vitro findings however, post treatment NAD+ and ATP levels in individual PDXs at this time point did not correlate with their sensitivity to OT-82 treatment (Supplementary Figure 5). Downstream of NAD+ depletion, in vivo PARP-1 inhibition was observed, but only convincingly in splenocytes from representative PDXs achieving PR, CR or MCR, while not in the Non-Responders (PD2) (Figure 4B).
Figure 4: OT-82 reduces NAD+, ATP levels and serum visfatin levels and inhibits PARP-1 in ALL PDXs in vivo.
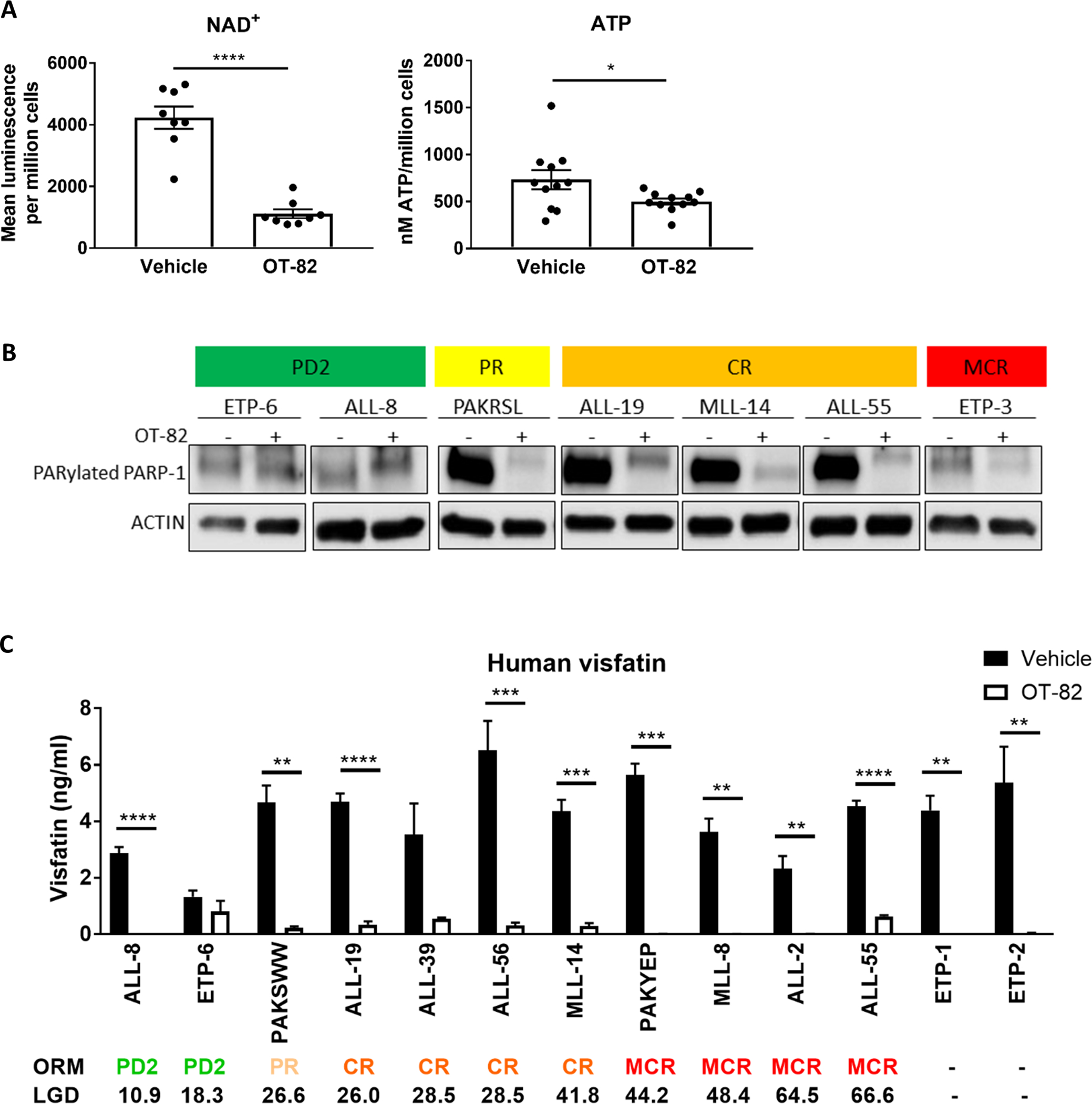
(A) Each dot point represents the mean NAD+ luminescence per million spleen mononuclear cells for one PDX (n=8 PDX, n=2 mice for each PDX) or the mean ATP concentration per million cells for one PDX (n=8, minimum 2 mice for each PDX). Mean NAD+ luminescence/million cells values or mean ATP levels/million cells between treatment groups were compared by Welch’s t-test. (B) Immunoblotting of spleen mononuclear cells from PDXs treated with OT-82 (+) or vehicle (−) for 3 consecutive days. (C) Mean serum levels of visfatin as measured in PDX mice treated with OT-82 or vehicle on 3 consecutive days for minimum 2 mice per condition. Mean serum visfatin levels between OT-82 and vehicle-treated mice were compared by t-tests. Spleen mononuclear cells and serum were harvested 4h after final administration of a 3-day treatment course with daily injections of vehicle or 40 mg/kg OT-82. *, P<0.05; **, P<0.01; ***, P<0.001; ****, P<0.0001.
Visfatin as a pharmacodynamic marker for OT-82 reaching its target in vivo
NAMPT exists in an intracellular (iNAMPT) and extracellular (eNAMPT) form, also known as visfatin. It is believed that visfatin is a secreted form of iNAMPT, and that both proteins share a common pool within the cell.45 Although the exact mechanisms by which visfatin is secreted by different cell types are yet to be fully elucidated, studies in adipocytes show that deacetylation of iNAMPT by the NAD+-dependent SIRT1 promotes the secretion of the protein.46 Visfatin and iNAMPT levels seem to be directly correlated and in theory, visfatin secretion reduces the iNAMPT pool.45, 47–49 As OT-82 inhibits iNAMPT functioning, thereby creating a significant metabolic strain, and reduces intracellular NAD+ levels needed by SIRT1 for the deacetylation of iNAMPT, we hypothesized that OT-82 treatment might reduce visfatin secretion by leukemia cells resulting in lower circulating serum levels of the protein.
To assess whether the serum levels of visfatin changed in response to OT-82 treatment and whether this could be used as a pharmacodynamic marker for OT-82 reaching its target in vivo, we measured human and mouse visfatin levels in serum samples from representative PDXs treated with either OT-82 or vehicle for 3 consecutive days. While baseline or changes in serum visfatin levels after this short-term treatment did not predict treatment response to OT-82 (Supplementary Figure 6 A–C), OT-82 treatment reduced human serum visfatin levels in all tested PDXs, although this failed to reach significance in 2/13 PDXs (Figure 4C). In addition, OT-82 significantly decreased mouse visfatin levels (Supplementary Figure 6D) providing indirect evidence that OT-82 inhibits mouse iNAMPT at applied effective doses. The change in serum visfatin levels may thus be useful as a non-invasive pharmacodynamic marker for OT-82 reaching its target in vivo.
