

Abstract
The prevention of the disease severity seems critical for reducing the mortality of Coronavirus (CoV) disease‐19. The neutrophils play a key role in the induction of severity. It is proposed here that inhibition of neutrophil activation and/or cascade reactions of complement, leading to this cell activation at the early phase of the disease, is a potential tool to inhibit aggravation of the disease. The need for appropriate timing in intervention is emphasized as follows. (1) Intervention at the very early stage of severe acute respiratory syndrome‐CoV‐2 infection may harm the defensive host response to the infection because of the critical function of neutrophils in this response, and (2) intervention at too late a stage will not stop the infiltration of fully activated neutrophils that produce large amounts of toxic substances.
Keywords: ARDS, COVID‐19, GPI‐80, infection immunity, neutrophil
Abbreviations
- ARDS
acute respiratory distress syndrome
- CAR‐T
chimeric antigen receptor‐transfected T cell
- C5a
complement factor 5a
- C5aR1
C5a receptor 1
- CoV
Coronavirus
- COVID‐19
CoV disease 2019
- CSS
cytokine storm syndrome
- CXC
C‐X‐C chemokine
- CXCR
CXC receptor
- CXCL
CXC ligand
- DAD
diffuse alveolar damage
- DAMP
damage‐associated molecular pattern
- ds
double‐stranded
- fMLP
N‐formyl‐l‐methionyl‐l‐leucyl‐phenylalanine
- G‐CSF
granulocyte‐colony stimulating factor
- GM‐CSF
granulocyte macrophage colony‐stimulating factor
- GPI
glycosylphosphatidylinositol
- IRF
IFN regulatory factor
- IKK
I kappa B kinase
- JAK
Janus tyrosine kinase
- LCN
lipocalin
- LDN
low density neutrophil
- MPO
myeloperoxidase
- NET
neutrophil extracellular trap
- PAMPs
pathogen‐associated molecular pattern
- PMA
phorbol 12‐myristate 13‐acetate
- PRR
pattern recognition receptor
- RETN
resistin
- RIG‐1
retinoic acid inducible gene‐1
- RLR
RIG‐1 like receptor
- ROS
reactive oxygen species
- SARS
severe acute respiratory syndrome
- scRNA‐seq
single cell RNA sequencing
- sGPI‐80
soluble form of GPI‐80
- ss
single‐stranded
- suPAR
soluble form of urokinase type plasminogen activator receptor
- TAK‐1
TGF‐β‐activated kinase‐1
INTRODUCTION
Coronavirus (CoV) disease‐19 (COVID‐19), induced by severe acute respiratory syndrome (SARS)‐CoV‐2, is spreading around the world. As of this writing, the number of people infected with SARS‐CovV‐2 is approximately 440 million, and the total number of deaths from COVID‐19 to date has reached around 6 million (WHO March 3, 2022)
The development of vaccines against COVID‐19 resulting in a reduction of SARS‐CoV‐2 infection has been changing the situation of pandemics of this communicable disease. 1 , 2 , 3 However, recent studies have revealed that emerging variants may escape from the protective immunity obtained through vaccination, since breakthrough infections after two full vaccinations have been observed in persons infected with variant viruses, especially the B.1.1.529 variant (Omicron). 4 , 5 , 6 , 7 , 8 These situations give us an impression that many problems remain to be resolved to gain long term protective immunity against SARS‐CoV‐2 infection. We believe that clarifying the possible mechanisms of the detailed pathogenesis is essential for the prevention and/or therapy of COVID‐19, since previous research on this disease suggests that interactions between SARS‐CoV‐2 and human hosts have a high variability depending on the individuals suffering from it and the phase of the disease. Symptoms of COVID‐19 vary from asymptomatic to severe such as dyspnea with pneumonia. We need to study the pathophysiology of each phase of the disease, and the reason why some people are asymptomatic and others have severe disease which finally ends in death. Otherwise, it seems difficult to find possible ways to prevent severe disease.
Many studies on the pathogenesis of COVID‐19 suggest that the host response plays a pivotal role in the determination of the disease direction. 9 , 10 , 11 Single cell RNA sequencing (scRNA‐seq) analysis of whole blood cells and those from bronchoalveolar lavage (BAL) suggest that various dysregulations of cytokine production is observed in severe COVID‐19 patients, which may result from an imbalance between pro‐inflammatory and anti‐inflammatory signatures in severe disease. We need to analyze the possible mechanisms of this imbalance in order to reach prevention and therapy of the disease through immunological intervention in the course towards severity. Understanding the sequence and tempo of the host response to SARS‐CoV‐2 infection seems most important, because cells involved in the inflammatory and the immune response, and the cytokines produced by these cells that play a key role in the response change enormously depending on the phase of the response. 9 , 12 , 13
Cell interaction which may determine the direction of the host response in the course of COVID‐19 from asymptomatic to the severe phase has been studied previously mainly with interaction between different classes of macrophages and lymphocytes, and various hypotheses have been proposed in consideration of these cells to explain the mechanisms that differentiate severe disease from mild or moderate. However, recent studies using scRNA‐seq suggests that neutrophils are also one of the main players in the host response to COVID‐19, 14 although these cells have been overlooked as an immunological coordinator in the host response to infections.
From the view point of the immunological manipulation of COVID‐19, we believe that neutrophils may be an important target because of their intensive function as an effector cell not only in the host defense, but also in damage to normal tissues. If we are able to manipulate the function of these cells, we may stop the worsening pathway toward severe COVID‐19. Considering that these cells are double‐edged swords in the host response, meaning that neutrophils play an important role for host protection against COVID‐19 in the very early phase of the disease and show a harmful effect in the late phase of the severe disease, we have to accurately select the timing of the manipulation. In this context, we will attempt to delve into the issue by finally focusing especially on studies of the dynamics of neutrophils in this disease that may directly lead to saving the lives of patients with COVID‐19.
THE KEY FACTORS FOR DISCRIMINATION OF SEVERE FROM MILD OR MODERATE COVID‐19 IN THE BEGINNING OF SARS‐COV‐2 INFECTION
The first line of defense against viral infection is mainly governed by inflammation and innate immunity. The recognition system of innate immunity has not been clearly presented scientifically for a long time, but molecular biological and immunogenetic studies in the past two decades have revealed mechanisms at the molecular level. 15 Thus, the primary host defense to viral infection is nowadays understood through pattern recognition receptors (PRRs), such as Toll‐like receptors (TLRs) and retinoic acid inducible gene‐1 (RIG‐1) like receptor (RLR), that recognize pathogen‐associated molecular patterns (PAMPs). Nucleic acids of viruses such as double‐stranded (ds) RNA, single‐stranded (ss) RNA, and DNA are recognized by PRRs, which is followed by the production of varying cytokines including IFN and inflammatory cytokines such as IL‐1, IL‐6, TNF‐α, and IL‐12. 16
Under this situation, the primary host response to SARS‐CoV‐2 infection which may proceed to severe disease of COVID‐19 is now analyzed from a viewpoint of PPRs and signaling pathways that are induced through interaction of PPRs with PAMPs. A typical dysregulation of cytokine production in SARS‐CoV‐2 infection is a delayed type‐I IFN (IFN‐I) response. 17 As IFN‐I production is the first host response to viral infection, its delayed response inhibits the subsequent host response, which results in an over proliferation of the virus. Recent studies show that various molecules in SARS‐Cov‐2 inhibit IFN production through attenuating PRR signaling pathways. SARS‐CoV‐2 nucleocapsid protein promotes hyperactivation of nuclear factor‐kappa B (NF‐κB) by enhancing the association between TGF‐β‐activated kinase‐1 (TAK‐1) and the I kappa B kinase (IKK) complex, which results in acceleration of the host inflammatory response. 18 Similarly, the main proteases of SARS‐Cov‐2, nsp 5, and nsp 12, attenuate IFN‐I production through inhibition of nuclear translocation of IFN regulatory factor (IRF) 3 that promotes IFN α/β production. 19 , 20 Furthermore, SARS‐CoV‐2 nsp 1,3,12,13,14, orf 3, orf 6, and M protein show a similar inhibition of the IFN response. 17 Very recently, it was shown that the transcriptional footprint of NF‐κB is essential for SARS‐CoV‐2 replication. 21
These in vitro findings suggest that in severe SARS‐CoV‐2 infection, the PRR signaling pathways, which lead to NF‐κB activation that induces inflammatory cytokine production, are dominant, while the IRF pathways, which induce IFN production, are inhibited. These results may correspond with the in vivo features of interaction of SARS‐CoV‐2 with the host response to this virus infection in that inflammation predominates in the innate immune response in COVID‐19, especially in its severe disease. In this context, to restore the imbalance between inflammation and anti‐inflammation to the proper condition with appropriate timing may be critical for prevention of the worsening of COVID‐19.
