

Abstract
The emergence of the severe acute respiratory syndrome coronavirus‐2 (SARS‐CoV‐2) with the most contagious variants, alpha (B.1.1.7), beta (B.1.351), delta (B.1.617.2), and Omicron (B.1.1.529) has continuously added a higher number of morbidity and mortality, globally. The present integrated bioinformatics–cheminformatics approach was employed to locate potent antiviral marine alkaloids that could be used against SARS‐CoV‐2. Initially, 57 antiviral marine alkaloids and two repurposing drugs were selected from an extensive literature review. Then, the putative target enzyme SARS‐CoV‐2 main protease (SARS‐CoV‐2‐Mpro) was retrieved from the protein data bank and carried out a virtual screening‐cum‐molecular docking study with all candidates using PyRx 0.8 and AutoDock 4.2 software. Further, the molecular dynamics (MD) simulation of the two most potential alkaloids and a drug docking complex at 100 ns (with two ligand topology files from PRODRG and ATB server, separately), the molecular mechanics/Poisson‐Boltzmann surface area (MM/PBSA) free energy, and contributions of entropy were investigated. Then, the physicochemical‐toxicity‐pharmacokinetics‐drug‐likeness profiles, the frontier molecular orbitals energies (highest occupied molecular orbital, lowest unoccupied molecular orbital, and ΔE), and structural–activity relationship were assessed and analyzed. Based on binding energy, 8‐hydroxymanzamine (−10.5 kcal/mol) and manzamine A (−10.1 kcal/mol) from all alkaloids with darunavir (−7.9 kcal/mol) and lopinavir (−7.4 kcal/mol) against SARS‐CoV‐2‐Mpro were recorded. The MD simulation (RMSD, RMSF, Rg, H‐bond, MM/PBSA binding energy) illustrated that the 8‐hydroxymanzamine exhibits a static thermodynamic feature than the other two complexes. The predicted physicochemical, toxicity, pharmacokinetics, and drug‐likeness profiles also revealed that the 8‐hydroxymanzamine could be used as a potential lead candidate individually and/or synergistically with darunavir or lopinavir to combat SARS‐CoV‐2 infection after some pharmacological validation.
Keywords: antiviral marine alkaloids, drug‐likeness profiles prediction, molecular docking simulation, severe acute respiratory syndrome coronavirus‐2‐Mpro
1. INTRODUCTION
Coronavirus disease‐19 (COVID‐19) has created a global health emergency with unexpected mortality since the end of 2019. 1 , 2 Besides, crisis from the newly emerged mutated SARS‐CoV‐2‐alpha (B.1.1.7), beta (B.1.351), gamma (P.1), theta (P.3), kappa (B.1.617.1), delta (B.1.617.2), delta‐plus (B.1.617.2), and Omicron (B.1.1.529) strains have been posing more challenges for the applied or proposed drugs and vaccines. 2 , 3 , 4 The discriminating infection and re‐infection (second and third waves of SARS‐CoV‐2) without confirmative symptoms and unexpected morbidity‐mortality rates in certain regions and communities without any potential therapy by the Food and Drug Administration (FDA), “added fuel to the fire” at this health emerging situation. 3 , 4 At present, re‐purposing anti‐viral, anti‐inflammatory, immune‐modulating, antiparasitic regimens, including natural/herbal regimens is a frontline approach to moderately control SARS‐CoV‐2 pathogenesis. 5 , 6 However, the efficacy of applied re‐purposing drugs and vaccines varies from patient to region. 7 , 8 Although, proposed drugs and vaccine candidates got permission for emergency use and few drugs were also recommended by FDA to control the pandemic situation. But we still need more potential drugs with higher efficacy and fewer side effects. 7 , 8 Therefore, the current situation directly indicated exploring and developing more therapeutic candidates at a priority basis for the ongoing drug development module to save the human community from highly pathogenic SARS‐CoV‐2 strains. 5 , 9
Therefore, locating potential natural products from existing resources is considered as ideal research to accelerate and/or offer other antiviral drug development research groups or pharmaceutical companies to take over the preliminary findings for further pharmacological investigation against SARS‐CoV‐2. 9 , 10 , 11 , 12 Prima facie, several natural secondary metabolites isolated from eukaryotic and prokaryotic origins are continuously investigated against SARS‐CoV‐2 experimental (in vitro) and computational (in silico) methods. Opportunistically some of the regimens are also entered into different stages of clinical trials. Thus, in an emergency, the computer‐aided drug design (CADD) platform can trace several potential leads and proceed with such recognized potent candidates for an experimental validation expected to be the cost‐effective or resource‐saving drug development strategy against SARS‐CoV‐2. 6 , 13 , 14 , 15 , 16 , 17
From the history of natural product‐based drug discovery, marine diversity is one of the most substantial contributors of potentially unique metabolites with a wide range of mainstream therapeutic applications, mainly against cancer and infectious diseases. 6 , 18 , 19 , 20 For example, aztreonam (antibiotic), brentuximab vedotin (anticancer), ecteinascidin‐743 (anti‐cancer), erythromycin (anti‐bacterial), imipenem (antimicrobial), tetracycline (anti‐bacterial), vancomycin (anti‐microbial), vidarabine (anti‐viral drug), rifampicin (anti‐tubercular) are some rational supplements to mainstream drug discovery from the marine resource. In particular reference to marine‐derived nitrogenous constituents belonging to the alkaloid class of compounds displayed potent anti‐viral activity against human immunodeficiency virus (HIV), herpes simplex virus‐I/II (HSV‐I/ II), tobacco mosaic virus (TMV), influenza subtype or swine flu (H1N1), coronavirus (CoV‐A95), and so on from previous experimental reports (see Table S1). Overall, the alkaloid class of compounds has a higher success rate than other natural products. Thus, selecting such broad‐spectrum antiviral marine alkaloids from previous experimental and using those alkaloids against SARS‐CoV‐2 could be a rational approach in the current drug development module.
Currently, the massive effort of worldwide scientific groups, crystallographic protein structures of newly emerged SARS‐CoV‐2 main protease or 3‐chymotrypsin‐like cysteine protease (Mpro or 3CLpro), papine‐like protease (PLpro), spike glycoprotein (S‐protein) and RNA‐dependent RNA polymerase (RdRp) provided structural insights for novel drug development. 21 , 22 Significantly, the Mpro is one of the most putative drug targets for antiviral drug development associated with viral–host interaction for assembly, formation, and replication of virus genomes in the host cell. 9 , 10 , 11 , 12 , 21 This work examined 57 antiviral marine alkaloids (see Table S1) followed by an advanced computational platform targeting SARS‐CoV‐2‐Mpro. Further, advanced molecular dynamics simulation (MD simulation) at 100 ns with comparative analysis two different topology files generated by the PRODRG and the automated force field topology builder (ATB) server, the molecular mechanics/Poisson‐Boltzmann surface area (MM/PBSA) binding energy calculation with drug‐ability‐toxicity‐pharmacokinetics prediction, structural activity relationship (SAR), and frontier molecular orbitals (FMOs) in a combined “biophysics‐quantum chemistry” approach using various bioinformatics and cheminformatics tools to locate the potential “lead” against SARS‐CoV‐2‐Mpro (see Figure 1).
FIGURE 1.
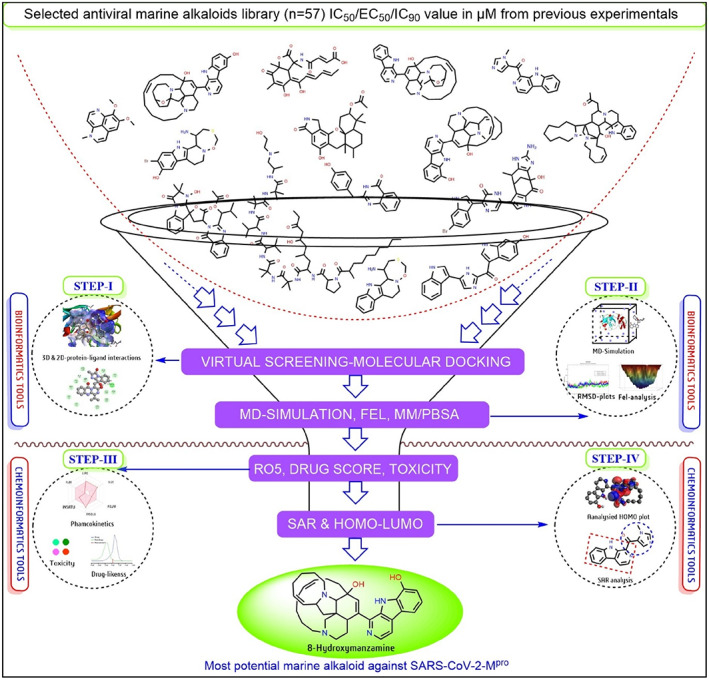
Schematic presentation of adopted systematic computational strategy to select potential anti‐SARS‐CoV‐2‐Mpro candidates from a library of antiviral marine alkaloids
2. MATERIALS AND METHODS
2.1. Preparation of ligand and target structure for molecular docking study
The entire computational work on a Linux‐Ubuntu 16.04 LTS workstation was carried out. According to the hypothesis, 57 antiviral marine alkaloids and two re‐purposing anti‐HIV drugs darunavir and lopinavir were retrieved for this study. Then, we have saved all chemical structures used as ligands were saved in (.pdb) file format for computational study. Parallelly, 306 amino acids consisting of x‐ray crystallographic of the SARS‐CoV‐2‐Mpro (PDB ID: 6Y2E) was extracted from the protein data bank and removed hetatm (water) before docking study. From literature and structural analysis with the BIOVIA Discovery Studio (BIOVIA DSB v4.5) software confirmed that the Mpro (6Y2E) protein structure is a homodimer protein structure present without any ligand and prior active site information. 16 , 23 From the source, it covers more than 11 cleavage sites with three domains, Domain I (10–99 residues), Domain II (100–182 residues), involved with viral RNA synthesis pathways and a globular cluster as Domain III (198–303 residues), regulating the dimerization of the Mpro. Accordingly, to identify ligand best interaction poses in a defined grid box in X‐, Y‐ and Z‐coordinates within dimensions (86 × 50 × 86) with a grid spacing of 0.675 Å mostly covering the catalytic residues using the virtual screening PyRx 0.8 software. 16 , 17 , 22 Again, we re‐dock the best candidates in AutoDock 4.2 software using the same grid size against SARS‐CoV‐2‐Mpro to analyze the reliability of docking score and protein–ligand interaction. 16 , 22 We have used the BIOVIA DSB v4.5 software to visualize intermolecular interactions of protein–ligand docking complexes in both two‐dimensional (2D) and three‐dimensional (3D) formats. 6 , 16
2.2. MD simulation of protein–ligand docking complexes
Based on a lower binding affinity/docking score (kcal/mol), the two most potential marine alkaloids manzamine A and 8‐hydroxymanzamine with one standard drug (darunavir) docking complexes were selected for MD simulation study with GROMACS 5.1.4 at a time scale of 100 ns using GROMOS96 54a7 force field. 16 , 24 For MD simulation, the required ligand topological files were generated by PRODRG as well as the ATB server for a comparative analysis based on ligand topology. Briefly, after analysis of PRODRG topology files, we have against generated the topology of two potential marine alkaloids from the ATB server to compare the topologies in the same way. The SPCE water‐cubic box model was employed and a total of 30 564, 30 573, and 30 563 solvent molecules were counterbalanced by adding 4 Na+ ions and performed the energy minimization using 50 000 steepest descent steps for each complex. 16 , 17 After minimizing the system, the NVT (number of particles, volume, and temperature) and NPT (number of particles, pressure, temperature) steps were performed for balancing all three systems on a 100 ps time scale, respectively. The final MD production was executed for the protein system at 100 ns with time steps of 2 fs (0.002 ps).
