

Abstract
Huntington’s disease (HD) is a devastating and fatal monogenic neurodegenerative disorder characterized by progressive loss of selective neurons in the brain and is caused by an abnormal expansion of CAG trinucleotide repeats in a coding exon of the huntingtin (HTT) gene. Progressive gene expression changes that begin at premanifest stages are a prominent feature of HD and are thought to contribute to disease progression. Increasing evidence suggests the critical involvement of epigenetic mechanisms in abnormal transcription in HD. Genomewide alterations of a number of epigenetic modifications, including DNA methylation and multiple histone modifications, are associated with HD, suggesting that mutant HTT causes complex epigenetic abnormalities and chromatin structural changes, which may represent an underlying pathogenic mechanism. The causal relationship of specific epigenetic changes to early transcriptional alterations and to disease pathogenesis require further investigation. In this article, we review recent studies on epigenetic regulation in HD with a focus on DNA and histone modifications. We also discuss the contribution of epigenetic modifications to HD pathogenesis as well as potential mechanisms linking mutant HTT and epigenetic alterations. Finally, we discuss the therapeutic potential of epigenetic-based treatments.
Keywords: Huntington’s disease (HD), Neurodegeneration, Epigenetic regulation, Transcription, DNA methyltransferases (DNMTs), Epigenetic-based therapy
1. Huntington’s disease
HD is a fatal autosomal dominant neurodegenerative disease caused by an inherited CAG trinucleotide repeat expansion in exon 1 of the huntingtin (HTT) gene, which produces a mutant protein with an abnormally elongated polyglutamine (polyQ) tract at its N-terminus (The Huntington’s Disease Collaborative Research Group, 1993). Mutant HTT exhibits improper folding and is prone to oligomerization and aggregation. The expression of mutant protein triggers pathological changes in the brain with progressive loss of selective neurons in the striatum and cortex, leading to movement, cognitive, and psychiatric disorders (Walker, 2007). HD has a prevalence of 10.6–13.7 in 100,000 persons in Western populations (McColgan and Tabrizi, 2017). It is one of the neurodegenerative disorders known as polyQ diseases, including spinal and bulbar muscular atrophy, spinal cerebellar ataxia types 1, 2, 3, 6, 7, and 17, and dentatorubral-pallidoluysian atrophy, which are caused by CAG repeat expansion in distinct polyQ proteins (Lieberman et al., 2019; Orr and Zoghbi, 2007). Although the mutation in HD was identified in 1993, how the toxic mutant protein drives neurodegeneration is still not fully understood, and no curative treatment currently exists. Thus, identification of molecular pathways that underlie or contribute to the pathogenesis of human HD is a priority.
The mutant HTT protein exhibits a toxic gain-of-function, and its expression triggers multiple detrimental events in the cell. Studies with cellular and animal models of HD have revealed that mutant protein disrupts neuronal function by several distinct cellular events, including transcriptional dysregulation, mitochondrial dysfunction, synaptic dysfunction, impairment of proteostasis, and deficits in vesicular transport, all of which subsequently contribute to neuronal dysfunction and death (Jimenez-Sanchez et al., 2017; Labbadia and Morimoto, 2013; Ross and Tabrizi, 2011). At present, which neuronal insults are primary or secondary or whether these cellular processes are interrelated or operate independently is unclear. Given that transcriptional alterations have been found in the early stages of HD progression (Cha, 2007; Seredenina and Luthi-Carter, 2012), these molecular changes may represent a potential unifying upstream mechanism for these divergent processes. Striatal medium spiny neurons (MSNs), the most vulnerable cell type in HD, exhibit progressive transcriptional changes more than a decade before the onset of motor symptoms in human HD (Seredenina and Luthi-Carter, 2012), suggesting a casual role of this change in subsequent neuronal dysfunction and death. Although the precise molecular mechanisms driving transcriptional dysregulation in HD remain unclear, many studies have pointed to the importance of epigenetic mechanisms in this process. In the past decade, advances in next-generation sequencing technologies have improved our understanding of the HD transcriptome and epigenome and have revealed that mutant HTT causes genome-wide alterations of several epigenetic marks in cellular and animal models of HD as well as in patient brains (Table 1). However, the functional importance of these changes, in particular the dominant epigenetic mechanism(s) leading to HD progression, remains to be elucidated.
Table 1.
Genome-wide DNA methylation and histone modification analyses in HD.
| DNA modifications | Samples | Reference |
|---|---|---|
|
| ||
| 5mC | Human HD (fibroblasts; with or without HDACi 4b treatment) | Jia et al. (2015) |
| Human HD (various brain regions: caudate nucleus, cingulate gyrus, cerebellum, hippocampus, parietal cortex, frontal lobe, occipital cortex, temporal cortex, midbrain, motor cortex, sensory cortex, and visual cortex) | Horvath et al. (2016) | |
| Human HD (cortex and liver) | De Souza et al. (2016) | |
| Human HD (blood) | Zadel et al. (2018) | |
| Human HD (blood, lymphoblastoid cells, and fibroblasts) | Lu et al. (2020) | |
| HD sheep (blood) | Lu et al. (2020) | |
| Q175 Htt KI mice (striatum, cerebellum, liver, cortex, and blood) | Lu et al. (2020) | |
| Mouse striatal cell lines, STHdhQ7 and STHdhQ111 | Ng et al. (2013) | |
| 5hmC | YAC128 HD mice (striatum and cortex) | Wang et al. (2013a) |
| Histone modifications | Samples | Reference |
|
| ||
| H3K9ac, H3K14ac, H4K12ac | N171-82Q HD mice (hippocampus and cerebellum) | Valor et al. (2013) |
| H3K9ac, H3K14ac | R6/2 HD mice (striatum, 12 wk) | McFarland et al. (2012) |
| H3K27ac | Human HD (caudate nucleus and cerebellum) | Merienne et al. (2019) |
| R6/1 HD mice (striatum, 30 wk of age) | Achour et al. (2015) | |
| R6/1 HD mice (striatum, 8 wk) and CHL2 (Hdh(CAG)150) heterozygous KI mice | Yildirim et al. (2019) | |
| H3K4me3 | Human HD (NeuN positive neurons from prefrontal cortex) | Dong et al. (2015) |
| Human HD (NeuN positive neurons from prefrontal cortex) | Bai et al. (2015) | |
| R6/2 HD mice (striatum and cortex, 8 and 12 wk) | Vashishtha et al. (2013) | |
| Mouse ESC and NPC (Htt KO, mutant Htt KI) | Biagioli et al. (2015) | |
| H3K9me3 | Human ESC, NPC (HTT knockdown) | Irmak et al. (2018) |
| Mouse striatal cell lines, STHdhQ7 and STHdhQ111 | Lee et al. (2013) | |
| R6/2 HD mice (striatum 10wk; with or without nogalamycin treatment) | Lee et al. (2017a) | |
| H3K27me3 | Mouse ESC and NPC (Htt KO, mutant Htt KI) | Biagioli et al. (2015) |
| H3K36me3 | Mouse ESC and NPC (Htt KO, mutant Htt KI) | Biagioli et al. (2015) |
| H2Aub | R6/2 HD mice (striatum, 12 wk) | McFarland et al. (2013) |
Details are discussed in the text. The age of mice is indicated for R6/1 and R6/2 models (wk = weeks). ESC, embryonic stem cells; NPC, neural progenitor cells; KI, knock-in; KO, knockout; ac, acetylation; me, methylation; ub, ubiquitination; nogalamycin, an anthracycline antibiotic.
The genetic defect of HD is an abnormal expansion of normally polymorphic CAG trinucleotide repeats in the HTT gene. Individuals with 40 or more CAG repeats develop HD (full penetrance) while those with repeat lengths between 36 and 39 may or may not be affected by HD (reduced penetrance), and individuals with repeat lengths below 36 are unaffected (non-pathological) (McColgan and Tabrizi, 2017). There is an inverse correlation between CAG-repeat length and the age at onset of motor symptoms, with longer CAG repeats generally leading to earlier onset (Gusella and MacDonald, 2009; Gusella et al., 2014). Although the length of CAG repeats in the HD allele is the primary determinant of age of onset, repeat length accounts for only about 56% of the variability in age at onset in adult-onset HD patients (Gusella et al., 2014). This indicates that the remaining variation in the age of onset could be attributable to genetic or environmental factors (Gusella and MacDonald, 2009; Gusella et al., 2014; Wexler et al., 2004). Recent human HD genome-wide association studies (GWAS) identified several modifier genes that affect the age of motor onset independent of HTT CAG repeat length, including genes involved in DNA repair and maintenance (Genetic Modifiers of Huntington’s Disease Consortium, 2015, 2019; Lee et al., 2017b; Moss et al., 2017). These findings provided human-validated, critical disease-modifying pathways that can potentially be targeted for HD therapy. Environmental factors may also influence age of onset by altering epigenetic signatures and gene expression and may, with increased understanding of the mechanisms, hold therapeutic potential.
In this article, we review recent advances in the epigenetic regulation in HD, with a selective focus on DNA methylation and histone modifications and their contributions to disease pathogenesis. We discuss mechanisms of epigenetic dysregulation and potential therapeutic interventions targeting epigenetic alterations for HD.
2. Mechanisms of transcriptional dysregulation in Huntington’s disease
Transcriptional dysregulation is a fundamental problem not only in HD but also in other neurodegenerative diseases, including Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS) and in the aging brain (Cha, 2007; Cooper-Knock et al., 2012; Lee et al., 2000; Lu et al., 2004; Seredenina and Luthi-Carter, 2012). Gene expression changes are an early molecular hallmark of HD and are thought to represent an underlying pathogenic mechanism for HD (Cha, 2007; Seredenina and Luthi-Carter, 2012). Gene expression profiling studies have identified robust RNA expression changes in cell and mouse models of HD as well as human HD brain tissues (Ament et al., 2017; Hodges et al., 2006; Kuhn et al., 2007; Labadorf et al., 2015; Langfelder et al., 2016; Lee et al., 2020; Luthi-Carter et al., 2000, 2002; Neueder and Bates, 2014; Pan et al., 2018; Seredenina and Luthi-Carter, 2012; Vashishtha et al., 2013). Genes typically dysregulated in HD and HD models are associated with important biological processes, including synaptic transmission, neurotrophin receptor signaling, calcium signaling and homeostasis, ion transport, G-protein coupled receptor signaling, transcription and chromatin remodeling, nervous system development, and metabolism (Hodges et al., 2006; Langfelder et al., 2016; Pan et al., 2018; Seredenina and Luthi-Carter, 2012; Vashishtha et al., 2013).
A previous transcriptomic study comparing the caudate of six different genetic HD mouse models, including HTT transgenic mice expressing full-length or exon 1 HTT proteins and homozygous HTT knock-in mice, as well as HD patients revealed a profile of altered gene expression with remarkable similarity among the different animal models and human HD (Kuhn et al., 2007). This study found that the effects of mutant HTT on striatal gene expression changes were similar among the models tested and did not differ with mutant HTT protein size (i.e. full-length versus exon 1 fragments) or wild-type (WT) HTT dosage, suggesting the polyQ-containing region of the mutant protein plays a critical role in triggering gene expression changes. Small mutant HTT exon 1 fragments containing expanded polyQ are highly pathogenic compared to the full-length protein. Interestingly, the toxic exon 1 fragment was found to be produced by aberrant splicing of mutant HTT RNA and expressed in postmortem human HD brains as well as HD knock-in mice carrying full-length mutant HTT alleles, providing an important new mechanism for HD pathogenesis (Gipson et al., 2013; Neueder et al., 2017; Sathasivam et al., 2013).
A large-scale, comprehensive transcriptomic study with 600 brain and peripheral tissue samples from HD allelic series knock-in mice identified that a consistent set of genes and networks are dysregulated in a CAG length- and age-dependent manner (Langfelder et al., 2016). The CAG repeat length-dependent transcriptional signature was most prominent in the striatum with altered expression of numerous genes including striatal MSN identity genes, such as Drd2, Drd1a, Ppp1r1b, and Gpr6, genes involved in cAMP signaling, cell death, and DNA repair, and protocadherin genes (Langfelder et al., 2016). The downregulation of these striatal MSN identity genes has also been observed in previous RNA expression studies with striatal samples from human HD and HD models (Hodges et al., 2006; Kuhn et al., 2007; Seredenina and Luthi-Carter, 2012; Vashishtha et al., 2013). These neuronal identity genes are important for neurons to maintain their neuronal identity, connectivity, and functions in vivo (Deneris and Hobert, 2014). Therefore, the downregulation of these striatal MSN specific genes may lead to dysfunction and degeneration of MSNs in HD.
Recent advances in cell-type specific RNA isolation techniques have enabled the identification of cell-type specific gene expression changes in vivo in human and mouse HD brain (Al-Dalahmah et al., 2020; Lee et al., 2020; Merienne et al., 2019). The transcriptomic data reveals a novel pathway, in which mutant HTT leads to the release of mitochondrial RNAs and activation of innate immune signaling, specifically in striatal spiny projection neurons, the most vulnerable neurons in HD (Lee et al., 2020). In addition, single nucleus RNA-seq analysis of human HD cingulate cortex identified various astrocytes states associated with HD (Al-Dalahmah et al., 2020). Astrocytes play important roles in maintaining an optimal microenvironment for neuronal function and have been suggested to contribute to HD pathogenesis (Gray, 2019; Palpagama et al., 2019). Understanding cell-type specific gene expression changes in these disease-relevant cell types in vivo will provide important insights into disease mechanisms.
How is gene expression dysregulated in the HD brain? Several potential mechanisms have been proposed, including altered activities of transcription factors, coactivators, or corepressors by aberrant binding to or sequestration by mutant HTT. These factors include specificity protein 1 (Spl), CREB-binding protein (CBP), TATA-binding protein (TBP), TBP-associated factor, 135 kD (TAFII-130), and the p53 tumor suppressor (Nucifora et al., 2001; Steffan et al., 2000); reviewed in (Cha, 2007; Kim and Kim, 2014; Sadri-Vakili and Cha, 2006; Seredenina and Luthi-Carter, 2012). Thus, mutant HTT interacts with a number of transcriptional regulators and impairs transcription. More recently, methyl-CpG binding protein 2 (MeCP2) was found as a direct binding partner of HTT, with higher binding affinity to mutant compared to WT HTT (McFarland et al., 2014). MeCP2 exhibits increased binding at brain-derived neurotrophic factor (Bdnf) promoter IV in mutant HTT-expressing cells, and MeCP2 knockdown reactivates Bdnf expression, suggesting that the MeCP2-mutant HTT interaction may contribute to Bdnf downregulation in HD (McFarland et al., 2014) (See a model in Fig. 1). Bdnf mRNA expression could also be dysregulated in HD by a transcriptional repressor, repressor element-1 silencing transcription factor/neuron-restrictive silencer factor (REST/NRSF). The differential binding affinity of WT versus mutant HTT to REST has been shown to contribute to the downregulation of Bdnf (at Bdnf promoter II) and potentially other neuronal genes containing REST/NRSF-binding sites (Buckley et al., 2010; Zuccato et al., 2003, 2007). In addition, accumulating evidence indicates that chromatin modifications and remodeling downstream of mutant HTT play an important role in transcriptional dysregulation in HD. These mechanisms may not be mutually exclusive and may cooperate to drive abnormal gene expression in HD.
Fig. 1.
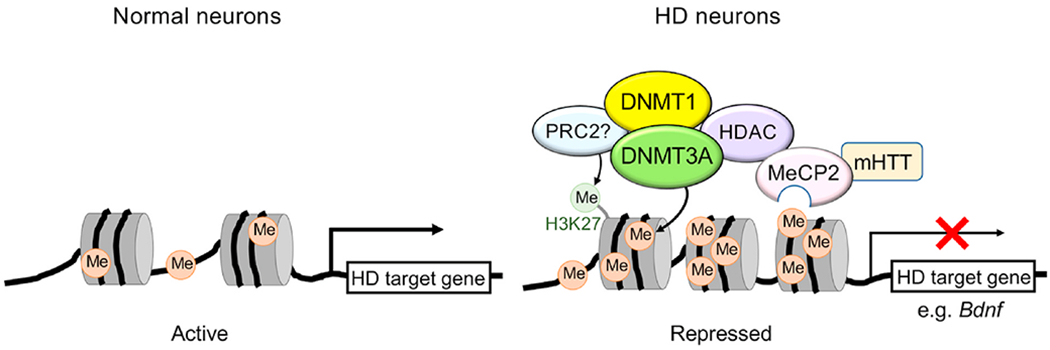
A model for DNA methylation-mediated gene repression in HD. Schematic shows potential players involved in promoter hypermethylation and transcriptional repression in HD neurons. Mutant HTT (mHTT) expression increases the levels of DNA methylation at specific gene promoters, including Bdnf exon IV promoter in cortical neurons (Pan et al., 2016). Dnmt1/DNMT3A-mediated promoter methylation contributes to transcriptional repression of genes important for neuronal function and survival, leading to neuronal dysfunction and death (Pan et al., 2016). Methyl-CpG-binding protein MeCP2 binds to methylated DNA and recruits epigenetic modifiers, including HDAC, to repress transcription. DNMT1 and DNMT3A may also interact with HDACs (Fuks et al., 2001; Rountree et al., 2000). mHTT directly binds to MeCP2 and enhances binding of MeCP2 to Bdnf exon IV promoter, leading to downregulation of Bdnf (McFarland et al., 2014). In cancer, Polycomb repressive complex 2 (PRC2) core subunit EZH2 recruits DNMTs to specific gene promoters via direct interaction (Vire et al., 2006). Whether this mechanism occurs in HD neurons remains to be tested. Me, methylation (5mC).