THE IMPORTANCE OF SEQUENCE AND TEMPO OF INFLAMMATORY AND IMMUNE RESPONSE DURING SARS‐COV‐2 INFECTION FOR UNDERSTANDING THE PATHWAY TO SEVERE COVID‐19
In the outbreak of COVID‐19, early studies have shown a correlation between viral load dynamics and disease severity, with a higher and longer lasting viral load in severe cases compared with mild ones. However, subsequent studies have shown that the severity of the disease does not necessarily depend only on the level of viral load. 22 Although the viral load is an essential factor in the severity of the disease, another factor that may contribute to the severity of the disease is the immunological dysregulation of the host.
The characteristic feature of dysregulation is a predominance of myeloid cell functions and, in contrast, down‐regulation of lymphocyte functions, resulting in systemic hyperinflammation. Dysfunction of the host response in severe disease starts in the very early stage of the infection with a functional change of monocytes, the first layer of inflammation/innate immunity, leading to neutrophil activation accompanied by a disturbance of coagulation. 23 , 24 , 25 , 26 , 27 In the sense that cytokines are important effector molecules responsible for inflammation/immunity, dysregulation of the host response in COVID‐19 has been discussed in terms of altered cytokine production and/or its release.
Type I/III IFN (IFN‐I, III) production as the first response to infection is reduced and delayed, in contrast, pro‐inflammatory cytokines such as TNF, IL‐6, IL‐8, and CCL3 are produced for a prolonged time in severe disease, compared with mild cases. On the other hand, production of IL‐1β and the response to IL‐1 is enhanced in critical patients, whereas expression of IL‐10 and IL‐7 is enhanced in noncritical patients. 28 , 29 , 30 However, the context of immune dysregulation in SARS‐CoV‐2 infection seems fairly complex. For example, production of IFN‐I, III in noncritical patients is inhibited, while the expression level of these IFNs in certain critical patients is up‐regulated. 31 A recent study demonstrated that signal transduction mechanisms of the detrimental IFN‐I production in critical patients are different from those of beneficial IFN‐I in the early protective phase of the infection. 32
A cytokine storm was first described in patients receiving treatment with chimeric antigen receptor‐transfected T cells (CAR‐T) showing a massive production of a wide range of cytokines. 33 Thereafter, the term “cytokine storm syndrome” (CSS) was utilized to demonstrate similar syndromes of hyperinflammation triggered by various conditions such as infections, cancers, and other secondary hemophagocytic lymphohistiocytosis. 34
When COVID‐19 surged and dysregulation of inflammation/immunity was recognized as a possible pathogenesis of this communicable disease, CSS was proposed to indicate the dysregulated feature of the host response, 35 , 36 , 37 , 38 since cytokine levels are increased in COVID‐19, clinical features are similar to those of other CSS, and symptoms of hyperinflammation are also observed in the severe phase of this disease. Although some features in the host response to this communicable disease differ from other CSS in that lymphocytopenia is an essential factor in COVID‐19, but not usually in other CSS, and the grade of cytokine production in this infection is less than that in other CSS, it seems reasonable to recognize that COVID‐19 lies within the cytokine storm umbrella term. 39 However, we may need to pay attention to a recent study showing that serum levels of certain cytokines such as IL‐8 and IL‐1 receptor antagonist clearly discriminate COVID‐19 from other CSS. 40
If a cytokine storm is a causative element of dysregulation in the host response of CSS including COVID‐19 that leads to disease severity and finally death, modulation of cytokine production and/or its release may help patients to recover and to live. Many trials mainly with IL‐1 or IL‐6 blockade and Janus‐kinase inhibition have been performed to see whether these treatments inhibit the severity of COVID‐19, resulting in positive or negative results with speculation on these results. 41 , 42 , 43 , 44 , 45 On the other hand, recent studies have indicated that treatment with anakinra, an IL‐1 α/β inhibitor, decreased the 28 day mortality only in patients with a plasma level higher than 6 ng/mL of soluble urokinase plasminogen activator receptor (suPAR), an early predictor of severe respiratory failure in COVID‐19 patients, 46 who received dexamethasone. 47 This result suggests that conditions of cytokine production induced by the infection is fairly complex in that the cell interactions involved in cytokine production sequentially change depending on many determinants such as viral dose, functional conditions of involved inflammatory/immune cells, and genetic factors of infected hosts. Under this situation, the immunological treatment of COVID‐19 by manipulating the cytokine concentration is not an easy task to accomplish.
The importance of the condition and timing in the immune modulation treatment of COVID‐19 was also shown in patients who received dexamethasone. Treatment with this corticosteroid was effective in reducing the 28‐day mortality in patients who received either invasive mechanical ventilation or oxygen alone, but was not effective without respiratory support. Furthermore, the treatment was effective when it was performed in patients who had had symptoms for more than 7 days, but not effective in patients having symptoms for less than 7 days. 48
DYSREGULATED MYELOID COMPARTMENT IN COVID‐19 SEVERITY
In order to understand the immunological mechanisms of severe COVID‐19 and to help in its prevention and treatment, it will be necessary not only to characterize the overproduction of inflammatory cytokines in patients with severe disease as described above, but also to analyze the abnormal cellular responses involved in the overproduction of these cytokines.
In the studies of COVID‐19 pathogenesis, an outstanding finding is that dysregulated hyperinflammation including infiltration of monocytes or macrophages and neutrophils to lung tissues is a causative factor for the change in the severity of this communicable disease. 49 In this sense, a dysregulated myeloid compartment has been analyzed precisely in the process of SARS‐CoV‐2 infection.
First, neutrophilia accompanied by lymphopenia had been demonstrated as a dominant symptom in early clinical studies of patients, and thereafter it was probed in a systemic review and meta‐analysis of studies of many patients in whom neutrophilia in conjunction with lymphopenia was an important and independent indicator of severe disease and fatality in patients. 50 , 51 , 52 Furthermore, the neutrophil to lymphocyte ratio (NLR) is indicative of inflammation, not only in COVID‐19, but in many other diseases such as cancers and cardiovascular diseases. 53 Although the possible mechanisms for these phenomena are complex and still remain to be clarified, it seems to give us an important and critical tool for prevention in patients. To date, a very large number of scientific papers have been published on the dysregulated myeloid compartment in the pathogenesis of severe COVID‐19.
Using single cell RNA sequencing and single cell proteomics, it was revealed that inflammatory HLADRhi, CD11chi CD14+ monocytes are predominant in mild COVID‐19, whereas dysfunctional HLA‐DRlow S100Ahi CD14+ monocytes predominate in severe disease. 49 These sequential changes in monocyte functions with different markers in the progress of COVID‐19 toward severe disease may be first induced by emergency myelopoiesis associated with SARS‐CoV‐2 infection as follows. To protect against the viral infection, hematopoietic stem cells in the bone marrow expand to release inflammatory and immune cells to peripheral tissues. In the process of this proper protection against infection, inflammatory and immune cells are produced in a balanced manner, while a dominance of inflammatory cell production is observable in severe viral infections through emergency myelopoiesis. 24 Under this situation, HLA‐DRlow S100Ahi CD14+ monocytes with immature and dysfunctional neutrophils (PD‐L1hi, CD62low) appear in the peripheral tissues, which result in dysregulation of these cell functions. Although the possible mechanisms of this imbalance remain to be clarified, dysregulation of the myeloid compartment may surely constitute a critical basis toward severe disease. Furthermore, when the myelopoiesis accompanying SARS‐Cov‐2 infection is too fast, a high expression of the cell cycle marker Ki‐67 is observed in the peripheral monocytes of the patients, especially in the severe disease 24 as demonstrated previously in infection with H1N1 influenza. 54 On the other hand, neutrophils in severe disease are so called low‐density neutrophils (LDNs) expressing alarmins (S100A8, S100A9). 24 , 55 Emergency myelopoiesis also produces PD‐L1+ neutrophils that inhibit T‐cell activation resulting in depressed acquired immunity in severe COVID‐19. Because it has already been shown that HLA‐DRlow monocytes 56 and dysfunctional neutrophils 57 appear with sepsis, the above dysregulation of immune cells in the severe infection is not unique to this disease, but should be regarded as a phenotype of severe inflammatory response syndromes such as sepsis, trauma, and possibly COVID‐19.
It should be noted that the features of a dysregulated myeloid compartment change depending on the time after the SARS‐CoV‐2 infection. For example, a reduction of inflammatory signatures such as demonstrated with lowered functions of inflammatory macrophages in severe COVID‐19 was observed before day 17 of symptom onset, but the inflammatory response was elevated in severe disease during the later phase, days 17–23. 58 Dysregulation of the myeloid compartment is also reflected in the recovery stage of COVID‐19. In the early recovery stage (ERS), an increase of CD14++ monocytes with high inflammatory gene expression is observed, while in the late recovery stage, the number of these cells decreases, which suggests that patients in the early days after hospital discharge are still likely to be sensitive to a severe change of the disease. 59
In conclusion, hyperinflammation induced mainly by dysregulated myeloid cells is a prerequisite for severity of COVID‐19, and therefore, its resolution may be a target for preventing disease severity.