2.3. Principal component analysis and free energy landscape investigation
Every nanosecond conformation of biomolecules corresponds to free energy contribution by computing through free energy landscape (FEL) analysis. Moreover, the FEL valley and hillside plots of MD simulated docking complexes with two different topology files of marine‐1 and marine‐2 separately were plotted using the resultant trajectory of the root means square fluctuation (RMSD), the root means square fluctuation (RMSF), the radius of gyration (Rg) and the hydrogen–bond interaction of each protein–ligand complex. Employed the Gromacs utility tools for the MD simulations trajectory analysis with in‐build Gromacs commands. The suitable statistical analysis, the principal component analysis (PCA) was also employed separately to spot the most high‐amplitude motion through respective eigenvectors of the covariance matrix of atomic fluctuations using the gmx_covar command in respect to the acquired charge of in two different topology files. 16 , 17 , 22 The PCA was mainly employed to calculate eigenvectors, eigenvalues, and associated projections (primarily the first two principal components: PC1 and PC2) with the gmx_anaeig command to understand the global motions of all system coordinates of each docking complex (protein and ligand).
2.4. MM/PBSA and binding free energy per residue analysis
To distinguish the dynamic behavior of ligand interaction in the form of the binding energy of two separately generated topology files for simulated protein–ligand docking complexes, the widely used MM/PBSA method for binding free energy (ΔGbind) with the g_mmpbsav5.12 suite in the GROMACS platform was performed. 16 , 17 , 22 The following equations was used to calculate Gibb's binding free energy (ΔGbind):
| (1) |
| (2) |
| (3) |
where ΔG = free energy; ΔGcomplex = free energy of protein–ligand complex, ΔGprotein = free energy of protein, and ΔGligand free energy of ligand in the solvent. TΔS = entropic contribution in vacuum for free energy, T = temperature, and S = entropy. ΔGsolv = solvation free energy, Gpolar = electrostatic, and Gnonpolar = nonelectrostatic contributions to the solvation free energy. ΔEMM is the vacuum potential energy of both bonded and nonbonded interactions.
Simultaneously, since entropy unlike free energies is dependent on the molecule's whole phase space, it cannot be derived as an average from an MD simulation like free energies can be. According to Schlitter, MD simulations of atomic fluctuations can be used to estimate an upper bound on a macromolecule's absolute entropy. 25 There have been applications for this approach in estimating the entropy of translations, rotations, and conformational changes. The quasi‐harmonic approach was presented by Levy et al., which calculated the atom's absolute entropy to the covariance matrix in Cartesian coordinates. 26 Thus, the entropy was calculated through both Schlitter's formula and quasi‐harmonic analysis. 25 , 26
2.5. Physicochemical and toxicity profiles prediction
Using generated simplified molecular‐input line‐entry system (SMILES) code of each ligand, the physicochemical properties, or the Lipinski rules of five (RO5) parameters such as molecular weight (MW), H‐bond donor (H‐bd), H‐bond acceptor (H‐ba), partition coefficient value (XLogP), topological polar surface area (tPSA) were recorded from Swiss‐ADME tool (http://www.swissadme.ch/). The possible toxicity profiles such as hepatotoxicity (HT), immunotoxicity (IT), mutagenicity (MG), toxicity class (TC), 50% lethal dose or LD50 (mg/kg) were predicted using the ProTox tool (http://tox.charite.de/protox_II/).
2.6. Pharmacokinetics and overall drug‐likeness profiles prediction
Using the same SMILES code, assessed the absorption, distribution, metabolism, excretion, and toxicity (ADME/T) with bioavailability overall known as pharmacokinetics profiles of ligands structure using the Swiss‐ADME tool. Finally, the drug‐likeness score for each alkaloid along with two anti‐HIV drugs was predicated using the tool, Molsoft (http://molsoft.com/mprop/).
2.7. FMOs and SAR analyses
We have analyzed the intermolecular reaction or kinetic outlines in the form of the highest occupied molecular orbital (HOMO), the lowest unoccupied molecular orbital (LUMO) plots, and energy gap (ΔE) of two potential marine alkaloids with two standard drugs using software, Avogadro‐ORCA 1.2.0 platform. 27 The Universal Force Field (UFF) with steepest descent algorithms for energy minimization of structure with the restricted Hartree–Fock (RHF) principle was employed with single point calculation method to compute the electron wave function in form of HOMO, LUMO, and ΔE orbital energy. Simultaneously, the most potential theoretical drug prospective approach in medicinal chemistry, the SAR study was employed to determine the structural configuration of potential ligands linked with their biological efficacy. 6 , 28 , 29 Mainly, the ChemDraw 18.2 software was used to display the ligand's chemical structure to represent the SAR results from the drug perspective point of view.
3. RESULTS
3.1. Preparation of ligand and target structure for molecular docking study
The molecular docking score of 57 marine‐alkaloids with two anti‐HIV drugs against SARS‐CoV‐2‐Mpro was recorded (Table 1). From which alkaloid no‐10 (bromotopsentin), 21 (dihydrodeoxybromotopsentin), 25 (dragmacidin F), 35 (manadomanzamine A), 36 (manadomanzamine B), 37 (manzamine A), 38 (manzamine E), 39 (manzamine F), 40 (manzamine X), 42 (neosartoryadin B), 45 (petrosin), 48 (quinadoline B), 56 (6‐deoxymanzamine X), 57 (8‐hydroxymanzamine) were recorded as most potential candidates against SARS‐CoV‐2‐Mpro with docking score >9.0 kcal/mol. The alkaloid no‐37 (manzamine A) with docking score −10.2 kcal/mol (His163, Glu166, Leu167, and His172) and alkaloid no‐57 (8‐hydroxymanzamine) with docking score −10.5 kcal/mol (Gln110, Thr111, Ile249, Phe294, and Val297), while darunavir and lopinavir displayed −7.9 kcal/mol (Tyr126, Trp207, Leu282, and Glu288) and −7.4 kcal/mol (Arg4, Lys137, Leu286, and Glu288) against SARS‐CoV‐2‐Mpro (Table 1). Overall, the 8‐hydroxymanzamine from alkaloids and darunavir from standard drugs were the most effective against SARS‐CoV‐2‐Mpro. Thus, SARS‐CoV‐2‐Mpro‐Marine‐1 (manzamine‐A), SARS‐CoV‐2‐Mpro‐Marine‐2 (8‐hydroxy manzamine), and SARS‐CoV‐2‐Mpro‐Drug (darunavir) docking complexes were selected for stability or physical movements of atoms and molecules in a specific computational environment related to the biological system by MD simulation at 100 ns.
TABLE 1.