3. Epigenetic modifications in adult brain
Chromatin is a complex of macromolecules composed of DNA and histone proteins. DNA is wrapped around histone octamers (2 copies each of the core histones H2A, H2B, H3, and H4) to form nucleosomes and is packaged into the nucleus in a highly organized fashion (Campos and Reinberg, 2009; Luger et al., 1997). Several epigenetic mechanisms, including DNA methylation, posttranslational modifications of histone proteins, non-coding RNAs, and chromatin remodeling factors, control gene expression by modulating chromatin structure and its accessibility to transcription factors. Epigenetic mechanisms regulate a number of important biological processes in developing and adult brains, ranging from neural development and neuronal differentiation to learning and memory and aging (Campbell and Wood, 2019; Qureshi and Mehler, 2018; Salinas et al., 2020; Shin et al., 2014). Epigenetic gene regulation is a cell-type specific mechanism, and the epigenetic state determines the phenotype of the cell. Epigenetic regulatory mechanisms are becoming increasingly recognized as important contributors to the pathogenesis of many neurodegenerative diseases, including AD, PD, ALS, and HD (Berson et al., 2018; Lardenoije et al., 2015). Our knowledge of neuronal epigenetics is, however, far less than that of other well-studied cell types, such as embryonic stem cells and cancer cells. We review recent literature on epigenetic regulation in HD with a focus on DNA and histone modifications. Excellent reviews for the role of non-coding RNAs in HD are found in (Kerschbamer and Biagioli, 2015; Riva et al., 2016; Wu and Kuo, 2020).
3.1. DNA methylation and demethylation
Methylation of the fifth carbon position of cytosine (5-methylcytosine or 5mC) in CpG dinucleotide is an important epigenetic mark that affects multiple biological processes, including embryonic development, genomic imprinting, X-chromosome inactivation, and diseases such as cancer (Smith and Meissner, 2013; Suzuki and Bird, 2008). The importance of DNA methylation in the nervous system has become increasingly clear in recent years, and DNA methylation-mediated transcriptional regulation is critically involved in the function of normal and diseased brains, including learning and memory, synaptic transmission, psychiatric and neurodegenerative disorders, and aging; reviewed in (Day et al., 2015; Heyward and Sweatt, 2015; Lu et al., 2013; Morris and Monteggia, 2014; Shin et al., 2014; Tognini et al., 2015).
The role of DNA methylation in gene promoters is well established, and generally, the transcriptional outcome of promoter hyper-methylation is repression. Promoter methylation can induce transcriptional repression by directly interfering with transcription factor binding (Campanero et al., 2000; Tate and Bird, 1993) or by recruiting chromatin remodeling co-repressors, such as histone deacetylase (HDAC), mediated through the binding of methyl-CpG-binding domain (MBD) proteins, to establish transcriptional repression (Fuks et al., 2003; Jones et al., 1998; Nan et al., 1998; Zhang et al., 1999). The 5mC modification is also found within gene bodies of actively transcribed genes (Lister et al., 2009), suggesting that it is not simply a mark of gene silencing. Methylation of DNA is catalyzed by members of the DNA methyl-transferase (DNMT) family of enzymes, including DNMT1, DNMT3A, and DNMT3B. DNMT1 has preferential activity towards hemi-methylated CpG and is essential for the maintenance of DNA methylation patterns in the newly synthesized strand during DNA replication in dividing cells, whereas DNMT3A and DNMT3B are required for de novo DNA methylation (Bird, 2002; Moore et al., 2013).
In addition to its essential role in embryonic development (Li et al., 1992; Okano et al., 1999), DNA methylation plays an important role in brain development and neuronal function. Disruption of DNMT1 or DNMT3A in prenatal stages causes neurodevelopmental defects and functionally impairs CNS neurons (Fan et al., 2001; Lavery et al., 2020). For example, conditional deletion of the Dnmt1 gene in mitotic CNS precursor cells in mice using the Nestin promoter-driven Cre (Nestin--Cre)-loxP system resulted in DNA hypomethylation in daughter cells and perturbed the function and survival of CNS neurons in postnatal animals (Fan et al., 2001). Interestingly, DNMTs are highly expressed in postnatal brain. Postmitotic neurons of the postnatal brain express two major DNMTs, DNMT1 and DNMT3A (Feng et al., 2005; Inano et al., 2000). A study in adult forebrain neurons using conditional knockout of DNMT1 and DNMT3A with the calcium/calmodulin-dependent protein kinase IIα promoter-driven Cre (CamKIIα-Cre)-loxP system revealed an important role for these enzymes in synaptic function; Dnmt1/Dnmt3a double conditional knockout mice, but not single Dnmt1 or Dnmt3a knockouts, showed abnormal long-term plasticity in the hippocampal CA1 region with deficits in learning and memory as well as decreased DNA methylation and deregulated gene expression, suggesting overlapping roles of these two DNMTs in maintaining DNA methylation and modulating neuronal gene expression in adult CNS neurons (Feng et al., 2010). These results also corroborated a previous study in which DNMT1 deficiency in postmitotic forebrain neurons in vivo had no effect on levels of global DNA methylation and neuronal survival during postnatal life (Fan et al., 2001).
Methylation of DNA is a dynamic and reversible process. DNA demethylation can occur by both passive and active mechanisms. Passive demethylation occurs by dilution of methylation marks through multiple rounds of cell division in the context of reduced DNMT activity or expression. DNA methylation in postmitotic neurons is dynamically regulated by active demethylation, which is achieved via ten-eleven translocase (TET)-mediated sequential oxidations of 5mC to 5-hydroxy-methylcytosine (5hmC) and to 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC), followed by the excision of 5fC and 5caC by thymine DNA glycosylase (TDG) via the base excision repair pathway (Guo et al., 2011; He et al., 2011; Ito et al., 2011; Wu and Zhang, 2017). No enzyme that directly converts 5mC to unmethylated C has been found. 5hmC, besides serving as a demethylation intermediate, is thought to have an independent role in regulating chromatin structure and gene expression. In brain, 5hmC is enriched in gene bodies and near genes; within gene bodies, coding exons are most enriched for 5hmC, and 5hmC levels correlate well with gene expression at these loci (Guo et al., 2014b; Mellen et al., 2012; Song et al., 2011a; Szulwach et al., 2011).
Interestingly, neurons have a unique methylome containing much higher levels of 5hmC and non-CpG methylation (mCpH, where H = A, C, or T) than non-neuronal cells (Guo et al., 2011, 2014a; Kriaucionis and Heintz, 2009; Song et al., 2011a); reviewed in (Clemens and Gabel, 2020; Jang et al., 2017; Wu and Zhang, 2017). Whether the neuronal abundance of mCpH and 5hmC marks plays a critical role in neurode-generative diseases is not yet known.
3.2. Posttranslational modifications of histones
Histone modifications, along with DNA methylation, control chromatin structure and help to define gene expression. Covalent posttranslational modifications of histone proteins, including methylation, acetylation, ubiquitination, and phosphorylation, modulate the degree of compaction of nucleosomes, thereby affecting chromatin accessibility to transcription factors and gene expression (Sadri-Vakili and Cha, 2006). The specific lysine (K) residues in the N-terminal histone tail can be modified by several key posttranslational modifications, including methylation, acetylation and ubiquitination. Methylation can also occur on an arginine residue. Transcriptional outcomes with these modifications depend upon the type of modification and the amino acid residue of histone that is modified. There are specific patterns of histone modification occupancy on the genome for each histone modification. For example, acetylation on histone H3 lysine 27 (H3K27ac) is associated with higher transcriptional activation and marks active enhancers; H3 lysine 4 trimethylation (H3K4me3) marks the transcriptional start sites (TSS) of actively transcribed genes. Comprehensive reviews for histone modifications can be found in the literature (Campos and Reinberg, 2009; Gates et al., 2017; Hyun et al., 2017; Sadri-Vakili and Cha, 2006). Histone modifications are regulated by two opposing families of enzymes and are reversible—for example, histone acetyl-transferases (HATs) and histone deacetylases (HDACs) for adding and removing an acetyl group, respectively, and histone methyltransferases (HMTs) and histone demethylases (HDMs) for adding and removing a methyl group, respectively. Alterations in histone modifications with subsequent gene expression changes have been implicated in the pathogenesis of cancer, neurodegenerative diseases, psychiatric disorders, learning and memory, and aging (Berson et al., 2018; Booth and Brunet, 2016; Comet et al., 2016; Graff and Tsai, 2013; Li et al., 2013; Tsankova et al., 2007). Therefore, agents that are able to modulate or reverse abnormal epigenetic changes might be beneficial for disease conditions with aberrant epigenetic regulation.
4. Epigenetic alterations in Huntington’s disease
Progressive and robust gene expression changes are a prominent feature of HD (Hervas-Corpion et al., 2018; Hodges et al., 2006; Kuhn et al., 2007; Labadorf et al., 2015; Langfelder et al., 2016; Luthi-Carter et al., 2000; Vashishtha et al., 2013). In the past decade, extensive research has focused on the identification of key epigenetic factors and chromatin mechanisms underlying transcriptional dysregulation and pathogenesis of HD. Altered patterns of several chromatin marks have been identified at the genome-wide level in human HD tissues and various models of HD, including DNA methylation, histone H3K9 and H3K14 acetylation (H3K9ac and H3K14ac), H3K27ac, H3K4me3, H3K9me3, and H3K27me3 (Table 1), indicating that mutant HTT appears to modulate chromatin structure via perturbations of epigenetic modifications. However, the mechanisms linking mutant HTT and altered chromatin modifications are poorly understood.
4.1. Alterations in DNA methylation and hydroxymethylation in HD
The first genome-wide DNA methylation analysis in a HD model was reported using striatal cell lines derived from mouse embryonic striatal progenitor cells from WT HTTQ7/Q7 or HD HTTQ111/Q111 knock-in mice (Ng et al., 2013). Reduced representation bisulfite sequencing (RRBS) and confirmatory methylated DNA immunoprecipitation-sequencing (MeDIP-seq) analyses found extensive DNA methylation changes in HD striatal cells compared to control WT cells. The changes are gene specific, not global, with some sites increased and others decreased in methylation, with the majority of the changes occurring in CpG-poor regions of the genome, compared to CpG-rich regions (Ng et al., 2013). A large fraction of the dysregulated genes in the mutant HTT-expressing cell line showed significant changes in DNA methylation, suggesting a potential causal effect of DNA methylation on gene changes. In addition, motif scanning and ChIP-seq analyses identified AP-1 and SOX2 as transcriptional regulators associated with DNA methylation changes, where loss of AP-1 and SOX2 binding is associated with increased DNA methylation, suggesting involvement of these transcriptional regulators in mutant HTT-induced methylation changes (Ng et al., 2013). The study highlighted a potential new mechanism for mutant HTT-induced transcriptional dysregulation. The functional importance of methylation changes in HD pathogenesis, however, remained unclear.
Several DNA methylation profiling studies have recently been performed using human HD tissues and blood (Table 1) (De Souza et al., 2016; Gutierrez et al., 2019; Horvath et al., 2016; Jia et al., 2015; Lu et al., 2020; Zadel et al., 2018). It has been established that DNA methylation levels at a subset of DNA methylation sites can be used to predict biological age across a broad spectrum of human tissues. The epigenetic measure of tissue age (referred to as the epigenetic clock) is estimated by DNA methylation levels at 353 CpG sites (Horvath, 2013). DNA methylation array analysis assessed the epigenetic age of various brain regions of human HD and control subjects and revealed that HD accelerates the epigenetic aging of human brain and disrupts DNA methylation levels (Horvath et al., 2016). The same group performed a comprehensive large-scale methylome study and characterized DNA methylation levels in six different tissues, including blood, from three mammalian species, i.e., a Htt knock-in mouse model, a transgenic HTT sheep model, and humans. The authors found profound DNA methylation changes associated with HD in all three species (Lu et al., 2020). Consistent with the epigenetic age acceleration observed in the postmortem HD brains in their earlier study (Horvath et al., 2016), blood of manifest HD cases showed accelerated epigenetic aging (Lu et al., 2020). The epigenome-wide association study (EWAS) of human blood revealed that HD mutation status is associated with 33 CpG sites, including the HTT gene (hypermethylated in HD group). The HTT/Htt associations were also reproduced in the mouse and sheep models. In addition, the hypomethylation of three loci (PEX14, GRIK4, and COX412) were associated with motor progression in manifest HD cases. This study provided new knowledge about epigenetic aging, the role of DNA methylation in HD, and epigenetic modifiers that may modulate disease progression in HD. Another array-based methylation study with the postmortem cortex samples from 7 HD cases and 6 controls showed minimal evidence of HD-associated DNA methylation changes (De Souza et al., 2016). Although HD-associated DNA methylation changes were not identified, site-specific differential DNA methylation in the promoter and intragenic regions of the HTT gene was found when matched human cortex and liver samples were compared. They identified a new differentially methylated binding site for the transcription factor CTCF, which is located in the HTT proximal promoter. Interestingly, this site showed decreased methylation and increased binding of CTCF in cortex compared to liver, consistent with higher expression of the HTT gene in cortex vs. liver, suggesting that DNA methylation may, in part, contribute to tissue-specific HTT expression via differential CTCF occupancy (De Souza et al., 2016).
Methylation levels of selected genes have also been investigated in human brain and HD models (Pan et al., 2016; Villar-Menendez et al., 2013). The expression of the adenosine A2a receptor (ADORA2A) gene, encoding a G-protein-coupled receptor highly expressed in basal ganglia, is severely downregulated in HD patients and various HD mouse models. The reduced ADORA2A expression in the putamen of HD patients was associated with increased levels of 5mC and decreased levels of 5hmC in the 5’UTR of ADORA2A (Villar-Menendez et al., 2013), suggesting that DNA methylation is a potential mechanism. This methylation change, however, was not observed in the striatum of HD transgenic mice (R6/1 and R6/2) and appeared to be differentially regulated between the species. BDNF is an important neurotrophic factor involved in various brain functions, including neuronal survival, differentiation, synaptic transmission, and learning and memory. The loss of BDNF mRNA and protein in the brain has been implicated in human HD pathogenesis (Zuccato et al., 2001). Lentiviral expression of mutant HTT exon 1 fragments in mouse primary cortical neurons increased the levels of 5mC in the Bdnf promoter IV, consistent with decreased Bdnf expression in the mutant HTT neurons compared to WT controls (Pan et al., 2016). Additionally, in the blood of human HD subjects, BDNF promoter IV methylation was elevated when compared to controls (Gutierrez et al., 2019). Interestingly, the increased DNA methylation levels at specific CpGs in the BDNF promoter of human HD blood were inversely correlated to anxiety and depression by Hospital Anxiety and Depression Scale (HADS) scores (Gutierrez et al., 2019). This suggests that methylation levels at specific loci in blood may represent a potential biomarker of psychiatric symptoms.
5hmC is thought to participate in transcriptional control and is found at higher levels in brain than in other tissues (Kriaucionis and Heintz, 2009; Song et al., 2011a). Genome-wide reduction of 5hmC marks has been found in the striatum and cortex of HD transgenic YAC128 mice carrying the HTT transgene with 128 CAG repeats (Wang et al., 2013a). Several differentially hydroxymethylated regions (DhMRs) in HD versus WT mice positively correlated with gene expression. Pathway analysis revealed that genes associated with DhMRs in the striatum (HD versus WT) are enriched in pathways involving neuronal development/differentiation (Wnt/β-catenin/Sox pathway and axonal guidance signaling pathway) and neuronal function/survival (glutamate receptor/calcium/CREB, GABA receptor signaling, and dopamine-DARPP32 feedback pathway) (Wang et al., 2013a). The reduction of 5hmC in the brain may be a critical feature of HD and requires verification in human brain in future studies.
