

Abstract
The CRISPR-Cas9 system has emerged as a powerful and efficient tool for genome editing. An important drawback of the CRISPR-Cas9 system is the constitutive endonuclease activity when Cas9 endonuclease and its sgRNA are co-expressed. This constitutive activity results in undesirable off-target effects that hinder studies using the system, such as probing gene functions or its therapeutic use in humans. Here, we describe a convenient method that allows temporal and tight control of CRISPR-Cas9 activity by combining transcriptional regulation of Cas9 expression and protein stability control of Cas9 in human stem cells. To achieve this dual control, we combined the doxycycline-inducible system for transcriptional regulation and FKBP12-derived destabilizing domain fused to Cas9 for protein stability regulation. We showed that approximately 5%–10% of Cas9 expression was observed when only one of the two controls was applied. By combining two systems, we markedly lowered the baseline Cas9 expression and limited the exposure time of Cas9 endonuclease in the cell, resulting in little or no undesirable on- or off-target effects. We anticipate that this dual conditional CRISPR-Cas9 system can serve as a valuable tool for systematic characterization and identification of genes for various pathological processes.
Keywords: MT: RNA/DNA editing, CRISPR-Cas9, conditional CRISPR-Cas9, gene editing, off-target effect, human stem cell
Graphical abstract
The CRISPR-Cas9 system is an efficient tool for genome editing. However, it has an undesirable off-target effect due to the constitutive Cas9 activity. We developed a dual conditional system to minimize the undesirable Cas9 activity. We anticipate that this system will serve as a valuable tool for various studies.
Introduction
The CRISPR-Cas9 system was derived from the bacterial endogenous adaptive immune system in which DNA sequences (clustered regularly interspaced short palindromic repeats, CRISPR) from invading viruses integrate into the bacterial genome.1,2 These CRISPR sequences in conjunction with Cas nucleases can be used to identify and cleave exogenous DNA sequences from subsequent infections with the same virus.3,4 The Cas9 nuclease is derived from Streptococcus pyogenes bacteria and is most extensively studied.5 Among the different types of Cas nucleases, the CRISPR-Cas9 system belongs to the type II category.6
The CRISPR-Cas9 system is a powerful gene-editing tool mainly due to its ease of designing single-guide RNA (sgRNA) that recognizes the target sequence. The ease of programming sgRNA, which guides Cas9 nuclease to the target site for cleavage, enables the use of the CRISPR-Cas9 system to study biological functions of certain genes.7,8 However, this system can cause undesirable mutations at off-target sites that have similar sequences to the on-target ones.9,10 These off-target effects may complicate experimental results and limit therapeutic uses of the technology.
To reduce genome-wide off-target mutations, various strategies have been exploited. Fu et al. showed that off-target mutations can be decreased up to several orders of magnitude by simply truncating several nucleotides from the 20-nucleotide sgRNA.11 In addition to manipulating sgRNA, another group developed a strategy that brings two separate Cas9 to the same locus by designing two sgRNAs. Both of these Cas9 proteins are converted to nickase enzymes to improve specificity and minimize off-target mutations.12 Kleinstiver et al. had a different approach and introduced three to four mutations into Cas9 to increase specificity and reduce off-target effects simultaneously.13 Alternatively, Kim et al. showed that a different Cas enzyme, Cpf1, appears to have a higher specificity than Cas9.14,15 Finally, several groups tried to decrease Cas9 activity or exposure time in cells after its on-target site had already been cleaved by using light-activated Cas9 variants,16 split Cas9 variants,17 small-molecule induction of Cas9,18 and an engineered allosterically regulated Cas9.19 The light-activated Cas9 system can induce oxidative stress to cells,20 while the primary drawback of both the split Cas9 and the small-molecule induction Cas9 systems is the high background expression of Cas9.21 Additionally, more convoluted systems can increase the size of Cas9 and related genes, which may complicate the delivery of the system. Recent approaches have taken advantage of RNA or protein platform to deliver the Cas9 protein or its derivatives to minimize exposure.22,23
An important drawback of the tetracycline-inducible system is the background expression.21 One way to overcome this problem is to develop a Cas9 enzyme whose activity can be post-translationally controlled by another layer of input. This approach has an advantage of avoiding background expression of Cas9.24 Here, we develop a novel method that allows temporal control of Cas9 activity by combining two conditional systems, one to regulate the transcription of Cas9 gene and another to control the stability of Cas9 protein. To regulate this dual conditional system, we combine the doxycycline-inducible system for transcriptional regulation and FKBP12-derived destabilizing domain fused to Cas9 for protein stability regulation. The doxycycline-inducible system depends on regulatory elements that regulate the activity of the tetracycline-resistance operon in bacteria and is used to control the activity of genes in eukaryotic cells. This system can be applied to various settings, varying from basic biological research to biotechnology and gene therapy applications.25 The FKBP12-derived destabilizing domain is used with a highly cell-permeable and nontoxic synthetic ligand, Shield1, which binds to the mutant FKBP12 tightly and prevents proteasome-induced degradation of the bound protein. This destabilized-domain system can be used with a variety of proteins to control diverse biological processes in vitro and in vivo.26 Since each conditional system by itself has a detectable level of Cas9 background expression,27,28 combining two conditional systems can dramatically decrease Cas9 background expression as well as off-target effects by reducing Cas9 exposure time in cells. Another advantage of this conditional system is that it can induce knockout of target genes only when it is desirable. Therefore, if Cas9 inactivation is required, for example, during stem cell differentiation or during certain developmental stages, we can temporally control Cas9 expression. We expect that this novel conditional CRISPR-Cas9 system will be a useful tool for studying genes during development or pathological processes.