OT-82 enhances the activity of established drugs used in the treatment of pediatric high-risk ALL
NAMPT inhibitors have been shown to synergize with DNA damage-inducing chemotherapeutics based on their inhibition of DNA damage repair by PARPs.33, 36, 50, 51 To further explore the clinical potential of OT-82, we tested the ability of OT-82 to enhance the cytotoxicity of established chemotherapeutic treatments used for high-risk pediatric ALL. In the highly chemotherapy-resistant MLLr-ALL PER-485 cell line52, OT-82 synergized in vitro with chemotherapeutics cytarabine (AraC, Figure 5A) and etoposide (VP16, Supplementary Figure 7A) as determined by Bliss analysis. Additionally, the combination of AraC and OT-82 enhanced accumulation of DNA damage (γH2AX) compared to single agent treatment (Figure 5A). When the combination of cytarabine and OT-82 was subsequently tested in vivo against two aggressive MLLr-ALL PDX models, therapeutic enhancement, defined as significantly greater activity for the combination than for either single agent, was observed in both PDXs (Figure 5B, Supplementary Figure 7B, Supplementary Table 5 and 6).
Figure 5: OT-82 potentiates currently used therapies for pediatric high-risk ALL.
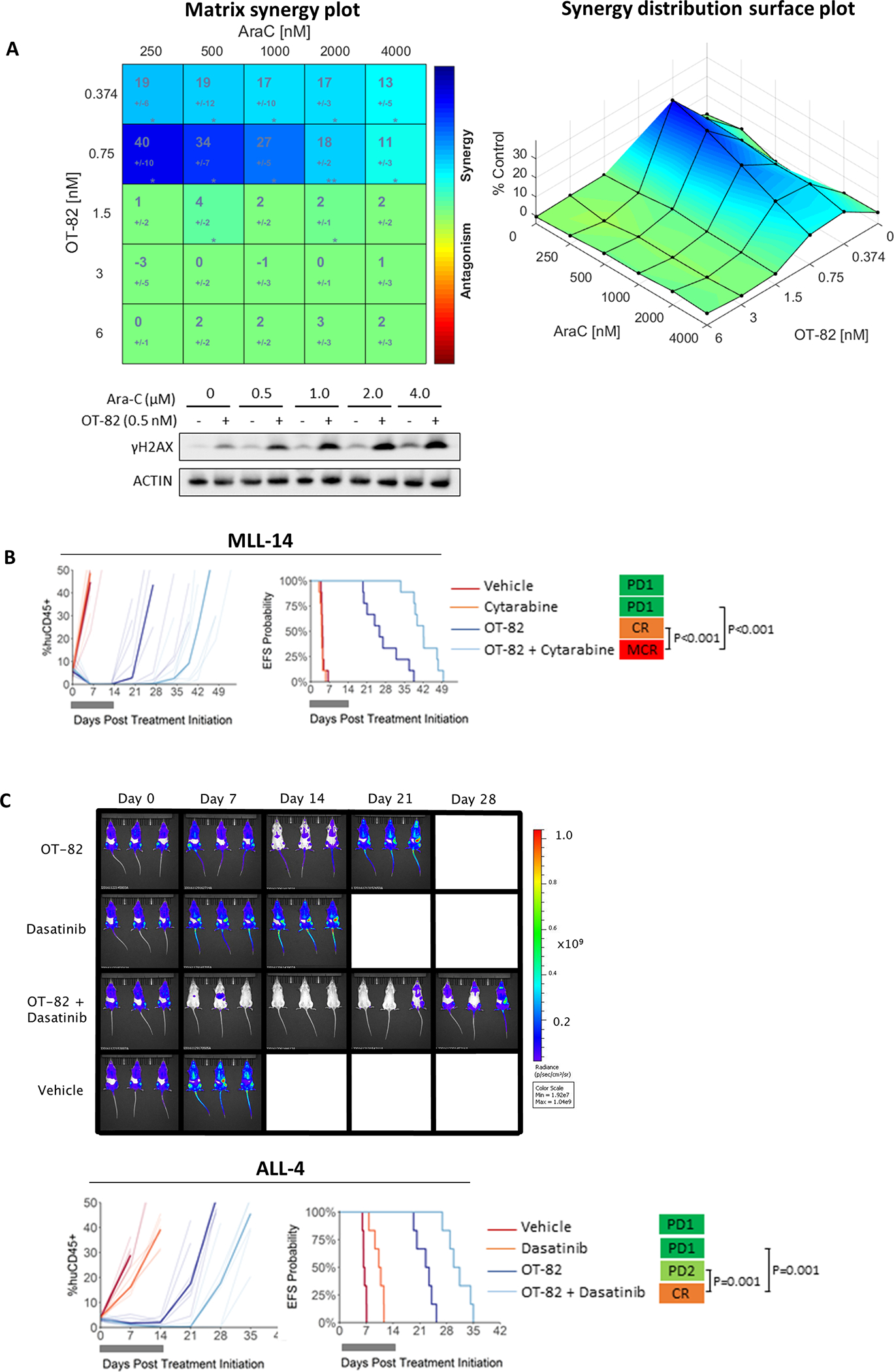
(A) PER-485 cells were treated with increasing doses of OT-82 combined with AraC in a 6×6 matrix format, at a fixed ratio with 2-fold incremental increases in drug concentrations. Cell viability was measured by resazurin reduction assays at 72 h and synergy was scored by Bliss and visualized by Combenefit. The matrix synergy plot (left panel) displays the synergy score for each combination +/− standard deviation as well as statistical significance of synergy for each combination (*, P<0.05; **, P<0.01) in 4 independent experiments. The right plot displays the synergy distribution according to Bliss. Representative immunoblotting on lysates from the PER-485 cell line treated with a sublethal dose of OT-82 or vehicle and a dose range of Ara-C for 48h (2 independent experiments except for 0.5 μM AraC: n=1). (B) Response of MLL-14 PDX treated with OT-82 (dark blue lines, 40 mg/kg p.o., 3 days on/4 days off, 2 weeks), Cytarabine (orange lines, 25 mg/kg i.p., 5 days/week, 2 weeks), OT-82+Cytarabine (light blue lines, same protocol as single agents) or vehicle control (red lines). (C) Response of ALL-4 PDX treated with OT-82 (dark blue lines) (40 mg/kg p.o., 3 days on/4 days off, 2 weeks), dasatinib (orange lines, 15 mg/kg, 5 days/week, 2 weeks), OT-82+dasatinib (light blue lines) or vehicle control (red lines). In (B-C) the left panels represent the % huCD45+ cells of individual mice over time (thin lines represent individual mice; bold lines represent group median). The right panels show the proportion of mice that remain event-free in a Kaplan-Meier plot. Gray bars represent the treatment period. Bioluminescent images of three randomly selected mice for each cohort are shown at weekly intervals. Images from the three additional mice are shown in Supplementary Figure 7C. MCR, Maintained Complete Response; CR, Complete Response; PR, Partial Response; PD1, Progressive Disease 1.
