

Abstract
Amid the current COVID‐19 pandemic, the emergence of several variants in a relatively high mutation rate (twice per month) strengthened the importance of finding out a chemical entity that can be potential for developing an effective medicine. In this study, we explored ethyl acetate (EtOAc) extract of a marine‐derived fungus Aspergillus cosatricaensis afforded three butenolide derivatives, butyrolactones I, VI and V (1–3), two naphtho‐γ‐pyrones, TMC‐256 A1 (4) and rubrofusarin B (5) and methyl p‐hydroxyphenyl acetate (6). Structure identification was unambiguously determined based on exhaustive spectral analyses including 1D/2D NMR and mass spectrometry. The isolated compounds (1–6) were assessed for their in vitro anti‐inflammatory, antiallergic, elastase inhibitory activities and in silico SARS‐CoV‐2 main protease (Mpro). Results exhibited that only butenolides (1 and 2) revealed potent activities similar to or more than reference drugs unlike butyrolactone V (3) suggesting them as plausible chemical entities for developing lead molecules.
Keywords: Aspergillus, butenolides, natural products, receptors, SARS-CoV-2 Mpro
Chromatographic exploration of ethyl acetate extract derived from Aspergillus cosatricaensis yielded three butenolide derivatives, butyrolactones I, VI and V (1–3), two naphtho‐γ‐pyrones, TMC‐256 A1 (4) and rubrofusarin B (5); and methyl p‐hydroxyphenyl acetate (6). Chemical structures of isolated compounds were unambiguously determined by 1D and 2D NMR spectroscopy. Among butenolides, only compounds 1 and 2 unlike 3 exhibited potent in vitro activities as anti‐inflammatory and antiallergic along with in silico SARS‐CoV‐2 main protease (Mpro) inhibitory activity.

Introduction
For about two years now, the entire world has been and is still combating the Coronavirus Disease 2019 pandemic (COVID‐19), a viral infection of zoonotic origin initiated by severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) first reported in December 2019 at Wuhan, China.[ 1 , 2 ] Over the following two years, researchers worldwide intensively collaborated in a race‐against‐time to attain an efficient vaccine protecting and/or preventing COVID‐19 infections or complications. In December 2020, this task was first successfully accomplished when Pfizer‐BioNTech vaccine was granted the first Emergency Use Authorization (EUA) by the US Food and Drug Administration (FDA) [3] and Oxford‐AstraZeneca vaccine was also licensed the emergency use to prevent COVID‐19 infections in subjects 16 years old or more in the United Kingdom. [4] From December 2019 to October 2020, SARS‐CoV‐2 was found to monthly acquire two mutations in the global population. Among these acquired mutants, four variants have been recognized at widely spaced localities and exhibited a noticeably higher spreadability compared to the genuine COVID‐19 virus. These four variants were trivially named as alpha (B.1.1.7) first reported in the United Kingdom, beta (B.1.351) in South Africa, gamma (P.1) in Japan and Brazil along with delta (B.1.617.2) in India.[ 5 , 6 ] The medical community is more concerned about alpha and delta variants emerged in the United Kingdom and in India, respectively, due to their considerable impact on severity and death cases compared to the other variants. The emergence of these new variants and their resulting uncontrolled human transmissions urged several countries to return to strict pandemic control measures including lockdown and social distancing. [5] Unexpectedly, fully vaccinated people were found to be infected with alpha and delta variants that also questioned the effectiveness of approved vaccines against emerging variants. Therefore, much interest has been directed toward discovering new or repurposing known pharmaceuticals that very recently succeeded in granting molnupiravir (Lagevrio® by Merck) and ritonavir (Paxlovid® by Pfizer) the Emergency Use Authorization (EUA) or on their ways to in UK and USA, respectively. [7]
However, there are still many concerns about their potential side effects that may occur based on their mechanisms of action and long‐term usage that are not yet investigated.