Results
Generation of a conditional CRISPR-Cas9 system
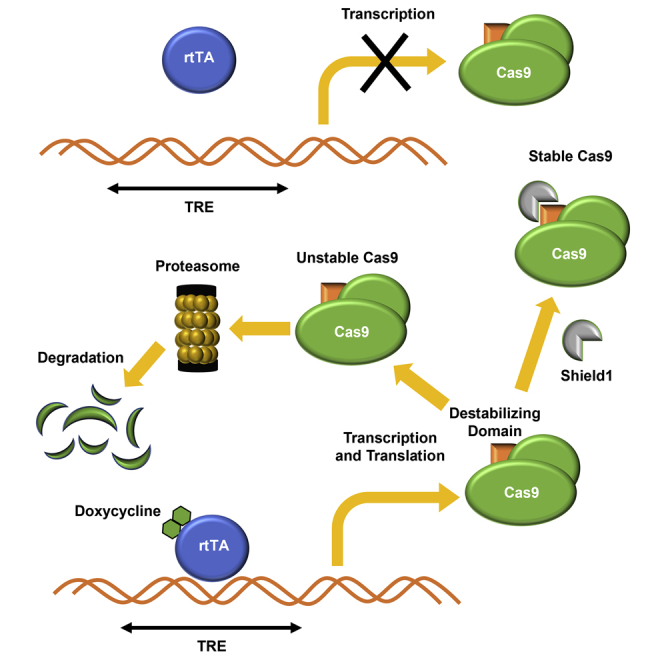
Although the CRISPR-Cas9 system is a convenient and powerful tool for genome editing, the constitutive endonuclease activity when Cas9 enzyme and its sgRNA are co-expressed is an important drawback of the CRISPR-Cas9 system. A conditional CRISPR-Cas9 system could compensate for such a drawback; however, conditional systems tend to be ‘leaky’ and often show background expression.27,28 Here, we develop a novel dual conditional system that allows control of transcription and protein stability of Cas9 endonuclease by combining two conditional systems: the doxycycline-inducible system for transcriptional regulation and the FKBP12-derived destabilizing domain fused to Cas9 for protein stability regulation. The Cas9 gene is transcribed upon doxycycline induction. The Cas9 protein is degraded rapidly when the Shield ligand does not bind to the FKBP12-derived destabilizing domain fused to the N terminus of the Cas9 protein (Figure 1A). The conditional CRISPR-Cas9 construct that we generated in this study comprises tetracycline response element (TRE) followed by the CMV minimal promoter. The FKBP12-derived destabilizing domain is fused to the N terminus of the Cas9 sequence which is followed by a Nanoluciferase reporter gene. Cas9 and Nanoluciferase are expressed as one polyprotein, and the intervening P2A peptide sequence allows auto-cleavage of the two proteins (Figure 1B). As the Nanoluciferase is no longer part of the Cas9 protein, it is not subjected to control by the destabilizing domain.
Figure 1.

Generation of a conditional CRISPR-Cas9 system
(A) Mechanism of a dual conditional CRISPR-Cas9 system. In the presence of doxycycline rtTA (reverse tetracycline transactivator) binds to TRE (tetracycline response element), which allows transcription of FKBP12-derived destabilizing domain fused Cas9. After translation, Shield1 ligand prevents Cas9 from degradation. (B) Schematic construction map for our novel conditional CRISPR-Cas9 system. Nanoluciferase activity was measured to assess Cas9 expression level. DD, destabilizing domain. (C and D) H9-Cas9 cells were seeded in white 96-well plates 24 h before doxycycline treatment. (C) The cells were incubated with 300 ng/mL doxycycline for 2, 6, and 24 h, and then Nanoluciferase activity and Cas9 mRNA levels were measured. Cas9 expression is induced after doxycycline addition. (D) Different concentrations of doxycycline were treated in H9-Cas9 cells followed by 24-h incubation. Nanoluciferase activity and Cas9 mRNA levels were measured. Cas9 expression is increased by doxycycline in a dose-dependent manner. Data are shown as mean values ±SEM of triplicate experiments.
The conditional CRISPR-Cas9 system was delivered by lentivirus into SW-induced pluripotent stem cells (iPSCs) or H9 embryonic stem cells (ESCs) and selected by G418 antibiotic. The selected pooled clones (SW-Cas9 or H9-Cas9) are then tested for their responsiveness to the two conditional systems. To confirm that transcriptional regulation was functional, the pooled clones were treated with doxycycline and Nanoluciferase activity and Cas9 mRNA level were measured. The Nanoluciferase activity and Cas9 mRNA level gradually increased over time (Figure 1C) and showed a dose-response curve to doxycycline induction (Figure 1D). Thus, our results indicate that our conditional CRISPR-Cas9 system responds to transcriptional regulation by the doxycycline-inducible system.
Dose-response control of Cas9 protein level by Shield1 ligand
To further validate our CRISPR-Cas9 system for protein stability regulation, we measured Cas9 protein level with different concentrations of Shield1. For this, H9-Cas9 pooled clones were treated with 300 ng/mL doxycycline and with 0–500 nM Shield1. Following Shield1 treatment, Cas9 protein level showed a dose-response increase, whereas the Cas9 mRNA level remained unchanged (Figure 2A). This result indicates that our conditional CRISPR-Cas9 system is under protein stability regulation by the FKBP12-derived destabilizing domain.
Figure 2.

Dose-response change of Cas9 protein level by Shield1 ligand
(A–C) H9-Cas9 cells were seeded in six-well plates 24 h before doxycycline and Shield1 ligand addition. (A) The cells were collected 24 h after 300 ng/mL doxycycline and different concentrations of Shield1 ligand addition. After protein and mRNA extraction, Cas9 protein level was measured by Wes assay, and mRNA level was determined by RT-qPCR. Cas9 protein level was regulated by Shield1 ligand in a dose-dependent manner, whereas Cas9 mRNA level was unchanged. (B) The cells were collected at different time points after 300 ng/mL doxycycline and 250 nM Shield1 ligand treatment. Twenty-four hours after doxycycline and Shield1 addition, the medium was replaced with fresh medium without doxycycline and Shield1. Removal of Shield1 results in decrease of Cas9 protein level. (C) Kinetic study for Cas9 protein level with or without doxycycline and/or Shield1 ligand. First row is the Cas9 protein level. Second row is the densitometry data for the first row Cas9 protein level. Third row is the Nanoluciferase activity, which represents the Cas9 mRNA level. Data are shown as mean values ±SEM of triplicate experiments.