Since NAMPT inhibitors have previously been reported to potentiate the effects of certain targeted therapies, we examined whether OT-82 could enhance the effects of the tyrosine kinase inhibitor, dasatinib, which is used in the treatment of Ph+ ALL.53–57 When the combination of OT-82 and dasatinib was tested against a Ph+ ALL PDX (ALL-4), decreased disease burden and therapeutic enhancement were observed (Figure 5C, Supplementary Table 5 and 6, Supplementary Figure 7C).22
Mutations in DNA damage response genes are associated with OT-82 susceptibility while high CD38 expression accompanies OT-82 resistance in vivo
Despite the high efficacy of OT-82 against most tested ALL PDXs, the response to OT-82 is variable with a small number of PDXs being unresponsive to the compound. To identify determinants of response to OT-82, we analyzed the baseline transcriptome of the panel of PDXs (RNA-sequencing data) in relation to in vivo OT-82 sensitivity. Differential expression analysis between PDXs that failed to achieve a significant leukemia growth delay or objective response (Non-Responders: PAKSWW, MLL-5, ETP-6, ALL-8) and Responder PDXs, revealed no genes with significant differential expression (FDR<0.05). In addition, previously reported determinants of response to NAMPT inhibition, namely the most important NAD+-producing enzymes NAMPT and NAPRT1, and the main NAD+-consuming enzymes CD38 and PARP-1, did not show up in the list of differentially expressed genes with a fold change in expression>2 and (unadjusted) P-value<0.05 (Supplementary Table 7).14, 15, 32, 34, 36, 57–61
As the unbiased gene expression analysis did not yield strong candidate markers of response, we next investigated whether the protein expression and enzyme activity of the major NAD+-producing and NAD+-consuming enzymes might predict responsiveness to OT-82 in high-risk pediatric ALL. We therefore firstly assessed whether the protein expression of NAMPT, NAPRT1, CD38 and the enzyme activity of PARP-1 in leukemia cell lines correlated with the variable sensitivity of these cell lines to OT-82 treatment in vitro (20-fold difference in IC50 for OT-82). While OT-82 IC50 did not correlate with baseline protein expression levels or activity of NAMPT, NAPRT1 or PARP-1 (Supplementary Figure 8), baseline expression of the NAD+-consuming enzyme CD38 significantly correlated with sensitivity to OT-82 in vitro (Figure 6A, Supplementary Figure 8).
Figure 6: Determinants of response to OT-82.
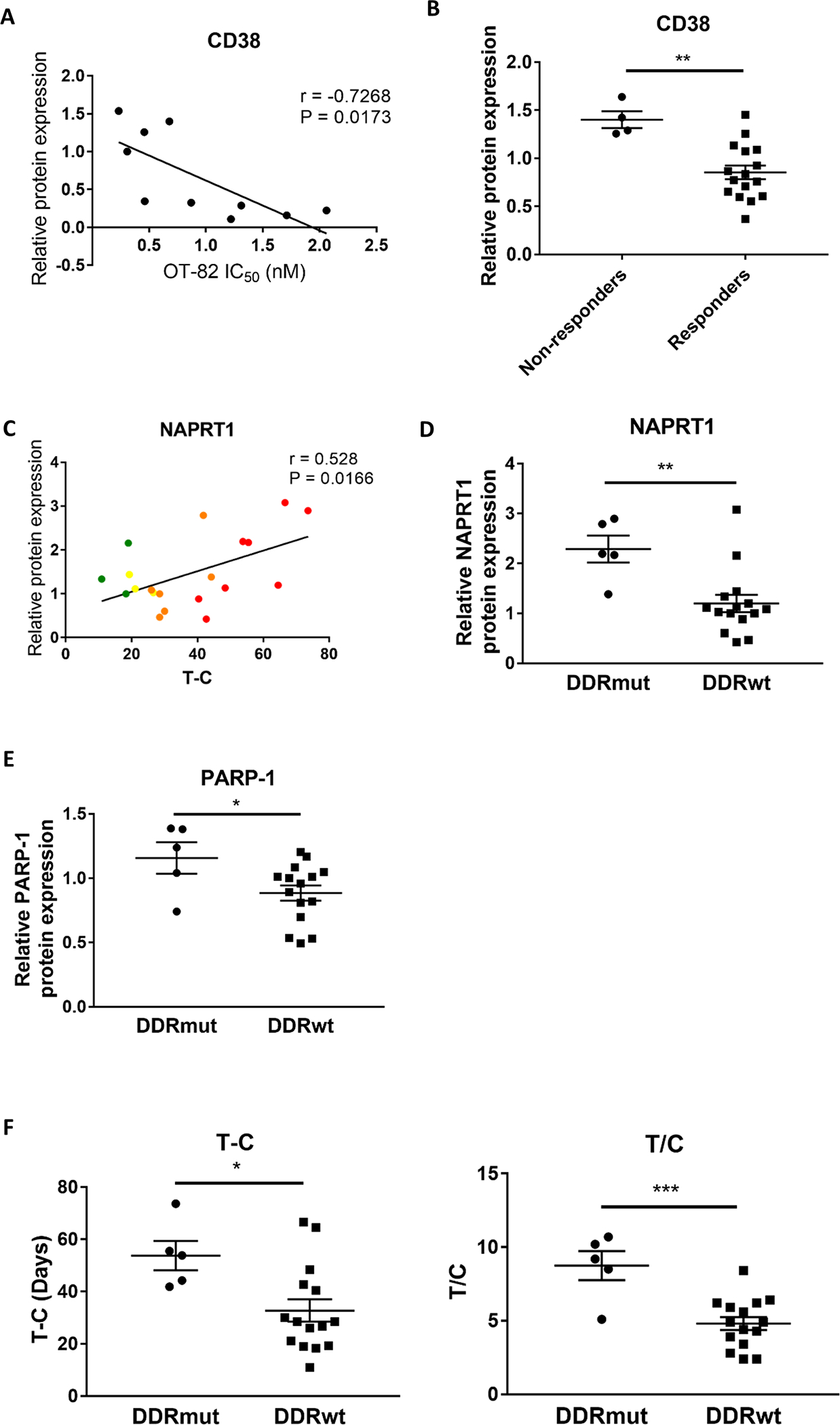
(A) Correlation analysis between mean OT-82 IC50 as determined in viability assays and relative baseline protein expression of CD38 in a panel of leukemia cell lines (as determined by densitometry of immunoblotting experiments described in Supplementary Figure 8) (Pearson). (B) Comparison of relative baseline expression levels of CD38 in the panel of PDXs (as determined by densitometry of immunoblotting experiments described in Supplementary Figure 9), stratified based on in vivo responsiveness to OT-82 (Non-responders: MLL-5, ALL-8, ETP-6, TGT-052) by t-test. (C) Correlation analysis between OT-82 T-C and relative baseline protein expression of NAPRT1 in the panel of PDXs (as determined by densitometry of immunoblotting experiments described in Supplementary Figure 9) (Pearson). (D-E) Comparison of relative baseline expression levels of NAPRT1 (D) and PARP-1 (E) in the DDRmut and DDRwt PDX groups (as determined by densitometry of immunoblotting experiments described in Supplementary Figure 9) based on t-tests. (F) Comparison of mean OT-82 T-C and T/C of the DDRmut and DDRwt PDX groups by t-test. *, P<0.05; **, P<0.01; ***, P<0.001.