4.2. The role and mechanisms of altered DNA methylation in HD
While a number of studies provided evidence for aberrant DNA methylation in HD, little is known about transcriptional consequences of such alterations and involvement in disease pathogenesis. Using a primary cortical neuron system, a recent cell-based unbiased epigenetic compound library screen identified a DNMT inhibitor, 5-aza-2’-deoxy-cytidine (decitabine) as an effective neuroprotective drug against mutant HTT-induced toxicity (Table 2) (Pan et al., 2016). Compounds in the library targeted writers, erasers, and readers of various epigenetic marks, but no hits were found with compounds targeting other epigenetic mechanisms in this screen. As verification, the structurally related DNMT inhibitor, 5-fluoro-2’-deoxycytidine (FdCyd) also exhibited a similar neuroprotective effect against mutant HTT, pointing to the essential role of DNMTs in mutant HTT-induced neurotoxicity. Mutant HTT-expressing cortical neurons showed elevated levels of DNA methylation in the Bdnf promoter IV, and inhibition of DNMTs in these neurons decreased the levels of DNA methylation and restored the expression of Bdnf. The effects of the pharmacological inhibitors were reproduced by RNA interference (RNAi)-mediated knockdown of DNMT1 or DNMT3A, verifying the role of DNMTs in mutant HTT-induced toxicity. DNMT inhibition could also rescue the expression of key striatal genes abnormally downregulated in HD, including Drd2, Penk, Adora2a, Ppp1r1b, and Rasd2 in mutant HTT-expressing primary striatal neurons and, importantly, in the striatum of HD mice in vivo (Pan et al., 2016). These findings highlight the causal role of DNA methylation in transcriptional alterations in HD (See a model in Fig. 1). Decitabine is an U.S. Food and Drug Administration (FDA)-approved drug for myelodysplastic syndromes (MDS) and has been used for treatment of hematologic malignancies in patients; FdCyd is being tested in clinical trials for cancer. However, these drugs have not been used clinically for neurodegenerative diseases, including HD. Whether these pharmacological inhibitors are beneficial to the neurodegeneration and pathogenesis of HD in vivo remains to be determined in future proof-of-concept preclinical studies with animal models. The deoxycytidine-analog DNMT inhibitors, including decitabine and FdCyd, need to be incorporated into DNA to exert their DNMT inhibitory activity (Creusot et al., 1982; Gnyszka et al., 2013; Lyko and Brown, 2005; Stresemann and Lyko, 2008). In dividing cells, drug incorporation occurs during DNA synthesis. But how these drugs can be incorporated into the DNA of non-dividing postmitotic neurons remains unclear, although it is possible that the base excision repair pathway contributes to this process. The exact mechanism of action of these drugs in postmitotic neurons remains to be determined.
Table 2.
Pharmacological and genetic inhibition of DNMTs in primary neuron and mouse models of HD and other neurodegenerative conditions.
| Neuronal insults and disease models | Methods targeting DNMTs | Beneficial effects | Reference |
|---|---|---|---|
| Primary neuron cultures | |||
| Mutant HTT-expressing primary cortical and striatal neurons | Decitabine, FdCyd; DNMT RNAi | Treatment with decitabine, FdCyd, or shRNAs targeting DNMT3A and DNMT1 attenuated mutant HTT (exon 1)-induced transcriptional changes and neurodegeneration in mouse primary cortical and striatal neuron systems. Decitabine treatment reduced the levels of mutant HTT aggregates in primary cortical neurons. | Pan et al. (2016) |
| Oxidative stress in primary cerebellar granule neurons | Decitabine | Hydrogen peroxide-induced neuronal death and reduction of Klotho protein levels were rescued by decitabine treament in mouse primary cerebellar granule neurons. | Xin et al. (2015) |
| Retinal photoreceptors from a mouse model of retinitis pigmentosa | Decitabine | Decitabine treatment reduced photoreceptor cell death in organotypic retinal explant cultures from a mouse model of retinitis pigmentosa. | Farinelli et al. (2014) |
| Mouse models in vivo | |||
| HD | FdCyd | Striatal genes downregulated in R6/2 HD mouse brain, including Drd2, Adora2a, Ppp1r1b, and Rosd2, were restored by ICV infusion of FdCdy by osmotic pump. | Pan et al. (2016) |
| SBMA | RG108 | ICV infusion of RG108 by osmotic pump improved motor function and survival of AR-97Q SBMA mice. RG108 treatment suppressed spinal motor neuron atrophy in AR-97Q mice. | Kondo et al. (2019) |
| Scitatic nerve avlusion | RG108 | Inhibition of DNMTs with RG108 blocked the increase in 5mC present in the cytoplasm and the nucleus and the apoptosis of motor neurons induced by sciatic nerve avulsion in adult mice. | Chestnut et al. (2011) |
| Ischemic brain injury | Decitabine; Dnmt1 heterozygous KO | ICV pre-administration of decitabine in WT mice reduced ischemic brain damage after mild focal ischemia (MCAo/reperfusion). Mutant mice heterozygous for a Dnmt1 gene deletion were resistant to mild ischemic damage. | Endres et al. (2000) |
| Conditional Dnmt1 heterozygous KO (CamKIIa-Cre) | Reduced levels of DNMT1, but not its complete deletion, in postmitotic neurons of the postnatal brain showed smaller infarcts following MCAo/reperfusion compared to control WT. | Endres et al. (2001) | |
ICV, intracerebroventricular; decitabine (5-aza-2′-deoxycytidine) and FdCyd (5-fluoro-2′-deoxycytidine), nucleoside analog DNMT inhibitors; RG108, non-nucleoside DNMT inhibitor; HD, Huntington’s disease; SBMA, spinal and bulbar muscular atrophy; AR, androgen receptor; MCAo, middle cerebral artery occlusion; KO, knockout.
Interestingly, in addition to mutant HTT-induced toxicity, DNMT inhibitors appeared to be neuroprotective for other neurological and neuronal stress conditions, including oxidative stress-induced neuronal death (Xin et al., 2015) and retinal photoreceptor degeneration (Farinelli et al., 2014) (Table 2). In contrast, decitabine enhanced apoptosis in cultured dopaminergic cell lines (Wang et al., 2013b). However, the cells used in the report were an immortalized, actively dividing cell line, and the DNMT inhibitor may have adverse effects on dividing cells. The beneficial effects of DNMT inhibitors (decitabine or non-nucleoside analog DNMT inhibitor RG108) or genetic reduction of DNMT have been shown in mouse models of different neurodegenerative and neurological disorders, including spinal and bulbar muscular atrophy (SBMA), a progressive neuromuscular disease caused by a polyQ expansion in androgen receptor (AR) (Kondo et al., 2019), sciatic nerve avulsion (Chestnut et al., 2011), and mild ischemic brain injury (Endres et al., 2000, 2001) (Table 2). These findings highlight the crucial role of DNA methylation in a broader range of neurodegenerative conditions.
How mutant HTT promotes aberrant DNA methylation at specific gene loci remains unknown, necessitating exploration in future studies. Possible mechanisms include mutant HTT expression enhancing the activity of DNMT enzymes and/or facilitating the recruitment of the DNA methylation machinery to specific genomic regions. These mechanisms may be mediated by aberrant protein-protein interactions and/or by posttranslational modifications of DNMTs in the context of mutant HTT. Since DNMT enzymes themselves do not have a strong sequence preference, it is reasonable to speculate that DNMT-interacting proteins capable of recognizing specific DNA motifs recruit DNMTs to particular loci. For example, previous studies have suggested the involvement of transcriptional repressor Polycomb repressive complex 2 (PRC2), a histone methyltransferase complex, in the regulation of DNA methylation (Vire et al., 2006). In cancer, extensive overlap has been observed between DNA methylation and the PRC2-mediated H3K27me3 mark, and PRC2 subunit, enhancer of zeste homolog 2 (EZH2) has been shown to recruit DNMTs to EZH2-target promoters via direct interaction (Schlesinger et al., 2007; Vire et al., 2006). Whether these interactions occur in neurons remains to be determined. It is also plausible that DNMT methylation patterns can be influenced by alterations of histone marks induced by mutant HTT. DNMT3A has been shown to bind specific histone modifications through its ATRX-DNMT3-DNMT3L (ADD) domain and proline-tryptophan-tryptophan-proline (PWWP) domain (ADD binding to unmodified H3K4 and PWWP binding to H3K36me2/3) (Dhayalan et al., 2010; Dukatz et al., 2019; Weinberg et al., 2019; Zhang et al., 2010); reviewed in (Castillo-Aguilera et al., 2017; Du et al., 2015; Jeltsch and Jurkowska, 2016; Rose and Klose, 2014), and therefore the mutant HTT-induced alterations of histone modification patterns may influence DNMT3A binding to specific genomic regions. Future studies are required to address these intriguing mechanistic possibilities.
4.3. Alterations in histone modifications in HD
Posttranslational modifications of histone proteins control chromatin dynamics and could affect gene expression. Genome-wide analyses identified altered patterns of several key histone marks in HD patients and various models of HD (Table 1) (Achour et al., 2015; Bai et al., 2015; Biagioli et al., 2015; Irmak et al., 2018; Lee et al., 2013; McFarland et al., 2012, 2013; Valor et al., 2013; Vashishtha et al., 2013). The altered modifications in HD include acetylated H3 lysine 9 (H3K9ac), H3K14ac, H4K12ac, H3K27ac, trimethylated H3K4 (H3K4me3), H3K9me3, and H3K27me3. The mechanisms underlying these chromatin changes and their consequences on transcription and disease pathogenesis in HD remain unclear. In this section, we focus on alterations and roles of two major histone modifications, acetylation and methylation in HD.
4.3.1. Histone acetylation
Among posttranslational modifications of histone proteins, histone acetylation and deacetylation have been most studied in HD, and these data are summarized in excellent reviews (Glajch and Sadri-Vakili, 2015; Sharma and Taliyan, 2015). Here we highlight more recent findings. Acetylation of H3K9, H3K14, and H4K12 are well characterized histone marks for transcriptional activation. In various models of HD and human HD samples, global reduction of histone acetylation (Chiu et al., 2011; Ferrante et al., 2003; Gardian et al., 2005; Giralt et al., 2012; Jiang et al., 2006; Lim et al., 2011; Stack et al., 2007) or the decreased levels of acetylation at specific genomic loci (McFarland et al., 2012; Sadri-Vakili et al., 2007; Thomas et al., 2008; Valor et al., 2013) have been found. Genome-wide studies identified altered patterns of histone acetylation in the striatum, hippocampus, and cerebellum of HD mice compared to control WT mice (McFarland et al., 2012; Valor et al., 2013). Although a number of hypoacetylated loci were associated with HD, histone acetylation and gene expression changes exhibited low correlation (McFarland et al., 2012; Valor et al., 2013). A different histone mark, H2A monoubiquitylation (H2Aub) at K119, traditionally associated with gene silencing, also showed limited correlation with transcriptional changes in a genome-wide study (McFarland et al., 2013). These modifications may cooperate with other chromatin modifications or mechanisms to exert the full range of transcriptional alterations associated with HD.
The mechanisms linking mutant HTT and loss of acetylation at specific genomic loci remain poorly understood. The aberrant patterns of histone acetylation could occur by the unbalanced action of two opposing enzymes, writers and erasers, i.e., histone acetyltransferases (HATs) and histone deacetylases (HDACs). Sequestration of CBP—a transcriptional co-activator with HAT activity—into polyQ aggregates is a potential mechanism for reduction of acetylation and was observed in cell and mouse models as well as human HD postmortem brain (Nucifora et al., 2001). CBP overexpression rescued cells from mutant HTT-induced toxicity, suggesting that inhibition of histone acetylation mediates cellular toxicity (Nucifora et al., 2001).
A study with Drosophila models of HD initially identified beneficial effects of HDAC inhibitors in HD neurodegeneration (Steffan et al., 2001). Many studies have corroborated the neuroprotective effects of HDAC inhibitors in mammalian models of HD. In mouse models, various HDAC inhibitors, including sodium butyrate, valproic acid (VPA), trichostatin A (TSA), suberoylanilide hydroxamic acid (SAHA), phenylbutyrate, HDACi 4b, RGFP966, and LBH589 (panobinostat), have been shown to ameliorate HD-related molecular, cellular, pathological, and/or behavioral phenotypes (Chiu et al., 2011; Chopra et al., 2016; Ferrante et al., 2003; Gardian et al., 2005; Giralt et al., 2012; Hockly et al., 2003; Jia et al., 2012a, 2012b, 2015, 2016; Mielcarek et al., 2011; Ryu et al., 2003; Sadri-Vakili et al., 2007; Siebzehnrubl et al., 2018; Thomas et al., 2008; Zadori et al., 2009).
LBH589, a hydroxamic acid-derived nonselective HDAC inhibitor, has recently been shown to be neuroprotective in mouse models of HD (Chopra et al., 2016). LBH589 induced histone hyperacetylation and improved pathological and behavioral phenotypes, i.e., prevented striatal neuronal atrophy and improved motor performance, in two different HD mouse models (Chopra et al., 2016). Another recent study showed beneficial effects of LBH589 on the early postnatal period in mouse and rat models of HD (Siebzehnrubl et al., 2018). Although HD typically manifests later in adulthood, emerging evidence from human and mouse HD has suggested that HD affects neurodevelopment (Barnat et al., 2020; HD iPSC Consortium, 2017; Siebzehnrubl et al., 2018). However, therapeutic approaches targeting early neurodevelopmental stages are currently lacking. This study showed that low-dose LBH589 treatment at preweaning age is able to reverse the early cellular, molecular, and behavioral phenotypes in HD rodent models (Siebzehnrubl et al., 2018). The study provided a proof-of-concept that neuro-developmental changes, including aberrant neuronal differentiation in HD, are reversible by an early therapeutic intervention targeting HDAC in vivo.
The HDAC superfamily consists of at least 18 members that are divided into two main families which are further ascribed into distinct classes: the classic HDAC family (Classes I, IIa, IIb, and IV) and the sirtuin family (Class III) (Sharma and Taliyan, 2015). Which HDAC(s) are relevant to HD pathogenesis? Addressing this question will lead to development of more specific therapeutics with selective HDAC inhibitors. Genetically engineered R6/2 HD mice with reduced levels of HDAC4 (Class IIa HDAC) showed extended lifespan, improved motor coordination, upregulated cortical Bdnf transcripts, and improved cortico-striatal synaptic function, pointing to the critical role of HDAC4 in HD pathogenesis (Mielcarek et al., 2013). Unexpectedly, however, although HDAC4 reduction rescued expression of several Bdnf transcripts, it did not affect global transcriptional dysregulation in R6/2 HD cortex by microarray profiling. HDAC4 colocalized in cytoplasmic mutant HTT inclusions, and HDAC4 reduction delayed the formation of mutant HTT cytoplasmic aggregates, but not nuclear aggregates, in HD mouse brain. Although HDAC4 traditionally acts as a transcriptional repressor via interactions with tissue specific transcription factors (Parra and Verdin, 2010), the beneficial effects of HDAC4 reduction in HD mice appeared to be independent of its traditional function on chromatin. The impact of genetic reduction or knockout of several other Hdac genes, including Hdac3, Hdac6, and Hdac7, has been tested in mouse models of HD. But HDAC4 is the only HDAC whose genetic reduction has so far been shown to improve HD phenotypes in mice (Benn et al., 2009; Bobrowska et al., 2011; Mielcarek et al., 2013; Moumne et al., 2012).
HDACi 4b, a HDAC inhibitor targeting HDACs 1 and 3, has been shown to ameliorate behavioral and pathological phenotypes and transcriptional abnormalities in HD mice (Jia et al., 2012a, 2012b; Thomas et al., 2008). Interestingly, treatment of HD mice with HDACi 4b induced alterations in DNA methylation and elicited transgenerational effects, in which the offspring (F1 generation) of HDAC inhibitor-treated HD male mice showed improved motor function and cognition compared with the offspring from vehicle-treated HD males (Jia et al., 2015). Alteration in sperm DNA methylation in the HDAC inhibitor treated HD male mice may mediate this transgenerational effect. This study provides important implications that ancestral HDAC inhibitor exposure may offer beneficial effects to offspring by mechanisms involving crosstalk between histone acetylation and DNA methylation (Jia et al., 2015).