POSSIBLE SEQUENTIAL INFLAMMATORY/IMMUNE RESPONSES IN RESPIRATORY TRACTS LEADING TO THE DEVELOPMENT OF SEVERE COVID‐19
All the information on inflammatory/immune dysregulation in severe COVID‐19 has been obtained mainly from peripheral blood, and represents the response of the whole body systemic immune system. However, pulmonary lesions in COVID‐19 leading to acute respiratory distress syndrome (ARDS) start with a local inflammatory/immune response in the respiratory tract that originates from SARS‐CoV‐2 infection into this tissue. Therefore, it is quite important to understand what is happening in the local inflammatory and immune responses, without which we cannot get a complete picture of the disease.
It has been shown that 75% of severe COVID‐19 induces respiratory failure with edema in both lungs. In order to understand the pathogenesis of the local lesions causing such clinical manifestations, it is necessary to refer to the findings of previous studies on early lesions with pulmonary edema leading to respiratory failure. Previous pathological studies have demonstrated that diffuse alveolar damage (DAD) associated with both alveolar epithelial and endothelial injury leading to accumulation of protein‐rich edematous fluid in the alveolar spaces is first observed in acute lung injury. The sequence of this process is as follows: first, increased permeability of liquid across the endothelium of perialveolar capillaries followed by interstitial edema formation is observed and, thereafter, translocation of fluid to the alveolar space associated with injury of alveolar epithelium occurs. 60 However, it should be noted that DAD is not necessarily observed in all early steps of ARDS, 61 which suggests that there are various entrance points in ARDS that usually associate with pulmonary lesions in COVID‐19. As a hallmark of alveolar edema formation, neutrophils are usually observed in the alveoli, and the role of these cells is clearly shown, especially in the induction of direct ARDS. 62 , 63
Considering these previous studies on the early phase of ARDS, lung edema in severe COVID‐19 may start with increased permeability of fluid from perialveolar capillaries as follows. First, infection of SARS‐CoV‐2 to alveolar epithelial cells induces pathogen‐associated molecular patterns (PAMPs) and/or damage‐associated molecular patterns (DAMPs) which will be recognized by activated alveolar macrophages. These cells then produce large amounts of inflammatory cytokines and chemokines. Thereafter, the neutrophils stimulated by these chemokines first infiltrate into the lung interstitial tissues, accompanied by protein‐rich fluid extravasation. Neutrophil infiltration seems to be a prerequisite for edema formation, since our previous finding and those of others show that in vivo neutrophil depletion completely inhibits the late phase subcutaneous edema formation induced by various inflammation inducing agents including TNF‐α. 64 , 65 , 66
As for the histopathology of the disease, it is extremely difficult to examine a biopsy specimen from live patients. In the early days of SARS‐CoV‐2 infection at Wuhan in China, two cases of histopathological findings of patients were accidentally obtained from lungs resected with lung cancer. Two persons showed no clinical signs of COVID‐19 at the time of lung resection, which suggests that the histopathology of their lungs was in the early phase of the disease. The identical histopathological findings of these two patients were pulmonary edema with prominent protein rich exudates and DAD, showing the onset of acute lung injury. 67
On the other hand, several severe cases with no DAD were reported and were characterized by lung septal capillary injury associated with luminal fibrin deposition and infiltration of neutrophils into alveolar spaces. In this type of COVID‐19, deposition of the terminal complements complex C5b‐9 and mannose binding lectin‐associated serine protease 2 in microvasculatures accompanied by microthrombosis was observed. These histopathological findings support a proposed synergistic function of complement and neutrophils for coagulopathy in the initiation of severe COVID‐19. 68 Recent findings suggest that this complement‐induced coagulopathy accompanied by widespread endothelial damage may be critical for the pathogenesis of COVID‐19 severity 69 in some patients. Pulmonary edema accompanied by DAD and/or microthrombosis in severe cases is usually observed in the early stages of the disease. In the last days of the late stages, however, pathological figures become more complex including type II alveolar epithelial cell hyperplasia, squamous metaplasia, and bronchopneumonia. 70 Furthermore, not only the pulmonary tract but other tissues are involved and multiple organ failure occurs in the late stage of the disease which is categorized as indirect ARDS. 60 Although many reports on immunological dysregulation of severe COVID‐19 are based on information from patients in the late stages of the disease, such as suffering from multiple organ failure, if the goal is to prevent disease aggravation, research should first be focused on the pathophysiology of the onset of the disease.
THE IMPORTANCE OF UNDERSTANDING NEUTROPHIL DYNAMICS FOR PLANNING COVID‐19 PREVENTION AND/OR TREATMENT STRATEGIES
Role of neutrophils for the pathogenesis of severe COVID‐19
Recent advance in omics including genomics, transcriptomics, and proteomics leads us to understand the pathogenesis of COVID‐19 from the omics point of view through mainly transcriptome analysis of blood and respiratory tissues, and many studies have demonstrated that neutrophils are the key player in the pathogenesis of this communicable disease as follows.
First, utilizing scRNA‐seq, tremendously large numbers of neutrophils compared with other inflammatory/immune cells were observed in the lungs of COVID‐19 patients, especially in the severe disease, indicating that neutrophils infiltrate to inflamed lung tissues. Moreover, these neutrophils are in the activated state, expressing up‐regulated IL1β, C‐X‐C chemokine (CXC) ligand 8 (CXCL8), and S100A12. 14 These results suggest that neutrophils play a pivotal role in inducing pathological lung lesions of COVID‐19 and its worsening. This notion may be strengthened by results that neutrophil extracellular trap (NET) formation that has been recognized as an indicator of severity of the disease, 71 was higher in the severe stage than in mild one. 72 , 73 , 74
Furthermore, we should clarify features of blood neutrophils in the patients in order to determine the possible systemic function of these cells in the pathogenesis of the disease. To this end, whole blood cells were employed for some experiments, because utilization of blood mononuclear cells alone misses information on a neutrophil containing granulocyte population. Through these studies, it was revealed that CD177, selectively expressed in human neutrophils, 75 was mostly up‐regulated in the blood of patients with severe COVID‐19. 76 Other molecules of granulocytes, such as neutrophil elastase, olfactomedin 4, myeloperoxidase (MPO), metalloprotease (MMP‐8, MMP‐9), and arginase were also more highly expressed in severe disease than in mild disease, suggesting that neutrophils work actively in the severe disease course. On the other hand, the genes of many lymphocyte‐associated molecules were down‐regulated in severe disease. 77 These results again suggest that severity is correlated with inflammation/immunity imbalance.
Recent studies on neutrophils utilizing omics technology have demonstrated the heterogeneity of neutrophils in hemostasis and infection. Neutrophil populations with distinct molecular signatures that consist of progenitors termed as pro‐neutrophils (pro‐Neu), neutrophil lineage committed pre‐neutrophils (pre‐Neu), immature neutrophils, and mature neutrophils were identified. 78 , 79 Based on these studies, using scRNA‐seq and single cell proteomics, low density neutrophils, usually observed in pathological conditions such as cancer and sepsis, were detected in the peripheral blood mononuclear cells of severe disease. These LDNs consist of mixtures of pro‐Neu, pre‐Neu, and mature neutrophils, demonstrating the existence of emergency granulopoiesis and dysfunctional peripheral neutrophils in patients with severe disease. 49 Further studies using whole blood leukocytes have demonstrated the existence of activation markers such as CD64 on the neutrophils of patients with severe disease. 79 All these results demonstrate that peripheral neutrophils in severe disease contain immature and dysfunctional fractions, different from those of flu‐like illness which mainly show markers of mature and highly active neutrophils. 80
Although the neutrophil dysregulation in COVID‐19, especially in severe disease, has been demonstrated through discussing various neutrophil markers, actual dysfunction of neutrophils was also observed. Reactive oxygen species (ROSs) production was profoundly impaired in severe disease, 49 and the formation of NETs which is observed in inflammation‐associated damage was immunohistopathologically detected in the lungs of persons who died from COVID‐19. 81 Furthermore, the formation of NETs induces the aggregation of neutrophils with platelets which leads to immunothrombosis, a critical factor for induction of cell death of the lung in severe disease. 82 Taken together, many studies from varying fields indicate that neutrophils play an important role in the pathophysiology of severe disease of COVID‐19. 14
Detection of primed neutrophils as a narrow but potential way for prevention and/or treatment of severe COVID‐19
As shown previously in this manuscript, the immunological treatment of COVID‐19 with modulation of cytokines is not an easy task. Under this situation, functional modulation of neutrophils, an important effector cell in severe conditions, may be another possible way for the prevention or treatment of severe cases of this communicable disease. If we want to prevent the progress from mild to severe COVID‐19 disease by modulation of neutrophil activity, it seems necessary to know the total neutrophil activation pathway, since we believe that it is almost impossible to stop the stream of flushed cascade reactions of fully activated neutrophils producing strong tissue damage factors such as ROSs, MPO, and NETs. Thus, we need to start possible modulation of neutrophils before these cascade reactions.