We subsequently evaluated whether the expression and activity of these enzymes correlated with in vivo sensitivity of the panel of ALL PDXs to OT-82. Surprisingly, PDXs that failed to achieve a significant leukemia growth delay or objective response (Non-Responders: PAKSWW, MLL-5, ETP-6, ALL-8) expressed significantly higher levels of CD38 than the Responder PDXs (Figure 6B, Supplementary Figure 9A), indicating that high CD38 levels were associated with resistance to OT-82, the opposite of our findings in vitro.
While no associations were observed in the ALL PDX panel between OT-82 T-C or T/C and baseline protein levels or activity of NAMPT and PARP-1 (Supplementary Figure 9A), higher baseline NAPRT1 protein expression was associated with greater sensitivity to OT-82 (Figure 6C). This result was unexpected as previous studies indicate that low NAPRT1 expression in cancer cells is associated with increased sensitivity to NAMPT inhibition due to a higher dependency of the cellular NAD+ reserve on NAMPT.59, 62–64 A recent study by Piacente et al. reported that ovarian cancer cells with defective homologous recombination DNA repair (or BRCAness), may use NAPRT1-mediated NAD+-production to provide energy for DNA repair, including PARP activity.64, 65 Therefore, we investigated whether the observed high NAPRT1 expression in our responsive ALL PDXs could be a marker of increased addiction of these PDXs to NAD+ due to mutations in the major DNA damage response (DDR) genes BRCA1/2, CHEK1/2, ATM and ATR.66 Five (4 MCR and 1 CR) out of 21 PDXs exhibited mutations in one of the major DDR genes (DDRmut), all of which were predicted to be pathogenic or likely pathogenic (Supplementary Table 2). In line with the hypothesis, the DDRmut PDXs expressed significantly higher levels of baseline NAPRT1 compared to the remaining DDRwt PDXs (Figure 6D), as well as higher levels of PARP-1 (Figure 6E). Moreover, a trend for a positive correlation was observed between baseline expression levels of NAPRT1 and PARP-1 in the PDX panel (Supplementary Figure 9B). Interestingly, the DDRmut PDXs had significantly longer leukemia growth delay values (Figure 6F) and a trend for a higher proportion of MCRs (Supplementary Figure 10) in response to OT-82 treatment compared to the DDRwt PDXs. This indicates that ALL PDXs with mutations in major DNA damage response genes are significantly more responsive to OT-82 treatment in vivo.
DISCUSSION
In this study we have shown that the novel NAMPT inhibitor OT-82 was well-tolerated and exhibited broad in vivo efficacy against a diverse panel of pediatric ALL PDXs, inducing significant disease regression in 86% of the PDXs tested. OT-82 was effective against PDXs derived from patients with aggressive and fatal disease and exerted synergistic in vivo efficacy with established drugs. Taken together, our findings indicate that OT-82 has potential as a novel therapeutic drug for the treatment of high-risk childhood ALL.
Our investigations into the cellular and molecular events downstream of OT-82 treatment indicated that the extent and rapidity of induced NAD+ and ATP decreases aligned with the in vitro sensitivity of the leukaemia cells to OT-82. However, the extent of NAD+ and ATP decreases induced by a 3-day OT-82 treatment of PDXs in vivo did not correlate with response to OT-82. There are several potential explanations for these findings. The NAD+ and ATP measurements were performed in splenocytes 4h after a 3-day daily treatment course with OT-82 and it is therefore possible that the reductions in NAD+ and subsequently ATP levels at a later time point are more significantly associated with treatment outcome. In addition, the in vivo NAD+ metabolism of an organism is more complex than that of a cell line in vitro and NAD+ and ATP levels in an in vivo setting are subject to circadian rhythms and can be influenced by food uptake and activity of the mouse.67 Measured NAD+ and ATP levels in isolated splenocytes could also be affected by the isolation process (handling time, stress on the cells) as well as storage of these cells prior to analysis. Moreover, it is conceivable that it is not only the extent of NAD+ and/or ATP reduction by itself that determines effectiveness of OT-82 in vivo, but the ‘addiction’ of the PDX to pathways downstream of NAD+ and ATP and/or the potential presence of intrinsic compensatory processes within the cell to allow the rescue of NAD+/ATP decreases. This is in line with a previous report on NAMPT inhibitor GNE-617, in which it was demonstrated that even though NAD+ was reduced in all tested non-small cell lung carcinoma cell lines upon treatment, there was a heterogeneous metabolic response to NAD+ depletion influenced by the underlying molecular and genetic framework of the cells.35 This potential explanation also finds further support in our observations of the absence of in vivo PARP inhibition in treated Non-Responder PDXs only, which warrants further investigation of PARP inhibition as a marker for therapeutic response to OT-82. In addition, our observed associations between baseline expression level of major NAD+-consumer CD38 and responsiveness to OT-82 provide further support that dependence of the cancer cell upon NAD+-dependent cellular processes determines responsiveness to OT-82 treatment.