Another important epigenetic modification, H3K27ac, a mark for active enhancers, was found to be altered in the R6/1 mouse model of HD at late disease stages in a genome-wide study (Achour et al., 2015). The downregulated genes controlling striatal neuronal identity and function in the striatum of R6/1 HD mice were associated with selective reduction of H3K27ac and RNA polymerase II (RNAPII) at super-enhancers, which are characterized by high levels and broad patterns of H3K27ac and RNAPII occupancy (Achour et al., 2015). The authors proposed that mutant HTT impairs the stabilization of striatal super-enhancers, and perturbation of histone acetyltransferase CBP contributes to this mechanism. The decreased H3K27ac mark at down-regulated neuronal identity genes in mice was validated in human caudate postmortem brain samples from HD vs. control subjects (Merienne et al., 2019). These studies provide new insights into the mechanism of transcriptional dysregulation in HD, in which selective reduction of H3K27ac at super enhancers drives downregulation of neuronal function genes in HD striatum (Achour et al., 2015; Merienne et al., 2019). Another study from the same group further explored the mechanism of altered striatal gene expression and found the extensive dysregulation of enhancer transcription in R6/1 HD mouse striatum (Le Gras et al., 2017). Transcription of non-coding RNAs transcribed from active enhancers, called enhancer RNAs (eRNAs), are involved in regulation of enhancer activity (Sartorelli and Lauberth, 2020). Their data suggest that decreased eRNAs transcription by reduced recruitment of RNAPII at super-enhancers leads to repression of striatal neuron identity genes (Le Gras et al., 2017). The study revealed a new epigenetic mechanism, which involves dysregulation of eRNAs and super enhancer activity, for striatal neuronal gene repression in HD mice in vivo. A recent genome-wide study with presymptomatic R6/1 HD mice identified aberrant transcription and changes in H3K27ac in the striatum of HD mice prior to the onset of overt disease symptoms (Yildirim et al., 2019). The analysis of the altered H3K27ac landscape led to identification of transcriptional factor Elk-1 as a potential regulator of early transcriptional changes in HD (Yildirim et al., 2019).
4.3.2. Histone methylation
Studies using HD models and human HD samples identified genomewide alterations in several histone methylation marks, including H3K4me3, H3K9me3, and H3K27me3 (Table 1) (Bai et al., 2015; Biagioli et al., 2015; Dong et al., 2015; Lee et al., 2013; Vashishtha et al., 2013), but little is known about the functional consequences of these alterations in HD in vivo. Methylation at these different lysine (K) residues has different transcriptional outcomes (Kimura, 2013); H3K4me3 marks the TSSs of actively transcribed genes; H3K9me3 and H3K27me3 are generally linked to transcriptional repression.
The cortex and striatum of R6/2 HD mice and human HD patients showed reduced H3K4me3 levels at promoters of selective down-regulated genes (Vashishtha et al., 2013). Integrated ChIP-seq and RNA-seq analyses further indicated that HD-associated changes in H3K4me3 patterns strongly correlate with transcriptional changes in the cortex and striatum of R6/2 HD mice (Vashishtha et al., 2013). Interestingly, genes downregulated in R6/2 HD mice exhibited a unique H3K4me3 signature with a broad peak downstream of the TSS in WT mice. These results suggest that the specific H3K4me3 signature determines a response to mutant HTT expression, providing insights into the mechanistic basis of transcriptional dysregulation in HD (Vashishtha et al., 2013). Importantly, reducing the levels of H3K4me3 demethylase SMCX/JARID1C (also known as KDM5C) reactivated several key neuronal genes that were downregulated by mutant HTT in primary neurons and was neuroprotective in a fly model of HD, suggesting that H3K4me3 changes may contribute to HD pathogenesis (Vashishtha et al., 2013). The role of these epigenetic changes in HD requires testing in mammalian models.
The contribution of H3K4me3 changes to transcriptional alterations in HD was further supported by a study using mouse embryonic stem cells (ESCs) and neural progenitor cells (NPCs) expressing mutant HTT with different sizes of CAG repeats (Biagioli et al., 2015). The study showed that mutant HTT expression causes CAG repeat-length dependent alterations in H3K4me3, which correlates with changes in gene expression (Biagioli et al., 2015). Furthermore, genome-wide H3K4me3 ChIP-seq studies using NeuN-positive neuronal nuclei from the prefrontal cortex of human HD and control subjects identified a number of differentially enriched H3K4me3 peaks between HD and controls (Bai et al., 2015; Dong et al., 2015). The differential H3K4me3 peaks were associated with genes implicated in neuronal development, synaptic functions, and neurodegeneration (Bai et al., 2015). Unlike the data from the mouse HD cortex (Vashishtha et al., 2013), integrative H3K4me3 ChIP-seq and RNA-seq analyses with human HD and control prefrontal cortex showed a poor correlation between HD-associated H3K4me3 changes and gene expression changes (Dong et al., 2015). In this human study, however, ChIP-seq was conducted with neuronal nuclei (NeuN positive cells) sorted from the prefrontal cortex, while RNA-seq was conducted with prefrontal cortical tissue homogenate containing multiple cell types. Integration of ChIP- and RNA-seq data from the same samples or similar preparations is needed to more accurately address the relationship between epigenetic and transcriptional changes in human HD. Mechanisms linking mutant HTT to aberrant H3K4me3 patterns remain to be addressed in future studies.
Hypermethylation of histone H3K9 leads to transcriptionally repressive, closed chromatin. Increased levels of H3K9 methylation (H3K9me2/3) and H3K9 methyltransferase ERG-associated protein with SET domain (ESET)/SETDB1 have been detected in the striatum of R6/2 HD mice and HD patients (Ferrante et al., 2004; Ryu et al., 2006). Treatment of R6/2 mice with mithramycin, a clinically approved antitumor antibiotic (anthracycline) that binds guanine-cytosine (CG)-rich DNA sequence, reduced the levels of H3K9me2/3 and SETDB1 and improved HD-like phenotypes as well as extended the life span of HD mice (Ferrante et al., 2004; Ryu et al., 2006). These observations were confirmed in studies using different HD models and related anthracyclines (Lee et al., 2017a; Stack et al., 2007). Importantly, deficiency of SETDB1 in a Drosophila model of HD attenuated mutant HTT-induced eye degeneration (Lee et al., 2017a). These findings suggest that increased H3K9me3 via upregulation of SETDB1 may contribute to HD pathogenesis. The role of SETDB1 in HD, however, needs further investigations using mammalian models. Several H3K9me2/3 methyltransferases are expressed in mammalian cells, and specific methyltransferase(s) involved in H3K9me3 changes in HD remain to be identified.
Interestingly, a recent study suggests a direct role of HTT in the regulation of H3K9me3 through its interaction with the ATF7IP, a regulator of SETDB1. The study showed that WT HTT inhibits the association of the ATF7IP-SETDB1 complex with other heterochromatin regulators and transcriptional repressors and thereby maintains low levels of H3K9me3 in human ESCs, while mutant HTT with an expanded polyQ stretch loses the ability to interact with the ATF7IP-SETDB1 complex and induces H3K9me3 in induced pluripotent stem cells (iPSC) from HD patients (Irmak et al., 2018).
To identify gene expression changes caused by dysregulation of H3K9me3, integrative analysis of H3K9me3 ChIP-seq and RNA-seq were performed using HD model striatal cell lines (Lee et al., 2013). 545 genes with increased promoter H3K9me3 and decreased mRNA levels were identified and were enriched in genes related to synaptic transmission, cell motility, and neuronal differentiation pathways (Lee et al., 2013). Another recent study from the same group identified changes in the H3K9me3 landscape in the striatum of R6/2 HD mice versus WT mice by ChIP-on-ChIP analysis; genes with increased H3K9me3 occupancy were associated with biological processes, including cellular protein metabolic process and intracellular signal transduction, and genes with decreased H3K9me3 occupancy were associated with sensory perception and neurological system processes in R6/2 mice compared to WT mice (Lee et al., 2017a). Nogalamycin, an anthracycline antibiotic and a chromatin remodeling drug, modulated HD-associated H3K9me3 landscape and showed beneficial effects in R6/2 HD mice, suggesting that targeting alterations in H3K9me3 occupancy in HD may be neuro-protective (Lee et al., 2017a).
A recent systematic genetic interaction study tested various lysine and arginine methyltransferases and demethylases in a Drosophila model of HD and identified H3K27me2/3 demethylase UTX (KDM6A) as a potential therapeutic target for HD, i.e., inhibition of UTX is neuro-protective (Song et al., 2018). Reducing the levels of UTX rescued HTT-induced pathology, while reducing the core components of H3K27 methyltransferase PRC2 led to more aggressive pathology (Song et al., 2018). In the following subsection, we further describe recent findings on PRC2 and its potential role in HD.
4.4. Potential role of PRC2 in HD
Several studies have suggested the involvement of PRC2, the enzyme complex that is responsible for methylation of H3K27, in transcriptional changes and neurodegeneration in HD (Biagioli et al., 2015; Dong et al., 2015; Seong et al., 2010; von Schimmelmann et al., 2016). PRC2 is an evolutionarily conserved, multisubunit protein complex containing core components—enhancer of zeste homolog 1 (EZH1) or EZH2 (the catalytic subunit), suppressor of zeste 12 (SUZ12), embryonic ectoderm development (EED), and RB binding protein (RBBP) 4/7 (also known as RbAp48/46)—with several sub-stoichiometric subunits (van Mierlo et al., 2019). PRC2 is capable of catalyzing mono-, di- and tri-methylation of H3K27 and the only methyltransferase responsible for H3K27 methylation, with H3K27me2/3 highly associated with repressed genes. PRC2 is essential for embryonic development, and has been implicated in diseases, such as cancer (Laugesen et al., 2016; Margueron and Reinberg, 2011; van Mierlo et al., 2019), but less is known about the role of PRC2 in the adult central nervous system.
Increasing evidence has implicated the involvement of PRC2 in transcriptional dysregulation in HD. HTT has been shown to interact with PRC2 and facilitate the PRC2 methyltransferase activity in a polyQ-length dependent manner (Seong et al., 2010), raising the possibility that mutant HTT with an expanded polyQ tract alters gene expression through modulating PRC2 activity. In support of this hypothesis, expression of mutant HTT with an expanded CAG tract in mouse ESC and NPC lines by knock-in led to alteration in genome-wide H3K27me3 patterns in both cell types compared to WT control lines (Biagioli et al., 2015). Interestingly, mutant HTT expression was also prominently associated with alteration in H3K4me3 at transcriptionally active loci (Biagioli et al., 2015). These findings suggest that mutant HTT may have broader functions in the HD epigenome. A study with human neurons isolated from the prefrontal cortex of HD and non-neurologic controls indicated PRC2 is depleted in HD-associated distal H3K4me3 peaks, suggesting the role of PRC2 inhibition is associated with increased H3K4me3 in HD (Dong et al., 2015). Together, mutant HTT appears to dysregulate PRC2 in HD via as-of-yet unknown mechanism.
A study using PRC2 conditional knockout mice revealed a link between PRC2 and neurodegeneration (von Schimmelmann et al., 2016). PRC2 represses genes that are detrimental to the function and survival of adult neurons, and PRC2 deficiency in adult striatal MSNs causes derepression of selected PRC2 target genes, including a large number of self-regulating transcription factors normally suppressed in these neurons, leading to progressive and fatal neurodegeneration in mice (von Schimmelmann et al., 2016). The study also pointed to the significant overlap in gene expression changes in adult PRC2-deficient neurons and HD brains from patients and mouse models, supporting the idea that loss of PRC2 may be involved in transcriptional alterations and neurodegeneration in HD (Song et al., 2018; von Schimmelmann et al., 2016). Interestingly, increased levels of H3K27me3 have also been linked to neurodegenerative disease. Ataxia-telangiectasia (A-T) is an autosomal recessive neurodegenerative disease caused by A-T mutated (ATM) protein deficiency. Loss of ATM has been shown to increase EZH2 stability as well as the levels of H3K27me3 in the cerebellum of human and mouse A-T (Li et al., 2013). EZH2 deficiency by lentiviral knockdown of EZH2 ameliorated Purkinje cell neurodegeneration and behavioral phenotypes in an ATM-deficient mouse model of A-T, suggesting that increased levels of H3K27me3 are deleterious (Li et al., 2013). The findings from these studies (Li et al., 2013; von Schimmelmann et al., 2016)—both loss and gain of H3K27me3 leading to neurodegeneration—indicate that PRC2 dysregulation may have a U-shaped effect in neurodegeneration and that proper balance of H3K27me3 is likely to be important to keep neurons healthy. It is still unclear whether PRC2 is abnormally activated or inactivated in HD neurons in vivo. Further investigations are needed to understand the role and mechanisms of PRC2 in HD neurodegeneration.
In addition to the potential connection between PRC2 and mutant HTT, wild-type HTT function may intersect with the PRC2 pathway. Genome-wide analysis of histone modifications using Htt knockout ESC and NPC lines showed that HTT is required for proper regulation of H3K27me3 at bivalent loci (Biagioli et al., 2015). In addition, in vivo mouse studies showed similar phenotypes in Htt knockout embryos and Polycomb Eed knockout embryos (Faust et al., 1995; Woda et al., 2005). HTT is essential for embryonic development as inactivation of Htt in mice is embryonic lethal (Dragatsis et al., 1998; Duyao et al., 1995; Nasir et al., 1995; Zeitlin et al., 1995). Htt null embryos showed early patterning defects, including abnormal streak development, lack of headfold formation, ectopic expression of T (Brachyury), Evx1, and Nodal, and disruption of anterior primitive streak mesoderm production (Woda et al., 2005). These features of Htt null embryos are reminiscent of phenotypes in embryos deficient in Eed (Faust et al., 1995; Woda et al., 2005).
5. Epigenetic regulation of DNA repair and genomic stability
Epigenetic regulation of chromatin structure plays a critical role in gene expression, but may also affect other key genomic processes, including DNA repair and genomic stability (Gorbunova et al., 2004; Jung and Bonini, 2007; Tini et al., 2002); reviewed in (Ding et al., 2019; Dion and Wilson, 2009; Huertas et al., 2009; Karakaidos et al., 2020) (Fig. 2). Eukaryotic genomes are packaged into nucleosomes and chromatin, and chromatin organization imposes structural constraints on DNA repair, suggesting a requirement of chromatin modifications and remodeling activities to open local chromatin structure to allow the repair machinery to access DNA lesions. In addition, epigenetic modifications may be involved in other steps during DNA repair, e.g., transcriptional repression to prevent the transcriptional machinery from interfering with the repair process as well as closing local chromatin structure to restore its original state after repair is complete (Ding et al., 2019). DNA repair is an important process to maintain DNA integrity and genomic function, and deficiency in DNA repair has been linked to several neurodegenerative disorders, including HD (Jeppesen et al., 2011). Comprehensive reviews for the role of chromatin modifications and remodeling in DNA repair are found in (Ding et al., 2019; Huertas et al., 2009; Karakaidos et al., 2020).
Fig. 2.
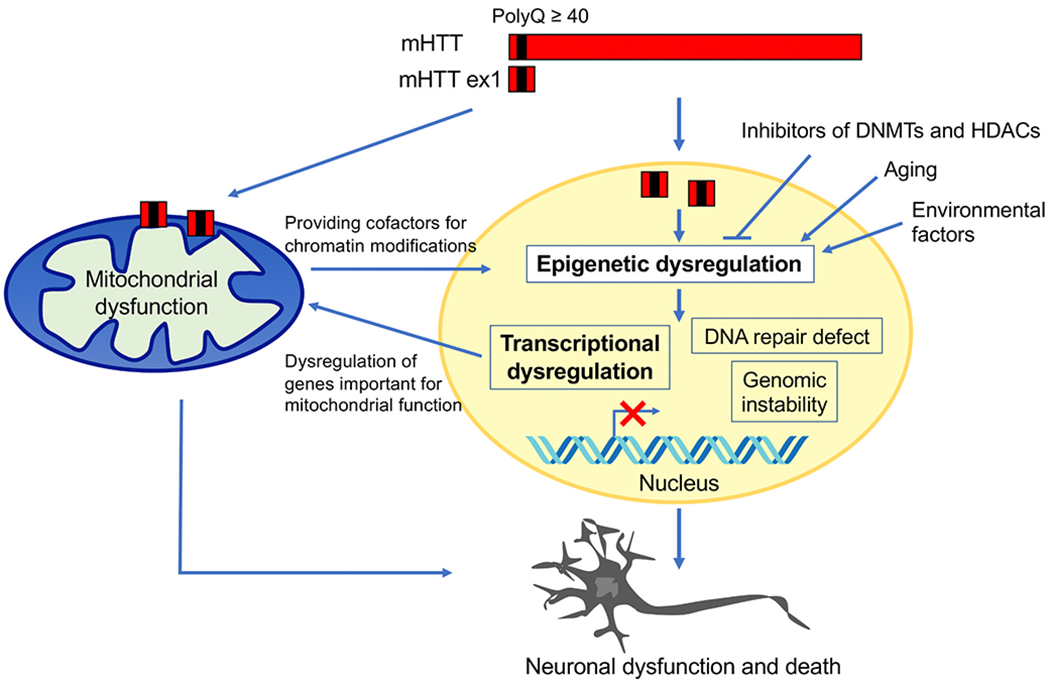
Epigenetic dysregulation leading to neurodegeneration in HD. N-terminal mutant HTT exon 1 (mHTT ex1) fragments generated by aberrant splicing are highly pathogenic. mHTT ex1 causes changes in DNA methylation and histone modifications and alters chromatin structure. These changes induce transcriptional dysregulation and could also affect DNA repair and genomic stability, leading to neuronal dysfunction and death in HD. Epigenetic and transcriptional dysregulation in HD could alter expression of nuclearly encoded mitochondrial proteins essential for mitochondrial function and energy metabolism, leading to mitochondrial dysfunction. The direct action of mHTT on mitochondria also causes toxicity. Metabolic changes in HD mitochondria may affect the availability of cofactors for chromatin modifying enzymes, causing epigenetic dysregulation. Aging and environmental factors could influence epigenetic mechanisms. Inhibitors of epigenetic modifiers, including DNMTs and HDACs, may reverse mutant HTT-induced epigenetic and transcriptional alterations and block neurodegeneration.