The activation status of neutrophils has been demonstrated to move from resting through primed to a fully activated one. 83 , 84 Priming is an augmentation of responsiveness to various neutrophil activators by priming agents such as microbial products, chemoattractants, and pro‐inflammatory cytokines (e.g., IL‐1, IL‐6, IL‐8, TNF‐α, and granulocyte macrophage colony‐stimulating factor [GM‐CSF]). Priming is observed in various neutrophil responses such as respiratory burst, granule release, adhesion, chemotaxis, and phagocytosis. Furthermore, some priming agents inhibit neutrophil apoptosis, which results in the prolongation of neutrophil survival. Priming in conjunction with subsequent neutrophil activation in vivo seems to be a prerequisite for full activation of neutrophils. For example, neutrophil activators (e.g., N‐formyl‐l‐methionyl‐l‐leucyl‐phenylalanine [fMLP]) alone do not induce a sufficient respiratory burst in vitro, but priming with TNF‐α accelerates the full response of the neutrophils to fMLP in this reaction. 85 Considering these results, neutrophil priming may inevitably be involved in the sequential in vivo response toward microbial killing or normal tissue damage in various infections, trauma, and sepsis. If we are able to dissect primed neutrophils from fully activated ones, various pathological lesions induced by neutrophil dysregulation may be inhibited through stopping the pathway from priming to activation of this cell.
The urokinase‐type plasminogen activator (uPA)/urokinase‐type plasminogen activator receptor (uPAR) system is a valuable biomarker to detect the activation state of neutrophils and the severity of illness as follows. First, it was revealed that resting neutrophils can be primed with TNF‐α and other pro‐inflammatory cytokines to release suPAR by subsequent activation with fMLP or IL‐8, 86 indicating that the uPA/uPAR system can be utilized to examine the condition of neutrophils from resting, priming, and to final activation. Second, surface uPAR expression of neutrophils is down‐regulated, and, in contrast, suPAR release is up‐regulated in various critical diseases, 87 suggesting that this system may be a predictor of disease severity.
Considering that pro‐inflammatory cytokines such as IL‐1β, TNF‐α, IL‐6, and IL‐8 are produced in the very early stage of severe COVID‐19, neutrophils should already be primed in vivo by these cytokines in the process towards severe disease. Indeed, phenomena have been reported that can be explained by the in vivo priming of neutrophils. When neutrophils from patients with obstructive jaundice were stimulated in vitro with fMLP alone, a high grade respiratory burst was observed in the absence of any priming event, but further pretreatment with TNF‐α or IL‐8 did not show any further priming effect on subsequent fMLP treatment in inducing respiratory burst production. 88 Furthermore, neutrophils from patients with ARDS showed a similar response in inducing oxidative burst by fMLP alone. 89 These results suggest that neutrophils can already be preprimed in vivo in various conditions.
Based on these previous findings, we may be able to discriminate primed neutrophils from resting ones in patients by examining the responsiveness in vitro to treatment with fMLP alone in suPAR production, since a certain level of suPAR may be released in vitro by treatment with fMLP alone, if neutrophils have already been primed in vivo by various cytokines in the early stage of the infection. Furthermore, since fully activated neutrophils in advanced ARDS spontaneously release suPAR even in the absence of a neutrophil activator, fMLP, 90 neutrophils from far advanced severe COVID‐19 may spontaneously release suPAR, which may identify fully activated neutrophils in certain patients with severe disease.
In an early study of neutrophil priming, mAbs, designated A17 and A27 was reported to recognize the epitope of human neutrophils primed with GM‐CSF and TNF‐α. 91 Interestingly, peripheral blood neutrophils from patients with exacerbated COPD showed an increased expression of A17 and A27 epitope, and this expression decreased after COPD treatment. 92 We may be able to utilize these mAbs to detect the neutrophil priming stage in COVID‐19 patients in order to find target patients for the prevention of severe disease through modulation of neutrophil functions.
On the other hand, in the context that neutrophils first infiltrate inflamed tissues in a defensive response to microbial infection, we should avoid inhibiting neutrophil activity in the very early days of SARS‐CoV‐2 infection, which may result in abrogation of the defensive response to the infection. We have to find the time point of the onset of the deterioration of COVID‐19 in which neutrophils work as critical effector cells. Considering these findings and the clinical data that lung edema in severe cases starts approximately 1 week after the onset of the disease, we should start to examine the status of neutrophil condition in the early days of disease onset (3–4 days), by which time the direction of inflammatory/immune responses to SARS‐CoV‐2 infection should already be determined as to whether they proceed to defensive immunological reactions or to deleterious inflammatory ones.
Potential utility of GPI‐80, as a marker of early neutrophil activation in the course toward severe disease
Although, as has been pointed out, suPAR production may be a good marker of neutrophil priming, the uPA/uPAR system is not selectively expressed in neutrophils, but it is expressed in many cell types. Thus, if possible, it may be better to find other indicator(s) to characterize the activating feature of neutrophils, since we are trying to prevent severe COVID‐19 through manipulation of the neutrophils themselves.
Since we are seeking to manipulate primed neutrophils, we need to identify the characteristics of primed neutrophils. Furthermore, a previous result showing that priming is a prerequisite for infiltration of neutrophils to the lung tissue, so playing a critical role for the pathogenesis of ARDS, 85 suggests the importance of the detection of primed neutrophils for the prevention of the severity of COVID‐19.
GPI‐80 was found by our group to be a modulator of neutrophil adherence and migration. Treatment of neutrophils with a mAb (3H9) to GPI‐80 sequentially augments and suppresses CD18 dependent adherence 93 and induces locomotion of these cells. 94 The crosslinking of GPI‐80 by 3H9 mAb up‐regulates CD11b/CD18 expression and induces shedding of CD62L, 95 which suggests that activation of GPI‐80 changes the status of neutrophils from a resting to a primed one. These functions of GPI‐80 seem to be downstream of integrin activation, since preactivation of CD18 is required for an increase in [Ca2+]in by 3H9 treatment. 94 With immune‐ and scanning electron microscopy, clusters of GPI‐80 were detected on the forward surfaces of transmigrating neutrophils, giving a morphological background of the possible role of GPI‐80 for neutrophil migration. 96 Furthermore, in vitro treatment of these cells with neutrophil priming agents, TNF‐α 97 or fMLP, 98 induces the release of a soluble form of GPI‐80 (sGPI‐80). It should be noted that since mobilization of secretory vesicles is the first sign of neutrophil priming of neutrophils 99 and GPI‐80 release is mainly mediated by this mobilization, 100 released sGPI‐80 may be a marker of the early priming of neutrophils. The biological characteristics of GPI‐80 as described above are very similar to those of uPAR, a regulator of integrin mediated neutrophil functions and two other glycosylphosphatidylinositol (GPI)‐anchored proteins on human leukocytes, CD16b, and CD14 also have the same functions. These results seem to indicate that GPI‐80 is a fourth member of this group of molecules.
Furthermore, the release of sGPI‐80 into sera was observed in patients with Wegener's granulomatosis depending on the disease activity, 101 and it was also detected in the synovial fluids of patients with rheumatoid arthritis. 98 These facts strongly suggest a potential clinical application of GPI‐80 to study the possible pathogenesis of severe COVID‐19, since sGPI‐80 may be detectable in plasma and other body fluids at an early stage of the neutrophil activation process toward severe disease in patients. The process of neutrophil activation and our hypothesis concerning GPI‐80 in COVID‐19 are summarized in Figure 1.
Figure 1.
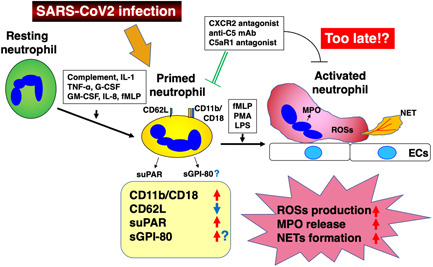
The process of neutrophil activation starting from resting, through primed, to activated states, and GPI‐80 as a possible marker of primed neutrophils. Resting neutrophils stimulated with various substances such as complement, IL‐1, TNF‐α, granulocyte‐colony stimulating factor (G‐CSF), granulocyte macrophage colony‐stimulating factor (GM‐CSF), IL‐8, and N‐formyl‐l‐methionyl‐l‐leucyl‐phenylalanine (fMLP) are primed to react with fMLP, phorbol 12‐myristate 13‐acetate (PMA), and LPS to be activated neutrophils which produce reactive oxygen species (ROSs), or neutrophil extracellular trap (NET) and release myeloperoxidase (MPO). We propose GPI‐80 as a marker of primed neutrophils as described in the text
On the other hand, as discussed previously, activation of the complement factor 5a (C5a)–C5a receptor 1 (C5aR1) axis in conjunction with subsequently occurring activation of neutrophils is involved in the early phase of aggravation of COVID‐19 leading to microthrombosis which is a critical lethal lesion in pathogenesis in some patients. Furthermore, complement activation is a neutrophil priming factor. 85 In this context, an increase in the plasma C5a level should be another marker of neutrophil priming and an early marker of disease progression. Therefore, we further propose to examine the C5a level in patients' plasma simultaneously with sGPI‐80. The important point is to examine sGPI‐80 and C5a serially in the course of COVID‐19 and to detect the earliest point at which there is sGPI‐80 positivity or an increase in C5a.