Knowing that our CRISPR-Cas9 system can be tightly regulated by the dual control system, we further tested how long Cas9 protein is present in cells with or without doxycycline and/or Shield1. First, we treated H9-Cas9 pooled clones with 300 ng/mL doxycycline and 250 nM Shield1 and measured Cas9 level at different time points. Cas9 protein level gradually increased from 6 h after addition of both ligands and declined following removal of them from the medium. Twenty-four hours after removal of both doxycycline and Shield1, Cas9 level became undetectable (Figure 2B). To further study the declining kinetics of Cas9 protein level upon withdrawal of the ligands, we tested four conditions: no withdrawal, withdrawal of both doxycycline and Shield1, withdrawal of doxycycline only, and withdrawal of Shield1 only. Even in the continued presence of both ligands, both Cas9 protein level and Nanoluciferase activity declined by the end of 72 h, probably because of the gradual degradation of the ligands. Upon withdrawal of both ligands, both proteins declined rapidly (Figure 2C). When doxycycline was removed, the Cas9 protein level decreased but still maintained a reasonable level at 72 h, whereas Nanoluciferase activity dropped rapidly, like the withdrawal of both ligands. Upon removal of Shield1, Cas9 protein level quickly declined and the Nanoluciferase activity decreased more slowly (Figure 2C). These findings are consistent with the transcriptional regulation (represented by Nanoluciferase activity) by doxycycline and Cas9 protein stability control by Shield1.
Dual conditional CRISPR-Cas9 system reduces undesirable on- or off-target effects
To validate whether our dual conditional CRISPR-Cas9 system reduces undesirable on- or off-target effects compared with other Cas9 systems such as the constitutive Cas9-expressing system,29 the doxycycline-inducible Cas9 system,30 the Shield1-regulated Cas9 system,26 and the Cas9 ribonucleoprotein delivery system,31 we generated H9-Cas9 pooled clones (bulk transformants) that targeted p53, EMX1, HBB, and FANCF, which have been used widely for off-target studies.10,32 To compare Cas9 expression level between different Cas9 systems, we isolated RNA and protein from each Cas9 system and performed qRT-PCR and Wes assay, respectively (Figure S1A). For the inducible systems, the cells were treated with doxycycline and/or Shield1 for 3 days followed by RNA/protein extraction and qRT-PCR/Wes assay. To test for gene-editing efficiencies, constitutive Cas9 cells were analyzed by TIDE (tracking of indels by decomposition) assay immediately after antibiotic selection. For the inducible systems, the cells were treated with doxycycline and/or Shield1 for 3 days followed by TIDE assay.
The ribonucleoprotein (RNP) delivery system showed the strongest gene-editing capacity for most of the target genes among all the Cas9 systems, but it also had the strongest off-target effect (Figure 3A). All the Cas9 systems except for the dual conditional system exhibited some off-target effects for most of the target genes. For EMX1 and HBB, both the constitutive system and the RNP delivery system have some off-target effects. The doxycycline-inducible system and Shield1-regulated system showed little off-target effects for p53, HBB, and FANCF. In addition to the off-target effects, both doxycycline-inducible and Shield1-regulated systems exhibited leaky on-target effects without doxycycline or Shield1 induction (Figure 3A). Especially, in the case of EMX1, our dual conditional system showed no leaky on-target effect, while the on-target efficiencies were comparable between our induced dual conditional system and the other two mono-inducible systems such as doxycycline-inducible and Shield1-regulated systems (dual on, 42.1%; dual leaky, 1.4%; dox on, 29.3%; dox leaky, 6.7%; Shield1 on, 56.7%; Shield1 leaky, 17.8%). Second off-target sites for the target genes were also tested, but the off-target gene editing efficiencies were quite low among the target genes (Figure S1B). This may be because one additional nucleotide mismatch significantly decreased off-target gene-editing efficiencies. In order to compare the off-target efficiencies more precisely between the Cas9 systems, we measured off/on-target ratios for the target genes (Figure S1C). When off/on ratios were measured, Cas9-induced groups were compared along with constitutive and RNP systems. Most of the off/on ratios did not show significant differences except for the HBB off/on ratio with off-target site 1. HBB off-target site 1 was the site where we detected overall the highest off-target gene-editing efficiency compared with the other genes. In HBB’s off/on ratio, constitutive and RNP systems were significantly higher than that of the dual Cas9 system. Thus, for some off-target sites, the dual system performed significantly better than other systems.
Figure 3.