Trinucleotide repeat instability is a key feature observed in human HD and other trinucleotide repeat disorders, including spinocerebellar ataxias (SCAs) and myotonic dystrophy, and repeat expansions in germline and somatic cells affect disease severity (Khristich and Mirkin, 2020). Mechanisms of trinucleotide repeat instability are not fully understood, but several mechanisms involving chromatin modifying enzymes, epigenetic marks, 3D chromatin domains, and DNA repair have been proposed (Dion et al., 2008; Dion and Wilson, 2009; Gorbunova et al., 2004; Jung and Bonini, 2007; Khristich and Mirkin, 2020; Sun et al., 2018). CBP, a HAT also known to regulate DNA repair (Dutto et al., 2018; Tini et al., 2002), has been shown to modulate repeat instability in Drosophila models of polyQ diseases, spinocerebellar ataxia type 3 (SCA3) and HD (Jung and Bonini, 2007). The germline cells in the fly models recapitulated key features of human CAG-repeat instability with strong expansion bias. Genetically reducing CBP in the fly models enhanced repeat instability of SCA3 and HD transgenes. In contrast, overexpression of CBP resulted in a reduced rate of instability—treatment of flies expressing the pathogenic polyQ protein with pharmacological HDAC inhibitor to normalize acetylation levels suppressed repeat instability (Jung and Bonini, 2007). Consistently, decreased CBP activity has been linked to polyQ diseases (Nucifora et al., 2001), and treatment of fly and mouse models of polyQ disease with HDAC inhibitor was protective against polyQ protein pathogenesis (Steffan et al., 2001). These results suggest that enhancing repeat instability is a toxic consequence of pathogenic polyQ protein. Genome-wide DNA demethylation has also been shown to destabilize trinucleotide repeat tracts (contractions or expansions) using DNMT inhibitors in two different cell lines, Chinese hamster ovary cells and fibroblasts from myotonic dystrophy patients, both carrying expanded trinucleotide repeats (Gorbunova et al., 2004). Reducing maintenance DNMT1 (heterozygous for a null allele of Dnmt1) promoted intergenerational CAG repeat expansion in the mouse germline at the murine spinocerebellar ataxia type 1 (Sea1) locus (Dion et al., 2008). Whether epigenetic mechanisms play an important role in somatic repeat instability in the context of postmitotic neurons remains to be investigated. A recent finding suggested the intriguing possibility that topological 3D chromatin structure could participate in somatic repeat instability observed in disease-associated short tandem repeats (Sun et al., 2018). Hi-C data indicated that the majority of disease-associated repeats, including HTT CAG repeats, are localized at 3D chromatin domain boundaries with markedly high CpG-island density, suggesting that the epigenetic and topological environment at the boundaries makes repeats unusually susceptible to instability (Sun et al., 2018).
DNA mismatch repair and nucleotide excision repair proteins have also been shown to be involved in trinucleotide repeat instability (Dion and Wilson, 2009; Dragileva et al., 2009; Flower et al., 2019; Jung and Bonini, 2007; Pinto et al., 2013; Tome et al., 2013). Recent GWAS data in patients with HD have implicated DNA repair pathways (with genes including FAN1, MLH1, PMS1, PMS2, MSH3, LIG1), in particular mismatch repair, as the most prominent modifiers of age of disease onset, highlighting the important contribution of repair pathways in the course of disease (Genetic Modifiers of Huntington’s Disease Consortium, 2015, 2019; Lee et al., 2017b; Moss et al., 2017). Inactivation of multiple mismatch repair genes, including MSH2, MSH3, MLH1, and MLH3, in HD mouse models, has been shown to suppress CAG expansion in HD (Schmidt and Pearson, 2016), suggesting a contribution of mismatch repair to HD repeat expansion in vivo. Single nucleotide polymorphisms (SNPs) of another repair factor, FAN1 also influences the age at onset in HD—knockout or knockdown of FAN1 in HD patient-derived iPSC and differentiated MSNs or HD mice increases somatic expansion of HTT CAG repeats (Goold et al., 2019; Kim et al., 2020; Loupe et al., 2020). Thus, the modulation of these repair proteins has therapeutic potential in HD. In addition, a recently characterized small molecule compound, naphthyridine-azaquinolone (NA) specifically binds the slipped-strand structure formed by expanded CAG repeats, resulting in specific contractions of the HD mutation both in HD cell models and in vivo in HD mouse striatum (Nakamori et al., 2020).
Many observations suggest that epigenetic alterations in HD affect genomic function beyond gene transcription. Whether mutant HTT-induced epigenetic dysregulation contributes to impairment of DNA repair and genome stability in neurons requires further investigation. Somatic trinucleotide repeat expansion of the mutant HTT gene is a hot target in current therapeutic development although the precise mechanisms of repeat expansion have yet to be elucidated. As mentioned above, epigenetic mechanisms, chromatin environment, and DNA mismatch repair are potential contributors to trinucleotide repeat expansion in HD. Whether these mechanisms are interrelated or work independently to drive repeat instability remains unclear. Improved understanding of mechanisms driving HTT CAG repeat expansion may lead to identify more effective strategies to prevent repeat expansion.
6. Mitochondria dysfunction and epigenetic dysregulation in Huntington’s disease
Accumulating evidence suggests that there is an interplay between mitochondrial function and chromatin dynamics (Fig. 2). Mitochondria metabolism plays an important role in chromatin regulation by providing intermediate metabolites for generation of chromatin modifications. Intermediate metabolites produced in mitochondria and the cytosol, such as acetyl-CoA (Ac-CoA), α-ketoglutarate (α-KG), and S-adenosyl methionine (SAM), serve as co-factors or substrates for chromatin modifying enzymes (Matilainen et al., 2017; van der Knaap and Verrijzer, 2016). For example, acetyl-CoA is used as an acetyl donor by histone acetyltransferases; α-KG generated in the TCA cycle is an essential cofactor of TET enzymes and Jumonji C domain-containing HDMs; SAM is used as a methyl donor by HMTs and DNMTs. Alterations in mitochondrial metabolism or metabolic state therefore affects the availability of cofactors and substrates for chromatin-modifying enzymes, leading to aberrant epigenetic signatures that alter chromatin accessibility and gene expression (Mohammed et al., 2020; van der Knaap and Verrijzer, 2016). Thus, mutant HTT-induced deficits in energy metabolism and mitochondrial dysfunction may contribute to epigenetic dysregulation (Fig. 2). It would be of interest to examine the levels of key intermediate metabolites that potentially contribute to chromatin modifications in HD. Additionally, a recently study showing the link between released mitochondrial RNA (a potent mitochondrial-derived innate immunogen) and the upregulation of innate immune genes in HD striatal spiny projection neurons suggests another potential point of crosstalk between mitochondrial dysfunction and epigenetic/transcriptional dysregulation (Lee et al., 2020).
In addition to the influence of mitochondrial dysfunction to epigenetic modifications, epigenetic and transcriptional dysregulation in HD could affect mitochondrial function (Fig. 2). 99% of the approximately 1500 mitochondrial proteins are encoded by the nuclear genome, and the expression of these nuclear encoded mitochondrial proteins could potentially be affected by mutant HTT-induced epigenetic alterations, leading to mitochondrial dysfunction. Mutant HTT has been shown to repress expression of a key transcriptional coactivator, PPAR gamma coactivator 1 alpha (PGC-1α), which regulates expression of genes involved in energy metabolism, leading to downregulation of mitochondrial biogenesis in HD (Chaturvedi and Beal, 2013; Cui et al., 2006; Kim et al., 2010; Weydt et al., 2006). Thus, mitochondria and the nucleus could influence each other, and potentially these interactions may exacerbate the function of both organelles. In addition, direct action of mutant HTT on mitochondria could cause mitochondrial defects (Orr et al., 2008; Panov et al., 2002; Song et al., 2011b; Yano et al., 2014; Yu et al., 2003). Therefore, targeting both epigenetic dysregulation and mitochondrial dysfunction may lead to more effective neuroprotective treatments for HD.
7. Therapeutic implications for epigenetic-based treatments for HD
Currently, no disease-modifying treatment exists to delay onset or slow disease progression in HD. Pharmacological or genetic inhibition of several epigenetic-modifying enzymes, including DNMTs and HDACs, in cell and animal models of HD, showed neuroprotection and ameliorated disease phenotype, suggesting their potential utility for HD. Inhibitors of DNMTs and HDACs were initially developed as cancer drugs and have been clinically used for treatment of cancers, but they have been gaining attention for the treatment of HD. The therapeutic potential of DNMT or HDAC inhibitors has also been suggested for several other neurodegenerative diseases, including polyQ disorder SBMA, AD, PD, and ALS (Kondo et al., 2019; Lu et al., 2013; Sharma and Taliyan, 2015; Shukla and Tekwani, 2020). The neuroprotective ability of DNMT inhibitors has been demonstrated in primary neuron models of HD (Pan et al., 2016), but it remains to be evaluated in vivo in proof-of-concept preclinical animal studies for HD. The efficacy of several broad-spectrum HDAC inhibitors, including sodium phenylbutyrate, SAHA, and TSA has been proven in preclinical studies with different HD animal models as described above. A phase II safety and tolerability trial for human HD (PHEND-HD, NCT00212316) with sodium phenylbutyrate had been completed (Hogarth et al., 2007). But most of the pharmacological inhibitors targeting HDACs or DNMTs used in HD preclinical studies are broad-spectrum and not inhibiting a specific enzyme. The use of a broad-spectrum inhibitors may face many challenges, such as unintended off-target effects and consequent toxicity and may fail to induce beneficial long-term response. Selective targeting of key chromatin modifier(s) is required and have higher potential to lead to effective clinical treatments with less toxicity.
The epigenetic-targeted strategies identified in other related neuro-degenerative diseases, which share some common molecular bases of neurodegeneration with HD, may also have therapeutic utility in HD. Additional strategies already proven to be effective in animal models of AD, such as the use of HAT activators (Chatterjee et al., 2013, 2018) or the use of epi-editors (epigenetic editing) focusing on particular identified target regulatory regions (Bustos et al., 2017) may have therapeutic relevance for HD.
Age is a well-known primary risk factor for HD and other late-onset neurodegenerative diseases and affects the epigenome. Environmental factors and experience, such as exercise, diet, microbiomes, and social interactions, could also impact the epigenome (Kanherkar et al., 2014). The interaction between the environment and the epigenome may cause beneficial changes in gene expression phenotypes of HD and could offer potential non-pharmacological strategies, although which factors have beneficial or harmful effects on the HD epigenome still requires careful study.
8. Conclusions and future perspectives
In the past decade, a number of studies have revealed that mutant HTT expression induces genome-wide changes in multiple epigenetic modifications in human HD tissues and cell and animal models (Table 1), suggesting that altered chromatin structure contributes to HD pathogenesis. Targeting these alterations represents a potential therapeutic strategy. Development of selective inhibitors targeting specific epigenetic regulator(s) involved in transcriptional alterations and neuronal death in HD in vivo will help to establish effective therapeutic strategies with fewer side effects and toxicity. Pharmacological inhibitors of chromatin-modifying enzymes, including HDACs and DNMTs, have been shown to be neuroprotective and beneficial to HD in disease models. Understanding the mechanisms of neuroprotection triggered by these epigenetic-based treatments is likely to provide additional new molecular targets for HD therapy. Environmental factors that counteract mutant HTT-induced epigenetic defects or aging may have beneficial effects on HD. As discussed, mitochondria may be another mechanism of epigenetic control (Fig. 2). Therapeutic strategies targeting the action of mutant HTT on both nucleus and mitochondria may represent a promising therapeutic approach for HD. Epigenetic dysregulation in HD may influence DNA repair and genomic stability, in addition to transcriptional activity (Fig. 2), and impairment of these genomic functions may contribute to HD pathogenesis. Improved molecular understanding of epigenetic regulation is expected to provide important foundational knowledge for the development of therapeutic strategies targeting epigenetic abnormalities in HD and potentially other neurodegenerative diseases.
Acknowledgments
The authors apologize for omitted references. This work was supported by the National Institutes of Health grants, R01 NS111014 (to H. Y.), R21 NS103509 (to H.Y.), R01 NS051255 (to A.H.K.), and a Pilot Project Award from the Hope Center for Neurological Disorders at Washington University (to H.Y.).
Declaration of competing interest
J.W.H. and H.Y. declare no conflicts of interest. A.H.K. is a consultant for Monteris Medical and has research grants from Stryker, Monteris Medical, and Collagen Matrix, which are not relevant to this study.
Footnotes
CRediT authorship contribution statement
Jae Wook Hyeon: Writing – original draft. Albert H. Kim: Writing – review & editing. Hiroko Yano: Writing – original draft, Writing – review & editing.