Molecular docking score (kcal/mol) against SARS‐CoV‐2‐Mpro with the drug‐likeness score, LD50 value and toxicity profiles of antiviral marine alkaloids and two anti‐HIV standard drugs
| Sl. No. | Drug‐ability prediction | Toxicity profile prediction | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mpro | DL | LD50 | BA | HT | CG | IT | MG | CT | TC | |
| 1. | −6.3 | −0.87 | 640 | 0.55 | MS | MR | HR | HR | HS | IV |
| 2. | −7.9 | 0.54 | 4000 | 0.55 | HS | MS | HR | MS | MS | V |
| 3. | −6.4 | 1.00 | 746 | 0.55 | HS | MR | MS | MS | MS | IV |
| 4. | −7.4 | 0.68 | 1600 | 0.17 | HS | MS | HR | HS | MS | IV |
| 5. | −7.1 | 0.65 | 1637 | 0.17 | HS | MS | HR | MS | MS | IV |
| 6. | −8.1 | 0.96 | 1190 | 0.17 | MR | MS | MS | HS | HS | IV |
| 7. | −5.9 | −0.75 | 960 | 0.55 | MS | MS | HS | MR | MS | IV |
| 8. | −6.1 | −0.66 | 1800 | 0.55 | MS | MS | HR | MR | HS | IV |
| 9. | −6.3 | −0.79 | 260 | 0.55 | MS | MS | HS | MS | HS | III |
| 10. | −9.1 | −0.71 | 1200 | 0.55 | MR | MS | MS | MS | MS | IV |
| 11. | −8.3 | −0.95 | 1000 | 0.55 | MR | MS | HS | MS | MS | IV |
| 12. | −5.8 | 1.17 | 1145 | 0.17 | HS | MS | HR | MS | HS | IV |
| 13. | −6.2 | 1.14 | 1145 | 0.17 | HS | MS | HR | MS | MS | IV |
| 14. | −5.9 | 1.28 | 1 | 0.17 | HS | MR | HR | MS | HS | I |
| 15. | −5.8 | 1.32 | 1 | 0.17 | HS | MS | HR | MS | HS | I |
| 16. | −5.9 | 1.32 | 1 | 0.17 | HS | MS | HR | MS | HS | I |
| 17. | −6.4 | −0.23 | 205 | 0.55 | MS | MR | HS | MR | MS | III |
| 18. | −6.9 | 0.92 | 1367 | 0.55 | HS | MR | HR | MS | MS | IV |
| 19. | −6.3 | 0.78 | 1807 | 0.55 | HS | MR | HS | MS | MS | IV |
| 20. | −7.5 | 0.91 | 850 | 0.55 | MS | HS | HR | MR | MS | IV |
| 21. | −9.2 | −0.44 | 410 | 0.55 | MS | HS | HS | MS | MS | IV |
| 22. | −7.6 | −0.01 | 450 | 0.55 | MS | MS | HR | HS | MS | IV |
| 23. | −8.0 | 0.27 | 2000 | 0.55 | HS | MS | HR | MS | MS | IV |
| 24. | −8.7 | −0.05 | 1000 | 0.17 | MR | MR | MR | MS | MS | IV |
| 25. | −9.8 | −0.07 | 6 | 0.17 | MS | MS | HR | MS | MS | II |
| 26. | −6.1 | −0.10 | 595 | 0.55 | MS | MR | MS | MR | MS | IV |
| 27. | −6.3 | 0.20 | 595 | 0.55 | MS | MR | HS | MR | MS | IV |
| 28. | −6.4 | −0.63 | 150 | 0.55 | MS | MR | HS | MR | MS | III |
| 29. | −5.9 | −0.38 | 150 | 0.55 | MS | MR | MS | MR | MS | III |
| 30. | −6.1 | 1.05 | 1 | 0.17 | HS | MS | HR | MS | HS | I |
| 31. | −8.4 | −0.54 | 480 | 0.55 | HS | MS | MR | HS | HS | IV |
| 32. | −7.4 | −0.38 | 501 | 0.55 | HS | MS | HS | HS | HS | IV |
| 33. | −5.9 | −0.45 | 1000 | 0.55 | MS | MS | HR | MR | MS | IV |
| 34. | −7.6 | −0.91 | 2498 | 0.17 | MS | MS | MS | MS | MS | II |
| 35. | −9.3 | 0.41 | 40 | 0.55 | HS | MS | MS | MS | MS | II |
| 36. | −9.4 | 0.41 | 40 | 0.55 | HS | MS | MS | MS | MS | II |
| 37. | −10.1 | 0.16 | 8 | 0.17 | HS | MS | MR | HS | HS | II |
| 38. | −9.9 | 0.36 | 9 | 0.55 | HS | MS | HR | MS | HS | II |
| 39. | −9.8 | 0.81 | 9 | 0.55 | HS | MS | HR | MS | MS | II |
| 40. | −9.8 | 0.27 | 8 | 0.55 | HS | MS | HR | MS | MS | II |
| 41. | −8.7 | 0.01 | 2000 | 0.55 | MS | MS | MS | MS | MS | IV |
| 42. | −9.1 | 0.01 | 2000 | 0.55 | MS | MS | HS | MS | MS | II |
| 43. | −8.7 | 0.90 | 1200 | 0.55 | MS | MS | MR | MS | MS | IV |
| 44. | −8.4 | −0.30 | 1100 | 0.55 | MS | MS | MS | MS | HS | IV |
| 45. | −9.1 | −0.27 | 338 | 0.55 | HS | MS | HS | MS | HS | IV |
| 46. | −8.2 | 0.47 | 189 | 0.17 | MS | MS | HS | HS | MS | II |
| 47. | −6.5 | 0.96 | 1 | 0.17 | HS | MS | HR | MS | HS | I |
| 48. | −9.1 | 0.52 | 290 | 0.55 | HS | MS | MS | MS | HS | III |
| 49. | −7.2 | −0.42 | 10 000 | 0.11 | MS | MS | HR | MS | MS | VI |
| 50. | −8.6 | 0.34 | 4000 | 0.55 | HS | MS | HR | MS | MS | V |
| 51. | −8.5 | −0.60 | 1200 | 0.55 | MR | MS | HR | MR | MS | IV |
| 52. | −7.4 | 0.46 | 2000 | 0.17 | MR | MS | HS | MS | MS | IV |
| 53. | −8.2 | −0.25 | 500 | 0.55 | MR | HR | MR | MR | MS | IV |
| 54. | −7.1 | 0.03 | 2000 | 0.55 | MS | MS | HS | MR | HS | IV |
| 55. | −6.1 | −0.55 | 1000 | 0.55 | MS | MS | HR | MR | MS | IV |
| 56. | −9.8 | 0.22 | 8 | 0.55 | HS | MS | HR | MS | HS | II |
| 57. | −10.5 | 0.47 | 68 | 0.55 | HS | MS | HR | MS | HS | II |
| 58 a | −7.9 | 0.60 | 500 | 0.17 | HR | MS | MS | MS | MS | III |
| 59 a | −7.4 | 1.10 | 5000 | 0.55 | HS | HS | HS | HS | HS | V |
Note: The toxicity profiles were represented in colors as follows: dark‐green (HS, highly safe), light‐green (MS, moderate safe), light‐pink (MR, moderate risk), red‐pink (HR, high risk) with lower toxicity class to higher toxicity class (I–VI) from lime‐green to dark brick‐red colors, respectively.
Abbreviations: BA, bioavailability score; CG, carcinogenicity; CT, cytotoxicity; DL, drug‐likeness; LD50, 50% lethal dose (mg/kg); HT, hepatotoxicity; IT, immunotoxicity; MG, mutagenicity; TC, toxicity class.
Standard drugs darunavir and lopinavir.
3.2. MD simulation of docked protein–ligand complexes
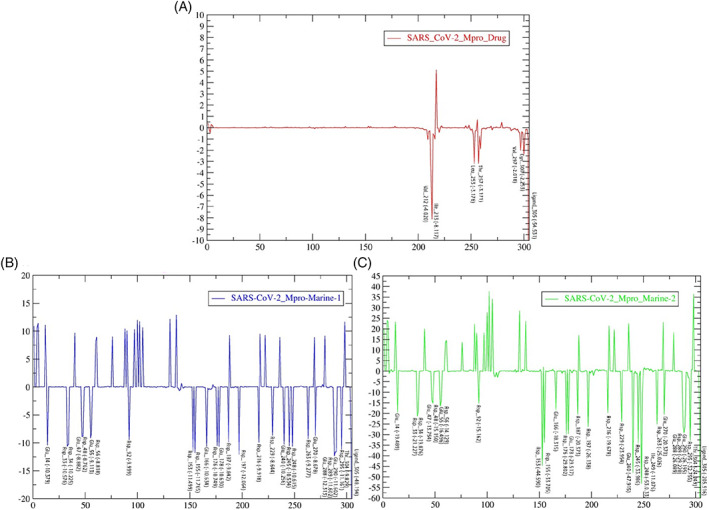
The RMSD‐protein backbone, RMSF‐C‐alpha and Rg‐score plots from MD simulations of two types of topology files of the same protein–ligand complexes at 100 ns were analyzed to demonstrate the structural stability. The RMSD plots of PRODRG‐derived SARS‐CoV‐2‐Mpro‐Marine‐1, SARS‐CoV‐2‐Mpro‐Marine‐2 and SARS‐CoV‐2‐Mpro‐Drug files confirmed that a constant deviation in backbone protein appeared during the 100 ns time scale. Nevertheless, after 70 ns, both alkaloids were found merely stable from the analyzed plot (Figure 2A). Notably, marine alkaloids showed stable deviations in between the range of 1.5–2.65 A0 and 0.5–1.5 A0, while the drug (darunavir) showed a more significant deviation at a range of 0.13–4.0 A0 in the protein backbone RMSD‐plot. At the same time, the ATB server topology file RMSD plots of SARS‐CoV‐2‐Mpro‐Marine‐1, SARS‐CoV‐2‐Mpro‐Marine‐2 also exhibited deviations throughout 100 ns in backbone protein, especially a higher deviation was observed in between 55 and 65 ns at a range of 0.2–0.45 A0 (Figure S1A). The RMSD‐plot for ligand defined as darunavir presented a more deviated state at a range of 0.09–0.43 A0, while both alkaloids were comparatively in a stable state within 100 ns. The RMSD‐plot of the marine‐2 was comparatively stable than the marine‐1 (manzamine‐A) within 100 ns in both topology files (Figures 2B and S1B). From generated RMSF plots, a higher binding affinity with less fluctuation in C‐alpha residues of both alkaloids than darunavir complex was observed in PRODRG‐derived topology files (Figures 2C and S1C). RMSF plots of marine‐1 and marine‐2 complexes deviated throughout 100 ns, but from minute observation marine‐1 is more deviated in PRODRG files and both complexes exhibited more relative deviation in MD simulation with ATB server derived ligand topology files (Figures 2C and S1C). Overall, in terms of the ligand RMSD, a substantial difference has been observed where the RMSD of both the marine alkaloids deviated from 0.1 nm to 0.25 nm, whereas using PRODRG topology, the RMSD of the two marine alkaloids seems stable ~0.15 nm. The difference has been notably observed due to the charge distribution pattern while generating the topology from the aforementioned servers. Based on the RMSF‐analyses, among three ligand complexes exhibited more relative deviations, while marine‐2 was comparatively more stable in PRODRG server‐generated topology files (Figures 2C and S1C). The overall compactness and the RMSF remain the same in both the marine‐1 and marine‐2 topologies. Some minute differences have been observed, which does not impact significantly the overall parametric calculations. Further, Rg‐plots displayed the compactness or solidity in marine‐2 with more squeezed Rg‐score than other complexes in both PRODRG and ATB‐derived topology files (Figures 2D and S1D).