Dual conditional CRISPR-Cas9 system reduces off-target effects
(A) H9 cells were used to generate pooled clonal cells with several CRISPR-Cas9 systems: our dual conditional Cas9 system (Dual), doxycycline-inducible Cas9 system (Dox), Shield1-regulated Cas9 system (Shield1), constitutive Cas9-expressing system (Const), and Cas9 ribonucleoprotein delivery system (RNP). For the inducible systems, the cells were treated with doxycycline and/or Shield1 for 3 days to knock out p53, EMX1, HBB, or FANCF gene. After DNA extraction, 500- to 1,000-bp sequences that comprise the knockout site were amplified by PCR and subsequent TIDE assay was performed. The data are expressed as indel frequency (%) (0% as no editing and 100% near-complete editing); p values were calculated by comparison with the uninduced dual conditional system (−/−). (B) SW-sgGFP cells (iPSC-Cas9-sgGFP) that were seeded in six-well plates were treated with or without doxycycline and/or Shield1 ligand. The cells were collected at 24 h, and Wes assay was performed. Dual control with doxycycline and Shield1 eliminates background Cas9 expression. (C) Dual control of Cas9 expression is measured by p53 promoter reporter assay. p53 promoter reporter plasmid was transfected in SW-sgp53 cells (iPSC-Cas9-sgp53) after 3 days of doxycycline and Shield1 treatment. Doxycycline and Shield1 were treated throughout the experiment to induce p53 gene knockout. Twenty-four hours after p53 promoter reporter plasmid transfection, 0.4 μg/mL doxorubicin was added to induce p53 expression; 48 h after transfection, the cells were measured for luciferase activity. SW-sgGFP (iPSC-Cas9-sgGFP), SW-Cas9 clones that express sgRNA targeting GFP gene; SW-sgp53 (iPSC-Cas9-sgp53), SW-Cas9 clones that express sgRNA targeting p53 gene. Data are shown as mean values ±SEM of triplicate experiments; ns, not significant. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Taken together, our dual conditional system showed the advantage of having few or no undesirable on-target or off-target effects for any of the four target genes we tested here. Although our dual conditional system had a lower on-target editing efficiency compared with constitutive and RNP systems, our system showed a comparable level of on-target efficiency with other inducible Cas9 systems such as the doxycycline-inducible system.
Additionally, to quantify off-target effects, we designed sgRNA targeting the Nanoluciferase gene and introduced a single nucleotide mutation at different locations in the 20-nucleotide sgRNA sequence. Over time, we expected a decrease in Nanoluciferase activity due to indel(s) caused by a Cas9 endonuclease double-stranded break at the target site. As expected, we observed that the Nanoluciferase level decreased progressively after induction of Cas9. The mutated sgRNAs also showed decreased Nanoluciferase activity over time, although the level of decline varied (Figure S2). More interestingly, in two of three mutated sgRNAs, Nanoluciferase levels began to decline between day 5 and day 7, whereas the on-target group began to decrease between day 1 and day 3 (Figures S2 and S3). Together, these results suggest that off-target effects can occur as long as both Cas9 and sgRNA are present in the cells; thus, regulating the exposure time of Cas9 may reduce off-target effects, as they arise later than the on-target effects.
Since both Cas9 and Nanoluciferase are expressed as a polyprotein via a P2A autoproteolytic cleavage sequence, we tested whether targeting Nanoluciferase sequence by Cas9 might interfere with the expression of the Cas9 gene. As shown in Figure 3B, Cas9 protein levels were similar among the different groups. This finding confirms that targeting Nanoluciferase does not affect the Cas9 expression or functionality.
Since the exposure time of Cas9 and sgRNA expression is an important factor for off-target effects, reducing constitutive background Cas9 expression is key to avoiding undesirable off-target effects. Therefore, to quantify the background Cas9 expression level, we introduced sgRNAs targeting GFP and p53 gene by lentivirus delivery and selected single clones that show regular cell morphology and high levels of induced Cas9 protein expression. As shown for the SW-SW-Cas9-sgGFP clone (iPSC-Cas9-sgGFP), Cas9 protein level was fully induced in the presence of doxycyclene and Shield1. When doxycycline was used without Shield1, a low but detectable level of Cas9 was observed, indicating that regulation by Shield1 was leaky (Figure 3B). As described above, we also noted that there was a low baseline transcriptional activity in the absence of doxycycline (Figures 1C, 1D).
To test the functional effect of Cas9 induction, SW-Cas9-sgp53 cells (iPSC-Cas9-sgp53) were initially transfected with the pGL4.38 [luc2P/p53 RE/Hygro] vector (p53 promoter reporter plasmid) and then treated with the p53 inducer doxorubicin. Each group was treated with or without doxycycline and/or Shield1. When the cells were treated with both doxycycline and Shield1, we observed a dramatic decrease of firefly luciferase activity that represents the on-target effect. If only doxycycline was used without Shield1, there seemed to be a slight decrease in firefly luciferase activity, but this decrease did not reach statistical significance (Figure 3C). Together, these results suggest that background Cas9 expression is present when either one of conditional systems is used individually. Therefore, combining the two conditional systems may provide a solution for an important drawback of CRISPR-Cas9 systems.
Dual conditional CRISPR-Cas9 system is efficient for gene knockout
Given that the dual conditional CRISPR-Cas9 system can reduce background Cas9 expression to an undetectable level that has a direct correlation to its off-target effects, it is important to validate that the dual conditional CRISPR-Cas9 is as efficient as other CRISPR-Cas9 systems. To evaluate the knockout efficiency of our CRISPR-Cas9 system, we performed a T7 endonuclease assay, a TIDE assay, and target protein level measurement. We observed that the dual CRISPR-Cas9 system was able to introduce mutations for all three target genes, p53, PTEN, and APC in an inducible manner (Figure 4). The editing efficiencies for the three genes with the pooled clones were 20%–40%, which were comparable to those in previous reports using human stem cells.33,34 Additionally, we confirmed that the dual inducible system edits p53 gene in a dose-dependent manner with Shield1 (Figure S4). Consistent with the presence of mutations in the p53 and PTEN genes, their protein levels were also lower. On the other hand, the APC protein level did not change much despite the presence of mutations by the other assays, the reason for which is not clear. Together, these results show that the dual conditional CRISPR-Cas9 system is efficient for gene knockout.
Figure 4.