References
- Achour M, Le Gras S, Keime C, Parmentier F, Lejeune FX, Boutillier AL, Neri C, Davidson I, Merienne K, 2015. Neuronal identity genes regulated by super-enhancers are preferentially down-regulated in the striatum of Huntington’s disease mice. Hum. Mol. Genet 24, 3481–3496. [DOI] [PubMed] [Google Scholar]
- Al-Dalahmah O, Sosunov AA, Shaik A, Ofori K, Liu Y, Vonsattel JP, Adorjan I, Menon V, Goldman JE, 2020. Single-nucleus RNA-seq identifies Huntington disease astrocyte states. Acta Neuropathol Commun 8, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ament SA, Pearl JR, Grindeland A, St Claire J, Earls JC, Kovalenko M, Gillis T, Mysore J, Gusella JF, Lee JM, et al. , 2017. High resolution time-course mapping of early transcriptomic, molecular and cellular phenotypes in Huntington’s disease CAG knock-in mice across multiple genetic backgrounds. Hum. Mol. Genet 26, 913–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai G, Cheung I, Shulha HP, Coelho JE, Li P, Dong X, Jakovcevski M, Wang Y, Grigorenko A, Jiang Y, et al. , 2015. Epigenetic dysregulation of hairy and enhancer of split 4 (HES4) is associated with striatal degeneration in postmortem Huntington brains. Hum. Mol. Genet 24, 1441–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnat M, Capizzi M, Aparicio E, Boluda S, Wennagel D, Kacher R, Kassem R, Lenoir S, Agasse F, Braz BY, et al. , 2020. Huntington’s disease alters human neurodevelopment. Science. 369, 787–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benn CL, Butler R, Mariner L, Nixon J, Moffitt H, Mielcarek M, Woodman B, Bates GP, 2009. Genetic knock-down of HDAC7 does not ameliorate disease pathogenesis in the R6/2 mouse model of Huntington’s disease. PloS One 4, e5747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berson A, Nativio R, Berger SL, Bonini NM, 2018. Epigenetic regulation in neurodegenerative diseases. Trends Neurosci. 41, 587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biagioli M, Ferrari F, Mendenhall EM, Zhang Y, Erdin S, Vijayvargia R, Vallabh SM, Solomos N, Manavalan P, Ragavendran A, et al. , 2015. Htt CAG repeat expansion confers pleiotropic gains of mutant huntingtin function in chromatin regulation. Hum. Mol. Genet 24, 2442–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird A, 2002. DNA methylation patterns and epigenetic memory. Genes Dev. 16, 6–21. [DOI] [PubMed] [Google Scholar]
- Bobrowska A, Paganetti P, Matthias P, Bates GP, 2011. Hdac6 knock-out increases tubulin acetylation but does not modify disease progression in the R6/2 mouse model of Huntington’s disease. PloS One 6, e20696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth LN, Brunet A, 2016. The aging epigenome. Mol. Cell 62, 728–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley NJ, Johnson R, Zuccato C, Bithell A, Cattaneo E, 2010. The role of REST in transcriptional and epigenetic dysregulation in Huntington’s disease. Neurobiol. Dis 39, 28–39. [DOI] [PubMed] [Google Scholar]
- Bustos FJ, Ampuero E, Jury N, Aguilar R, Falahi F, Toledo J, Ahumada J, Lata J, Cubillos P, Henriquez B, et al. , 2017. Epigenetic editing of the Dlg4/PSD95 gene improves cognition in aged and Alzheimer’s disease mice. Brain : J. Neurol 140, 3252–3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanero MR, Armstrong MI, Flemington EK, 2000. CpG methylation as a mechanism for the regulation of E2F activity. Proc. Natl. Acad. Sci. U. S. A 97, 6481–6486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell RR, Wood MA, 2019. How the epigenome integrates information and reshapes the synapse. Nat. Rev. Neurosci 20, 133–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos EI, Reinberg D, 2009. Histones: annotating chromatin. Annu. Rev. Genet 43, 559–599. [DOI] [PubMed] [Google Scholar]
- Castillo-Aguilera O, Depreux P, Halby L, Arimondo PB, Goossens L, 2017. DNA methylation targeting: the DNMT/HMT crosstalk challenge. Biomolecules 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha JH, 2007. Transcriptional signatures in Huntington’s disease. Prog. Neurobiol 83, 228–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S, Mizar P, Cassel R, Neidl R, Selvi BR, Mohankrishna DV, Vedamurthy BM, Schneider A, Bousiges O, Mathis C, et al. , 2013. A novel activator of CBP/p300 acetyltransferases promotes neurogenesis and extends memory duration in adult mice. J. Neurosci 33, 10698–10712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S, Cassel R, Schneider-Anthony A, Merienne K, Cosquer B, Tzeplaeff L, Haider Sinha S, Kumar M, Chaturbedy P, Eswaramoorthy M, et al. , 2018. Reinstating plasticity and memory in a tauopathy mouse model with an acetyltransferase activator. EMBO Mol. Med 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi RK, Beal MF, 2013. Mitochondria targeted therapeutic approaches in Parkinson’s and Huntington’s diseases. Molecular and cellular neurosciences 55, 101–114. [DOI] [PubMed] [Google Scholar]
- Chestnut BA, Chang Q, Price A, Lesuisse C, Wong M, Martin LJ, 2011. Epigenetic regulation of motor neuron cell death through DNA methylation. J. Neurosci 31, 16619–16636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu CT, Liu G, Leeds P, Chuang DM, 2011. Combined treatment with the mood stabilizers lithium and valproate produces multiple beneficial effects in transgenic mouse models of Huntington’s disease. Neuropsychopharmacology 36, 2406–2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopra V, Quinti L, Khanna P, Paganetti P, Kuhn R, Young AB, Kazantsev AG, Hersch S, 2016. LBH589, A hydroxamic acid-derived HDAC inhibitor, is neuroprotective in mouse models of Huntington’s disease. J. Huntingt. Dis 5, 347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens AW, Gabel HW, 2020. Emerging insights into the distinctive neuronal methylome. Trends Genet. 36, 816–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comet I, Riising EM, Leblanc B, Helin K, 2016. Maintaining cell identity: PRC2-mediated regulation of transcription and cancer. Nat. Rev. Cane 16, 803–810. [DOI] [PubMed] [Google Scholar]
- Cooper-Knock J, Kirby J, Ferraiuolo L, Heath PR, Rattray M, Shaw PJ, 2012. Gene expression profiling in human neurodegenerative disease. Nat. Rev. Neurol 8, 518–530. [DOI] [PubMed] [Google Scholar]
- Creusot F, Acs G, Christman JK, 1982. Inhibition of DNA methyltransferase and induction of Friend erythroleukemia cell differentiation by 5-azacytidine and 5-aza-2’-deoxycytidine. J. Biol. Chem 257, 2041–2048. [PubMed] [Google Scholar]
- Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D, 2006. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 127, 59–69. [DOI] [PubMed] [Google Scholar]
- Day JJ, Kennedy AJ, Sweatt JD, 2015. DNA methylation and its implications and accessibility for neuropsychiatric therapeutics. Annu. Rev. Pharmacol. Toxicol 55, 591–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Souza RA, Islam SA, McEwen LM, Mathelier A, Hill A, Mah SM, Wasserman WW, Kobor MS, Leavitt BR, 2016. DNA methylation profiling in human Huntington’s disease brain. Hum. Mol. Genet 25, 2013–2030. [DOI] [PubMed] [Google Scholar]
- Deneris ES, Hobert O, 2014. Maintenance of postmitotic neuronal cell identity. Nat. Neurosci 17, 899–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhayalan A, Rajavelu A, Rathert P, Tamas R, Jurkowska RZ, Ragozin S, Jeltsch A, 2010. The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J. Biol. Chem 285, 26114–26120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding N, Maiuri AR, O’Hagan HM, 2019. The emerging role of epigenetic modifiers in repair of DNA damage associated with chronic inflammatory diseases. Mutat. Res 780, 69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dion V, Wilson JH, 2009. Instability and chromatin structure of expanded trinucleotide repeats. Trends Genet. 25, 288–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dion V, Lin Y, Hubert L Jr., Waterland RA, Wilson JH, 2008. Dnmt1 deficiency promotes CAG repeat expansion in the mouse germline. Hum. Mol. Genet 17, 1306–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Tsuji J, Labadorf A, Roussos P, Chen JF, Myers RH, Akbarian S, Weng Z, 2015. The role of H3K4me3 in transcriptional regulation is altered in Huntington’s disease. PloS One 10, e0144398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragatsis I, Efstratiadis A, Zeitlin S, 1998. Mouse mutant embryos lacking huntingtin are rescued from lethality by wild-type extraembryonic tissues. Development 125, 1529–1539. [DOI] [PubMed] [Google Scholar]
- Dragileva E, Hendricks A, Teed A, Gillis T, Lopez ET, Friedberg EC, Kucherlapati R, Edelmann W, Lunetta KL, MacDonald ME, et al. , 2009. Intergenerational and striatal CAG repeat instability in Huntington’s disease knock-in mice involve different DNA repair genes. Neurobiol. Dis 33, 37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Johnson LM, Jacobsen SE, Patel DJ, 2015. DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Biol 16, 519–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dukatz M, Holzer K, Choudalakis M, Emperle M, Lungu C, Bashtrykov P, Jeltsch A, 2019. H3K36me2/3 binding and DNA binding of the DNA methyltransferase DNMT3A PWWP domain both contribute to its chromatin interaction. J. Mol. Biol 431, 5063–5074. [DOI] [PubMed] [Google Scholar]
- Dutto I, Scalera C, Prosperi E, 2018. CREBBP and p300 lysine acetyl transferases in the DNA damage response. Cell. Mol. Life Sci 75, 1325–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duyao MP, Auerbach AB, Ryan A, Persichetti F, Barnes GT, McNeil SM, Ge P, Vonsattel JP, Gusella JF, Joyner AL, et al. , 1995. Inactivation of the mouse Huntington’s disease gene homolog Hdh. Science 269, 407–410. [DOI] [PubMed] [Google Scholar]
- Endres M, Meisel A, Biniszkiewicz D, Namura S, Prass K, Ruscher K, Lipski A, Jaenisch R, Moskowitz MA, DirnagJ U, 2000. DNA methyltransferase contributes to delayed ischemic brain injury. J. Neurosci 20, 3175–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endres M, Fan G, Meisel A, Dirnagl U, Jaenisch R, 2001. Effects of cerebral ischemia in mice lacking DNA methyltransferase 1 in post-mitotic neurons. Neuroreport 12, 3763–3766. [DOI] [PubMed] [Google Scholar]
- Fan G, Beard C, Chen RZ, Csankovszki G, Sun Y, Siniaia M, Biniszkiewicz D, Bates B, Lee PP, Kuhn R, et al. , 2001. DNA hypomethylation perturbs the function and survival of CNS neurons in postnatal animals. J. Neurosci 21, 788–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farinelli P, Perera A, Arango-Gonzalez B, Trifunovic D, Wagner M, Carell T, Biel M, Zrenner E, Michalakis S, Paquet-Durand F, et al. , 2014. DNA methylation and differential gene regulation in photoreceptor cell death. Cell Death Dis. 5, e1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust C, Schumacher A, Holdener B, Magnuson T, 1995. The eed mutation disrupts anterior mesoderm production in mice. Development 121, 273–285. [DOI] [PubMed] [Google Scholar]
- Feng J, Chang H, Li E, Fan G, 2005. Dynamic expression of de novo DNA methyltransferases Dnmt3a and Dnmt3b in the central nervous system. J. Neurosci. Res 79, 734–746. [DOI] [PubMed] [Google Scholar]
- Feng J, Zhou Y, Campbell SL, Le T, Li E, Sweatt JD, Silva AJ, Fan G, 2010. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat. Neurosci 13, 423–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante RJ, Kubilus JK, Lee J, Ryu H, Beesen A, Zucker B, Smith K, Kowall NW, Ratan RR, Luthi-Carter R, et al. , 2003. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J. Neurosci 23, 9418–9427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante RJ, Ryu H, Kubilus JK, D’Mello S, Sugars KL, Lee J, Lu P, Smith K, Browne S, Beal MF, et al. , 2004. Chemotherapy for the brain: the antitumor antibiotic mithramycin prolongs survival in a mouse model of Huntington’s disease. J. Neurosci 24, 10335–10342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flower M, Lomeikaite V, Ciosi M, Cumming S, Morales F, Lo K, Hensman Moss D, Jones L, Holmans P, Investigators T-H, et al. , 2019. MSH3 modifies somatic instability and disease severity in Huntington’s and myotonic dystrophy type 1. Brain : J. Neurol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuks F, Burgers WA, Godin N, Kasai M, Kouzarides T, 2001. Dnmt3a binds deacetylases and is recruited by a sequence-specific repressor to silence transcription. EMBO J. 20, 2536–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuks F, Hurd PJ, Wolf D, Nan X, Bird AP, Kouzarides T, 2003. The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J. Biol. Chem 278, 4035–4040. [DOI] [PubMed] [Google Scholar]
- Gardian G, Browne SE, Choi DK, Klivenyi P, Gregorio J, Kubilus JK, Ryu H, Langley B, Ratan RR, Ferrante RJ, et al. , 2005. Neuroprotective effects of phenylbutyrate in the N171-82Q transgenic mouse model of Huntington’s disease. J. Biol. Chem 280, 556–563. [DOI] [PubMed] [Google Scholar]
- Gates LA, Foulds CE, O’Malley BW, 2017. Histone marks in the ’driver’s seat’: functional roles in steering the transcription cycle. Trends Biochem. Sci 42, 977–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gipson TA, Neueder A, Wexler NS, Bates GP, Housman D, 2013. Aberrantly spliced HTT, a new player in Huntington’s disease pathogenesis. RNA Biol. 10, 1647–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giralt A, Puigdellivol M, Carreton O, Paoletti P, Valero J, Parra-Damas A, Saura CA, Alberch J, Gines S, 2012. Long-term memory deficits in Huntington’s disease are associated with reduced CBP histone acetylase activity. Hum. Mol. Genet 21, 1203–1216. [DOI] [PubMed] [Google Scholar]
- Glajch KE, Sadri-Vakili G, 2015. Epigenetic mechanisms involved in Huntington’s disease pathogenesis. J. Huntingt. Dis 4, 1–15. [DOI] [PubMed] [Google Scholar]
- Genetic Modifiers of Huntington’s Disease Consortium, 2015. Identification of genetic factors that modify clinical onset of Huntington’s disease. Cell 162, 516–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genetic Modifiers of Huntington’s Disease Consortium, 2019. CAG repeat not polyglutamine length determines timing of Huntington’s disease onset. Cell 178, 887–900 e814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnyszka A, Jastrzebski Z, Flis S, 2013. DNA methyltransferase inhibitors and their emerging role in epigenetic therapy of cancer. Anti cancer Res. 33, 2989–2996. [PubMed] [Google Scholar]
- Goold R, Flower M, Moss DH, Medway C, Wood-Kaczmar A, Andre R, Farshim P, Bates GP, Holmans P, Jones L, et al. , 2019. FAN1 modifies Huntington’s disease progression by stabilizing the expanded HTT CAG repeat. Hum. Mol. Genet 28, 650–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbunova V, Seluanov A, Mittelman D, Wilson JH, 2004. Genome-wide demethylation destabilizes CTG.CAG trinucleotide repeats in mammalian cells. Hum. Mol. Genet 13, 2979–2989. [DOI] [PubMed] [Google Scholar]
- Graff J, Tsai LH, 2013. Histone acetylation: molecular mnemonics on the chromatin. Nat. Rev. Neurosci 14, 97–111. [DOI] [PubMed] [Google Scholar]
- Gray M, 2019. Astrocytes in Huntington’s disease. Adv. Exp. Med. Biol 1175, 355–381. [DOI] [PubMed] [Google Scholar]
- Guo JU, Su Y, Zhong C, Ming GL, Song H, 2011. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 145, 423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JU, Su Y, Shin JH, Shin J, Li H, Xie B, Zhong C, Hu S, Le T, Fan G, et al. , 2014a. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat. Neurosci 17, 215–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JU, Szulwach KE, Su Y, Li Y, Yao B, Xu Z, Shin JH, Xie B, Gao Y, Ming GL, et al. , 2014b. Genome-wide antagonism between 5-hydroxymethylcytosine and DNA methylation in the adult mouse brain. Front. Biol 9, 66–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusella JF, MacDonald ME, 2009. Huntington’s disease: the case for genetic modifiers. Genome Med. 1, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusella JF, MacDonald ME, Lee JM, 2014. Genetic modifiers of Huntington’s disease. Movement disorders, official journal of the Movement Disorder Society 29, 1359–1365. [DOI] [PubMed] [Google Scholar]
- Gutierrez A, Corey-Bloom J, Thomas EA, Desplats P, 2019. Evaluation of biochemical and epigenetic measures of peripheral brain-derived neurotrophic factor (BDNF) as a biomarker in Huntington’s disease patients. Front. Mol. Neurosci 12, 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HD iPSC Consortium, 2017. Developmental alterations in Huntington’s disease neural cells and pharmacological rescue in cells and mice. Nat. Neurosci 20, 648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, et al. , 2011. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333, 1303–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hervas-Corpion I, Guiretti D, Alcaraz-Iborra M, Olivares R, Campos-Caro A, Barco A, Valor LM, 2018. Early alteration of epigenetic-related transcription in Huntington’s disease mouse models. Sci. Rep 8, 9925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyward FD, Sweatt JD, 2015. DNA methylation in memory formation: emerging insights. Neuroscientist: a review journal bringing neurobiology, neurology and psychiatry 21, 475–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockly E, Richon VM, Woodman B, Smith DL, Zhou X, Rosa E, Sathasivam K, Ghazi-Noori S, Mahal A, Lowden PA, et al. , 2003. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proc. Natl. Acad. Sci. U. S. A 100, 2041–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges A, Strand AD, Aragaki AK, Kuhn A, Sengstag T, Hughes G, Elliston LA, Hartog C, Goldstein DR, Thu D, et al. , 2006. Regional and cellular gene expression changes in human Huntington’s disease brain. Hum. Mol. Genet 15, 965–977. [DOI] [PubMed] [Google Scholar]