FIGURE 2.
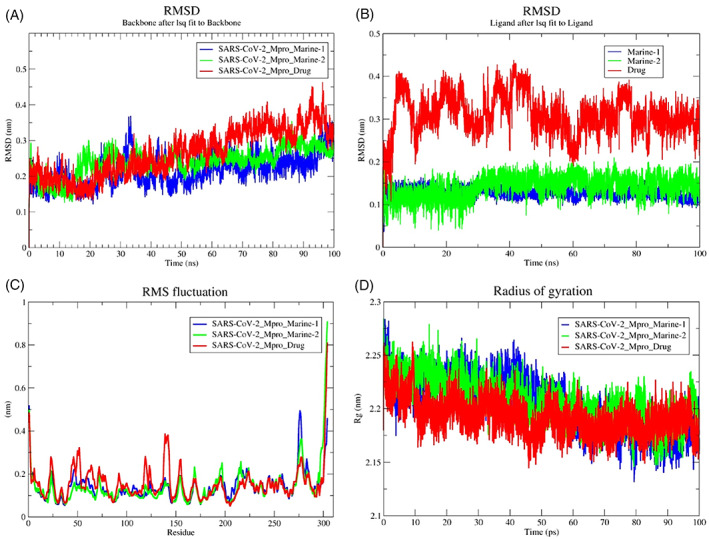
Conformational stability of PRODGR‐derived topology files in the form of RMSD‐plots at 100 ns MD simulation in individual color plots; (A), overlayed RMDS‐plots of SARS‐CoV‐2‐Mpro‐Drug (darunavir), SARS‐CoV‐2‐Mpro‐Marine‐1 (manzamine A) and SARS‐CoV‐2‐Mpro‐Marine‐2 (8‐hydroxymanzamine); (B), overlayed RMDS‐plots of individual ligand stability in the docked complexes; (C), overlayed RMSF‐plots of SARS‐CoV‐2‐Mpro‐drug (darunavir), SARS‐CoV‐2‐Mpro‐Marine‐1 (manzamine A) and SARS‐CoV‐2‐Mpro‐Marine‐2 (8‐hydroxymanzamine) and (D), overlayed Rg‐plots of SARS‐CoV‐2‐Mpro‐Drug (darunavir), SARS‐CoV‐2‐Mpro‐Marine‐1 (manzamine A) and SARS‐CoV‐2‐Mpro‐Marine‐2 (8‐hydroxymanzamine)
Apart from that, H‐bond interaction analysis states that marine alkaloids have more H‐bonds than darunavir (Figure 3). More often, marine‐2 ligand (set in green color) high propensity to H‐bonds in comparison to marine‐1 (set in blue color) and even darunavir (set in red color) throughout MD simulation within 100 ns time scale (Figure 3). Sametime, the ATB‐derived topology files showed some different results, as marine‐1 displayed higher numbers of compact H‐bond interactions than marine‐2 (Figure S2). Therefore, after molecular docking, MD‐simulation is the most imposing tool in current CAAD modules to study the stability/behavior of an individual complex at the atomic level, 16 , 17 , 22 , 27 which is helpful toward the selection of potential lead candidates for further experimental study. Nevertheless, the diverse outputs according to topology files indicated that the generation of the most accurate topology especially by adding atomic charges plays an important role and/or needs more expertise on topology‐generated servers with forcefield selection to get more reliable outputs from MD.
FIGURE 3.
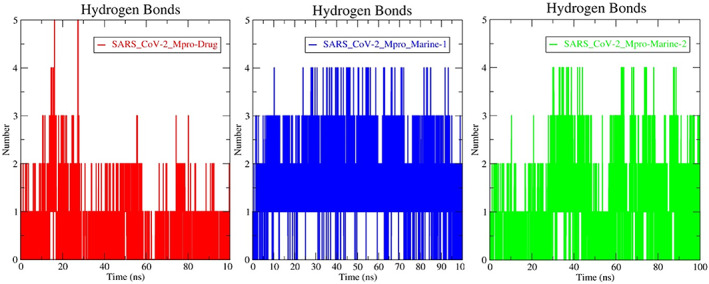
Molecular stability based on H‐bond interactions analyses of PRODRG‐derived topology files SARS‐CoV‐2‐Mpro‐Drug (darunavir) in red color, SARS‐CoV‐2‐Mpro‐Marine‐1 (manzamine A) in blue color and SARS‐CoV‐2‐Mpro‐Marine‐2 (8‐hydroxymanzamine) in green color at 100 ns
3.3. PCA and FEL investigation
Both PCA and FEL analysis come under the essential MD simulation that displays most meta‐stable macromolecule conformations or crucial dynamics of a system at time intervals concerning low‐dimensional free energy. The PCA‐based FEL‐2D and ‐3D plots for both topologies files were generated separately using the software Mathematica (student version), showing the minimal energy point for every three SARS‐CoV‐2‐Mpro complexes system (Figures 4 and S3). In the elucidated FEL plot, the middle position displayed the lowest minima of the energy scale in the violet part and dumped the structure at that position using MD trajectories. Notably, the SARS‐CoV‐2‐Mpro‐Marine‐2 complex exhibited the stable dynamic motion of interactions than the marine‐1 and darunavir complexes graphically (Figures 4 and S3).
FIGURE 4.
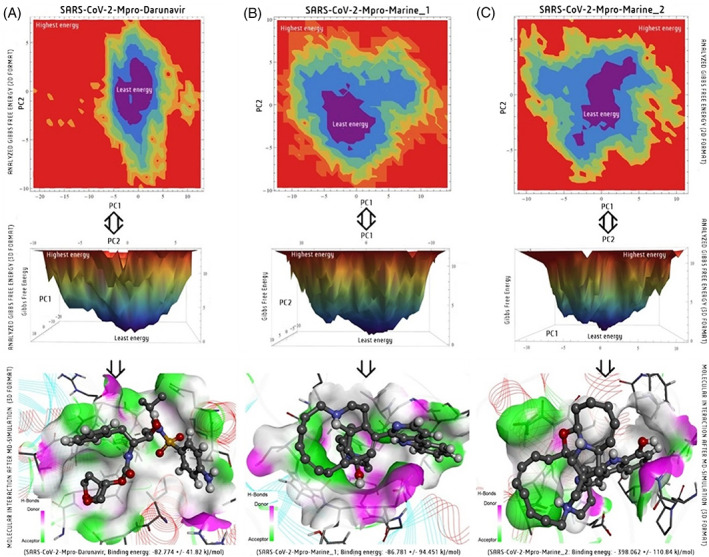
Free energy landscape (FEL) analysis of PRODRG‐derived topology files in 2D and 3D graphical presentation corresponding to minimize energy structure; (A), SARS‐CoV‐2‐Mpro‐Drug (darunavir); (B), SARS‐CoV‐2‐Mpro‐Marine‐1 (manzamine A) and (C), SARS‐CoV‐2‐Mpro‐Marine‐2 (8‐hydroxy manzamine) after MD‐simulation at 100 ns
Parallelly, we have analyzed the docking pattern interaction in pre‐MD and post‐MD to know the conservative nature of interaction toward solid binding efficacy. This pre‐ and post‐MD interaction analysis confirmed that darunavir loses its stability with the target by reducing the H‐bond interaction, van der Waal, and other exchanges in 100 ns (Figure 5A). The manzamine A also reduces the p‐alkyl interactions but maintains the two H‐bond interactions in distinct residues with the target enzyme from pre‐and post‐simulation analyses (Figure 5B). Significantly, the 8‐hydroxymanzamine maintained their strength with the target through several H‐bond interactions and mostly established a salt bridge (combining noncovalent interactions, hydrogen bonding, and ionic bonding), as a solid interaction with crucial residues with SARS‐CoV‐2‐Mpro at the end of the 100 ns in PRODRG‐derived topology file (Figure 5C). However, the pre and post analyses with ATB‐derived files were observed slightly different results, the hydrogen‐bond formation amino acid GLU166 and another covalently bonded amino acid LEU167 were found stable but found broken in chemical space at end of the 100 ns. Nevertheless, without any hydrogen interaction, most of the interactions were found in both pre‐and pro‐MD stages marine‐2 (Figure S4). Interestingly, the PRO294 residue formed a hydrogen‐bond interaction in the pre‐MD stage, while it formed Pi‐Pi‐T‐shaped interaction with the target enzyme in the post‐MD stage (Figure S4). Overall, marine‐2 interactions are comparatively more stable than marine‐1 with the SARS‐CoV‐2‐Mpro target enzyme and could be a suitable lead candidate to inhibit the viral life cycle. Both PCA and FEL are associated advanced programs to analyze the dynamic motion and least free energy interaction sites of MD simulation results with an added texture of visualization in current drug development modules. 13 , 17
FIGURE 5.