Trials to prevent initiation of the cascade response leading to severe COVID‐19 with inhibition of neutrophil migration in combination with down‐regulation of C5a–C5aR1 axis
In order to propose a way of neutrophil manipulation for the prevention and/or treatment of severe COVID‐19, we need to refer to previous trials utilizing neutrophils as therapeutic targets. Many trials in experiments and in human preclinical studies in enhancing or inhibiting various neutrophil functions have been performed with both effective and noneffective results. 102 Of particular note for successful modulation of neutrophil functions is the timing of the artificial neutrophil manipulation. As already discussed, since neutrophils first infiltrate inflamed tissues as a defensive response to microbial infections, we should avoid inhibiting neutrophil activity in the very early days of infection, which may result in abrogation of the defensive response to infection. In contrast, since the functions of neutrophils are depressed in the severe stage of certain infections such as severe sepsis, 103 inhibition of neutrophil activity may be harmful for disease control in such a stage of the disease. How to determine the appropriate timing of neutrophil manipulation is critical for the effective prevention of diseases, but it seems not easy to determine. Among the many clinical trials for the inhibition of neutrophil function, blocking neutrophil extravasation through targeting CXC receptor (CXCR)1/CXCR2, the most important chemokine receptor for CXC chemokines such as CXCL8 (IL‐8), gave certain positive effects. A CXCR2 antagonist, SB‐656933, reduced sputum neutrophils and elastase in patients with cystic fibrosis, while no change of pulmonary function or respiratory symptoms was detected in this clinical trial. 104 Another CXCR2 antagonist, MK‐7123, induced a significant improvement of the forced expiratory volume 1.0 (FEV1) in COPD patients. 105 Neutrophil elastase (NE) is an important effector molecule in tissue damage by activated neutrophils. A NE inhibitor, AZD9668, improved FEV1 and reduced sputum IL‐6 and RANTES as well as plasma IL‐8 in a clinical trial of patients with bronchiectasis, 106 while many preclinical studies with this and other NE inhibitors did not show beneficial effects in improving the pulmonary functions of patients. 107 Blockers of signaling pathways in neutrophils were also targeted as therapeutic tools. Janus tyrosine kinase (JAK) inhibitors such as the JAK1/JAK3 inhibitor, tofacitinib, were approved for the treatment of rheumatoid arthritis, psoriatic arthritis, and ulcerative colitis. 108 , 109
On the other hand, not only in clinical trials on disease therapy targeting neutrophils, 102 but in many other clinical trials, the evaluation of various therapies is now performed by statistical analyses with a large number of cohorts that contain groups of many different conditions. These analyses seem to be important for authorizing each trial among many preclinical studies. However, we wonder whether the method of data analysis mentioned above is the most suitable for analyzing the results of therapeutic trials based on immunological host response, since the inflammatory/immune response at a certain time point is determined through the collaboration of many factors such as those mainly involved immune cells, released cytokines, and involved inflamed tissues, and thus, it develops distinct features depending on the timing examined. Considering this situation, the first and most important point of discussion in immunoprevention is to observe how the host response shifts depending on the collaboration of each determinant but not to summarize data from various different conditions.
As for the immunotherapy of COVID‐19, polyclonal convalescent plasma, mAbs to spike proteins, corticosteroids, kinase inhibitors, anti‐cytokine treatment (anti‐IL1, anti‐IL6), anti‐complement treatment, IFNs, and kallikrein–kinin inhibitors have been tried, and some beneficial results have been obtained. 110 Considering the inhibition of severe COVID‐19 through neutrophil manipulation, anti‐cytokine, complement treatment, and JAK inhibitors may be utilized as candidates for the trial. Furthermore, CXCR1/CXCR2 antagonists may be another hopeful one. Actually, the rationale for CXCR2 antagonists for the treatment was described recently. 111 As we repeatedly emphasize in this manuscript, the most important point for immunoprevention or therapy of COVID‐19 is in the timing of artificial control of the host response. We may exacerbate the disease with inappropriate intervention. In other words, previous preclinical trials might have resulted in better results if more appropriate timing of intervention was selected. In, fact, as already discussed, IL1 antagonist and corticosteroid therapy were effective only in certain conditions. 47 , 48
As a plausible trial for the prevention of worsening of COVID‐19 toward severe disease, we would like to choose inhibition of neutrophil extravasation in combination with activation of the C5a–C5aR1 axis to target, since these seem to be initial entrance points of the cascade response to ARDS, the main cause of lung edema. As for the rationale for targeting neutrophil extravasation, recent literature proposed treatment with CXCR2 antagonists for blocking inflammation. 111 AZD5069 and other CXCR2 antagonists are under clinical investigation for the therapy of lung diseases. Inhibition of neutrophil infiltration to alveoli at a proper time point by CXCR2 antagonists may facilitate the prevention of severity of the disease. Activation of the C5a–C5aR1 axis leading to neutrophil extravasation is currently regarded as a critical driver of initiation of coagulopathy associated with severity. 69 Inhibition of this axis may be another potential tool for the manipulation of dysregulated inflammatory/immune response in the early phase of the disease from the viewpoint of how to modulate the response, since complement activation is positioned in the very early point of inflammatory/immune response. Treatment of the patients with monoclonal antibodies to C5a or C5aR1 antagonists are undergoing clinical research. 69
Considering intervention timing for the host response mediated prevention or treatment of severe COVID‐19, the first point to be achieved seems to be to analyze the actual situation of the inflammatory/immune response at the time of intervention in targeted patients. We propose to examine the serum concentration of suPAR and sGPI‐80 in the process of the infection, since the early expression of these markers seems to indicate priming of neutrophils as discussed previously. Furthermore, examination of the in vitro release of these GPI‐anchored proteins by patients' leukocytes in response to stimulation with neutrophil stimulating agents such as TNF‐α and fMLP may also be important to determine the activation status of neutrophils, resting, primed, or fully activated. As mentioned in the previous part of this manuscript, immunological prevention of severe disease in patients with fully activated neutrophils through manipulation of neutrophil functions may be almost impossible to perform, and thus, to dissect primed from fully activated neutrophils may be critical to successfully accomplish these trials. We know well that this discrimination is not so easy to perform, but an increase in serum NE, histone‐DNA, MPO‐DNA, free dsDNA may be predictors of fully activated neutrophils. 112 , 113
After we select the patients with primed neutrophils as candidates for recipients of this disease immunoprevention, if possible, it is better to differentiate them into two groups that consist of patients who will recover from the infection and those who will worsen toward severe disease. As for this point, a definitely beneficial result has been obtained recently in that predictive plasma biomarkers were detected that distinguish critically ill and noncritically ill patients. 114 Using scRNA‐seq, many plasma biomarkers related with immunological response were examined in COVID‐19 patients. To determine the relationships between biomarkers and disease severity, the patients were divided into three groups, asymptomatic nonhospitalized (control), patients treated in a medical intensive care unit (ICU), and those treated on standard medical floors (non‐ICU). The plasma from patients on day 1 after hospitalization was used for examination. To see the specific plasma proteins that may affect the disease severity, a certain prediction model was employed. The top five biomarkers predicting severity were proteins related to neutrophil activation. Resistin (RETN), lipocalin‐2 (LCN‐2), and hepatocyte growth factor (HGF) were the top three of the five proteins released from secondary granules upon neutrophil activation. The next two predictors were granulocyte‐colony stimulating factor (G‐CSF) and IL‐8, a granulopoiesis stimulator and a neutrophil chemoattractant, respectively. The relative protein levels of RETN, LCN2, and HGF differed significantly among each of the three groups (ICU > non‐ICU > control), which suggests that these biomarkers can be utilized as a predictor of severity. Importantly, through examining the elevation of neutrophil activation biomarkers on day 1 of hospitalization, we can detect noncritically ill patients who are at risk of becoming critically ill. Thus, we can select target patients for immunoprevention and/or therapy through examination of the levels of plasma proteins of neutrophil activation before they become critically ill.
We propose here a trial to inhibit severe COVID‐19 through restoring dysregulated neutrophil functions (abbreviated as neutrophil therapy) as follows: (1) to select patients with primed neutrophils by consecutive measurement of the plasma concentration of suPAR and sGPI‐180, both being markers of neutrophil priming, (2) to identify target patients in the selected group for neutrophil therapy by picking out persons for whom the plasma concentration of either RETN, LCN‐2, and HGF is high as predictable markers of patients of severe disease in the future, (3) to start treatment with CXCR2 antagonists in conjunction with C5a antagonists to inhibit an initial entrance point of hyperinflammation proceeding toward severity (Figure 2). This proposal just shows a rationale for alteration of neutrophil functions as a target for immunomodulatory inhibition of severe COVID‐19. Further studies are required to establish concrete conditions for the selection of patients whose neutrophils are primed and schedules for CXCR2 and complement antagonist administration.
Figure 2.