Another strategy can be followed in combating the pandemic is to find out treatment alternatives aiming at ameliorating the complications of COVID‐19 variants’ infections in high‐risk population particularly the intense respiratory symptoms caused, among others, by the intracellular enzyme neutrophil elastase (NE) stored in azurophilic granules of polymorphonuclear neutrophils (PMNs).[ 8 , 9 ] More research interests have been directed toward exploring natural products for their potential NE inhibitory activities. Despite that the main function of NE is to degrade functional pathogenic proteins, it also initiates pathologic effects on elastin‐rich connective tissue, such as in lungs, leading to acute respiratory distress syndrome (ARDS), acute lung injury (ALI) or chronic obstructive pulmonary disorder (COPD).[ 9 , 10 ]
To fulfill this task and in the course of our running research aiming at exploring fungal secondary metabolites and their potential bioactivities, in this study, we investigated those isolated from a marine‐associated fungus Aspergillus costaricaensis. All isolated compounds were tested in vitro for anti‐inflammatory and antiallergic properties. In addition, isolated compounds were subjected to in silico molecular modelling and molecular dynamics studies against NE and SARS‐CoV‐2 main protease (Mpro) as a plausible target for antiviral drug design. In the present study, we purified and identified three fungal butenolides, butyrolactones I, VI and V (1–3), together with two naphtho‐γ‐pyrones (4 and 5) and methyl p‐hydroxyphenyl acetate (6). In addition, we discuss the results of in vitro bioactivity and in silico molecular modelling and molecular dynamics simulation along with structure activity relationships (SARs).
Results and Discussion
Purification and Identification of Secondary Metabolites in Fungal Extract
A thorough chromatographic exploration of the fungal extract, applying different separation procedures, yielded (Figure 1) three butenolide derivatives, butyrolactones I,[ 11 , 12 , 13 ] VI [14] and V[ 15 , 16 ] (1–3) along with two naphtho‐γ‐pyrones namely, TMC‐256A1 (4),[ 17 , 18 , 19 , 20 ] rubrofusarin B (5)[ 20 , 21 , 22 ] and methyl p‐hydroxyphenyl acetate (6).[ 23 , 24 ] Chemical structures of isolated compounds were determined based on mass spectrometry, exhaustive 1D, 2D NMR analyses and by comparison with reported literature (see Supporting Information).
Figure 1.

Chemical structures of 1–6.
Compounds (1–3) were obtained as amorphous colourless solids whose UV spectra revealed almost similar two absorption maxima at 220 and 310 nm indicating that these three compounds are related derivatives and possibly sharing a common chromophore. The molecular formulae of 1–3 were established to be C24H24O7, C24H26O9 and C24H24O8 according to their HRESIMS, respectively (see Supporting Information, Figures S3, S10 and S17). The molecular formulae showed clear differences between three compounds as two additional hydroxyl groups in 3 and an oxygen atom in 2 compared to 1, respectively. 1H and 13C NMR spectral data of 1–3 clearly explained the differences noticed in molecular formulae (see Supporting Information, Tables S1–S3). By comparing 1D NMR spectra of compounds (1–3) along with their molecular formulae and weights with the reported literature, they were unambiguously determined to be butyrolactones I,[ 11 , 12 , 13 ] VI [14] and V,[ 15 , 16 ] respectively. The stereochemistry of C‐4 in compounds (1–3) was determined to have R‐configuration based on their 1D/2D NMR spectral data including 1H, 13C NMR and NOESY in addition to the comparison with reported literature.[ 12 , 14 , 15 , 16 ] The stereochemistry of C‐8” in 2 was not possible to determine based on the available 2D NMR spectral data including NOESY spectrum as also reported in literature. [15] However, the stereochemistry of C‐8” in 3 was depicted to be S‐configuration based on the close similarity between the measured 1H/13C NMR spectral data and those reported in literature.[ 14 , 15 , 16 ]
Compounds 4 and 5 exhibited quite similar UV spectra with maximal absorption peaks (λmax) at 240, 275 and 330 nm that suggested the similar chromophore group in their structures. By comparing 1H and 13C NMR spectral data of 4 and 5 (see Supporting Information, Tables S4 and S5), they displayed a close similarity in both chemical shifts and coupling constant values in 1H NMR and carbon resonances in 13C NMR spectra. The main difference was the existence of two methoxy groups in 5 instead of one methoxy group in 4. The second methoxy group in 5 showed a singlet methyl resonance at δH 3.91 nm in 1H NMR spectrum that was correlated to a carbon peak at δC 56.0 as revealed by HMQC spectrum. Based on spectral data for 4 and 5, and by searching the reported literature, they were determined to be naphtho‐γ‐pyrone derivatives namely, TMC‐256A1[ 17 , 18 , 19 , 20 ] and rubrofusarin B,[ 20 , 21 , 22 ] respectively.