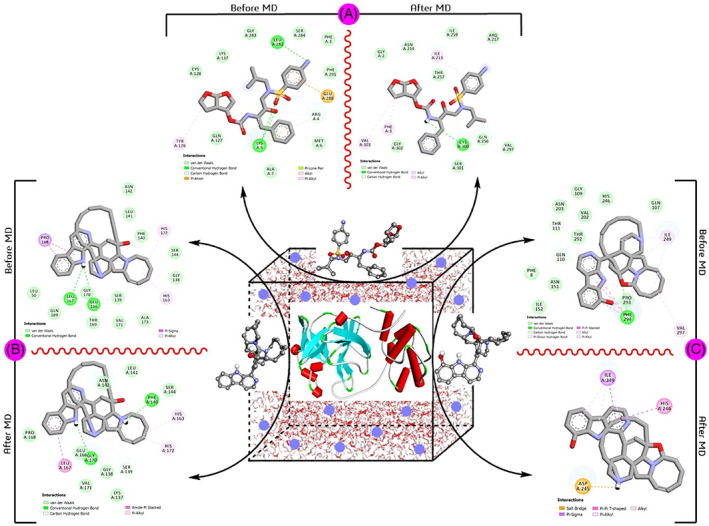
Comparative pre‐ and post‐MD‐simulation analyses of PRODRG‐derived topology files; (A), SARS‐CoV‐2‐Mpro‐Drug (darunavir); (B), SARS‐CoV‐2‐Mpro‐Marine‐1 (manzamine A) and (C), SARS‐CoV‐2‐Mpro‐Marine‐2 (8‐hydroxy manzamine) at 100 ns
3.4. MM/PBSA and binding free energy per residue analysis
Further, the MM/PBSA calculation of both PRODRG and ATB ligands topology files were analyzed for all three selected docking complexes binding energy during the MD simulation. Binding affinity with other energies such as E, MM, G polar, and G nonpolar was calculated between 80 and 100 ns at a 0.1 ns time step. PRODRG‐derived topology file of the SARS‐CoV‐2‐Mpro‐darunavir presented lower binding energy −82.774 kJ/mol and the other two complexes, SARS‐CoV‐2‐Mpro‐manzamine A with −86.781 kJ/mol and SARS‐CoV‐2‐Mpro‐8‐hydroxymanzamine exhibited the most significant binding energy −398.062 kJ/mol (Table 2). Parallelly, the van der Waal energy, electrostatic energy, polar solvation energy, and solvent accessible surface area (SASA) energy were too calculated for each complex (Table 2). The ATB‐server‐derived topology file's binding energies of marine‐1 and marine‐2 were documented, separately. Mainly the atomic charged ATB‐files showed their energy in a lower value as −93.070 ± 0.925 kJ/mol binding energy for marine‐1 complex and −143.9 ± 0.985 kJ/mol for the marine‐2 complex (Table S2). According to another energy was also recorded in a higher range (Table S2). Overall, from both topology file analyses, the 8‐hydroxymanzamine was more potential than against SARS‐CoV‐2‐Mpro as per the energy per binding energies and residues calculation in a graph format within 100 ns (Figure 6). There is a maximum number of common residues with comparatively lower binding energy in marine‐2 than marine‐1 and darunavir within 100 ns. Thus, the MM/PBSA is widely accepted in the current CADD module to calculate absolute binding affinity per residue during ligand integration with the target molecule. 17 , 30
TABLE 2.
The binding free energy and other associated energy value of SARS‐CoV‐2‐Mpro‐Darunavir, SARS‐CoV‐2‐Mpro‐Marine‐1 and SARS‐CoV‐2‐Mpro‐Marine‐2 MD‐simulated complexes ligand topology derived from the PRODRG server using the MM/PBSA method
| Energy parameter (kJ/mol) | SARS‐CoV‐2‐Mpro‐Darunavir | SARS‐CoV‐2‐Mpro‐Marine‐1 | SARS‐CoV‐2‐Mpro‐Marine‐2 |
|---|---|---|---|
| Binding energy | −82.774 ± 41.82 | −86.781 ± 94.451 | −398.062 ± 110.84 |
| Electrostatic energy | −0.876 ± 11.47 | −101.019 ± 105.112 | −588.658 ± 154.81 |
| Polar solvation energy | −65.76 ± 44.38 | 31.453 ± 65.758 | −339.163 ± 94.57 |
| van der Waal energy | −135.300 ± 12.27 | −15.790 ± 47.509 | −34.020 ± 3.476 |
| SASA energy | −12.364 ± 2.624 | −1.424 ± 4.810 | −14.224 ± 4.316 |
Abbreviation: MM/PBSA, molecular mechanics/Poisson‐Boltzmann surface area.
FIGURE 6.
Energy per residue of SARS‐CoV‐2‐Mpro‐drug (darunavir in red color), SARS‐CoV‐2‐Mpro‐Marine‐1 (manzamine A in blue color) and SARS‐CoV‐2‐Mpro‐Marine‐2 (8‐hydroxymanzamine in green color) docking complexes have been recorded through the MM/PBSA binding energy calculation method with PRODRG‐derived topology files at 100 ns MD simulation
3.5. Topology‐based comparison and contributions of entropy analyses
As mentioned earlier, the topology‐based analysis of the two marine alkaloids has been performed using ATB and compared with PRODRG to depict the parametric difference, for example, RMSD, RMSF, Rg, and so on. The analysis revealed a substantial difference in RMSD deviation notably at 50–60 ns time point where the protein backbone RMSD was estimated to be ~0.5 nm. After 70 ns, it attains a stable RMSD between ~0.4 and 0.5 nm which is comparable with simulation performed using PRODRG topology. Both the marine alkaloids behave in a similar pattern throughout the 100 ns simulation. In terms of the ligand RMSD, a substantial difference has been observed where the RMSD of both the marine alkaloids deviated from 0.1 nm to 0.25 nm, whereas using PRODRG topology, the RMSD of the two marine alkaloids seems stable ~0.15 nm. The difference has been notably observed due to the charge distribution pattern while generating the topology from the aforementioned servers. The overall compactness and the RMSF remain similar types of features in both the topologies. Minute differences have been observed, which does not impact the overall parametric calculations.
In addition, Schlitter's formula and quasi‐harmonic analyses were evaluated against the two most potential marine alkaloids that reproduce translational, rotational, and vibrational entropies. For marine‐1, the entropy is 35493.5 J/mol K followed by the Schlitter formula and 725 129 J/mol K from the Quasi‐Harmonic analysis. Similarly, for marine‐2, the entropy is 35526.1 J/mol K followed by the Schlitter formula and 726 670 J/mol K from the quasi‐harmonic analysis. Overall, the topology particularly the addition of atomic charges via the ATB server comparatively diversified the MD result, the binding energy of PRODRG files. PCA and FEL analyses also exhibited some diversified results of the same docking complexes (Figures S3 and S4). The reason behind choosing the ATB server in contrast to the PRODRG is because of charge distribution as PRODRG works with the concept of charge groups, which are defined as a group of bonded atoms with an integer charge whereas ATB works with the concept of quantum mechanical (QM) calculations combined with a knowledge‐based approach where charge groups (including partial charges) are assigned automatically with parameters consistent with GROMOS forcefield (GROMOS96 54A7 forcefield) and provide the choice of user selection in case if any ambiguity arises. 24 Theoretically, biophysical processes can be better understood by measuring the solute molecules' configurational entropy. It is vital to examine the entropy of solute molecules while investigating various biophysical processes, such as ligand binding. 31 , 32 The limitation of translational, rotational, and conformational motion that occurs when a ligand or protein binds to protein results in a considerable loss of entropy. As a result, precise calculations of absolute and relative entropies are of great importance. It is critical to know how ligands and substrates themselves contribute to total binding or activation entropies, even if solvent entropy effects dominate. Inaccuracies in the computation of entropies are frequently many orders of magnitude bigger than errors in the calculation of the free energy.
3.6. Physicochemical and toxicity profiles prediction
The physicochemical/RO5 properties of all marine‐alkaloids and reference anti‐HIV drugs were recorded (Table S3). Briefly, 23 marine‐alkaloids did not follow the standardized molecular weight (≤500 g/mol), 17 did not follow the logp value (≤5), 9 did not follow the ideal tPSA value (≤142 Å2), and 1 does not attend the perfect number of H‐bond acceptors (≤10). Overall, 26 marine alkaloids have followed the ideal drug parameters proposed under the RO5. Darunavir and lopinavir were also violating the RO5 standard parameters. The RO5 prediction suggested that most potential alkaloids displayed in a higher range than standardized parameters due to large chemical structure but not that much to disrupt the oral bioavailability (Table S3). Nevertheless, RO5 is a standardized parameter to locate bioactive oral drugs with some ideal metabolism for the current drug development modules. 33
From the predicted toxicity profiles, marine‐alkaloids and anti‐HIV drugs exhibited comparatively moderate hepatotoxicity, carcinogenicity, and mutagenicity profiles with adverse immunotoxicity profiles tabulated in Table 1. Lopinavir is safer than darunavir from prediction from toxicity profiles. Alkaloids are under toxicity class‐II and IV from toxicity class prediction, while selected potential marine alkaloids, manzamine A and 8‐hydroxymanzamine are class‐2. Alkaloid no. 14 (crambescidin 826), 15 (crambescidin 830), 16 (crambescidin 844), 30 (fromiamycalin), and 47 (ptilomycalin A) exhibited higher toxicity as overall predicted toxicity class‐I category. Overall, alkaloid no. 2 (acetylstachyflin), 34 (lamellarin‐α‐20‐sulfate), 50 (stachyflin) with lopinavir drug were safer candidates with toxicity profiles class‐VI (Table 1).