Dual conditional CRISPR-Cas9 system is efficient for gene knockout
(A) Using H9-Cas9 clones that express sgRNAs targeting p53, PTEN, and APC genes, T7 endonuclease assay was performed to test knockout efficiency. Each H9-Cas9 clone was treated with doxycycline and Shield1 for 5 days to knock out p53, PTEN, and APC, respectively. After DNA extraction, 500- to 1,000-bp sequences that comprise the knockout site were amplified by PCR, and subsequent T7 endonuclease assay was performed. (B) TIDE assay was also performed for the same cell clones. The cells were treated with doxycycline and Shield1 for 10 days followed by DNA extraction and PCR amplification for the sequences including the knockout site. (C) For the same cell clones, doxycycline and Shield1 were added for 5 or 10 days continuously. Then, Wes assay was performed after protein extraction. Wes assay showed Cas9 protein levels with or without CRISPR-Cas9-induced knockout. The assays were repeated with triplicate experiments.
Conditional CRISPR-Cas9 system and human stem cell hepatocyte differentiation
Knowing that the dual CRISPR-Cas9 system is functional at the stem cell stage, we evaluated whether this system also works during hepatocyte or macrophage differentiation. We found that, with the original doxycycline-induced promoter containing the CMV minimal promoter, Cas9 protein expression was dramatically reduced upon differentiation of the Cas9-expressing stem cell clones. Not surprisingly, complete transgene silencing is often observed during differentiation of stem cells.35,36 Therefore, we replaced the original CMV minimal promoter and tested the efficiency of several different minimal promoters to determine the best alternative that would maintain the highest promoter activity following differentiation. We tested two different minimal promoter sequences, the EF1α and human albumin promoters, in the dual conditional system (Figure 5A).
Figure 5.

Conditional CRISPR-Cas9 system and human stem cell hepatocyte differentiation
(A) Diagram of conditional CRISPR-Cas9 system with various promoter systems. (B) H9-Cas9 clones that were replaced with EF1α and human albumin (hALB) promoters were differentiated into hepatocyte-like cells. Level of sgRNA targeting p53 gene was measured by RT-qPCR during hepatocyte differentiation. (C) CMV, EF1α, and hALB promoter activities in the conditional CRISPR-Cas9 system, as shown by the Nanoluciferase activities, were compared during hepatocyte differentiation of SW-Cas9 (iPSC) and H9-Cas9 (ESC) clones. Data are shown as mean values ±SEM of triplicate experiments; ns, not significant. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
First, we evaluated sgp53 RNA levels in the H9-Cas9-sgp53 clones during differentiation to assess whether sgp53 RNA expression is being silenced during hepatic differentiation (Figure 5B). The level of sgp53 RNA showed an approximateyl 2-fold decrease during differentiation, but transcription of Cas9 gene was strongly silenced and failed to show any induction by doxycycline (Figure 5C). Therefore, the main reason for the loss of knockout efficiency is silencing of Cas9 expression.
Since Cas9 and Nanoluciferase are expressed as one polyprotein, we measured Nanoluciferase activity to quantify Cas9 gene silencing during differentiation. All the promoter constructs showed substantial silencing during hepatocyte or macrophage differentiation (Figures 5C and S5). However, the promoter activity varies depending on the distinct stage of stem cell differentiation. For example, the human albumin promoter appears to be the optimum promoter during hepatocyte differentiation. As shown in Figure 5C, inducible Nanoluciferase activity remained after hepatocyte differentiation for 15 days. Alternatively, if the transgene is required to be active during the endodermal differentiation, the EF1α-based promoter works well because of its inducible editing activity at both the stem cell and definitive endoderm stages (Figure S6). As cells underwent hepatic differentiation, the activity of the EF1α-based promoter became silenced (Figure 5C). Although we observed some remaining transcriptional activity after hepatic differentiation with human albumin promoter, we were not able to detect gene editing after hepatic differentiation. We suspect that the Cas9 protein expression was not high enough to carry out efficient gene editing after differentiation.
Discussion
The CRISPR-Cas9 system has offered unprecedented convenient and efficient genome editing in many settings. However, its constitutive endonuclease activity poses problems such as genome-wide off-target effects and unexpected effects during developmental studies. To overcome the drawbacks of constitutive CRISPR-Cas9 systems, many conditional CRISPR-Cas9 systems have been developed for conditional Cas9 expression to reduce exposure time of Cas9 endonucleases in the cells.
An important drawback of many conditional systems is the background expression of the system.21 The Tet system is one of the best-known inducible systems that has been employed in many CRISPR-Cas9 studies.18,30,34,37 However, like many other conditional systems, background expression is also a problem for the Tet system.21 We observed about 5%–10% of background transcriptional activity in the absence of doxycycline based on the Nanoluciferase assay. To improve the Tet system, we combined it with another conditional system, the FKBP12-derived destabilizing domain, to regulate Cas9 protein stability control. This protein stability control itself has background expression like the other conditional systems in the absence of Shield1 ligand, which stabilizes the destabilizing domain (Figure 2A, red arrow; Figure 3A). When we combined the Tet system and the destabilizing domain protein stability control system, the background Cas9 expression was virtually undetectable (Figure 2A, blue arrow). Therefore, this dual conditional CRISPR-Cas9 system has the major advantage of eliminating background expression of Cas9.
To validate whether our conditional CRISPR-Cas9 system reduces off-target effects compared with other Cas9 systems, we used p53 and three previously known genes, EMX1, HBB, and FANCF, for off-target studies.10,32 This experiment suggests that the RNP delivery system shows the strongest gene editing among the Cas9 systems above, but at the same time it also has the strongest off-target effects for most of the target genes we tested (Figure 3A). Besides the RNP delivery system, the constitutive system also exhibits strong on- and off-target effects for all four target genes. The doxycycline-inducible and Shield1-regulated Cas9 systems shows leaky on-target effects as well as off-target effects for certain genes (Figure 3A). Given that our dual conditional system did not show any off-target or leaky on-target effects, we believe that this dual conditional system is highly advantageous. It should be noted that, although the T7 endonuclease and TIDE assays that we used for determination of on- and off-target effects are cost-effective, they display a relatively low sensitivity. Many other off-target detection methods with higher sensitivity have been developed and should be utilized in the future to further confirm these findings. For example, deep sequencing is known for precision, but it is biased and misses potential off-target sites elsewhere in the genome.38 On the contrary, GUIDE-Seq is unbiased and detects off-target frequencies as low as 0.12%, but false negatives are present and it is limited by chromatin accessibility.10 Digenome-Seq is also unbiased, with sensitivity as low as 0.1%.39 Application of these assays to fully quantify the off-target effects of our system would be important.