Compound 6 was easily identified based on its 1H and 13C NMR spectral data (see Supporting Information Table S6) that suggested the presence of 1,4‐disubstituted aromatic ring via the presence of two proton peaks each integrated for a pair of magnetically equivalent protons at δH 6.72 and at δH 7.07 with a similar coupling constant (J value) of 8.4 Hz and were correlated via HMQC spectrum to two carbon resonances at δC 116.3 and δC 131.3, respectively. In addition, a singlet methylene and a singlet‐oxygenated methyl were also identified at δH 3.53 and at δH 3.66 ppm, respectively. Based on the obtained data and by comparing with the reported literature,[ 23 , 24 ] compound 6 was found to be methyl p‐hydroxyphenyl acetate.
Anti‐Inflammatory Activity
Anti‐inflammatory activity assay was conducted in human neutrophils by determining fMLF/CB‐induced elastase release and the data of 2 and 4–6 were compared with literature 1 and 3 (Table 1). [25] The previous study revealed that butyrolactone I (1) significantly and dose‐dependently inhibited elastase release with IC50 2.30 μM, while butyrolactone V (3) was inactive. [25] The current results further demonstrated that butyrolactone VI (2) was also active, but with lower potency of IC50 5.25 μM. Compounds 4–6 were inactive. The data indicated importance of prenyl group in 1 but also presence of vicinal dihydroxy group in 2 for the activity, while the presence of oxirane ring in 3 indicated inactivity. Compounds 3 and 6 did not show significant effects. The cell viability was not affected by neither 1, [25] 2 or 3 [25] at 10 μM (Table 1).
Table 1.
Effects of compounds (1–6) on elastase release, viability and elastase enzyme activity in vitro.
|
Compound |
Elastase Release, Human Neutrophils[a] |
Cell Viability, Human Neutrophils[c] |
Elastase Enzymatic Activity (Cell‐Free)[d] |
|---|---|---|---|
|
IC50 (μM) [b] |
(% at 10 μM) |
IC50 (μM)[b] |
|
|
Butyrolactone I (1) [f] |
2.30±0.27 [25] |
94.13±2.31 [25] |
16.70±2.64 [25] |
|
Butyrolactone VI (2) |
5.25±0.38 |
97.52±2.26 |
12.61±0.25[e] |
|
Butyrolactone V (3) [f] |
>10 [25] |
98.25±1.77 [25] |
>30 [25] |
|
TMC‐256 A1 (4) |
>10 |
n.t. |
n.t. |
|
Rubrofusarin B (5) |
>10 |
n.t. |
n.t. |
|
Methyl p‐hydroxyphenyl acetate (6) |
>10 |
n.t. |
n.t. |
[a] Inhibition of fMLF/cytochalasin B (CB)‐induced elastase release in human neutrophils. Values marked as “>10” are considered as inactive. Genistein inhibited elastase release with an IC50 value 32.67±1.45 [25] [b] Concentration required for 50 % inhibition (IC50). The results are presented as mean±S.E.M. (n=3). [c] Percentage of cell viability (%) at 10 μM. The results are based on the lactate dehydrogenase release and presented as mean±S.E.M. (n=3); n.t.: not tested. [d] Sivelestat was used as a positive control and inhibited elastase enzyme with an IC50 value 17.92±4.66 nM; [25] n.t.: not tested. [e] Butyrolactone VI (2) inhibited elastase by 69.65 % at 30 μM. [f] Data shown in Molecules 2021, 26, 3354, 10.3390/molecules26113354 [25]
Antiallergic Activity
The antiallergic activity of samples 2 and 4–6 was evaluated by β‐hexosaminidase release assay in RBL‐2H3 cells using calcium ionophore A23187 and antigen (DNP‐BSA plus anti anti‐DNP IgE) as inducers (Table S7). The cells were first treated with samples to find their non‐toxic effects using MTT assay (over 90 % viability at 100 μM). Compounds 2 and 4–6 didn't show significant effects (see Supporting Information, Table S7). However, previous publication revealed significant and dose‐dependent activity of butyrolactone I (1) on both A23187 (IC50=39.7 μM) and antigen‐induced (IC50 41.6 μM) degranulation, while butyrolactone V (3) was inactive. [25] Thus, the presence of prenyl group may be an important factor for antiallergic activity of butanolides.