3.7. Pharmacokinetics and overall drug‐likeness profile prediction
Possible pharmacokinetic profiles of marine‐alkaloids and anti‐HIV drugs were recorded (Table S4 and Figure S5). From prediction, including darunavir and 15 marine candidates exhibited lower gastrointestinal absorption (GI‐abs). However, 17 candidates may cross the blood–brain barrier (BBB) as per prediction, while both standard drugs do not have this property. On the other hand, 18 compounds do not have p‐glycoprotein (p‐gp)‐substrate ability as a plasma membrane protein associated with drug transport in the human body. Briefly, the overall pharmacokinetics properties are associated with the physiochemical and pH of the drug candidate. As a result, most of RO5 violated candidates bearing poor pharmacokinetics properties. Expectantly, darunavir and 8‐hydroxymanzaminein synergism could be an alternative treatment option with balanced pharmacokinetic profiles against SARS‐CoV‐2.
The drug‐likeness score for each marine‐alkaloid was recorded in Table 1. Among 57 candidates, 33 candidates displayed positive drug‐ability scores. Notably, alkaloid no. 3 (batzelladine C), 12 (crambescidin 800), 13 (crambescidin 816), 14 (crambescidin 826), 15 (crambescidin 830), 16 (crambescidin 844), and 30 (fromiamycalin) displayed higher drug‐ability values (>1), while manzamine A, 8‐hydroxymanzamine, darunavir, and lopinavir also displayed positive values such as 0.16, 0.47, 0.60, and 1.10, respectively (Table 1; Figure S6). However, the >2 scores are also not good ideal as per the predicted tool. Mainly, the score within 0–2 is the more preferred score for a candidate under the predefined bell‐shaped curve for identification of drug or nondrug category by Molsoft. To conclude, drugability is the sum of all drug‐parameter scores as per the chemical composition of a candidate. 17 , 33 , 34 Thus, the predicted drug‐likeness score could be another crucial parameter used to select lead candidates, where marine alkaloids displayed potential drug‐ability scores like a mainstream drug.
3.8. FMOs and SAR analyses
In drug development, the concept of FMOs energy can analyze the flexibility (hardness/softness) of lead candidates in terms of HOMO to LUMO and their energy gap (ΔE) between the ground state and the excited state. The HOMO and LUMO chemical space distribution (in red color for positive electron density and blue color for negative electron density), were identified and calculated the energy gap of two alkaloids and drugs was presented (Figure 7). From FMOs energy gap displayed as follows; manzamine A (9.453 eV), 8‐hydroxymanzamine (9.453 eV), darunavir (11.179 eV), and lopinavir (12.041 eV). The HOMO represents the electron donor site associated with ionization potential (IP), while LUMO means space to an electron acceptor, corresponding to the electron affinity (EA). The energy gap indicated optical and electronic properties and herein both alkaloids exhibited similar to darunavir and lopinavir orbital electron energy gap as all are presented in an excited state with more interactive modes. The higher energy gap also indicated has low chemical reactivity and high kinetic stability. Thus, both 8‐hydroxymanzamine showed more interactive action with Mpro, comparatively less than lopinavir and darunavir. Overall, FMO is a drug investigation tool using the concept of electronic orbital energy of a molecule. 27
FIGURE 7.
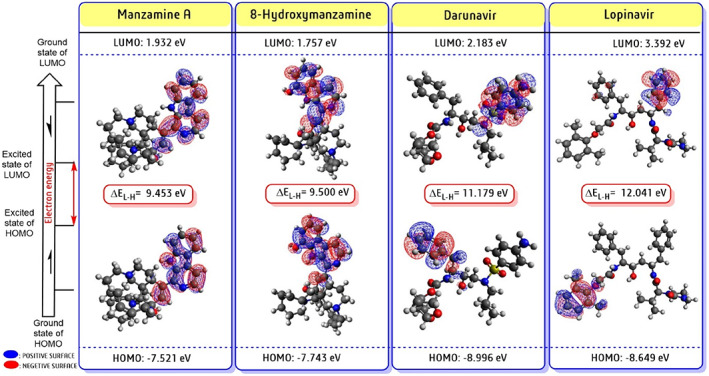
Frontier molecular orbitals (FMOs) analysis in the form of LUMO, HOMO, and their energy gap (ΔE in kcal/mol) were predicted for two potent candidates along with two repurposing anti‐HIV drugs in Avogadro‐ORCA planform. The red color spear represents positive electron density and the blue color for negative electron density spear
Parallelly, we found that magazine A like manzamine‐E, ‐F, ‐X, manadomanzamine A‐B, xestomanzamine A, 6‐deoxymanzamine X, 8‐hydroxymanzamine derivatives exhibited potential‐cum‐similar types of docking scores as they belong to a similar class of polycyclic alkaloids, mostly isolated from a marine sponge. Thus, we have conducted a SAR study taking this group of compounds (Figure 8). First, beta‐carboline (C11H8N2), a combined unit of indole (C8H7N) and piperidine (C5H11N), is the parental skeleton for the above alkaloid starting from xestomanzamine A. In the chemical dismemberment view, several supporting side‐chain functional groups and chemical moieties, namely, hydroxy (—OH), carbonyl (C═O), octa‐hydro isoquinoline (C9H15N), pyrrolidine (C4H9N), azonane (C8H17N), butanone (CH3C(O)CH2CH3), azetidine (C3H7N), and other attached cyclononane (C9H18), cyclodecane (C10H20) ring makes the alkaloid unique and complex too. From the SAR point of view, the side‐chain position of any small functional group can change the activity, other physicochemical nature, and even toxicity profiles. For example, β‐carboline with an imidazole‐carbaldehyde derivative attached group in xestomanzamine A exhibited a docking score of −7.1 kcal/mol. At the same time, 8‐hydroxymanzamine is similar to manzamine E (C36H44N4O2) but displayed a higher docking score of −10.5 as, manzamine E (−9.9 kcal/mol) contains one hydroxy and one carbonyl group and 8‐hydroxymanzamine has two hydroxy groups. As a result, manzamine E slightly reduces the binding affinity. The manzamine A has displayed a −10.1 kcal/mol docking score and is composed of beta‐carboline pharmacophore with azonane, octahydroisoquinoline, pyrrolidine with a hydroxy group. Similarly, manzamine X (C36H44N4O3) exhibited the same binding affinity −9.8 kcal/mol as the most similar chemical composition to manzamine E. Thus, as per the SAR analyses, the hydroxy (‐OH) group influenced the binding affinity as well as interaction stability. Overall, SAR is the most versatile approach in medicinal chemistry for novel drug‐chemistry analysis of a series of bioactive chemicals based on their chemical structure before any experimental study. 34
FIGURE 8.
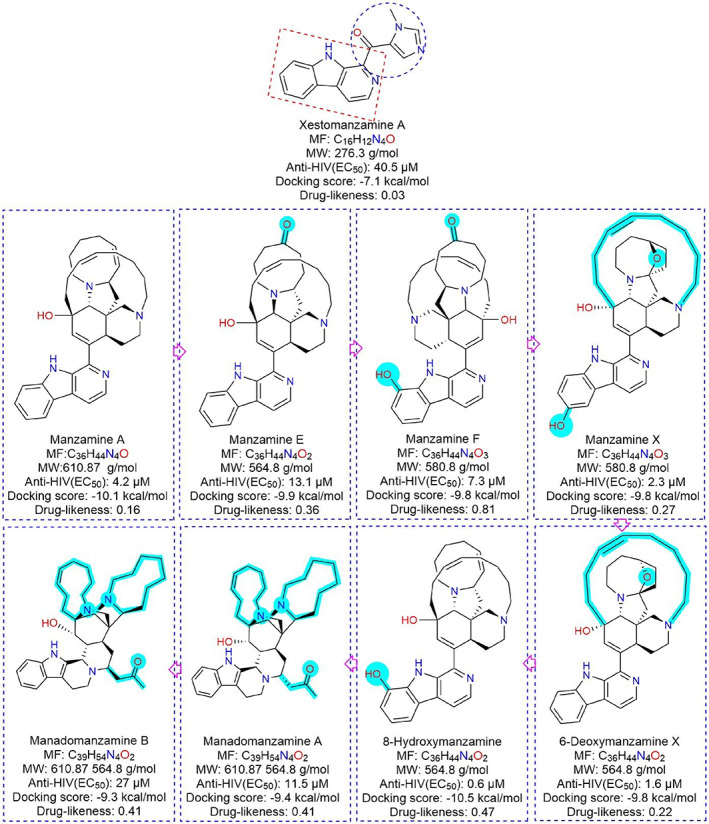
The overall computational prediction associated with structural activity relationships of potential manzamine alkaloid class of derivatives against SARS‐CoV‐2‐Mpro
As per the prediction, inappropriate drug‐likeness profiles could be overcome through advanced nano‐formulation and structural modification approaches later. 35 , 36 , 37 , 38 Nevertheless, exploring more bioactive compounds from different ethnomedical‐natural sources is one of the potential directions in the primary drug discovery module. 11 , 18 , 19 , 39 Simultaneously, the use of advanced artificial intelligence programs accommodating to find out the potential candidates (herein marine alkaloids) against specific diseases (herein SARS‐CoV‐2 targeting Mpro enzyme) with drug‐chemistry analysis (RMSD‐RMSF‐Rg‐H‐bond interaction, free‐energy calculation with RO5‐toxicity‐pharmacokinetics profiles) at the primary stage of the drug development approach. 16 , 30 , 40 , 41 , 42
4. DISCUSSION
The marine ecosystem produces a plethora of bioactive metabolites that are recognized from their previous contribution, namely, streptomycin, rifamycin, vancomycin, and so on. 16 , 19 , 43 From the anti‐viral drug development point of view, previous experimental records and the present computational investigation indicated that the marine sponge is the most suitable source of anti‐viral alkaloids (see Table S1). In contrast, plant genera also provide several alkaloid classes of drug namely quinine, morphine, nicotine, vinblastine, vincamine and emetine, and so on, to mainstream medicine. However, the structural composition and complexity have differentiated the uniqueness of marine alkaloid and plant alkaloid metabolites. 18 , 44 , 45 Therefore, alkaloids class of metabolites are always a most potent source of mainstream drug development and herein marine alkaloids also showed potential activity for SARS‐CoV‐2‐Mpro, computationally.