- Hogarth P, Lovrecic L, Krainc D, 2007. Sodium phenylbutyrate in Huntington’s disease: a dose-finding study. Mov. Disord. : official journal of the Movement Disorder Society 22, 1962–1964. [DOI] [PubMed] [Google Scholar]
- Horvath S, 2013. DNA methylation age of human tissues and cell types. Genome Biol. 14, R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S, Langfelder P, Kwak S, Aaronson J, Rosinski J, Vogt TF, Eszes M, Faull RL, Curtis MA, Waldvogel HJ, et al. , 2016. Huntington’s disease accelerates epigenetic aging of human brain and disrupts DNA methylation levels. Aging (Albany NY) 8, 1485–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huertas D, Sendra R, Munoz P, 2009. Chromatin dynamics coupled to DNA repair. Epigenetics 4, 31–42. [DOI] [PubMed] [Google Scholar]
- Hyun K, Jeon J, Park K, Kim J, 2017. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med 49, e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inano K, Suetake I, Ueda T, Miyake Y, Nakamura M, Okada M, Tajima S, 2000. Maintenance-type DNA methyltransferase is highly expressed in post-mitotic neurons and localized in the cytoplasmic compartment. Journal of biochemistry 128, 315–321. [DOI] [PubMed] [Google Scholar]
- Irmak D, Fatima A, Gutierrez-Garcia R, Rinschen MM, Wagle P, Altmuller J, Arrigoni L, Hummel B, Klein C, Frese CK, et al. , 2018. Mechanism suppressing H3K9 trimethylation in pluripotent stem cells and its demise by polyQ-expanded huntingtin mutations. Hum. Mol. Genet 27, 4117–4134. [DOI] [PubMed] [Google Scholar]
- Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y, 2011. Tet proteins can convert 5-methylcytosine to 5-formyl cytosine and 5-carboxylcytosine. Science 333, 1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang HS, Shin WJ, Lee JE, Do JT, 2017. CpG and non-CpG methylation in epigenetic gene regulation and brain function. Genes 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeltsch A, Jurkowska RZ, 2016. Allosteric control of mammalian DNA methyltransferases-a new regulatory paradigm. Nucleic Acids Res. 44, 8556–8575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeppesen DK, Bohr VA, Stevnsner T, 2011. DNA repair deficiency in neurodegeneration. Prog. Neurobiol 94, 166–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia H, Kast RJ, Steffan JS, Thomas EA, 2012a. Selective histone deacetylase (HDAC) inhibition imparts beneficial effects in Huntington’s disease mice: implications for the ubiquitin-proteasomal and autophagy systems. Hum. Mol. Genet 21, 5280–5293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia H, Pallos J, Jacques V, Lau A, Tang B, Cooper A, Syed A, Purcell J, Chen Y, Sharma S, et al. , 2012b. Histone deacetylase (HDAC) inhibitors targeting HDAC3 and HDAC1 ameliorate polyglutamine-elicited phenotypes in model systems of Huntington’s disease. Neurobiol. Dis 46, 351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia H, Morris CD, Williams RM, Loring JF, Thomas EA, 2015. HDAC inhibition imparts beneficial transgenerational effects in Huntington’s disease mice via altered DNA and histone methylation. Proc. Natl. Acad. Sci. U. S. A 112, E56–E64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia H, Wang Y, Morris CD, Jacques V, Gottesfeld JM, Rusche JR, Thomas EA, 2016. The effects of pharmacological inhibition of histone deacetylase 3 (HDAC3) in Huntington’s disease mice. PloS One 11, e0152498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Poirier MA, Liang Y, Pei Z, Weiskittel CE, Smith WW, DeFranco DB, Ross CA, 2006. Depletion of CBP is directly linked with cellular toxicity caused by mutant huntingtin. Neurobiol. Dis 23, 543–551. [DOI] [PubMed] [Google Scholar]
- Jimenez-Sanchez M, Licitra F, Underwood BR, Rubinsztein DC, 2017. Huntington’s disease: mechanisms of pathogenesis and therapeutic strategies. Cold Spring Harb Perspect Med 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP, 1998. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet 19, 187–191. [DOI] [PubMed] [Google Scholar]
- Jung J, Bonini N, 2007. CREB-binding protein modulates repeat instability in a Drosophila model for polyQ disease. Science 315, 1857–1859. [DOI] [PubMed] [Google Scholar]
- Kanherkar RR, Bhatia-Dey N, Csoka AB, 2014. Epigenetics across the human lifespan. Front Cell Dev Biol 2, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakaidos P, Karagiannis D, Rampias T, 2020. Resolving DNA damage: epigenetic regulation of DNA repair. Molecules 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerschbamer E, Biagioli M, 2015. Huntington’s disease as neurodevelopmental disorder: altered chromatin regulation, coding, and non-coding RNA transcription. Front. Neurosci 9, 509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khristich AN, Mirkin SM, 2020. On the wrong DNA track: molecular mechanisms of repeat-mediated genome instability. J. Biol. Chem 295, 4134–4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Kim KT, 2014. Therapeutic approaches for inhibition of protein aggregation in Huntington’s disease. Experimental neurobiology 23, 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Moody JP, Edgerly CK, Bordiuk OL, Cormier K, Smith K, Beal MF, Ferrante RJ, 2010. Mitochondrial loss, dysfunction and altered dynamics in Huntington’s disease. Hum. Mol. Genet 19, 3919–3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KH, Hong EP, Shin JW, Chao MJ, Loupe J, Gillis T, Mysore JS, Holmans P, Jones L, Orth M, et al. , 2020. Genetic and functional analyses point to FAN1 as the source of multiple Huntington disease modifier effects. Am. J. Hum. Genet 107, 96–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H, 2013. Histone modifications for human epigenome analysis. J. Hum. Genet 58, 439–445. [DOI] [PubMed] [Google Scholar]
- Kondo N, Tohnai G, Sahashi K, Iida M, Kataoka M, Nakatsuji H, Tsutsumi Y, Hashizume A, Adachi H, Koike H, et al. , 2019. DNA methylation inhibitor attenuates polyglutamine-induced neurodegeneration by regulating Hes5. EMBO Mol. Med 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriaucionis S, Heintz N, 2009. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 324, 929–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn A, Goldstein DR, Hodges A, Strand AD, Sengstag T, Kooperberg C, Becanovic K, Pouladi MA, Sathasivam K, Cha JH, et al. , 2007. Mutant huntingtin’s effects on striatal gene expression in mice recapitulate changes observed in human Huntington’s disease brain and do not differ with mutant huntingtin length or wild type huntingtin dosage. Hum. Mol. Genet 16, 1845–1861. [DOI] [PubMed] [Google Scholar]
- Labadorf A, Hoss AG, Lagomarsino V, Latourelle JC, Hadzi TC, Bregu J, MacDonald ME, Gusella JF, Chen JF, Akbarian S, et al. , 2015. RNA sequence analysis of human Huntington disease brain reveals an extensive increase in inflammatory and developmental gene expression. PloS One 10, e0143563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbadia J, Morimoto RI, 2013. Huntington’s disease: underlying molecular mechanisms and emerging concepts. Trends Biochem. Sci 38, 378–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Cantle JP, Chatzopoulou D, Wang N, Gao F, Al-Ramahi I, Lu XH, Ramos EM, El-Zein K, Zhao Y, et al. , 2016. Integrated genomics and proteomics define huntingtin CAG length-dependent networks in mice. Nat. Neurosci 19, 623–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lardenoije R, Iatrou A, Kenis G, Kompotis K, Steinbusch HW, Mastroeni D, Coleman P, Lemere CA, Hof PR, van den Hove DL, et al. , 2015. The epigenetics of aging and neurodegeneration. Prog. Neurobiol 131, 21–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laugesen A, Hojfeldt JW, Helin K, 2016. Role of the polycomb repressive complex 2 (PRC2) in transcriptional regulation and cancer. Cold Spring Harb Perspect Med 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavery LA, Ure K, Wan YW, Luo C, Trostle AJ, Wang W, Jin H, Lopez J, Lucero J, Durham MA, et al. , 2020. Losing Dnmt3a dependent methylation in inhibitory neurons impairs neural function by a mechanism impacting Rett syndrome. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Gras S, Keime C, Anthony A, Lotz C, De Longprez L, Brouillet E, Cassel JC, Boutillier AL, Merienne K, 2017. Altered enhancer transcription underlies Huntington’s disease striatal transcriptional signature. Sci. Rep 7, 42875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CK, Weindruch R, Prolla TA, 2000. Gene-expression profile of the ageing brain in mice. Nat. Genet 25, 294–297. [DOI] [PubMed] [Google Scholar]
- Lee J, Hwang YJ, Shin JY, Lee WC, Wie J, Kim KY, Lee MY, Hwang D, Ratan RR, Pae AN, et al. , 2013. Epigenetic regulation of cholinergic receptor M1 (CHRM1) by histone H3K9me3 impairs Ca(2+) signaling in Huntington’s disease. Acta Neuropathol. 125, 727–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Hwang YJ, Kim Y, Lee MY, Hyeon SJ, Lee S, Kim DH, Jang SJ, Im H, Min SJ, et al. , 2017a. Remodeling of heterochromatin structure slows neuropathological progression and prolongs survival in an animal model of Huntington’s disease. Acta Neuropathol. 134, 729–748. [DOI] [PubMed] [Google Scholar]
- Lee JM, Chao MJ, Harold D, Abu Elneel K, Gillis T, Holmans P, Jones L, Orth M, Myers RH, Kwak S, et al. , 2017b. A modifier of Huntington’s disease onset at the MLH1 locus. Hum. Mol. Genet 26, 3859–3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Fenster RJ, Pineda SS, Gibbs WS, Mohammadi S, Davila-Velderrain J, Garcia FJ, Therrien M, Novis HS, Gao F, et al. , 2020. Cell type-specific transcriptomics reveals that mutant huntingtin leads to mitochondrial RNA release and neuronal innate immune activation. Neuron 107, 891–908 e898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E, Bestor TH, Jaenisch R, 1992. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 69, 915–926. [DOI] [PubMed] [Google Scholar]
- Li J, Hart RP, Mallimo EM, Swerdel MR, Kusnecov AW, Herrup K, 2013. EZH2-mediated H3K27 trimethylation mediates neurodegeneration in ataxia-telangiectasia. Nat. Neurosci 16, 1745–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman AP, Shakkottai VG, Albin RL, 2019. Polyglutamine repeats in neurodegenerative diseases. Annu. Rev. Pathol 14, 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S, Chesser AS, Grima JC, Rappold PM, Blum D, Przedborski S, Tieu K, 2011. D-beta-hydroxybutyrate is protective in mouse models of Huntington’s disease. PloS One 6, e24620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, et al. , 2009. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462, 315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loupe JM, Pinto RM, Kim KH, Gillis T, Mysore JS, Andrew MA, Kovalenko M, Murtha R, Seong I, Gusella JF, et al. , 2020. Promotion of somatic CAG repeat expansion by Fanl knock-out in Huntington’s disease knock-in mice is blocked by Mlh1 knock-out. Hum. Mol. Genet 29, 3044–3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, Yankner BA, 2004. Gene regulation and DNA damage in the ageing human brain. Nature 429, 883–891. [DOI] [PubMed] [Google Scholar]
- Lu H, Liu X, Deng Y, Qing H, 2013. DNA methylation, a hand behind neurodegenerative diseases. Front. Aging Neurosci 5, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu AT, Narayan P, Grant MJ, Langfelder P, Wang N, Kwak S, Wilkinson H, Chen RZ, Chen J, Simon Bawden C, et al. , 2020. DNA methylation study of Huntington’s disease and motor progression in patients and in animal models. Nat. Commun 11, 4529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ, 1997. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389, 251–260. [DOI] [PubMed] [Google Scholar]
- Luthi-Carter R, Strand A, Peters NL, Solano SM, Hollingsworth ZR, Menon AS, Frey AS, Spektor BS, Penney EB, Schilling G, et al. , 2000. Decreased expression of striatal signaling genes in a mouse model of Huntington’s disease. Hum. Mol. Genet 9, 1259–1271. [DOI] [PubMed] [Google Scholar]
- Luthi-Carter R, Strand AD, Hanson SA, Kooperberg C, Schilling G, La Spada AR, Merry DE, Young AB, Ross CA, Borchelt DR, et al. , 2002. Polyglutamine and transcription: gene expression changes shared by DRPLA and Huntington’s disease mouse models reveal context-independent effects. Hum. Mol. Genet 11, 1927–1937. [DOI] [PubMed] [Google Scholar]
- Lyko F, Brown R, 2005. DNA methyltransferase inhibitors and the development of epigenetic cancer therapies. J. Natl. Cancer Inst 97, 1498–1506. [DOI] [PubMed] [Google Scholar]
- Margueron R, Reinberg D, 2011. The Polycomb complex PRC2 and its mark in life. Nature 469, 343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matilainen O, Quiros PM, Auwerx J, 2017. Mitochondria and epigenetics-crosstalk in homeostasis and stress. Trends Cell Biol. 27, 453–463. [DOI] [PubMed] [Google Scholar]
- McColgan P, Tabrizi SJ, 2017. Huntington’s disease: a clinical review. Eur. J. Neurol [DOI] [PubMed] [Google Scholar]
- McFarland KN, Das S, Sun TT, Leyfer D, Xia E, Sangrey GR, Kuhn A, Luthi-Carter R, Clark TW, Sadri-Vakili G, et al. , 2012. Genome-wide histone acetylation is altered in a transgenic mouse model of Huntington’s disease. PloS One 7, e41423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland KN, Das S, Sun TT, Leyfer D, Kim MO, Xia E, Sangrey GR, Kuhn A, Luthi-Carter R, Clark TW, et al. , 2013. Genome-wide increase in histone H2A ubiquitylation in a mouse model of Huntington’s disease. J. Huntingt. Dis 2, 263–277. [DOI] [PubMed] [Google Scholar]
- McFarland KN, Huizenga MN, Darnell SB, Sangrey GR, Berezovska O, Cha JH, Outeiro TF, Sadri-Vakili G, 2014. MeCP2: a novel Huntingtin interactor. Hum. Mol. Genet 23, 1036–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellen M, Ayata P, Dewell S, Kriaucionis S, Heintz N, 2012. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell 151, 1417–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merienne N, Meunier C, Schneider A, Seguin J, Nair SS, Rocher AB, Le Gras S, Keime C, Faull R, Pellerin L, et al. , 2019. Cell-type-specific gene expression profiling in adult mouse brain reveals normal and disease-state signatures. Cell Rep. 26, 2477–2493 e2479. [DOI] [PubMed] [Google Scholar]
- Mielcarek M, Benn CL, Franklin SA, Smith DL, Woodman B, Marks PA, Bates GP, 2011. SAHA decreases HDAC 2 and 4 levels in vivo and improves molecular phenotypes in the R6/2 mouse model of Huntington’s disease. PloS One 6, e27746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mielcarek M, Landles C, Weiss A, Bradaia A, Seredenina T, Inuabasi L, Osborne GF, Wadel K, Touller C, Butler R, et al. , 2013. HDAC4 reduction: a novel therapeutic strategy to target cytoplasmic huntingtin and ameliorate neurodegeneration. PLoS Biol. 11, e1001717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammed SA, Ambrosini S, Luscher T, Paneni F, Costantino S, 2020. Epigenetic control of mitochondrial function in the vasculature. Front Cardiovasc Med 7, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore LD, Le T, Fan G, 2013. DNA methylation and its basic function. Neuropsychopharmacology 38, 23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris MJ, Monteggia LM, 2014. Role of DNA methylation and the DNA methyltransferases in learning and memory. Dialogues Clin. Neurosci 16, 359–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss DJH, Pardinas AF, Langbehn D, Lo K, Leavitt BR, Roos R, Durr A, Mead S, investigators T-H, investigators R, et al. , 2017. Identification of genetic variants associated with Huntington’s disease progression: a genome-wide association study. Lancet Neurol 16, 701–711. [DOI] [PubMed] [Google Scholar]
- Moumne L, Campbell K, Howland D, Ouyang Y, Bates GP, 2012. Genetic knockdown of HDAC3 does not modify disease-related phenotypes in a mouse model of Huntington’s disease. PloS One 7, e31080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamori M, Panigrahi GB, Lanni S, Gall-Duncan T, Hayakawa H, Tanaka H, Luo J, Otabe T, Li J, Sakata A, et al. , 2020. A slipped-CAG DNA-binding small molecule induces trinucleotide-repeat contractions in vivo. Nat. Genet 52, 146–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A, 1998. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 393, 386–389. [DOI] [PubMed] [Google Scholar]
- Nasir J, Floresco SB, O’Kusky JR, Diewert VM, Richman JM, Zeisler J, Borowski A, Marth JD, Phillips AG, Hayden MR, 1995. Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell 81, 811–823. [DOI] [PubMed] [Google Scholar]