Molecular modelling studies
In this study, docking study was used to predict possible binding poses of tested compounds with the human neutrophil elastase (NE). We used the reported NE crystal structure (PDB: 1H1B) for our study. [26] The co‐crystalized ligand, a pyrrolidine‐based inhibitor called GW475151, was found to form the crucial hydrogen bond with SER 195 that is involved in the binding and recognition. [27] We have reported earlier a docking procedure validation in which we removed the co‐crystalized ligand and then redock it in the active site. AutoDock Vina was able to predict the same crystal structure pose with RMSD of 1.317 and a docking score of −6.9 kcal/mol. [26]
Here, we docked the tested compounds (1–6) in the pocket of the human neutrophil elastase (PDB ID: 1H1B). Out of the tested compounds, two butyrolactones have shown docking scores superior or similar to GW475151, our control ligand (Table 2). These compounds include butyrolactone I (1) and butyrolactone VI (2). Both butyrolactones formed hydrogen bonds with SER 195 as seen in GW475151. It is noticeable that these two compounds, 1 and 2, were able to inhibit NE in in vitro assay with IC50 of 2.30 and 5.25 μM, respectively. In addition to SER 195, 1 and 2 were able to form additional interactions with arginine residues in the active site including ARG 147 and ARG 177. Figure 2 shows binding poses of both compounds and their interaction with amino acids in the pocket of the human neutrophil elastase.
Table 2.
Predicted binding affinities (kcal/mol) of isolated tested natural products (1–6) with human neutrophil elastase and SARS‐CoV‐2 viral main protease (Mpro). Important interacting residues are also shown. Data shown represents the least energy‐binding mode (first binding pose) unless otherwise mentioned.
|
Isolated Natural Product/Ligand |
1H1B (Elastase) |
6LU7 (Mpro) |
||
|---|---|---|---|---|
|
Binding Affinity (kcal/mol) |
Interacting Residues |
Binding Affinity (kcal/mol) |
Interacting Residues |
|
|
Butyrolactone I (1)[a][25] |
−7.3 |
Ser195‐Arg147 |
−7.3 |
Gly143‐Ser144‐His163‐Glu166 |
|
Butyrolactone VI (2) |
−6.9 |
Ser195‐Arg177 |
−8.1 |
Thr26‐Gly143‐Ser144‐Glu166 |
|
Butyrolactone V (3)[a][25] |
−6.9 (2nd pose) |
Ser195‐Val216 |
−7.2 |
His163‐His164‐Arg188‐Gln189 |
|
TMC‐256 A1 (4) |
−6.4 |
Ser195‐Phe41‐Gly193 |
−6.8 |
Gly143‐Glu166‐His41 |
|
Rubrofusarin B (5) |
−6.4 |
Ser195‐Phe41‐Val216‐Gly193 |
−7.0 |
Leu141‐Gly143‐Glu166 |
|
Methyl p‐hydroxyphenyl acetate (6) |
−4.9 |
Ser195 |
−4.8 |
Glu166 |
|
−6.9 |
Ser195 |
– |
– |
|
|
N3 |
– |
– |
−7.1 (3rd pose) |
Phe140, Gly143, His163, His164, Glu166, Gln189, Thr190 |
[a] Data shown in Molecules 2021, 26, 3354, 10.3390/molecules26113354. [25]
Figure 2.

Docking of tested compounds against human NE. a. Hydrogen bond interactions of 1 (blue). b. Docking pose of 1 in target active site represented as a surface colored according to electrostatic potential. c. Hydrogen bond interactions of 2 (yellow). d. Docking pose of 2 in target active site represented as a surface colored according to electrostatic potential.
The current COVID‐19 pandemic has encouraged several researchers to investigate potential roles of natural products to compete SARS‐CoV‐2 targets. Based on that, we decided to check if our isolated compounds could play a role against SARS‐CoV‐2 main protease (Mpro) using docking. This target was among the first targets investigated for this virus since the emergence of the pandemic. For the purpose of this study, we used the reported crystal structure of main protease (Mpro) (PDB: 6LU7) which is co‐crystalized with an inhibitor called N3.[ 26 , 28 ] We have used the same crystal structure earlier to investigate other natural products and reported docking validation where the co‐crystalized ligand showed docking score of −7.1 kcal/mol. [26] Docking results of the tested compounds were interesting where all three butyrolactones were found to show docking scores better than the co‐crystalized ligand.