In the present scenario, the CADD platform is a time and cost‐saving method in the ongoing drug discovery module to select potential lead candidates at an early stage. Mainly, the computational screening method locates any bioactive candidate based on lower binding energy known as docking scores represented in kilocalorie per mole unit and recently several natural anti‐SARS‐CoV‐2 products were also highlighted/recognized through the computational program. 13 , 15 , 16 , 17 Furthermore, before synthesis and expensive experimental validation, primary molecular interaction studies with molecular docking‐simulation for biological activity and other drug‐likeness predictions directly help to reduce resources and time compared to ongoing traditional drug selection procedures. 13 , 15 , 16 , 17 , 22 Thus, the CADD/artificial intelligence is a promising platform for the rapid selection of most potential anti‐CoV candidates from various conventional/existing sources for further pharmacological/clinical validation. 15 , 16 , 17
The toxicity profile is a significant obstacle for any therapeutic agent. Accordingly, the maximum number of lead drug candidates are withdrawing in different clinical trial stages due to side effects. In alkaloid cases, nitrogen atoms in the alkaloid moiety possess higher toxicity than other natural chemical classes such as terpenoids and flavonoids, primarily in higher concentrations. 6 , 11 Nevertheless, most alkaloids showed potential biological activities at a lower concentration. 18 , 44 , 45 Drugs such as rifampicin, tigecycline, vancomycin, darunavir and lopinavir have been approved by the FDA despite violations of the RO5. For example, rifampicin, tigecycline, vancomycin, even darunavir, and lopinavir. Sometimes, strict implementation of the RO5 opposes specific bioactive compounds where opportunities exist. Therefore, a negotiation between the biological activity and physiological profile of any bioactive molecules should be needed during selection or undergoes further formulation toward fulfilling the ideal pharmacokinetics. 46 , 47 Overall, the cost‐effective computational RO5 prediction in the early stage is more helpful for current drug development modules, but selection only through RO5 is challenging. 47 , 48
Simultaneously, the pharmacokinetics profile also plays a crucial role in the dose formulation of a newer drug, which is associated with physicochemical property or RO5. Briefly, the transport protein present in the GI tract resides is fully responsible for facilitating the transport of nutrition/drugs across the intestine. 6 , 49 , 50 Similarly, p‐gp is another factor associated with drug distribution across the cell membrane/efflux‐membrane transporter, directly linked to optimal drug delivery. 51 , 52 Overall, the computational prediction has reduced the resource and cost of selecting ideal lead candidates by filtering the essential drug parameters. 52 , 53 , 54 , 55 Concludingly, this cost‐effective, resource‐saving, and highly efficient computational screening is helpful to locate several repurposing candidates like marine alkaloids toward control of gruesome SARS‐CoV in this health emergency.
5. CONCLUSIONS
In the present scenario, exploring natural regimens like marine alkaloids based on previous pharmacological reports to combat SARS‐CoV‐2 is provisionally an ideal approach. Herein, 57 antiviral marine‐alkaloids were examined through bioinformatics and cheminformatics tools followed by a step‐by‐step procedure in the CADD platform. Primarily, based on docking scores, two marine alkaloids manzamine A (−10.2 kcal/mol) and 8‐hydroxymanzamine (−10.5 kcal/mol) were the most potential candidates than drug darunavir (−7.9 kcal/mol) and lopinavir (−7.4 kcal/mol) against SARS‐CoV‐2‐Mpro. Biophysical analyses as stability of protein–ligand complex by MD simulation at 100 ns (RMSD, RMSF, Rg and H‐bond interaction profiles) confirmed that 8‐hydroxymanzamine was comparatively most active and stable against SARS‐CoV‐2‐Mpro. Notably, the observation of two ligand topology files for the same docking complex with diverse results is also an interesting feature of this article indicated to select more appropriate topology and force fields to get reliable outputs. From the previous pharmacological record, 8‐hydroxymanzamine was also reported as a potential anti‐HIV candidate with an EC50 value of 0.6 μM and in some way supports the current computational investigation against SARS‐CoV‐2‐Mpro. Additionally, projected physicochemical parameters, pharmacokinetics‐toxicity profiles, and positive drug‐likeness‐bioavailability score confirmed that the manzamine class of alkaloids could be used against SARS‐CoV‐2 individually or in synergistic formulation with darunavir/lopinavir‐like repurposing drug. Strategically, CADD could be a promising platform to recognize possible natural “lead” drug candidates against SARS‐CoV‐2 at a primary level than the traditional hit‐and‐trial to accelerate anti‐CoV drug discovery.
CONFLICT OF INTEREST
The authors declare no competing interests.
AUTHOR CONTRIBUTIONS
Shasank S. Swain, Satya R. Singh, and Alaka Sahoo conceptualized the research design; Shasank S. Swain, Alaka Sahoo, and Pritam Kumar Panda carried out the advanced computation assessments; Shasank S. Swain drafted the MS and Tahziba Hussain edited and commented on it. Sanghamitra Pati provided overall support. All authors approved the MS before submission.
Supporting information
Appendix S1 Supporting Information
ACKNOWLEDGMENTS
This work is acknowledged to the ‘ICMR‐Post Doctoral Research Grant’ awarded to Dr. Shasank S. Swain (No. 3/1/3/PDF (21)/HRD‐2019‐2), from the Indian Council of Medical Research, Department of Health Research, Govt. of India.
Swain SS, Singh SR, Sahoo A, Panda PK, Hussain T, Pati S. Integrated bioinformatics–cheminformatics approach toward locating pseudo‐potential antiviral marine alkaloids against SARS‐CoV‐2‐Mpro . Proteins. 2022;90(9):1617‐1633. doi: 10.1002/prot.26341
Shasank S. Swain and Satya R. Singh contributed equally to this study.
Funding information Indian Council of Medical Research
DATA AVAILABILITY STATEMENT
All data are openly available in public repository sources, cited adequately, and complete information is available in the reference section or by request to the corresponding author for the full paper.