With the goal of findings markers that can predict responsiveness to OT-82 and thereby identify patients most likely to benefit from treatment before treatment initiation, we sought insight into baseline determinants of response to this small molecule drug. Apparently conflicting data were generated regarding the contribution of CD38 expression in determining responsiveness to OT-82, with a positive correlation noted between CD38 expression in leukemia cell lines and in vitro OT-82 sensitivity, while Non-Responder PDXs in vivo presented with higher expression levels of CD38 compared to Responders. As in vitro sensitivity of our ALL PDXs to OT-82 correlated with in vivo OT-82 sensitivity of the PDXs, this discrepancy is likely not the consequence of differences in in vivo and in vitro regulation of NAD+ metabolism. Our findings are interesting in light of the already existing controversy within the field regarding CD38 expression as a determinant of response to NAMPT inhibition. In a study in pancreatic cancer cell lines, silencing of CD38 decreased in vitro susceptibility to the NAMPT inhibitor FK866.58 Similarly, Takao et al. reported that in a small panel of B-ALL cell lines, in vitro sensitivity to a dual PAK4/NAMPT inhibitor positively correlated with mRNA expression levels of CD38.15 The findings of both these in vitro cell line studies are consistent with our results in leukemia cell lines, suggesting that high NAD+ consumption by increased expression of CD38 in cancer cell lines results in a higher sensitivity to NAMPT inhibition. In contrast, a study in primary cells derived from chronic lymphocytic leukemia (CLL) patients demonstrated that patient samples positive for CD38 expression were more resistant to in vitro FK866 treatment, which is consistent with our findings for OT-82 in ALL PDXs in vivo.36 Since positive CD38 status in CLL is a prognostic factor for poor response to chemotherapy, the authors of this study hypothesized that high CD38-expressing CLL cells were representative for a more aggressive cancer type that is more difficult to treat and thus more resistant to NAMPT inhibition.36 From the apparently conflicting data on the value of CD38 expression as a predictor of response to NAMPT inhibition, it appears likely that CD38 may play different roles in the metabolism of long-term cultured cell lines versus short-term cultured PDXs or primary cells. This is conceivable as CD38 is a multifunctional enzyme that catalyzes the metabolism of two distinct Ca2+ messengers, cyclic ADP-ribose and nicotinic acid adenine dinucleotide, and therefore contributes to several cellular processes including calcium signaling, cell adhesion and signal transduction which might be differentially regulated in high-passage versus low-passage cancer cells.68, 69
In the interpretation of these data it is important to note that our investigations into OT-82 response markers that are based on in vitro responsiveness of cell lines, are limited by a narrow dynamic range in in vitro sensitivities (IC50 ranging from 0.3 to 2.0 nM). However, for the PDXs, a similarly narrow range in ex vivo sensitivity to OT-82 is noted (IC50 ranging from 0.4 to 3.6 nM). This difference in ex vivo sensitivity is however clearly translated into an in vivo difference in responsiveness to OT-82 as indicated by the observed correlation between ex vivo and in vivo sensitivity of the ALL PDXs to this compound. The PDXs with highest IC50 (ALL-7, PAKSWW, PAKRSL) were clearly less responsive in vivo than the PDXs with the lowest IC50 (MLL-7, ALL-55). Taken together this indicates that a narrow range in in vitro sensitivity may very well be translated into a clinically relevant difference in response to OT-82 and therefore the analysis of the differential presence of markers relative to in vitro OT-82 sensitivity is relevant and sheds more light onto the reported controversies on response markers for NAMPT inhibitors.
Another striking finding in our study was the observed positive correlation between baseline NAPRT1 expression and OT-82 sensitivity in vivo. This correlation is opposite to what was expected based on several studies linking decreased NAPRT1 expression with increased sensitivity of cancer cells to NAMPT inhibition.59, 62–64 In our ALL PDX panel, no NAPRT1 gene amplifications or IDH1/2 mutations were detected that could explain the observed variability in NAPRT1 protein expression levels as reported for other cancer types.59, 62–64 Our findings that ALL PDXs with mutations in the major DNA damage response genes had higher protein levels of NAPRT1 and PARP-1 and were more sensitive to OT-82 treatment, are consistent with the hypothesis that elevated protein expression of NAPRT1 is a metabolic adaptation of the cell to fuel a higher NAD+ demand due to mutations in DNA damage response genes. OT-82 thus appears to have a pronounced effect on ALL PDXs that have a higher dependency on NAD+-mediated DNA repair, reminiscent of the synthetic lethality of PARP inhibitors in BRCA1/2 mutated cancers.70 Our data thus suggest caution when assessing NAPRT1 expression level as a predictive marker for susceptibility to NAMPT inhibition, highlighting that a contiguous assessment of mutations in DNA damage response genes might be warranted in this context.59, 62–64
Collectively, our findings suggest that in high-risk pediatric ALL patients, high baseline CD38 protein expression in their leukemic cells might predict resistance to NAMPT inhibition by OT-82, while the presence of mutations in DDR genes might identify patients who are more likely to respond. In addition, our finding of decreased PARylated PARP-1 levels only in Responder PDXs following OT-82 treatment suggests that reduced PARP-1 activity in samples from patients being treated with OT-82 might predict treatment response. It should be noted that due to the high efficacy of OT-82, our conclusions regarding markers of susceptibility and therapeutic response to OT-82 are limited by the small number of Non-Responder PDXs, which may be promising for the future use of OT-82 in the clinic, where these conclusions will need to be validated. It should also be noted that not all PDXs achieving MCR or CR possess mutations in DNA damage response genes and that sensitivity to OT-82 is highly likely to be influenced by several other factors, which is in line with the diverse roles of NAD+ in a cell.71, 72 In addition, follow-up studies are needed to further investigate whether the observed associations between OT-82 sensitivity and expression levels of CD38 and NAPRT1 are based on functional links.
The previously reported disappointing results of NAMPT inhibitors in clinical trials have raised concerns about the presence of a clinically relevant therapeutic window for NAMPT inhibitors. In Korotchkina et al., we showed that bone marrow mononuclear cells isolated from leukemia patients were significantly more sensitive to OT-82 than those isolated from healthy controls, implying a therapeutic window for NAMPT inhibition by OT-82. In rigorous toxicological studies conducted in mice and non-human primates, OT-82 showed no cardiac or retinal toxicities observed with previously developed NAMPT inhibitors.13, 73, 74 In this study, OT-82 was very well tolerated across 21 ALL PDXs and 170 leukemia-engrafted mice treated with the compound for three weeks, with only 3/170 mice exhibiting toxic death. As OT-82 was shown to inhibit mouse NAMPT with similar potency as human NAMPT (Korotchkina L. et al.) and based on our findings that OT-82 also reduced serum mouse visfatin levels, our study thus provides additional evidence for the safety of the compound at effective doses.
As part of the Pediatric Preclinical Testing Consortium, the PDX models of high-risk childhood ALL used in this study have been employed extensively for preclinical testing and prioritization of novel drugs for childhood malignancies.20 In our hands, OT-82 has proven to be one of the more broadly active compounds tested so far in this PDX panel of high-risk pediatric ALL.75 OT-82 thus appears to be a promising anti-cancer drug for the treatment of a broad range of high-risk and aggressive pediatric ALL subtypes for which novel therapeutic options are urgently needed.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by grants from the National Cancer Institute (CA199222 and CA199000), The National Health and Medical Research Council of Australia (NHMRC Fellowships APP1059804 and APP1157871), Cancer Australia and Kids’ Cancer Project (Priority-driven Collaborative Cancer Research Scheme APP1164865), Anthony Rothe Memorial Trust, Cancer Council NSW (PG16-01), Tenix Foundation, ISG Foundation, the Children’s Leukemia & Cancer Research Foundation (Perth) and Australian Postgraduate Awards from the Australian Government Department of Education and Training.
Children’s Cancer Institute is affiliated with the UNSW Sydney and the Sydney Children’s Hospital Network. The authors would like to thank Raymond Yung and Lisa McDermott (CCI) for their help with experiments.