Amongst three compounds, butyrolactone VI (2) exhibited the best docking score (−8.1 kcal/mol). All three butyrolactones have shown several hydrogen bonds among which Gly143 and Glu166 are common between the three butyrolactones and co‐crystalized ligand (see Table 2). The binding modes of three butyrolactones are very similar and are overlapping with the same positions taken by the co‐crystallized ligand. Figure 3 shows the binding pose and interactions of the best butyrolactone VI (2). It also shows the overlapped pose with both N3 and other butyrolactones as predicted by docking. These results suggest that butyrolactones isolated here might possess promising inhibitory effects on SARS‐CoV‐2 main protease (Mpro) and encourage for further investigations of this class of compounds.
Figure 3.

Docking of tested compounds with the main protease (Mpro) of SARS‐CoV‐2 (PDB ID: 6LU7). a. Interactions of butyrolactone VI (2) with residues in the active site. b. Predicted binding pose of butyrolactone VI (2). c. Docking pose of 2 superimposed on the co‐crystalized ligand N3 (blue). d. Predicted binding pose of 2 overlapped with that of butyrolactone I (1, green) showing similar binding poses. e. Structure of the co‐crystallized ligand (N3) in 6LU7 pdf file.
Molecular dynamic simulation
To better understand the potential interaction between our isolated butyrolactones and SARS‐CoV‐2 main protease, we decided to run a molecular dynamics (MD) simulation for four different complexes for a duration of 100 ns production run. These complexes are the apoprotein main protease (Mpro) and the complexes with co‐crystalized ligand, butyrolactone I (1) and VI (2). For the purpose of our study, all complexes were prepared based on the docking poses and were subjected to canonical ensemble (NVT) followed by isothermal‐isobaric ensemble (NPT) equilibration.
This was followed by a production run of 100 ns. The produced trajectories were then analyzed to investigate the stability of the assessed complexes. Results of the trajectory analysis are shown in Figure 4 and Table S8. Stable root mean square deviation (RMSD) of the investigated protein indicate stable dynamics run. Figure 4a shows that the RMSD of protein of all the studied complexes is stable with minimal fluctuation. The standard deviation of RMSD was less than 0.6 Å in all the studied complexes.
Figure 4.

Molecular dynamics plots in the active site of Mpro. a. RMSD of protein. b. Radius of Gyration (Rg) of protein. c. RMSD of ligand heavy atoms. d. Average count of hydrogen bonds between tested compound and target.
The fluctuation in case of butyrolactones was less than that of the co‐crystalized ligand as seen in Table S8. Radius of gyration (Rg) is another parameter that reflects compactness of the protein. Stable Rg indicate preferred ligand binding. Figure 4b shows that the insertion of ligands did not affect Rg indicating stable binding between tested ligands and the main protease. Low RMSD of the ligands indicate tight binding between that ligand and the protein. Figure 4c shows that the fluctuation of butyrolactone VI (2) was the least among the tested ligands. RMSD of butyrolactone VI was 0.358±0.076 nm which is less than that of the co‐crystalized ligand (0.445±0.156 nm) and butyrolactone I (0.488±0.115 nm).
This suggests that butyrolactone VI might be a good inhibitor of SARS‐CoV‐2 Mpro and requires further investigation. This is also supported by the average number of hydrogen bonds (Figure 4d and Table S8). The average number of hydrogen bonds between butyrolactone VI and the main protease (Mpro) during the 100 ns production run was found to be 3.43 hydrogen bonds. This is higher than that of the co‐crystalized ligand (3.09 hydrogen bonds) and butyrolactone I (2.26 hydrogen bonds). All these results support plausible potential inhibition of SARS‐CoV‐2 main protease (Mpro) by butyrolactone VI and encourage its further investigation.
Elastase enzymatic activity
Human neutrophil elastase plays an important role in the development of many inflammatory syndromes accompanying several acute and chronic respiratory disorders. In the cell‐free system, butyrolactone I (1) previously revealed a dose‐dependent direct inhibitory effect on the enzymatic activity of elastase. [25] According to current study, butyrolactone VI (2) also inhibited elastase enzymatic activity (Figure 5) in the dose‐dependent manner (Table 1) with even more potent activity (IC50 12.61 μM) in comparison to 1 (IC50 16.70 μM). The data are in accordance to the in silico molecular binding data. Based on these results, the anti‐inflammatory effects of 2 were, at least partly, attributed to its interaction with elastase enzyme, and by comparing the potency with 1, [25] the proportion of elastase enzyme inhibitory activity was higher in 2.