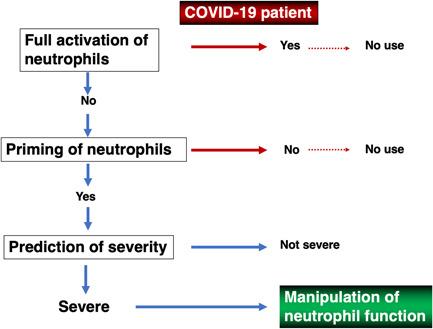
Diagram of prevention of severe disease by regulation of neutrophil function in CoV disease 2019 (COVID‐19). Full neutrophil activation is detected by examining serum neutrophil elastase, histone DNA, myeloperoxidase (MPO)‐DNA, and free dsDNA. Neutrophil priming is detected as follows: release of soluble form of urokinase type plasminogen activator receptor (suPAR) or soluble form of GPI‐80 (sGPI‐80) from neutrophils of COVID‐19 patients in the presence of N‐formyl‐l‐methionyl‐l‐leucyl‐phenylalanine (fMLP) is compared with that of healthy controls. When these release levels are significantly higher in patients than controls, neutrophils are recognized as already primed in vivo. Patients are considered to be critically ill when proteins involved in neutrophil activation (resistin [RETN[, lipocalin‐2 [LCN‐2], hepatocyte growth factor [HGF]) are significantly higher in serum
A recent study raising a model of neutrophil development and activation driving pathogenesis of severe COVID‐19 114 gives us a story in which neutrophils may be a driving force to induce severe disease as follows. First, G‐CSF releases developing neutrophils from the bone marrow, and then IL‐8 with the help of other cytokines induce infiltration of neutrophils into inflamed tissues. Extravasated and then activated neutrophils play a pivotal role in evoking a serial inflammatory/immune response toward severe COVID‐19. This model may be applicable to other diseases inducing pulmonary tissue damage. Under this situation, neutrophil therapy may be a possible tool for the prevention of these diseases.
CONCLUDING REMARKS
We propose the prevention of severe COVID‐19 through the modulation of the early host response of SARS‐CoV‐2 infection leading to ARDS placing pinpoint focus on inhibiting neutrophil and/or complement activation at the sites of local inflammatory/immune response. It may not be easy for these trials to be introduced, because determining the timing of the intervention differs from patient to patient and the thresholds of titers of sGPI‐80 or an increase of C5a to determine whether patients are further treated with some manipulation is unknown before the start of the trial. However, retrospective studies on patients may give us an answer. Finally, we emphasize again that for the successful prevention of severe disease by immunological manipulation, we need to understand the sequence and tempo of the inflammatory/immune response and to target accurately the onset of disease aggravation, because this response is a double‐edged sword that could protect or aggravate COVID‐19.
AUTHOR CONTRIBUTION
Fujiro Sendo, Hiroshi Yoshitake, and Yoshihiko Araki wrote the manuscript; Hiroshi Yoshitake and Yoshihiko Araki created the illustrations shown in Figures.
DISCLOSURE
Authors state no conflicts of interest.
ACKNOWLEDGMENTS
The excellent secretarial assistance of Ms Keiko Fukuda is gratefully acknowledged. This work was supported in part by Grants‐in Aid for Scientific Research Nos. 21K09503/21H03386, and for Fostering Joint International Research No. 18KK0256 from the JSPS.
Sendo F, Yoshitake H, Araki Y. Targeting of neutrophil activation in the early phase of the disease for prevention of Coronavirus disease‐19 severity. Microbiol Immunol. 2022;66:264–276. 10.1111/1348-0421.12978
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were reported or analyzed in this study.
REFERENCES
- 1. Polack FP, Thomas SJ, Kitchin N, et al. Safety and efficacy of the BNT162b2 mRNA COVID‐19 vaccine. N Engl J Med. 2020;383:2603–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Voysey M, Clemens SAC, Madhi SA, et al. Safety and efficacy of the ChAdOx1 nCoV‐19 vaccine (azd1222) against SARS‐COV‐2: an interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. Lancet. 2021;397:99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pritchard E, Matthews PC, Stoesser N, et al. Impact of vaccination on new SARS‐Cov‐2 infections in the United Kingdom. Nat Med. 2021;27:1370–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hacisuleyman E, Hale C, Saito Y, et al. Vaccine breakthrough infections with SARS‐Cov‐2 variants. N Engl J Med. 2021;384:2212–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chau NVV, Ngoc NM, Nguyet LA, et al. An observational study of breakthrough SARS‐Cov‐2 Delta variant infections among vaccinated healthcare workers in Vietnam. EClinicalMedicine. 2021;41:101143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Antonelli M, Penfold RS, Merino J, et al. Risk factors and disease profile of post‐vaccination SARS‐Cov‐2 infection in UK users of the COVID symptom study app: a prospective, community‐based, nested, case‐control study. Lancet Infect Dis. 2022;22:43–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mlcochova P, Kemp SA, Dhar MS, et al. SARS‐Cov‐2 B.1.617.2 Delta variant replication and immune evasion. Nature. 2021;599:114–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. WHO Newsletter . Classification of Omicron (B.1.1.529): SARS‐CoV‐2 Variant of Concern. 2021. 2[cited 2021 November 6]. Available from: https://www.who.int/news/item/26-11-2021-classification-of-omicron-(b.1.1.529)-sars-cov-2-variant-of-concern
- 9. Ong EZ, Chan YFZ, Leong WY, et al. A dynamic immune response shapes COVID‐19 progression. Cell Host Microbe. 2020;27:879–82.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Su Y, Chen D, Yuan D, et al. Multi‐omics resolves a sharp disease‐state shift between mild and moderate COVID‐19. Cell. 2020;183:1479–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lucas C, Wong P, Klein J, et al. Longitudinal analyses reveal immunological misfiring in severe COVID‐19. Nature. 2020;584:463–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim KD, Zhao J, Auh S, et al. Adaptive immune cells temper initial innate responses. Nat Med. 2007;13:1248–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cicchese JM, Evans S, Hult C, et al. Dynamic balance of pro‐ and anti‐inflammatory signals controls disease and limits pathology. Immunol Rev. 2018;285:147–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Reusch N, De Domenico E, Bonaguro L, et al. Neutrophils in COVID‐19. Front Immunol. 2021;12:652470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. [DOI] [PubMed] [Google Scholar]
- 16. Kawai T, Akira S. Toll‐like receptor and RIG‐I‐like receptor signaling. Ann N Y Acad Sci. 2008;1143:1–20. [DOI] [PubMed] [Google Scholar]
- 17. Lei X, Dong X, Ma R, et al. Activation and evasion of type I interferon responses by SARS‐Cov‐2. Nat Commun. 2020;11:3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wu Y, Ma L, Cai S, et al. RNA‐induced liquid phase separation of SARS‐CoV‐2 nucleocapsid protein facilitates NF‐κB hyper‐activation and inflammation. Signal Transduct Target Ther. 2021;6:167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fung SY, Siu KL, Lin H, Yeung ML, Jin DY. SARS‐CoV‐2 main protease suppresses type I interferon production by preventing nuclear translocation of phosphorylated IRF. Int J Biol Sci. 2021;17:1547–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang W, Zhou Z, Xiao X, et al. SARS‐Cov‐2 nsp12 attenuates type I interferon production by inhibiting IRF3 nuclear translocation. Cell Mol Immunol. 2021;18:945–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nilsson‐Payant BE, Uhl S, Grimont A, et al. The NF‐κB transcriptional footprint is essential for SARS‐Cov‐2 replication. J Virol. 2021;95:e0125721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dadras O, Afsahi AM, Pashaei Z, et al. The relationship between COVID‐19 viral load and disease severity: a systematic review. Immun Inflamm Dis. 2022;10:e580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Song CY, Xu J, He JQ, Lu YQ. Immune dysfunction following COVID‐19, especially in severe patients. Sci Rep. 2020;10:15838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mann ER, Menon M, Knight SB, et al. Longitudinal immune profiling reveals key myeloid signatures associated with COVID‐19. Sci. Immunol. 2020;5:eabd6197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liao M, Liu Y, Yuan J, et al. Single‐cell landscape of bronchoalveolar immune cells in patients with COVID‐19. Nat Med. 2020;26:842–4. [DOI] [PubMed] [Google Scholar]
- 26. Li J, Guo M, Tian X, et al. Virus‐host interactome and proteomic survey reveal potential virulence factors influencing SARS‐CoV‐2 pathogenesis. Med (N Y). 2021;2:99–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vanderbeke L, Van Mol P, Van Herck Y, et al. Monocyte‐driven atypical cytokine storm and aberrant neutrophil activation as key mediators of COVID‐19 disease severity. Nat Commun. 2021;12:4117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Arunachalam PS, Wimmers F, Mok CKP, et al. Systems biological assessment of immunity to mild versus severe COVID‐19 infection in humans. Science. 2020;369:1210–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Del Valle DM, Kim‐Schulze S, Huang HH, et al. An inflammatory cytokine signature predicts COVID‐19 severity and survival. Nat Med. 2020;26:1636–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Galani IE, Rovina N, Lampropoulou V, et al. Untuned antiviral immunity in COVID‐19 revealed by temporal type I/III interferon patterns and flu comparison. Nat Immunol. 2021;22:32–40. [DOI] [PubMed] [Google Scholar]