Footnotes
Competing Interests statement: A.V. Gudkov is a consultant of and O. Chernova, M. Gupta and L. Korotchkina are employed by Oncotartis, Inc. which developed and holds the IP on OT-82. This research was supported by grants from the National Cancer Institute (CA199222 and CA199000), The National Health and Medical Research Council of Australia (NHMRC Fellowships APP1059804 and APP1157871), Cancer Australia and Kids’ Cancer Project (Priority-driven Collaborative Cancer Research Scheme APP1164865), Anthony Rothe Memorial Trust, Cancer Council NSW (PG16-01), Tenix Foundation, ISG Foundation, the Children’s Leukemia & Cancer Research Foundation (Perth) and Australian Postgraduate Awards from the Australian Government Department of Education and Training.
Supplementary information is available at Leukemia’s website
REFERENCES
- 1.Hunger SP, Mullighan CG. Redefining ALL classification: toward detecting high-risk ALL and implementing precision medicine. Blood. 2015;125(26):3977–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pui CH, Carroll WL, Meshinchi S, Arceci RJ. Biology, risk stratification, and therapy of pediatric acute leukemias: an update. J Clin Oncol. 2011;29(5):551–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hunger SP, Lu X, Devidas M, Camitta BM, Gaynon PS, Winick NJ, et al. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the children’s oncology group. J Clin Oncol. 2012;30(14):1663–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arico M, Schrappe M, Hunger SP, Carroll WL, Conter V, Galimberti S, et al. Clinical outcome of children with newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia treated between 1995 and 2005. J Clin Oncol. 2010;28(31):4755–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pui CH, Chessells JM, Camitta B, Baruchel A, Biondi A, Boyett JM, et al. Clinical heterogeneity in childhood acute lymphoblastic leukemia with 11q23 rearrangements. Leukemia. 2003;17(4):700–6. [DOI] [PubMed] [Google Scholar]
- 6.Ko RH, Ji L, Barnette P, Bostrom B, Hutchinson R, Raetz E, et al. Outcome of patients treated for relapsed or refractory acute lymphoblastic leukemia: a Therapeutic Advances in Childhood Leukemia Consortium study. J Clin Oncol. 2010;28(4):648–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith MA, Seibel NL, Altekruse SF, Ries LA, Melbert DL, O’Leary M, et al. Outcomes for children and adolescents with cancer: challenges for the twenty-first century. J Clin Oncol. 2010;28(15):2625–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith M, Arthur D, Camitta B, Carroll AJ, Crist W, Gaynon P, et al. Uniform approach to risk classification and treatment assignment for children with acute lymphoblastic leukemia. J Clin Oncol. 1996;14(1):18–24. [DOI] [PubMed] [Google Scholar]
- 9.Linabery AM, Ross JA. Trends in childhood cancer incidence in the U.S. (1992–2004). Cancer. 2008;112(2):416–32. [DOI] [PubMed] [Google Scholar]
- 10.Ness KK, Armenian SH, Kadan-Lottick N, Gurney JG. Adverse effects of treatment in childhood acute lymphoblastic leukemia: general overview and implications for long-term cardiac health. Expert Rev Hematol. 2011;4(2):185–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Discov. 2011;10(9):671–84. [DOI] [PubMed] [Google Scholar]
- 12.Garten A, Schuster S, Penke M, Gorski T, de Giorgis T, Kiess W. Physiological and pathophysiological roles of NAMPT and NAD metabolism. Nat Rev Endocrinol. 2015;11(9):535–46. [DOI] [PubMed] [Google Scholar]
- 13.Sampath D, Zabka TS, Misner DL, O’Brien T, Dragovich PS. Inhibition of nicotinamide phosphoribosyltransferase (NAMPT) as a therapeutic strategy in cancer. Pharmacol Ther. 2015;151:16–31. [DOI] [PubMed] [Google Scholar]
- 14.Zhao G, Green CF, Hui YH, Prieto L, Shepard R, Dong S, et al. Discovery of a Highly Selective NAMPT Inhibitor That Demonstrates Robust Efficacy and Improved Retinal Toxicity with Nicotinic Acid Coadministration. Mol Cancer Ther. 2017;16(12):2677–88. [DOI] [PubMed] [Google Scholar]
- 15.Takao S, Chien W, Madan V, Lin DC, Ding LW, Sun QY, et al. Targeting the vulnerability to NAD(+) depletion in B-cell acute lymphoblastic leukemia. Leukemia. 2018;32(3):616–625. [DOI] [PubMed] [Google Scholar]
- 16.Matheny CJ, Wei MC, Bassik MC, Donnelly AJ, Kampmann M, Iwasaki M, et al. Next-generation NAMPT inhibitors identified by sequential high-throughput phenotypic chemical and functional genomic screens. Chem Biol. 2013;20(11):1352–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dong G, Chen W, Wang X, Yang X, Xu T, Wang P, et al. Small Molecule Inhibitors Simultaneously Targeting Cancer Metabolism and Epigenetics: Discovery of Novel Nicotinamide Phosphoribosyltransferase (NAMPT) and Histone Deacetylase (HDAC) Dual Inhibitors. J Med Chem. 2017;60(19):7965–83. [DOI] [PubMed] [Google Scholar]
- 18.Palacios DS, Meredith E, Kawanami T, Adams C, Chen X, Darsigny V, et al. Structure based design of nicotinamide phosphoribosyltransferase (NAMPT) inhibitors from a phenotypic screen. Bioorg Med Chem Lett. 2017. Dec 18. pii:S0960-894X(17)31210-6. [DOI] [PubMed] [Google Scholar]
- 19.Estoppey D, Hewett JW, Guy CT, Harrington E, Thomas JR, Schirle M, et al. Identification of a novel NAMPT inhibitor by CRISPR/Cas9 chemogenomic profiling in mammalian cells. Sci Rep. 2017;7:42728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Houghton PJ, Morton CL, Tucker C, Payne D, Favours E, Cole C, et al. The pediatric preclinical testing program: description of models and early testing results. Pediatr Blood Cancer. 2007;49(7):928–40. [DOI] [PubMed] [Google Scholar]
- 21.Liem NL, Papa RA, Milross CG, Schmid MA, Tajbakhsh M, Choi S, et al. Characterization of childhood acute lymphoblastic leukemia xenograft models for the preclinical evaluation of new therapies. Blood. 2004;103(10):3905–14. [DOI] [PubMed] [Google Scholar]