Figure 5.
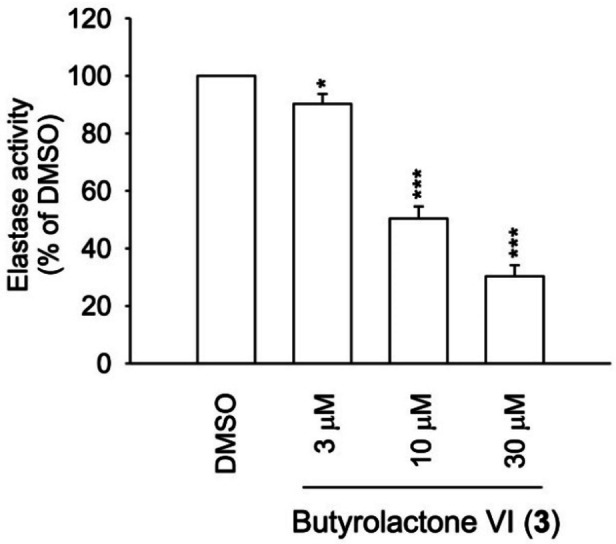
Elastase inhibitory activity of butyrolactone VI (2) in the cell‐free system. The results are shown as mean±S.E.M. (n=3). * p < 0.05, *** p < 0.001 compared with 0.1 % DMSO control.
Protective effects against human coronavirus 229E infection
Human coronavirus 229E (HCoV‐229E) is a strain of coronavirus family viruses, that causes infection of upper respiratory tract with symptoms of common cold. [29] The data of 1 and 3 were previously reported. [25] We further assessed compound 2 in vitro protective effect against the human coronavirus 229E (HCoV‐229E) (see Supporting Information, Figure S2). None of the compounds exhibited a cellular protective effect against HCoV‐229E infection. It is important to note, that the cytopathic effects may not be observed at lower doses used in both studies (10 μM) and the differences between the strains should be considered. Thus, the absence of in vitro cytoprotective activity against viral infection does not necessarily contradict the in silico SARS‐CoV‐2 main protease (Mpro) inhibitory activity that implies in the final stage of assembly of the new viruses.
Over the past year, many researchers recognized SARS‐CoV‐2 Mpro as a crucial target for new drug design and repurposing to defend against COVID‐19 pandemic. Secondary metabolites from different chemical categories and/or sources have been explored as a rich source of inhibitors with many positive examples belonging to flavonoids, alkaloids, peptides, terpenoids, coumarins, and tannins. [30] Flavonoids presented many hit compounds as Mpro inhibitors including flavonoids (quercetin, apigenin and luteolin with IC50 of 20–200 μM) and bisflavonoids (amentoflavone with IC50=8.3 μM). [31]
For more understanding of the Mpro inhibitory activity, in silico molecular modelling and molecular dynamics, simulation studies were performed.[ 26 , 32 , 33 ] In this study, our results enriched the pipeline of potential Mpro inhibitors with additional examples supported by the obtained results regarding their binding poses together with being proved in vitro as inhibitors of human elastase release. In summary, the obtained results also support the plausible efficiency of butyrolactones I (1) and VI (2) in defending the respiratory distress symptoms caused by SARS‐CoV‐2 infection inflammatory responses.
Conclusion
Six different compounds including three butenolides, butyrolactones I, VI and V (1–3), two naphtho‐γ‐pyrones, TMC‐256 A1 (4) and rubrofusarin B (5), together with methyl p‐hydroxyphenyl acetate (6) were purified from EtOAc extract of a marine‐associated fungus Aspergillus costaricaensis. Amongst the isolated compounds, only butyrolactones I (1) and VI (2) revealed potent activities in the in vitro human neutrophil elastase release and cell‐free antielastase assays with IC50 values ranging from 2.30 to 16.70 μM, sometimes exceeding those of the used reference drugs (IC50 values of 32.67 and17.92 μM, respectively). The inhibitory activities of 1 and 2 are of particular importance for improving the debilitating effects of SARS‐CoV‐2 infection on pulmonary system as elastin‐rich organ. In an attempt to understand these results, molecular docking and molecular dynamics simulation studies were carried out to determine the binding modes of butyrolactones to target receptors and the possible inhibitory activity(ies) of 1–3 against SARS‐CoV‐2 main protease, a crucial enzyme for producing the viral functional proteins. Butyrolactone VI (2) proved to be a promising candidate for further investigation to develop a lead compound that might become a nucleus for a new pharmaceutical against SARS‐CoV‐2.