Applying this conditional CRISPR-Cas9 system to a developmental system, such as stem cell differentiation, the possibility of promoter silencing needs to be considered. We observed that Cas9 expression was gradually silenced over time, as hepatocyte or macrophage differentiation was initiated for Cas9-expressing cell clones. Promoter silencing was also reported in many other studies.36,40,41 Introduction of chromatin insulator element has been proposed to avoid promoter silencing of transgenes;42,43 however, the efficiency of this insulator is still controversial in stem cells. Switching promoters is another way to minimize the promoter-silencing problem, as we have done in this study. Careful selection of the optimal promoter based on empirical studies is an alternative method to avoid promoter silencing.
For in vivo application, additional development for our dual conditional system is needed. Since our system depends on doxycycline-inducible regulation, it needs to be delivered by an AAV vector to carry the tetracycline response element. Another reason for using AAV vector is that it can avoid permanent integration of transgenes, which is a feature of lentiviral delivery.44 Because of the small packaging capacity of AAV (∼2 kb), it is difficult to package SpCas9 (∼4 kb) into an AAV vector. However, we can instead deliver a miniature Cas12f1 nuclease (∼1.5 kb). Kim et al. recently reported that they could deliver the compact Cas12f1 into an AAV vector and achieved high gene-editing efficiency by modifying guide RNAs.45
In summary, our work demonstrates that combining two conditional systems maintains CRISPR-Cas9’s efficient genome-editing activity and reduces off-target effects by minimizing background expression and controlling exposure time to Cas9 endonuclease. We expect that this dual conditional CRISPR-Cas9 system can provide a valuable platform for studying the systematic characterization and identification of genes involved in developmental processes.
Materials and methods
Plasmid construction and lentivirus production
We constructed a novel inducible CRISPR-Cas9 system by combining the doxycycline-inducible system and FKBP12-derived destabilizing domain fused to Cas9. Lenti-iCas9-neo (Addgene: 85400) was used as a backbone, and the destabilizing domain (a gift from Dr. Raffaella Sordella, Cold Spring Harbor Laboratory, Huntington, NY, USA) was inserted into the N terminus of spCas9 sequence.26,30 Commercially synthesized P2A-Nanoluciferase sequence (GenScript, Piscataway, NJ, USA) was added to the C terminus of spCas9 sequence. For improved Cas9 expression, SV40 PolyA signal was integrated after the Nanoluciferase sequence. To generate several CRISPR-Cas9 systems for comparison of on- and off-target efficiencies, the constitutive Cas9-expressing system for LentiCRISPR v2 (Addgene: 52961) and the doxycycline-inducible Cas9 system for Lenti-iCas9-neo (Addgene: 85400) were used. The Shield1-regulated Cas9 system was a gift from Dr. Raffaella Sordella. The wild-type Cas9 sequence in the LentiCRISPR v2 (containing a neomycin resistance gene) was replaced with the destabilizing domain-containing Cas9 sequence from the Shield1-regulated Cas9 system to generate the construct for subsequent experiments. The complete nucleotide information of our dual conditional Cas9 plasmids has been submitted to the NCBI GenBank. The accession numbers are MW651968 (CMV promoter), MW651969 (EF1α promoter), and MW651970 (human albumin promoter). The Cas9 RNP delivery system (TrueCut Cas9 Protein v2) was obtained from Thermo Fisher Scientific (Waltam, MA, USA). For sgRNA generation, LentiGuide-Puro (Addgene: 52963) was used.29 The sequence information for sgRNAs targeting p53, PTEN, APC, and GFP were provided by Dr. Wen Xue (University of Massachusetts Medical School, Worcester, MA, USA) (Table S1). The sequence information for sgRNAs targeting EMX1, HBB, and FANCF were reported in the previous studies.10,32
For lentivirus production, the above constructed plasmids, pMD2.G (Addgene: 12259), and psPAX2 (Addgene: 12260) were cotransfected using FuGENE 6 transfection reagent (Promega, Madison, WI, USA) into HEK293T cells. The medium was changed 24 h after transfection. 48 h after transfection, the medium was collected and concentrated using ultracentrifugation for 1.5 h at 25,000 rpm. The concentrated lentivirus was resuspended in 200 μL of PBS buffer and stored at −80°C until infection for ESCs or iPSCs. To generate pooled clonal cells, stem cells in a six-well plate format were infected with 50 μL of recombinant lentivirus and 24 h later was treated with 0.5 mg/mL G418 (Cat#: 10131035, Thermo Fisher Scientific) for 5 days. The constitutive and Shield1-regulated Cas9 systems, which contain the sgRNA expression cassette, need to undergo only one antibiotic selection. Shield1 was obtained from Cheminpharma LLC (Woodbridge, CT, USA). For the dual conditional and the doxycycline-inducible system, an additional selection step was needed because these systems are two-vector systems (Cas9 in one construct and sgRNA in another construct). For the second selection, previously selected cells were infected with recombinant sgRNA-expressing lentivirus and treated with 5 μg/mL puromycin (Cat#: A1113803, Thermo Fisher Scientific) for 3 days. The selected cells were then used for experiments.