- 22.Jones L, Richmond J, Evans K, Carol H, Jing D, Kurmasheva RT, et al. Bioluminescence Imaging Enhances Analysis of Drug Responses in a Patient-Derived Xenograft Model of Pediatric ALL. Clin Cancer Res. 2017;23(14):3744–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Somers K, Chudakova DA, Middlemiss SM, Wen VW, Clifton M, Kwek A, et al. CCI-007, a novel small molecule with cytotoxic activity against infant leukemia with MLL rearrangements. Oncotarget. 2016;7(29):46067–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khaw SL, Suryani S, Evans K, Richmond J, Robbins A, Kurmasheva RT, et al. Venetoclax responses of pediatric ALL xenografts reveal sensitivity of MLL-rearranged leukemia. Blood. 2016;128(10):1382–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Somers K, Wen VW, Middlemiss SMC, Osborne B, Forgham H, Jung M, et al. A novel small molecule that kills a subset of MLL-rearranged leukemia cells by inducing mitochondrial dysfunction. Oncogene. 2019. Jan 22. Epub ahead of print: doi: 10.1038/s41388-018-0666-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao W, Sachsenmeier K, Zhang L, Sult E, Hollingsworth RE, Yang H. A New Bliss Independence Model to Analyze Drug Combination Data. J Biomol Screen. 2014;19(5):817–21. [DOI] [PubMed] [Google Scholar]
- 27.Bliss CI. The calculation of microbial assays. Bacteriol Rev. 1956;20(4):243–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Di Veroli GY, Fornari C, Wang D, Mollard S, Bramhall JL, Richards FM, et al. Combenefit: an interactive platform for the analysis and visualization of drug combinations. Bioinformatics. 2016;32(18):2866–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Suryani S, Carol H, Chonghaile TN, Frismantas V, Sarmah C, High L, et al. Cell and molecular determinants of in vivo efficacy of the BH3 mimetic ABT-263 against pediatric acute lymphoblastic leukemia xenografts. Clin Cancer Res. 2014;20(17):4520–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kees UR, Ford J, Watson M, Murch A, Ringner M, Walker RL, et al. Gene expression profiles in a panel of childhood leukemia cell lines mirror critical features of the disease. Mol Cancer Ther. 2003;2(7):671–7. [PubMed] [Google Scholar]
- 31.Lock RB, Liem N, Farnsworth ML, Milross CG, Xue C, Tajbakhsh M, et al. The nonobese diabetic/severe combined immunodeficient (NOD/SCID) mouse model of childhood acute lymphoblastic leukemia reveals intrinsic differences in biologic characteristics at diagnosis and relapse. Blood. 2002;99(11):4100–8. [DOI] [PubMed] [Google Scholar]
- 32.Watson M, Roulston A, Belec L, Billot X, Marcellus R, Bedard D, et al. The small molecule GMX1778 is a potent inhibitor of NAD+ biosynthesis: strategy for enhanced therapy in nicotinic acid phosphoribosyltransferase 1-deficient tumors. Mol Cell Biol. 2009;29(21):5872–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan M, Gravel M, Bramoulle A, Bridon G, Avizonis D, Shore GC, et al. Synergy between the NAMPT inhibitor GMX1777(8) and pemetrexed in non-small cell lung cancer cells is mediated by PARP activation and enhanced NAD consumption. Cancer Res. 2014;74(21):5948–54. [DOI] [PubMed] [Google Scholar]
- 34.Xiao Y, Elkins K, Durieux JK, Lee L, Oeh J, Yang LX, et al. Dependence of tumor cell lines and patient-derived tumors on the NAD salvage pathway renders them sensitive to NAMPT inhibition with GNE-618. Neoplasia. 2013;15(10):1151–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xiao Y, Kwong M, Daemen A, Belvin M, Liang X, Hatzivassiliou G, et al. Metabolic Response to NAD Depletion across Cell Lines Is Highly Variable. PLoS One. 2016;11(10):e0164166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gehrke I, Bouchard ED, Beiggi S, Poeppl AG, Johnston JB, Gibson SB, et al. On-target effect of FK866, a nicotinamide phosphoribosyl transferase inhibitor, by apoptosis-mediated death in chronic lymphocytic leukemia cells. Clin Cancer Res. 2014;20(18):4861–72. [DOI] [PubMed] [Google Scholar]
- 37.Cerna D, Li H, Flaherty S, Takebe N, Coleman CN, Yoo SS. Inhibition of nicotinamide phosphoribosyltransferase (NAMPT) activity by small molecule GMX1778 regulates reactive oxygen species (ROS)-mediated cytotoxicity in a p53- and nicotinic acid phosphoribosyltransferase1 (NAPRT1)-dependent manner. J Biol Chem. 2012;287(26):22408–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schmitz M, Breithaupt P, Scheidegger N, Cario G, Bonapace L, Meissner B, et al. Xenografts of highly resistant leukemia recapitulate the clonal composition of the leukemogenic compartment. Blood. 2011;118(7):1854–64. [DOI] [PubMed] [Google Scholar]
- 39.Clappier E, Gerby B, Sigaux F, Delord M, Touzri F, Hernandez L, et al. Clonal selection in xenografted human T cell acute lymphoblastic leukemia recapitulates gain of malignancy at relapse. J Exp Med. 2011;208(4):653–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anderson K, Lutz C, van Delft FW, Bateman CM, Guo Y, Colman SM, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature. 2011;469(7330):356–61. [DOI] [PubMed] [Google Scholar]
- 41.Coustan-Smith E, Mullighan CG, Onciu M, Behm FG, Raimondi SC, Pei D, et al. Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol. 2009;10(2):147–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van der Veer A, Waanders E, Pieters R, Willemse ME, Van Reijmersdal SV, Russell LJ, et al. Independent prognostic value of BCR-ABL1-like signature and IKZF1 deletion, but not high CRLF2 expression, in children with B-cell precursor ALL. Blood. 2013;122(15):2622–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Szymanska B, Wilczynska-Kalak U, Kang MH, Liem NL, Carol H, Boehm I, et al. Pharmacokinetic modeling of an induction regimen for in vivo combined testing of novel drugs against pediatric acute lymphoblastic leukemia xenografts. PLoS One. 2012;7(3):e33894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kotecha RS, Gottardo NG, Kees UR, Cole CH. The evolution of clinical trials for infant acute lymphoblastic leukemia. Blood Cancer J. 2014;4:e200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grolla AA, Travelli C, Genazzani AA, Sethi JK. Extracellular nicotinamide phosphoribosyltransferase, a new cancer metabokine. Br J Pharmacol. 2016;173(14):2182–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yoon MJ, Yoshida M, Johnson S, Takikawa A, Usui I, Tobe K, et al. SIRT1-Mediated eNAMPT Secretion from Adipose Tissue Regulates Hypothalamic NAD+ and Function in Mice. Cell Metab. 2015;21(5):706–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Audrito V, Manago A, Zamporlini F, Rulli E, Gaudino F, Madonna G, et al. Extracellular nicotinamide phosphoribosyltransferase (eNAMPT) is a novel marker for patients with BRAF-mutated metastatic melanoma. Oncotarget. 2018;9(27):18997–9005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Revollo JR, Korner A, Mills KF, Satoh A, Wang T, Garten A, et al. Nampt/PBEF/Visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metab. 2007;6(5):363–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reddy PS, Umesh S, Thota B, Tandon A, Pandey P, Hegde AS, et al. PBEF1/NAmPRTase/Visfatin: a potential malignant astrocytoma/glioblastoma serum marker with prognostic value. Cancer Biol Ther. 2008;7(5):663–8. [DOI] [PubMed] [Google Scholar]