Supporting Information Summary
Supplementary data associated with this article can be found in the online version at doi: XXXXX A detailed description of the applied experimental methods along with full spectroscopic data of compounds (1–6; Figures S3‐S38) are included in Supporting Information File. Tables S1‐S6 are describing comparisons of both measured 1H/13C NMR and those reported in literature of compounds (1–6), respectively.
Author Contributions
S.S.E., W.L. and B.K.: conceptualization, writing, reviewing and editing the manuscript; I.S.U., S.S.E. and B.K.: methodology, purification, and isolation of pure metabolites; I.S.U., S.S.E. and B.K.: NMR analysis and data curation; A.A. and B.S.A.: molecular modelling; M.K. bioactivity assays analysis. All authors have read and agreed to the published version of the manuscript.
Conflict of interest
The authors declare that there is no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
S.S.E. acknowledges The Scientific and Technological Research Council of Turkey (TÜBITAK) for financially supporting him via a visiting scientist fellowship. We are grateful to Prof. Tsong‐Long Hwang, Bing‐Hung Chen and Jim‐Tong Horng for bioactivity assays support.
I. S. Uras, M. Korinek, A. Albohy, B. S. Abdulrazik, W. Lin, S. S. Ebada, B. Konuklugil, ChemistrySelect 2022, 7, e202200130.
Contributor Information
Prof. Sherif S. Ebada, Email: sherif_elsayed@pharma.asu.edu.eg.
Prof. Belma Konuklugil, Email: konuklug@pharmacy.ankara.edu.tr, Email: belma.konuklugil@lokmanhekim.edu.tr.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Rothan H. A., Byrareddy S. N., J. Autoimmun. 2020, 108, 102433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. da Silva P. G., Mesquita J. R., Nascimento M. D. J., Ferriera V. A. M., Sci. Total Environ. 2021, 750, 141483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pfizer-BioNtech COVID-19 Vaccine. Available online: https://www.fda.gov/emergency-preparedness-and-response/coronavirus-disease-2019-covid-19/pfizer-biontech-covid-19-vaccine (accessed on 4 January 2021).
- 4.COVID-19: Oxford-AstraZeneca Vaccine Approved for Use in UK. Available online: https://www.bbc.com/news/health-55280671 (accessed on 4 January 2021).
- 5.Risk Assessment: Risk Related to Spread of New SARS-CoV-2 Variants of Concern in the EU/EEA. Available online: https://www.ecdc.europa.eu/en/publications-data/covid-19-risk-assessment-spread-new-sars-cov-2-variants-eueea (accessed on 4 January 2021).
- 6. Harvey W. T., Carabelli A. M., Jackson B., Gupta R. K., Thomson E. C., Harrison E. M., Ludden C., Reeve R., Rambaut A., Peacock S. J., Robertso D. L., Nat. Rev. Microbiol. 2021, 19, 409–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Imran M., Arora M. K., Asdaq S. M. B., Khan S. A., Alaqel S. I., Alshammari M. K., Alshehri M. M., Alshrari A. S., Ali A. M., Al-shammeri A. M., Alhazmi B. D., Harshan A. A., Alam M. T., Abida, Molecules 2021, 26, 5795.34641339 [Google Scholar]
- 8. Henriksen P. A., Curr. Opin. Hematol. 2014, 21, 23–28. [DOI] [PubMed] [Google Scholar]
- 9. Hoenderdos K., Condliffe A., Am. J. Respir. Cell Mol. Biol. 2013, 48, 531–539. [DOI] [PubMed] [Google Scholar]
- 10. Moraes T. J., Chow C. W., Downey G. P., Crit. Care Med. 2003, 31, S189–S194. [DOI] [PubMed] [Google Scholar]
- 11. Niu X., Dahse H.-M., Menzel K.-D., Lozach O., Walther G., Meijer L., Grabley S., Sattle I., J. Nat. Prod. 2008, 71, 689–692. [DOI] [PubMed] [Google Scholar]