Nanoluciferase assay
Cas9-expressing cell clones were seeded in a 96-well white microplate and induced by doxycycline (Cat#: D9891, Sigma-Aldrich) for Nanoluciferase expression several hours or 24 h before Nanoluciferase assay. The Nanoluciferase assay was performed according to the manufacturer’s instructions from the Nano-Glo Luciferase Assay System (Cat#: N1120, Promega).
Protein extraction and Wes assay
Cells or ground tissue samples were lysed with RIPA lysis and extraction buffer (Thermo Fisher Scientific) and protease inhibitor cocktail (Roche, Basel, Switzerland) for 10 min at room temperature. Then, the samples were centrifuged for 10 min at 12,000 rpm at 4°C. The supernatants were collected and further processed using a Pierce BCA protein assay kit (Thermo Fisher Scientific) for protein quantification.
For target protein measurement, automated quantitative western blot (Wes assay) was performed on a Wes instrument (ProteinSimple, San Jose, CA, USA) according to the manufacturer’s instruction. The following antibodies were purchased commercially: anti-Cas9 (Cat#: NBP2-36440; Novus Biologicals, Centennial, CO, USA), anti-p53 (Cat#: NBP2-29453, Novus Biologicals), anti-PTEN (Cat#: AF847-SP, Novus Biologicals), anti-APC (Cat#: NB100-91662SS, Novus Biologicals), and anti-actin (Cat#: ab8227, Abcam, Cambridge, UK).
RNA extraction and qRT-PCR
Total intracellular RNA was extracted using TRIzol Reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions. Then, mRNA levels were quantified by quantitative real-time qRT-PCR using a Verso 1-step RT-qPCR Kit (Cat#: AB4106A, Thermo Fisher Scientific). The reaction condition was followed per the manufacturer’s instructions. Primer sequences for Cas9 and GAPDH are listed in Table S2.
DNA extraction and TIDE and T7 endonuclease assays
Total intracellular DNA was extracted using a Promega Wizard Genomic DNA Purification Kit (Cat#: A1120) according to the manufacturer’s instructions. To measure Cas9-induced mutation for the target sites, both T7 endonuclease and TIDE assays were used. For both assays, the sequences comprising the target sites were amplified by primers listed in Table S2. PCR reactions were performed using CloneAmp HiFi PCR Premix (Cat#: 639298; Takara, Kusatsu, Japan) according to the manufacturer’s instructions. Then, the amplified products were isolated by gel purification method using NucleoSpin Gel and PCR Clean-Up (Cat#: 740609, Takara). For the TIDE assay, the purified samples were read by conventional Sanger sequencing method and further processed by the open-source software on the website (https://tide.nki.nl/). For the T7 endonuclease assay, the purified samples were further processed by T7 Endonuclease I (Cat#: M0302; New England BioLabs, Ipswich, MA, USA) according to the manufacturer’s instructions.
CRISPR-Cas9 RNP delivery
H9 ESCs were dissociated by Accutase at 37°C for 5 min. The detached cells were pelleted by centrifugation at 1,500 rpm for 3 min and resuspended to a density of 2 × 107 cells/mL in Resuspension Buffer R (Neon Transfection System 10 μL Kit, Cat#: MPK1025, Thermo Fisher Scientific). At the same time, 625 ng (3.75 pmol) of Cas9 protein (TrueCut Cas9 Protein v2, Cat#: A36496, Thermo Fisher Scientific) and 120 ng (3.75 pmol) of gRNAs (Invitrogen TrueGuide sgRNA Modified, Custom, Cat#: A35534, Thermo Fisher Scientific: gRNA for p53 GAAUGAGGCCUUGGAACUCA, gRNA for EMX1 GAGUCCGAGCAGAAGAAGAA, gRNA for HBB CUUGCCCCACAGGGCAGUAA, gRNA for FANCF GGAAUCCCUUCUGCAGCACC) were prepared in 10 μL of Resuspension Buffer R and incubated at room temperature for 20 min. After incubation, the Cas9 and gRNA mixture was supplemented by 5 μL of resuspended H9 ESCs. Then, 10 μL of cell-Cas9-gRNA mixture was aspirated by Neon pipette and inserted into the Neon tube containing 3.5 mL of Electrolytic Buffer E in the Neon pipette station. Cells were pulsed twice with a voltage of 1,050 (V) and a width of 30 (ms). After the pulses, cells were quickly transferred into pre-warmed mTeSR1 medium-containing plates.
The p53 promoter reporter assay
The p53 knockout efficiency caused by the Cas9-induced target site mutation was measured by firefly luciferase assay. pGL4.38[luc2P/p53 RE/Hygro] vector (Cat#: E3651, Promega) was transfected in SW iPSC clones expressing Cas9 and sgRNAs targeting p53 gene or GFP, which was used for control purposes. Twenty-four hours after transfection, 0.4 μg/mL doxorubicin was treated to induce p53 expression; 48 h after transfection, the cells were processed using the Luciferase Assay System (Cat#: E1500, Promega) based on the manufacturer’s instructions.