- Neueder A, Bates GP, 2014. A common gene expression signature in Huntington’s disease patient brain regions. BMC Med. Genom 7, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neueder A, Landles C, Ghosh R, Howland D, Myers RH, Faull RLM, Tabrizi SJ, Bates GP, 2017. The pathogenic exon 1 HTT protein is produced by incomplete splicing in Huntington’s disease patients. Sci. Rep 7, 1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng CW, Yildirim F, Yap YS, Dalin S, Matthews BJ, Velez PJ, Labadorf A, Housman DE, Fraenkel E, 2013. Extensive changes in DNA methylation are associated with expression of mutant huntingtin. Proc. Natl. Acad. Sci. U. S. A 110, 2354–2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nucifora FC Jr., Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, Troncoso J, Dawson VL, et al. , 2001. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science 291, 2423–2428. [DOI] [PubMed] [Google Scholar]
- Okano M, Bell DW, Haber DA, Li E, 1999. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–257. [DOI] [PubMed] [Google Scholar]
- Orr HT, Zoghbi HY, 2007. Trinucleotide repeat disorders. Annu. Rev. Neurosci 30, 575–621. [DOI] [PubMed] [Google Scholar]
- Orr AL, Li S, Wang CE, Li H, Wang J, Rong J, Xu X, Mastroberardino PG, Greenamyre JT, Li XJ, 2008. N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J. Neurosci 28, 2783–2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palpagama TH, Waldvogel HJ, Faull RLM, Kwakowsky A, 2019. The role of microglia and astrocytes in Huntington’s disease. Front. Mol. Neurosci 12, 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Daito T, Sasaki Y, Chung YH, Xing X, Pondugula S, Swamidass SJ, Wang T, Kim AH, Yano H, 2016. Inhibition of DNA methyltransferases blocks mutant huntingtin-induced neurotoxicity. Sci. Rep 6, 31022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Zhu Y, Yang W, Tycksen E, Liu S, Palucki J, Zhu L, Sasaki Y, Sharma MK, Kim AH, et al. , 2018. The role of Twist1 in mutant huntingtin-induced transcriptional alterations and neurotoxicity. J. Biol. Chem 293, 11850–11866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panov AV, Gutekunst CA, Leavitt BR, Hayden MR, Burke JR, Strittmatter WJ, Greenamyre JT, 2002. Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nat. Neurosci 5, 731–736. [DOI] [PubMed] [Google Scholar]
- Parra M, Verdin E, 2010. Regulatory signal transduction pathways for class Ha histone deacetylases. Curr. Opin. Pharmacol 10, 454–460. [DOI] [PubMed] [Google Scholar]
- Pinto RM, Dragileva E, Kirby A, Lloret A, Lopez E, St Claire J, Panigrahi GB, Hou C, Holloway K, Gillis T, et al. , 2013. Mismatch repair genes Mlh1 and Mlh3 modify CAG instability in Huntington’s disease mice: genome-wide and candidate approaches. PLoS Genet. 9, e1003930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi IA, Mehler MF, 2018. Epigenetic mechanisms underlying nervous system diseases. Handb. Clin. Neurol 147, 43–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riva P, Ratti A, Venturin M, 2016. The long non-coding RNAs in neurodegenerative diseases: novel mechanisms of pathogenesis. Curr. Alzheimer Res 13, 1219–1231. [DOI] [PubMed] [Google Scholar]
- Rose NR, Klose RJ, 2014. Understanding the relationship between DNA methylation and histone lysine methylation. Biochim. Biophys. Acta 1839, 1362–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross CA, Tabrizi SJ, 2011. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 10, 83–98. [DOI] [PubMed] [Google Scholar]
- Rountree MR, Bachman KE, Baylin SB, 2000. DNMT1 binds HDAC2 and a new corepressor, DMAP1, to form a complex at replication foci. Nat. Genet 25, 269–277. [DOI] [PubMed] [Google Scholar]
- Ryu H, Lee J, Olofsson BA, Mwidau A, Dedeoglu A, Escudero M, Flemington E, Azizkhan-Clifford J, Ferrante RJ, Ratan RR, 2003. Histone deacetylase inhibitors prevent oxidative neuronal death independent of expanded polyglutamine repeats via an Sp1-dependent pathway. Proc. Natl. Acad. Sci. U. S. A 100, 4281–4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu H, Lee J, Hagerty SW, Soh BY, McAlpin SE, Cormier KA, Smith KM, Ferrante RJ, 2006. ESET/SETDB1 gene expression and histone H3 (K9) trimethylation in Huntington’s disease. Proc. Natl. Acad. Sci. U. S. A 103, 19176–19181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadri-Vakili G, Cha JH, 2006. Mechanisms of disease: histone modifications in Huntington’s disease. Nat. Clin. Pract. Neurol 2, 330–338. [DOI] [PubMed] [Google Scholar]
- Sadri-Vakili G, Bouzou B, Benn CL, Kim MO, Chawla P, Overland RP, Glajch KE, Xia E, Qiu Z, Hersch SM, et al. , 2007. Histones associated with downregulated genes are hypo-acetylated in Huntington’s disease models. Hum. Mol. Genet 16, 1293–1306. [DOI] [PubMed] [Google Scholar]
- Salinas RD, Connolly DR, Song H, 2020. Invited review: epigenetics in neurodevelopment. Neuropathol. Appl. Neurobiol 46, 6–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartorelli V, Lauberth SM, 2020. Enhancer RNAs are an important regulatory layer of the epigenome. Nat. Struct. Mol. Biol 27, 521–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathasivam K, Neueder A, Gipson TA, Landles C, Benjamin AC, Bondulich MK, Smith DL, Faull RL, Roos RA, Howland D, et al. , 2013. Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc. Natl. Acad. Sci. U. S. A 110, 2366–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlesinger Y, Straussman R, Keshet I, Farkash S, Hecht M, Zimmerman J, Eden E, Yakhini Z, Ben-Shushan E, Reubinoff BE, et al. , 2007. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat. Genet 39, 232–236. [DOI] [PubMed] [Google Scholar]
- Schmidt MHM, Pearson CE, 2016. Disease-associated repeat instability and mismatch repair. DNA Repair 38, 117–126. [DOI] [PubMed] [Google Scholar]
- Seong IS, Woda JM, Song JJ, Lloret A, Abeyrathne PD, Woo CJ, Gregory G, Lee JM, Wheeler VC, Walz T, et al. , 2010. Huntingtin facilitates polycomb repressive complex 2. Hum. Mol. Genet 19, 573–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seredenina T, Luthi-Carter R, 2012. What have we learned from gene expression profiles in Huntington’s disease? Neurobiol. Dis 45, 83–98. [DOI] [PubMed] [Google Scholar]
- Sharma S, Taliyan R, 2015. Transcriptional dysregulation in Huntington’s disease: the role of histone deacetylases. Pharmacol. Res 100, 157–169. [DOI] [PubMed] [Google Scholar]
- Shin J, Ming GL, Song H, 2014. DNA modifications in the mammalian brain. Philos. Trans. R. Soc. Lond. B Biol. Sci 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla S, Tekwani BL, 2020. Histone deacetylases inhibitors in neurodegenerative diseases, neuroprotection and neuronal differentiation. Front. Pharmacol 11, 537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siebzehnrubl FA, Raber KA, Urbach YK, Schulze-Krebs A, Canneva F, Moceri S, Habermeyer J, Achoui D, Gupta B, Steindler DA, et al. , 2018. Early postnatal behavioral, cellular, and molecular changes in models of Huntington disease are reversible by HDAC inhibition. Proc. Natl. Acad. Sci. U. S. A 115, E8765–E8774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ZD, Meissner A, 2013. DNA methylation: roles in mammalian development. Nat. Rev. Genet 14, 204–220. [DOI] [PubMed] [Google Scholar]
- Song CX, Szulwach KE, Fu Y, Dai Q, Yi C, Li X, Li Y, Chen CH, Zhang W, Jian X, et al. , 2011a. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat. Biotechnol 29, 68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Chen J, Petrilli A, Liot G, Klinglmayr E, Zhou Y, Poquiz P, Tjong J, Pouladi MA, Hayden MR, et al. , 2011b. Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related protein-1 and increases its enzymatic activity. Nat. Med 17, 377–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Zsindely N, Farago A, Marsh JL, Bodai L, 2018. Systematic genetic interaction studies identify histone demethylase Utx as potential target for ameliorating Huntington’s disease. Hum. Mol. Genet 27, 649–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stack EC, Del Signore SJ, Luthi-Carter R, Soh BY, Goldstein DR, Matson S, Goodrich S, Markey AL, Cormier K, Hagerty SW, et al. , 2007. Modulation of nucleosome dynamics in Huntington’s disease. Hum. Mol. Genet 16, 1164–1175. [DOI] [PubMed] [Google Scholar]
- Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu YZ, Gohler H, Wanker EE, Bates GP, Housman DE, Thompson LM, 2000. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl. Acad. Sci. U. S. A 97, 6763–6768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, et al. , 2001. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature 413, 739–743. [DOI] [PubMed] [Google Scholar]
- Stresemann C, Lyko F, 2008. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int. J. Cane 123, 8–13. [DOI] [PubMed] [Google Scholar]
- Sun JH, Zhou L, Emerson DJ, Phyo SA, Titus KR, Gong W, Gilgenast TG, Beagan JA, Davidson BL, Tassone F, et al. , 2018. Disease-associated short tandem repeats Co-localize with chromatin domain boundaries. Cell 175, 224–238 e215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki MM, Bird A, 2008. DNA methylation landscapes: provocative insights from epigenomics. Nat. Rev. Genet 9, 465–476. [DOI] [PubMed] [Google Scholar]
- Szulwach KE, Li X, Li Y, Song CX, Wu H, Dai Q, Irier H, Upadhyay AK, Gearing M, Levey AI, et al. , 2011. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat. Neurosci 14, 1607–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate PH, Bird AP, 1993. Effects of DNA methylation on DNA-binding proteins and gene expression. Curr. Opin. Genet. Dev 3, 226–231. [DOI] [PubMed] [Google Scholar]
- The Huntington’s Disease Collaborative Research Group, 1993. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72, 971–983. [DOI] [PubMed] [Google Scholar]
- Thomas EA, Coppola G, Desplats PA, Tang B, Soragni E, Burnett R, Gao F, Fitzgerald KM, Borok JF, Herman D, et al. , 2008. The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington’s disease transgenic mice. Proc. Natl. Acad. Sci. U. S. A 105, 15564–15569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tini M, Benecke A, Um SJ, Torchia J, Evans RM, Chambon P, 2002. Association of CBP/p300 acetylase and thymine DNA glyeosylase links DNA repair and transcription. Mol. Cell 9, 265–277. [DOI] [PubMed] [Google Scholar]
- Tognini P, Napoli D, Pizzorusso T, 2015. Dynamic DNA methylation in the brain: a new epigenetic mark for experience-dependent plasticity. Front. Cell. Neurosci 9, 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tome S, Manley K, Simard JP, Clark GW, Slean MM, Swami M, Shelbourne PF, Tillier ER, Monckton DG, Messer A, et al. , 2013. MSH3 polymorphisms and protein levels affect CAG repeat instability in Huntington’s disease mice. PLoS Genet. 9, e1003280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsankova N, Renthal W, Kumar A, Nestler EJ, 2007. Epigenetic regulation in psychiatric disorders. Nat. Rev. Neurosci 8, 355–367. [DOI] [PubMed] [Google Scholar]
- Valor LM, Guiretti D, Lopez-Atalaya JP, Barco A, 2013. Genomic landscape of transcriptional and epigenetic dysregulation in early onset polyglutamine disease. J. Neurosci 33, 10471–10482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Knaap JA, Verrijzer CP, 2016. Undercover: gene control by metabolites and metabolic enzymes. Genes Dev. 30, 2345–2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Mierlo G, Veenstra GJC, Vermeulen M, Marks H, 2019. The complexity of PRC2 subcomplexes. Trends Cell Biol. 29, 660–671. [DOI] [PubMed] [Google Scholar]
- Vashishtha M, Ng CW, Yildirim F, Gipson TA, Kratter IH, Bodai L, Song W, Lau A, Labadorf A, Vogel-Ciernia A, et al. , 2013. Targeting H3K4 trimethylation in Huntington disease. Proc. Natl. Acad. Sci. U. S. A 110, E3027–E3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villar-Menendez I, Blanch M, Tyebji S, Pereira-Veiga T, Albasanz JL, Martin M, Ferrer I, Perez-Navarro E, Barrachina M, 2013. Increased 5-methylcytosine and decreased 5-hydroxymethylcytosine levels are associated with reduced striatal A2AR levels in Huntington’s disease. NeuroMolecular Med. 15, 295–309. Valor, 2013 #98}. [DOI] [PubMed] [Google Scholar]
- Vire E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, Morey L, Van Eynde A, Bernard D, Vanderwinden JM, et al. , 2006. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 439, 871–874. [DOI] [PubMed] [Google Scholar]
- von Schimmelmann M, Feinberg PA, Sullivan JM, Ku SM, Badimon A, Duff MK, Wang Z, Lachmann A, Dewell S, Ma’ayan A, et al. , 2016. Polycomb repressive complex 2 (PRC2) silences genes responsible for neurodegeneration. Nat. Neurosci 19, 1321–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker FO, 2007. Huntington’s disease. Lancet 369, 218–228. [DOI] [PubMed] [Google Scholar]
- Wang F, Yang Y, Lin X, Wang JQ, Wu YS, Xie W, Wang D, Zhu S, Liao YQ, Sun Q, et al. , 2013a. Genome-wide loss of 5-hmC is a novel epigenetic feature of Huntington’s disease. Hum. Mol. Genet 22, 3641–3653. [DOI] [PubMed] [Google Scholar]
- Wang Y, Wang X, Li R, Yang ZF, Wang YZ, Gong XL, Wang XM, 2013b. A DNA methyltransferase inhibitor, 5-aza-2’-deoxycytidine, exacerbates neurotoxicity and upregulates Parkinson’s disease-related genes in dopaminergic neurons. CNS Neurosci. Ther. 19, 183–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg DN, Papillon-Cavanagh S, Chen H, Yue Y, Chen X, Rajagopalan KN, Horth C, McGuire JT, Xu X, Nikbakht H, et al. , 2019. The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature 573, 281–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wexler NS, Lorimer J, Porter J, Gomez F, Moskowitz C, Shackell E, Marder K, Penchaszadeh G, Roberts SA, Gayan J, et al. , 2004. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington’s disease age of onset. Proc. Natl. Acad. Sci. U. S. A 101, 3498–3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weydt P, Pineda VV, Torrence AE, Libby RT, Satterfield TF, Lazarowski ER, Gilbert ML, Morton GJ, Bammler TK, Strand AD, et al. , 2006. Thermoregulatory and metabolic defects in Huntington’s disease transgenic mice implicate PGC-1alpha in Huntington’s disease neurodegeneration. Cell Metabol. 4, 349–362. [DOI] [PubMed] [Google Scholar]
- Woda JM, Calzonetti T, Hilditch-Maguire P, Duyao MP, Conlon RA, MacDonald ME, 2005. Inactivation of the Huntington’s disease gene (Hdh) impairs anterior streak formation and early patterning of the mouse embryo. BMC Dev. Biol 5, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YY, Kuo HC, 2020. Functional roles and networks of non-coding RNAs in the pathogenesis of neurodegenerative diseases. J. Biomed. Sci 27, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Zhang Y, 2017. TET-mediated active DNA demethylation: mechanism, function and beyond. Nat. Rev. Genet 18, 517–534. [DOI] [PubMed] [Google Scholar]
- Xin YJ, Yuan B, Yu B, Wang YQ, Wu JJ, Zhou WH, Qiu Z, 2015. Tet1-mediated DNA demethylation regulates neuronal cell death induced by oxidative stress. Sci. Rep 5, 7645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano H, Baranov SV, Baranova OV, Kim J, Pan Y, Yablonska S, Carlisle DL, Ferrante RJ, Kim AH, Friedlander RM, 2014. Inhibition of mitochondrial protein import by mutant huntingtin. Nat. Neurosci 17, 822–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildirim F, Ng CW, Kappes V, Ehrenberger T, Rigby SK, Stivanello V, Gipson TA, Soltis AR, Vanhoutte P, Caboche J, et al. , 2019. Early epigenomic and transcriptional changes reveal Elk-1 transcription factor as a therapeutic target in Huntington’s disease. Proc. Natl. Acad. Sci. U. S. A 116, 24840–24851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu ZX, Li SH, Evans J, Pillarisetti A, Li H, Li XJ, 2003. Mutant huntingtin causes context-dependent neurodegeneration in mice with Huntington’s disease. J. Neurosci 23, 2193–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zadel M, Maver A, Kovanda A, Peterlin B, 2018. DNA methylation profiles in whole blood of Huntington’s disease patients. Front. Neurol 9, 655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zadora D, Geist A, Vamps E, Vesey L, Klivenyi P, 2009. Valproate ameliorates the survival and the motor performance in a transgenic mouse model of Huntington’s disease. Pharmacol. Biochem. Behav 94, 148–153. [DOI] [PubMed] [Google Scholar]
- Zeitlin S, Liu JP, Chapman DL, Papaioannou VE, Efstratiadis A, 1995. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington’s disease gene homologue. Nat. Genet 11, 155–163. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Ng HH, Erdjument-Bromage H, Tempst P, Bird A, Reinberg D, 1999. Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev. 13, 1924–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Jurkowska R, Soeroes S, Rajavelu A, Dhayalan A, Bock I, Rathert P, Brandt O, Reinhardt R, Fischle W, et al. , 2010. Chromatin methylation activity of Dnmt3a and Dnmt3a/3L is guided by interaction of the ADD domain with the histone H3 tail. Nucleic Acids Res. 38, 4246–4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, et al. , 2001. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science 293, 493–498. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Tartari M, Crotti A, Goffredo D, Valenza M, Conti L, Cataudella T, Leavitt BR, Hayden MR, Timmusk T, et al. , 2003. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat. Genet 35, 76–83. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Belyaev N, Conforti P, Ooi L, Tartari M, Papadimou E, MacDonald M, Fossale E, Zeitlin S, Buckley N, et al. , 2007. Widespread disruption of repressor element-1 silencing transcription factor/neuron-restrictive silencer factor occupancy at its target genes in Huntington’s disease. J. Neurosci 27, 6972–6983. [DOI] [PMC free article] [PubMed] [Google Scholar]