- 12. Rao K. V., Sadhukhan A. K., Veerender M., Ravikumar V., Mohan E. V. S., Dhanvantri S. D., Sitaramkumar M., Babu J. M., Vyas K., Reddy G. O., Chem. Pharm. Bull. 2000, 48, 559–562. [DOI] [PubMed] [Google Scholar]
- 13. Kiriyama N., Nitta K., Sakaguchi Y., Taguchi Y., Yamamoto Y., Chem. Pharm. Bull. 1977, 25, 2593–2601. [Google Scholar]
- 14. Liu M., Zhou Q., Wang J., Liu J., Qi C., Lai Y., Zhu H., Xue Y., Hu Z., Zhang Y., RSC Adv. 2018, 8, 13040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lin T., Lu C., Shen Y., Nat. Prod. Res. 2009, 23, 77–85. [DOI] [PubMed] [Google Scholar]
- 16. Haritakun R., Rachtawee P., Chanthaket R., Boonyuen N., Isaka M., Chem. Pharm. Bull. 2010, 58, 1545–1548. [DOI] [PubMed] [Google Scholar]
- 17. Bouras N., Mathieu F., Coppel Y., Lebrihi A., Nat. Prod. Res. 2005, 19, 653–659. [DOI] [PubMed] [Google Scholar]
- 18. Priestap H. A., Mag. Res. Chem. 1986, 24, 875–878. [Google Scholar]
- 19. Priestap H. A., Tetrahedron 1984, 40, 3617–3624. [Google Scholar]
- 20. Sakurai M., Kohno J., Yamamoto K., Okuda T., Nishio M., Kawano K., Ohnuki T., J. Antibiot. 2002, 55, 685–692. [DOI] [PubMed] [Google Scholar]
- 21. Shaaban M., Shaaban K. A., Abdel-Aziz M. S., Org. Med. Chem. Lett. 2012, 2, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Graham J. G., Zhang H., Pendland S. L., Santarsiero B. D., Mesecar A. D., Cabieses F., Farnsworth N. R., J. Nat. Prod. 2004, 67, 225–227. [DOI] [PubMed] [Google Scholar]
- 23. Shen S., Li W., Wang J., Nat. Prod. Res. 2013, 27, 2286–2291. [DOI] [PubMed] [Google Scholar]
- 24. Bose D. S., Narsaiah A. V., Bioorg. Med. Chem. 2005, 13, 627–630. [DOI] [PubMed] [Google Scholar]
- 25. Uras I. S., Ebada S. S., Korinek M., Albohy A., Abdulrazik B. S., Wang Y.-H., Chen B.-H., Horng J.-T., Lin W., Hwang T.-L., Konuklugil B., Molecules 2021, 26, 3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ebada S. S., Al-Jawabri N. A., Youssef F. S., El-Kashef D. H., Knedel T.-O., Albohy A., Korinek M., Hwang T.-L., Chen B.-H., Lin G.-H., Lin C.-Y., Aldalaien S. M., Disi A. M., Janiak C., Proksch P., RSC Adv. 2020, 10, 32128–38141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Macdonald S. J. F., Dowle M. D., Harrison L. A., Clarke G. D. F., Inglis G. G. A., Johnson M. R., Shah P., Smith R. A., Amour A., Fleetwood G., Humphreys D. C., Molloy C. R., Dixon M., Godward R. E., Wonacott A. J., Singh O. M. P., Hodgson S. T., Hardy G. W., J. Med. Chem. 2002, 45, 3878–3890. [DOI] [PubMed] [Google Scholar]
- 28. Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R., Science 2020, 368, 409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zumla A., Chan J. F., Azhar E. I., Hui D. S., Yuen K. Y., Nat. Rev. Drug Discovery 2016, 15, 327–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sayed A. M., Khattab A. R., AboulMagd A. M., Hassan H. M., Rateb M. E., Zaid H., Abdelmohsen U. R., RSC Adv. 2020, 10, 19790–19802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ryu Y. B., Jeong H. J., Kim J. H., Kim Y. M., Park J. Y., Kim D., Nguyen T. T., Park S. J., Chang J. S., Park K. H., Rho M.-C., Lee W.-S., Bioorg. Med. Chem. 2010, 18, 7940–7947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Joshi R. S., Jagdale S. S., Bansode S. B., Shankar S. S., Tellis M. B., Pandya V. K., Chugh A., Giri A. P., Kulkarni M. J., J. Biomol. Struct. Dyn. 2021, 39, 3099–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Prasanth D. S. N. B. K., Murahari M., Chandramohan V., Panda S. P., Atmakuri L. R., Guntupalli C., J. Biomol. Struct. Dyn. 2021, 39, 4618–4632. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.