- 50.Moore Z, Chakrabarti G, Luo X, Ali A, Hu Z, Fattah FJ, et al. NAMPT inhibition sensitizes pancreatic adenocarcinoma cells to tumor-selective, PAR-independent metabolic catastrophe and cell death induced by beta-lapachone. Cell Death Dis. 2015;6:e1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bi TQ, Che XM, Liao XH, Zhang DJ, Long HL, Li HJ, et al. Overexpression of Nampt in gastric cancer and chemopotentiating effects of the Nampt inhibitor FK866 in combination with fluorouracil. Oncol Rep. 2011;26(5):1251–7. [DOI] [PubMed] [Google Scholar]
- 52.Cruickshank MN, Ford J, Cheung LC, Heng J, Singh S, Wells J, et al. Systematic chemical and molecular profiling of MLL-rearranged infant acute lymphoblastic leukemia reveals efficacy of romidepsin. Leukemia. 2017;31(1):40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leoni V, Biondi A. Tyrosine kinase inhibitors in BCR-ABL positive acute lymphoblastic leukemia. Haematologica. 2015;100(3):295–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bajrami I, Kigozi A, Van Weverwijk A, Brough R, Frankum J, Lord CJ, et al. Synthetic lethality of PARP and NAMPT inhibition in triple-negative breast cancer cells. EMBO Mol Med. 2012;4(10):1087–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cea M, Soncini D, Fruscione F, Raffaghello L, Garuti A, Emionite L, et al. Synergistic interactions between HDAC and sirtuin inhibitors in human leukemia cells. PLoS One. 2011;6(7):e22739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zoppoli G, Cea M, Soncini D, Fruscione F, Rudner J, Moran E, et al. Potent synergistic interaction between the Nampt inhibitor APO866 and the apoptosis activator TRAIL in human leukemia cells. Exp Hematol. 2010;38(11):979–88. [DOI] [PubMed] [Google Scholar]
- 57.Nahimana A, Aubry D, Breton CS, Majjigapu SR, Sordat B, Vogel P, et al. The anti-lymphoma activity of APO866, an inhibitor of nicotinamide adenine dinucleotide biosynthesis, is potentialized when used in combination with anti-CD20 antibody. Leuk Lymphoma. 2014;55(9):2141–50. [DOI] [PubMed] [Google Scholar]
- 58.Chini CC, Guerrico AM, Nin V, Camacho-Pereira J, Escande C, Barbosa MT, et al. Targeting of NAD metabolism in pancreatic cancer cells: potential novel therapy for pancreatic tumors. Clin Cancer Res. 2014;20(1):120–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tateishi K, Wakimoto H, Iafrate AJ, Tanaka S, Loebel F, Lelic N, et al. Extreme Vulnerability of IDH1 Mutant Cancers to NAD+ Depletion. Cancer Cell. 2015;28(6):773–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cea M, Zoppoli G, Bruzzone S, Fruscione F, Moran E, Garuti A, et al. APO866 activity in hematologic malignancies: a preclinical in vitro study. Blood. 2009;113(23):6035–7; author reply 7–8. [DOI] [PubMed] [Google Scholar]
- 61.Barraud M, Garnier J, Loncle C, Gayet O, Lequeue C, Vasseur S, et al. A pancreatic ductal adenocarcinoma subpopulation is sensitive to FK866, an inhibitor of NAMPT. Oncotarget. 2016;7(33):53783–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee J, Kim H, Lee JE, Shin SJ, Oh S, Kwon G, et al. Selective Cytotoxicity of the NAMPT Inhibitor FK866 Toward Gastric Cancer Cells With Markers of the Epithelial-Mesenchymal Transition, Due to Loss of NAPRT. Gastroenterology. 2018;155(3):799–814 e13. [DOI] [PubMed] [Google Scholar]
- 63.Shames DS, Elkins K, Walter K, Holcomb T, Du P, Mohl D, et al. Loss of NAPRT1 expression by tumor-specific promoter methylation provides a novel predictive biomarker for NAMPT inhibitors. Clin Cancer Res. 2013;19(24):6912–23. [DOI] [PubMed] [Google Scholar]
- 64.Piacente F, Caffa I, Ravera S, Sociali G, Passalacqua M, Vellone VG, et al. Nicotinic Acid Phosphoribosyltransferase Regulates Cancer Cell Metabolism, Susceptibility to NAMPT Inhibitors, and DNA Repair. Cancer Res. 2017;77(14):3857–69. [DOI] [PubMed] [Google Scholar]
- 65.Konstantinopoulos PA, Spentzos D, Karlan BY, Taniguchi T, Fountzilas E, Francoeur N, et al. Gene expression profile of BRCAness that correlates with responsiveness to chemotherapy and with outcome in patients with epithelial ovarian cancer. J Clin Oncol. 2010;28(22):3555–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Minchom A, Aversa C, Lopez J. Dancing with the DNA damage response: next-generation anti-cancer therapeutic strategies. Ther Adv Med Oncol. 2018;10:1758835918786658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Peek CB, Affinati AH, Ramsey KM, Kuo HY, Yu W, Sena LA, et al. Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science. 2013;342(6158):1243417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chini EN. CD38 as a regulator of cellular NAD: a novel potential pharmacological target for metabolic conditions. Curr Pharm Des. 2009;15(1):57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee HC. Structure and enzymatic functions of human CD38. Mol Med. 2006;12(11–12):317–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481(7381):287–94. [DOI] [PubMed] [Google Scholar]
- 71.Di Stefano G, Manerba M, Vettraino M. NAD metabolism and functions: a common therapeutic target for neoplastic, metabolic and neurodegenerative diseases. Curr Top Med Chem. 2013;13(23):2918–29. [DOI] [PubMed] [Google Scholar]
- 72.Canto C, Menzies KJ, Auwerx J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015;22(1):31–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zabka TS, Singh J, Dhawan P, Liederer BM, Oeh J, Kauss MA, et al. Retinal toxicity, in vivo and in vitro, associated with inhibition of nicotinamide phosphoribosyltransferase. Toxicol Sci. 2015;144(1):163–72. [DOI] [PubMed] [Google Scholar]
- 74.Misner DL, Kauss MA, Singh J, Uppal H, Bruening-Wright A, Liederer BM, et al. Cardiotoxicity Associated with Nicotinamide Phosphoribosyltransferase Inhibitors in Rodents and in Rat and Human-Derived Cells Lines. Cardiovasc Toxicol. 2017;17(3):307–18. [DOI] [PubMed] [Google Scholar]
- 75.Jones L, Carol H, Evans K, Richmond J, Houghton PJ, Smith MA, et al. A review of new agents evaluated against pediatric acute lymphoblastic leukemia by the Pediatric Preclinical Testing Program. Leukemia. 2016;30(11):2133–41. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.