REFERENCES
- 1. WHO‐coronavirus‐19 (COVID1‐9) dashboard . Assessed June 15, 2021. https://covid19.who.int/
- 2. Kirby T. New variant of SARS‐CoV‐2 in the UKcauses surge of COVID‐19. Lancet Respir Med. 2021;9:e20‐e21. doi: 10.1016/S2213-2600(21)00005-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. About Variants of the Virus that Causes COVID‐19 . Assessed June 15, 2021 https://www.cdc.gov/coronavirus/2019-ncov/variants/variant.html
- 4. Lauring AS, Hodcroft EB. Genetic variants of SARS‐CoV‐2‐what do they mean? JAMA. 2021;325:529‐531. [DOI] [PubMed] [Google Scholar]
- 5. Gatti M, De Ponti F. Drug repurposing in the COVID‐19 era: insights from case studies showing pharmaceutical peculiarities. Pharmaceutics. 2021;13:302. doi: 10.3390/pharmaceutics13030302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sahoo A, Fuloria S, Swain SS, et al. Potential of marine terpenoids against SARS‐CoV‐2: an in silico drug development approach. Biomedicine. 2021;9(11):1505. doi: 10.3390/biomedicines9111505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ahn DG, Shin HJ, Kim MH, et al. Current status of epidemiology, diagnosis, therapeutics, and vaccines for novel coronavirus disease 2019 (COVID‐19). J Microbiol Biotechnol. 2020;30:313‐324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang C, Horby PW, Hayden FG, Gao GF. A novel coronavirus outbreak of global health concern. Lancet. 2020;395:470‐473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mirtaleb MS, Mirtaleb AH, Nosrati H, et al. Potential therapeutic agents to COVID‐19: an update review on antiviral therapy, immunotherapy, and cell therapy. Biomed Pharmacother. 2021;138:111518. doi: 10.1016/j.biopha.2021.111518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Verma S, Twilley D, Esmear T, et al. Anti‐SARS‐CoV natural products with the potential to inhibit SARS‐CoV‐2 (COVID‐19). Front Pharmacol. 2020;11:561334. doi: 10.3389/fphar.2020.561334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Swain SS, Panda SK, Luyten W. Phytochemicals against SARS‐CoV as potential drug leads. Biom J. 2020;S2319‐4170:30229‐30228. doi: 10.1016/j.bj.2020.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Babaei F, Mirzababaei M, Nassiri‐Asl M, Hosseinzadeh H. Review of registered clinical trials for the treatment of COVID‐19. Drug Dev Res. 2021;82:474‐493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mishra CB, Pandey P, Sharma RD, et al. Identifying the natural polyphenol catechin as a multi‐targeted agent against SARS‐CoV‐2 for the plausible therapy of COVID‐19: an integrated computational approach. Brief Bioinform. 2021;22:1346‐1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Luan B, Huynh T. Crystal‐structures‐guided design of fragment‐based drugs for inhibiting the main protease of SARS‐CoV‐2. Proteins. 2021;90(5):1081‐1089. doi: 10.1002/prot.26260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Maiti S, Banerjee A. Epigallocatechin gallate and theaflavin gallate interaction in SARS‐CoV‐2 spike‐protein central channel with reference to the hydroxychloroquine interaction: bioinformatics and molecular docking study. Drug Dev Res. 2021;82:86‐96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Swain SS, Singh SR, Sahoo A, Hussain T, Pati S. Anti‐HIV‐drug and phyto‐flavonoid combination against SARS‐CoV‐2: a molecular docking‐simulation base assessment. J Biomol Struct Dyn. 2021;1‐14. doi: 10.1080/07391102.2021.1885495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bharadwaj S, Dubey A, Yadava U, et al. Exploration of natural compounds with anti‐SARS‐CoV‐2 activity via inhibition of SARS‐CoV‐2 Mpro . Brief Bioinform. 2021;22:1361‐1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Swain SS, Padhy RN, Singh PK. Anticancer compounds from cyanobacterium Lyngbya species: a review. Antonie Van Leeuwenhoek. 2015;108:223‐265. [DOI] [PubMed] [Google Scholar]
- 19. Swain SS, Paidesetty SK, Padhy RN. Antibacterial, antifungal and antimycobacterial compounds from cyanobacteria. Biomed Pharmacother. 2017;90:760‐776. [DOI] [PubMed] [Google Scholar]
- 20. Bhatt A, Arora P, Prajapati SK. Can algal‐derived bioactive metabolites serve as potential therapeutics for the treatment of SARS‐CoV‐2 like viral infection? Front Microbiol. 2020;11:596374. doi: 10.3389/fmicb.2020.596374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Arya R, Kumari S, Pandey B, et al. Structural insights into SARS‐CoV‐2 proteins. J Mol Biol. 2021;433:166725. doi: 10.1016/j.jmb.2020.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Swain SS, Paidesetty SK, Dehury B, et al. Computer‐aided synthesis of dapsone‐phytochemical conjugates against dapsone‐resistant Mycobacterium leprae . Sci Rep. 2020;10:6839. doi: 10.1038/s41598-020-63913-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang L, Lin D, Sun X, et al. Crystal structure of SARS‐CoV‐2 main protease provides a basis for design of improved α‐ketoamide inhibitors. Science. 2020;368(6489):409‐412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schmid N, Eichenberger AP, Choutko A, et al. Definition and testing of the GROMOS force‐field versions 54A7 and 54B7. Eur Biophys J. 2011;40:843‐856. [DOI] [PubMed] [Google Scholar]
- 25. Schlitter J. Estimation of absolute and relative entropies of macromolecules using the covariance matrix. Chem Phy Let. 1993;215(6):617‐621. [Google Scholar]
- 26. Levy RM, Karplus M, Kushick J, Perahia D. Evaluation of the configurational entropy for proteins: application to molecular dynamics simulations of an alfa‐helix. Macromolecules. 1984;17:1370‐1374. https://pubs.acs.org/doi/10.1021/ma00137a013 [Google Scholar]
- 27. Hagar M, Ahmed HA, Aljohani G, Alhaddad OA. Investigation of some antiviral N‐heterocycles as covid 19 drug: molecular docking and DFT calculations. Int J Mol Sci. 2020;21:3922. doi: 10.3390/ijms21113922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McKinney JD, Richard A, Waller C, et al. The practice of structure activity relationships (SAR) in toxicology. Toxicol Sci. 2000;56:8‐17. [DOI] [PubMed] [Google Scholar]
- 29. Fichez J, Soulie C, Le Corre L, et al. Discovery, SAR study and ADME properties of methyl 4‐amino‐3‐cyano‐1‐(2‐benzyloxyphenyl)‐1H‐pyrazole‐5‐carboxylate as an HIV‐1 replication inhibitor. RSC Med Chem. 2020;11:577‐582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Huang K, Luo S, Cong Y, et al. An accurate free energy estimator: based on MM/PBSA combined with interaction entropy for protein‐ligand binding affinity. Nanoscale. 2020;12:10737‐10750. [DOI] [PubMed] [Google Scholar]
- 31. Ngo ST, Tam NM, Pham MQ, Nguyen TH. Benchmark of popular free energy approaches revealing the inhibitors binding to SARS‐CoV‐2 Mpro. J Chem Inf Model. 2021;61(5):2302‐2312. [DOI] [PubMed] [Google Scholar]
- 32. Schafer H, Mark AE, van Gunsteren WF. Absolute entropies from molecular dynamics simulation trajectories. J Chem Phys. 2000;113(18):7809‐7817. [Google Scholar]
- 33. Lipinski CA. Lead‐ and drug‐like compounds: the rule‐of‐five revolution. Drug Discov Today Technol. 2004;1:337‐341. [DOI] [PubMed] [Google Scholar]
- 34. Itoh H, Inoue M. Comprehensive structure‐activity relationship studies of macrocyclic natural products enabled by their total syntheses. Chem Rev. 2019;119:10002‐10031. [DOI] [PubMed] [Google Scholar]
- 35. Ehrenworth AM, Peralta‐Yahya P. Accelerating the semi‐synthesis of alkaloid‐based drugs through metabolic engineering. Nat Chem Biol. 2017;13:249‐258. [DOI] [PubMed] [Google Scholar]
- 36. Mirhadi E, Rezaee M, Malaekeh‐Nikouei B. Nano strategies for berberine delivery, a natural alkaloid of Berberis. Biomed Pharmacother. 2018;104:465‐473. [DOI] [PubMed] [Google Scholar]
- 37. Abdifetah O, Na‐Bangchang K. Pharmacokinetic studies of nanoparticles as a delivery system for conventional drugs and herb‐derived compounds for cancer therapy: a systematic review. Int J Nanomedicine. 2019;14:5659‐5677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Swain SS, Paidesetty SK, Padhy RN, Hussain T. Isoniazid‐phytochemical conjugation: a new approach for potent and less toxic anti‐TB drug development. Chem Biol Drug des. 2020;96:714‐730. [DOI] [PubMed] [Google Scholar]
- 39. Xian Y, Zhang J, Bian Z, et al. Bioactive natural compounds against human coronaviruses: a review and perspective. Acta Pharm Sin B. 2020;10:1163‐1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chan HCS, Shan H, Dahoun T, Vogel H, Yuan S. Advancing drug discovery via artificial intelligence. Trends Pharmacol Sci. 2019;40:801. doi: 10.1016/j.tips.2019.06.004 [DOI] [PubMed] [Google Scholar]
- 41. Amin SA, Banerjee S, Gayen S, Jha T. Protease targeted COVID‐19 drug discovery: what we have learned from the past SARS‐CoV inhibitors? Eur J Med Chem. 2021;215:113294. doi: 10.1016/j.ejmech.113294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yang Y, Zhu Z, Wang X, et al. Ligand‐based approach for predicting drug targets and for virtual screening against COVID‐19. Brief Bioinform. 2021;22:1053‐1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jose PA, Jha B. New dimensions of research on actinomycetes: quest for next generation antibiotics. Front Microbiol. 2016;7:1295. doi: 10.3389/fmicb.2016.01295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Thawabteh A, Juma S, Bader M, et al. The biological activity of natural alkaloids against herbivores, cancerous cells and pathogens. Toxins (Basel). 2019;11:656. doi: 10.3390/toxins11110656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Saeed AFUH, Su J, Ouyang S. Marine‐derived drugs: recent advances in cancer therapy and immune signaling. Biomed Pharmacother. 2021;134:111091. doi: 10.1016/j.biopha.2020.111091 [DOI] [PubMed] [Google Scholar]
- 46. Zheng X, Wu F, Lin X, et al. Developments in drug delivery of bioactive alkaloids derived from traditional Chinese medicine. Drug Deliv. 2018;25:398‐416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Benet LZ, Hosey CM, Ursu O, Oprea TI. BDDCS, the rule of 5 and drug‐ability. Adv Drug Deliv Rev. 2016;101:89‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Doak BC, Kihlberg J. Drug discovery beyond the rule of 5 ‐ opportunities and challenges. Expert Opin Drug Discov. 2017;12:115‐119. [DOI] [PubMed] [Google Scholar]
- 49. Dahlgren D, Lennernäs H. Intestinal permeability and drug absorption: predictive experimental, computational and in vivo approaches. Pharmaceutics. 2019;11:411. doi: 10.3390/pharmaceutics11080411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kiela PR, Ghishan FK. Physiology of intestinal absorption and secretion. Best Pract Res Clin Gastroenterol. 2016;30:145‐159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Amin ML. P‐glycoprotein inhibition for optimal drug delivery. Drug Target Insights. 2013;7:27‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chen C, Lee MH, Weng CF, Leong MK. Theoretical prediction of the complex p‐glycoprotein substrate efflux based on the novel hierarchical support vector regression scheme. Molecules. 2018;23:1820. doi: 10.3390/molecules23071820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Subramanian G, Kitchen DB. Computational approaches for modeling human intestinal absorption and permeability. J Mol Model. 2006;12:577‐589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tao L, Zhang P, Qin C, et al. Recent progresses in the exploration of machine learning methods as in‐silico ADME prediction tools. Adv Drug Deliv Rev. 2015;86:83‐100. [DOI] [PubMed] [Google Scholar]
- 55. Yuce M, Cicek E, Inan T, et al. Repurposing of FDA‐approved drugs against active site and potential allosteric drug‐binding sites of COVID‐19 main protease. Proteins. 2021;89:1425‐1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Supporting Information
Data Availability Statement
All data are openly available in public repository sources, cited adequately, and complete information is available in the reference section or by request to the corresponding author for the full paper.