- 32. Domizio JD, Gulen MF, Saidoune F, et al. The cGAS‐STING pathway drives type I IFN immunopathology in COVID‐19. Nature. 2022;603:145–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fajgenbaum DC, June CH. Cytokine storm. N Engl J Med. 2020;383:2255–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ. COVID‐19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395:1033–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Moore JB, June CH. Cytokine release syndrome in severe COVID‐19. Science. 2020;368:473–4. [DOI] [PubMed] [Google Scholar]
- 37. Mangalmurti N, Hunter CA. Cytokine storms: understanding COVID‐19. Immunity. 2020;53:19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gao YM, Xu G, Wang B, Liu BC. Cytokine storm syndrome in coronavirus disease 2019: a narrative review. J Intern Med. 2021;289:147–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mehta P, Fajgenbaum DC. Is severe COVID‐19 a cytokine storm syndrome: a hyperinflammatory debate. Curr Opin Rheumatol. 2021;33:419–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kessel C, Vollenberg R, Masjosthusmann K, et al. Discrimination of COVID‐19 from inflammation‐induced cytokine storm syndromes using disease‐related blood biomarkers. Arthritis Rheumatol. 2021;73:1791–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hermine O, Mariette X, Tharaux PL, Resche‐Rigon M, Porcher R, Ravaud P. Effect of tocilizumab vs usual care in adults hospitalized with COVID‐19 and moderate or severe pneumonia: a randomized clinical trial. JAMA Intern Med. 2021;181:32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Domingo P, Mur I, Mateo GM, et al. Association between administration of IL‐6 antagonists and mortality among patients hospitalized for COVID‐19: a meta‐analysis. JAMA. 2021;326:499–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Caricchio R, Abbate A, Gordeev I, et al. Effect of canakinumab vs placebo on survival without invasive mechanical ventilation in patients hospitalized with severe COVID‐19: a randomized clinical trial. JAMA. 2021;326:230–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jones SA, Hunter CA. Is IL‐6 a key cytokine target for therapy in COVID‐19? Nat Rev Immunol. 2021;21:337–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cron RQ, Caricchio R, Chatham WW. Calming the cytokine storm in COVID‐19. Nat Med. 2021;27:1674–5. [DOI] [PubMed] [Google Scholar]
- 46. Rovina N, Akinosoglou K, Eugen‐Olsen J, Hayek S, Reiser J, Giamarellos‐Bourboulis EJ. Soluble urokinase plasminogen activator receptor (suPAR) as an early predictor of severe respiratory failure in patients with COVID‐19 pneumonia. Crit Care. 2020;24:187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kyriazopoulou E, Poulakou G, Milionis H, et al. Early treatment of COVID‐19 with anakinra guided by soluble urokinase plasminogen receptor plasma levels: a double‐blind, randomized controlled phase 3 trial. Nat Med. 2021;27:1752–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. RECOVERY Collaborative G, Horby P, Lim WS, et al. Dexamethasone in hospitalized patients with COVID‐19. N Engl J Med. 2021;384:693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schulte‐Schrepping J, Reusch N, Paclik D, et al. Severe COVID‐19 is marked by a dysregulated myeloid cell compartment. Cell. 2020;182:1419–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Liu Y, Du X, Chen J, et al. Neutrophil‐to‐lymphocyte ratio as an independent risk factor for mortality in hospitalized patients with COVID‐19. J Infect. 2020;81:e6–e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liu J, Liu Y, Xiang P, et al. Neutrophil‐to‐lymphocyte ratio predicts critical illness patients with 2019 coronavirus disease in the early stage. J Transl Med. 2020;18:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Henry B, Cheruiyot I, Vikse J, et al. Lymphopenia and neutrophilia at admission predicts severity and mortality in patients with COVID‐19: a meta‐analysis. Acta Biomed. 2020;91:e2020008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Song M, Graubard BI, Rabkin CS, Engels EA. Neutrophil‐to‐lymphocyte ratio and mortality in the United States general population. Sci Rep. 2021;11:464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cole SL, Dunning J, Kok WL, et al. M1‐like monocytes are a major immunological determinant of severity in previously healthy adults with life‐threatening influenza. JCI Insight. 2017;2:e91868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wilk AJ, Rustagi A, Zhao NQ, et al. A single‐cell atlas of the peripheral immune response in patients with severe COVID‐19. Nat Med. 2020;26:1070–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Venet F, Demaret J, Gossez M, Monneret G. Myeloid cells in sepsis‐acquired immunodeficiency. Ann N Y Acad Sci. 2021;1499:3–17. [DOI] [PubMed] [Google Scholar]
- 57. Darcy CJ, Minigo G, Piera KA, et al. Neutrophils with myeloid derived suppressor function deplete arginine and constrain T cell function in septic shock patients. Crit Care. 2014;18:R163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Liu C, Martins AJ, Lau WW, et al. Time‐resolved systems immunology reveals a late juncture linked to fatal COVID‐19. Cell. 2021;184:1836–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wen W, Su W, Tang H, et al. Immune cell profiling of COVID‐19 patients in the recovery stage by single‐cell sequencing. Cell Discov. 2020;6:31. Erratum in:(2020) Cell Discov. 6 :41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Matthay MA, Zemans RL, Zimmerman GA, et al. Acute respiratory distress syndrome. Nat Rev Dis Primers. 2019;5:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Magro C, Mulvey JJ, Berlin D, et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID‐19 infection: a report of five cases. Transl Res. 2020;220:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Grommes J, Soehnlein O. Contribution of neutrophils to acute lung injury. Mol Med. 2011;17:293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Williams AE, Chambers RC. The mercurial nature of neutrophils: still an enigma in ARDS? Am J Physiol Lung Cell Mol Physiol. 2014;306:L217–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sekiya S, Yamashita T, Sendo F. Suppression of late phase enhanced vascular permeability in rats by selective depletion of neutrophils with a monoclonal antibody. J Leukoc Biol. 1990;48:258–65. [DOI] [PubMed] [Google Scholar]
- 65. Abe Y, Sekiya S, Yamasita T, Sendo F. Vascular hyperpermeability induced by tumor necrosis factor and its augmentation by IL‐1 and IFN‐gamma is inhibited by selective depletion of neutrophils with a monoclonal antibody. J Immunol. 1990;145:2902–7. [PubMed] [Google Scholar]
- 66. Suo J, Linke B, Meyer dos Santos S, et al. Neutrophils mediate edema formation but not mechanical allodynia during zymosan‐induced inflammation. J Leukoc Biol. 2014;96:133–42. [DOI] [PubMed] [Google Scholar]
- 67. Tian S, Hu W, Niu L, Liu H, Xu H, Xiao SY. Pulmonary pathology of early‐phase 2019 novel coronavirus (COVID‐19) pneumonia in two patients with lung cancer. J Thorac Oncol. 2020;15:700–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Carvelli J, Demaria O, Vély F, et al. Association of COVID‐19 inflammation with activation of the c5a‐c5aR1 axis. Nature. 2020;588:146–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Perico L, Benigni A, Casiraghi F, Ng LFP, Renia L, Remuzzi G. Immunity, endothelial injury and complement‐induced coagulopathy in COVID‐19. Nat Rev Nephrol. 2021;17:46–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Calabrese F, Pezzuto F, Fortarezza F, et al. Pulmonary pathology and COVID‐19: lessons from autopsy. The experience of European pulmonary pathologists. Virchows Arch. 2020;477:359–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Borges L, Pithon‐Curi TC, Curi R, Hatanaka E. COVID‐19 and neutrophils: the relationship between hyperinflammation and neutrophil extracellular traps. Mediators Inflamm. 2020;2020:8829674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Barnes BJ, Adrover JM, Baxter‐Stoltzfus A. Targeting potential drivers of COVID‐19: neutrophil extracellular traps. J Exp Med. 2020;217:e20200652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Tomar B, Anders HJ, Desai J, Mulay SR. Neutrophils and neutrophil extracellular traps drive necroinflammation in COVID‐19. Cells. 2020;9:1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ackermann M, Anders HJ, Bilyy R, et al. Patients with COVID‐19: in the dark‐nets of neutrophils. Cell Death Differ. 2021;28:3125–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bai M, Grieshaber‐Bouyer R, Wang J, et al. CD177 modulates human neutrophil migration through activation‐mediated integrin and chemoreceptor regulation. Blood. 2017;130:2092–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Aschenbrenner AC, Mouktaroudi M, Krämer B, et al. Disease severity‐specific neutrophil signatures in blood transcriptomes stratify COVID‐19 patients. Genome Med. 2021;13:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kwok I, Becht E, Xia Y, et al. Combinatorial single‐cell analyses of granulocyte‐monocyte progenitor heterogeneity reveals an early uni‐potent neutrophil progenitor. Immunity. 2020;53:303–18. [DOI] [PubMed] [Google Scholar]
- 78. Xie X, Shi Q, Wu P, et al. Single‐cell transcriptome profiling reveals neutrophil heterogeneity in homeostasis and infection. Nat Immunol. 2020;21:1119–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hoffmann JJ. Neutrophil CD64: a diagnostic marker for infection and sepsis. Clin Chem Lab Med. 2009;47:903–16. [DOI] [PubMed] [Google Scholar]
- 80. Camp JV, Jonsson CB. A role for neutrophils in viral respiratory disease. Front Immunol. 2017;8:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Radermecker C, Detrembleur N, Guiot J, et al. Neutrophil extracellular traps infiltrate the lung airway, interstitial, and vascular compartments in severe COVID‐19. J Exp Med. 2020;217: e20201012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Middleton EA, He XY, Denorme F, et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID‐19 acute respiratory distress syndrome. Blood. 2020;136:1169–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Vogt KL, Summers C, Chilvers ER, Condliffe AM. Priming and de‐priming of neutrophil responses in vitro and in vivo. Eur J Clin Invest. 2018;48(Suppl 2):e12967. [DOI] [PubMed] [Google Scholar]
- 84. Pillay J, Ramakers BP, Kamp VM, et al. Functional heterogeneity and differential priming of circulating neutrophils in human experimental endotoxemia. J Leukoc Biol. 2010;88:211–20. [DOI] [PubMed] [Google Scholar]
- 85. Miralda I, Uriarte SM, McLeish KR. Multiple phenotypic changes define neutrophil priming. Front Cell Infect Microbiol. 2017;7:217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Pliyev BK. Activated human neutrophils rapidly release the chemotactically active D2D3 form of the urokinase‐type plasminogen activator receptor (uPAR/CD87). Mol Cell Biochem. 2009;321:111–22. [DOI] [PubMed] [Google Scholar]
- 87. Gussen H, Hohlstein P, Bartneck M, et al. Neutrophils are a main source of circulating suPAR predicting outcome in critical illness. J Intensive Care. 2019;7:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Jiang WG, Puntis MC, Hallett MB. Neutrophil priming by cytokines in patients with obstructive jaundice. HPB Surg. 1994;7:281–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Juss JK, House D, Amour A, et al. Acute respiratory distress syndrome neutrophils have a distinct phenotype and are resistant to phosphoinositide 3‐kinase inhibition. Am J Respir Crit Care Med. 2016;194:961–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Pliyev BK, Menshikov MY. Release of the soluble urokinase‐type plasminogen activator receptor (suPAR) by activated neutrophils in rheumatoid arthritis. Inflammation. 2010;33:1–9. [DOI] [PubMed] [Google Scholar]
- 91. Koenderman L, Kanters D, Maesen B, et al. Monitoring of neutrophil priming in whole blood by antibodies isolated from a synthetic phage antibody library. J Leukoc Biol. 2000;68:58–64. [PubMed] [Google Scholar]
- 92. Oudijk EJ, Gerritsen WB, Nijhuis EH, et al. Expression of priming‐associated cellular markers on neutrophils during an exacerbation of COPD. Respir Med. 2006;100:1791–9. [DOI] [PubMed] [Google Scholar]
- 93. Suzuki K, Watanabe T, Sakurai S, et al. A novel glycosylphosphatidyl inositol‐anchored protein on human leukocytes: a possible role for regulation of neutrophil adherence and migration. J Immunol. 1999;162:4277–84. [PubMed] [Google Scholar]
- 94. Suzuki H, Takei H, Ohtake K, Watanabe T, Sendo F. External calcium‐dependent, F‐actin‐independent and pertussis toxin‐insensitive novel neutrophil locomotion induced by a mAb. Int Immunol. 1997;9:763–9. [DOI] [PubMed] [Google Scholar]
- 95. Yoshitake H, Takeda Y, Nitto T, Sendo F. Cross‐linking of GPI‐80, a possible regulatory molecule of cell adhesion, induces up‐regulation of CD11b/CD18 expression on neutrophil surfaces and shedding of l‐selectin. J Leukoc Biol. 2002;71:205–11. [PubMed] [Google Scholar]
- 96. Nakamura‐Sato Y, Sasaki K, Watanabe H, Araki Y, Sendo F. Clustering on the forward surfaces of migrating neutrophils of a novel GPI‐anchored protein that may regulate neutrophil adherence and migration. J Leukoc Biol. 2000;68:650–4. [PubMed] [Google Scholar]
- 97. Nitto T, Araki Y, Takeda Y, Sendo F. Pharmacological analysis for mechanisms of GPI‐80 release from tumour necrosis factor‐alpha‐stimulated human neutrophils. Br J Pharmacol. 2002;137:353–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Huang J, Takeda Y, Watanabe T, Sendo F. A sandwich ELISA for detection of soluble GPI‐80, a glycosylphosphatidyl‐inositol (GPI)‐anchored protein on human leukocytes involved in regulation of neutrophil adherence and migration – its release from activated neutrophils and presence in synovial fluid of rheumatoid arthritis patients. Microbiol Immunol. 2001;45:467–71. [DOI] [PubMed] [Google Scholar]
- 99. Sengeløv H, Follin P, Kjeldsen L, Lollike K, Dahlgren C, Borregaard N. Mobilization of granules and secretory vesicles during in vivo exudation of human neutrophils. J Immunol. 1995;154:4157–65. [PubMed] [Google Scholar]
- 100. Dahlgren C, Karlsson A, Sendo F. Neutrophil secretory vesicles are the intracellular reservoir for GPI‐80, a protein with adhesion‐regulating potential. J Leukoc Biol. 2001;69:57–62. [PubMed] [Google Scholar]
- 101. Yoshitake H, Nitto T, Ohta N, et al. Elevation of the soluble form GPI‐80, a β2 integrin‐associated glycosylphosphatidylinositol anchored protein, in the serum of patients with Wegener's granulomatosis. Allergol Int. 2005;54:299–303. [Google Scholar]
- 102. Németh T, Sperandio M, Mócsai A. Neutrophils as emerging therapeutic targets. Nat Rev Drug Discov. 2020;19:253–75. [DOI] [PubMed] [Google Scholar]
- 103. Sônego F, Alves‐Filho JC, Cunha FQ. Targeting neutrophils in sepsis. Expert Rev Clin Immunol. 2014;10:1019–28. [DOI] [PubMed] [Google Scholar]
- 104. Moss RB, Mistry SJ, Konstan MW, et al. Safety and early treatment effects of the CXCR2 antagonist SB‐656933 in patients with cystic fibrosis. J Cyst Fibros. 2013;12:241–8. [DOI] [PubMed] [Google Scholar]
- 105. Rennard SI, Dale DC, Donohue JF, et al. CXCR2 antagonist MK‐7123. A phase 2 proof‐of‐concept trial for chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2015;191:1001–11. [DOI] [PubMed] [Google Scholar]
- 106. Stockley R, De Soyza A, Gunawardena K, et al. Phase II study of a neutrophil elastase inhibitor (AZD9668) in patients with bronchiectasis. Respir Med. 2013;107:524–33. [DOI] [PubMed] [Google Scholar]
- 107. Polverino E, Rosales‐Mayor E, Dale GE, Dembowsky K, Torres A. The role of neutrophil elastase inhibitors in lung diseases. Chest. 2017;152:249–62. [DOI] [PubMed] [Google Scholar]
- 108. O'Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, Laurence A. The JAK‐STAT pathway: impact on human disease and therapeutic intervention. Annu Rev Med. 2015;66:311–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Genovese MC, Kremer J, Zamani O, et al. Baricitinib in patients with refractory rheumatoid arthritis. N Engl J Med. 2016;374:1243–52. [DOI] [PubMed] [Google Scholar]
- 110. van de Veerdonk FL, Giamarellos‐Bourboulis E, Pickkers P, et al. A guide to immunotherapy for COVID‐19. Nat Med. 2022;28:39–50. [DOI] [PubMed] [Google Scholar]
- 111. Koenig LM, Boehmer DFR, Metzger P, Schnurr M, Endres S, Rothenfusser S. Blocking inflammation on the way: rationale for CXCR2 antagonists for the treatment of COVID‐19. J Exp Med. 2020;217:e20201342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Kodama T, Yukioka H, Kato T, Kato N, Hato F, Kitagawa S. Neutrophil elastase as a predicting factor for development of acute lung injury. Intern Med. 2007;46:699–704. [DOI] [PubMed] [Google Scholar]
- 113. Guéant JL, Guéant‐Rodriguez RM, Fromonot J, et al. Elastase and exacerbation of neutrophil innate immunity are involved in multi‐visceral manifestations of COVID‐19. Allergy. 2021;76:1846–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Meizlish ML, Pine AB, Bishai JD, et al. A neutrophil activation signature predicts critical illness and mortality in COVID‐19. Blood Adv. 2021;5:1164–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were reported or analyzed in this study.