Stem cell culture
SW iPSCs were kindly provided by Dr. Cynthia E. Dunbar (NIH, Bethesda, MD, USA). Non-colony monolayer-type culture for human ESCs and iPSCs have been described previously.46,47 Briefly, SW iPSCs or H9 ESCs were maintained on growth factor reduced Matrigel matrix (0.4 mg/mL) (Corning, NY, USA) in mTeSR1 medium (STEMCELL Technologies, Vancouver, BC, Canada). Cells were routinely passaged in 1:3 or 1:4 dilutions every 2–3 days with Accutase (Thermo Fisher Scientific) and reseeded with 10 μM or Rock Inhibitor Y-27632 (STEMCELL Technologies) as described previously.46,47
Hepatocyte-like cell (HLC) differentiation
Monolayer-type iPSCs or ESCs were resuspended using Accutase, and 2 million cells were seeded into one well in six-well plates coated with growth factor reduced Matrigel matrix (0.125 mg/mL, Corning) 1 day before beginning of the differentiation. From the next day, the cells were then cultured with a STEMdiff Definitive Endoderm Kit (STEMCELL Technologies) for definitive endoderm induction, following the manufacturer’s instructions. For the first day of definitive endoderm, the cells were cultured in STEMdiff definitive endoderm basal medium with supplements A and B. For the next 3 days, the cells were given the STEMdiff definitive endoderm basal medium with supplement B only, with daily medium change. For hepatic specification, the cells were then resuspended using Accutase and seeded with a 79,000 cells/cm2 concentration in neμw plates that were coated with growth factor reduced Matrigel matrix (0.125 mg/mL) (Corning). The basal medium from hepatic specification stage contained high-glucose Dulbecco’s modified Eagle’s medium (DMEM), F12, 10% KnockOut serum replacement (KOSR), 1% non-essential amino acids (NEAA), 1% glutamine, and 1% penicillin-streptomycin (Thermo Fisher Scientific). For differentiation, the basal medium was supplemented with 100 ng/mL human growth factor (HGF; Peprotech, Rocky Hill, NJ, USA), 1% dimethyl sulfoxide (DMSO, Sigma-Aldrich, St. Louis, MO, USA), and 10 uM Rock Inhibitor Y-27632 (STEMCELL Technologies). During the next 7 days, the cells were cultured in differentiation medium containing 100 ng/mL HGF and 1% DMSO, with daily medium change. After hepatic specification stage, the hepatoblast-like cells were then matured in differentiation medium containing 10−7 M dexamethasone (Sigma-Aldrich) for 3 days. Differentiated HLCs were then maintained in William’s E medium containing 10% fetal bovine serum (FBS), 0.17 μM human insulin (Sigma-Aldrich), 10 μM hydrocortisone 21-hemisuccinate (Sigma-Aldrich), 1.8% DMSO, and 1% penicillin-streptomycin, with medium changed 2–3 times per week.
Macrophage differentiation
The protocol for macrophage differentiation was adapted from Alasoo et al.48 The hiPSCs were first converted from monolayer culture to colony culture by using vitronectin (STEMCELL Technologies) as a basement membrane. The cells were allowed to acclimate for at least 1 week in the new culture format. Following acclimation, hiPSC colonies were grown to 80% confluence before being split into small clumps by using 0.5 mM EDTA and manual dissociation. The clumps were seeded in six-well low-adherence plates at a 1:1 dilution and cultured in KSR medium [Advanced DMEM-F12 (Thermo Fisher Scientific), 20% Knockout Serum Replacement (Thermo Fisher Scientific), 2 mM glutamine, 1% penicillin-streptomycin, and 50 μM 2β-mercaptoethanol (Sigma-Aldrich)] for 7 days, changing the medium every 2 days in order to form embryoid bodies. The embryoid bodies were then seeded onto gelatin-coated six-well plates at a density of 10–20 embryoid bodies per well in myeloid progenitor medium (X Vivo 15 Serum-Free Hematopoietic Cell Medium [Lonza, Basel, Switzerland], 2 mM glutamine, 1% penicillin-streptomycin, 50 μM 2β-mercaptoethanol]) supplemented with 25 ng/mL IL3 (R&D Systems, Minneapolis, MN, USA) and 50 ng/mL MCSF (R&D Systems). These plates were maintained for 4–6 months, changing the medium every 7 days. In order to differentiate myeloid progenitors into macrophages, the progenitors were collected from the culture medium and seeded at a density of 9,000 cells/cm2 onto tissue culture plastic in myeloid progenitor medium supplemented with 100 ng/mL MCSF for 24 h. The following day, the medium was changed to macrophage differentiation medium (RPMI 1640 [Thermo Fisher Scientific], 10% FBS, 2 mM glutamine, 1% penicillin-streptomycin) supplemented with 100 ng/mL MCSF and cultured for 7 days without medium change. The Nanoluciferase activity was measured at various points throughout the differentiation including the hiPSC stage, the embryoid body stage, the myeloid progenitor stage, and the final macrophage differentiation stage in order to assess promoter silencing throughout the differentiation. At each stage, cells were cultured for 24 h with or without doxycycline before Nanoluciferase activity was assessed according to the manufacturer’s protocol. The experiment was performed in technical triplicate, and Nanoluciferase activity was normalized to protein concentration.
Statistical analysis
Student’s unpaired two-tailed t tests were performed with GraphPad Prism software (GraphPad Software, La Jolla, CA, USA). Data are presented as means ± SEM. Two-sided p values, adjusted for multiple comparison (Tukey or Dunnett) whenever appropriate, were used in analyses and p values < 0.05 were considered to be statistically significant: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
Acknowledgments
This work was supported by the Intramural Research program of the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, USA. We wish to thank Dr. Wen Xue of UMass Medical School and Dr. Raffaella Sordella of Cold Spring Harbor Laboratory for providing various sgRNA and Cas9 constructs and advice on the CRSPR-Cas9 system.
Author contributions
S.B.P. performed most of the experiments, prepared figures, and edited the manuscript. T.U., S.T., C.D.M., and M.L. assisted experiments and cell culturing. Z.H. provided expertise in troubleshooting. M.E. and S.G.H. provided advice. T.J.L. conceived and supervised the project and critically reviewed and edited the manuscript.
Declaration of interests
The authors declare no competing interest.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2022.04.013.
Contributor Information
Seung Bum Park, Email: seungbum.park@nih.gov.
T. Jake Liang, Email: jakel@bdg10.niddk.nih.gov.
Supplemental information
References
- 1.Bolotin A., Quinquis B., Sorokin A., Ehrlich S.D. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology. 2005;151:2551–2561. doi: 10.1099/mic.0.28048-0. [DOI] [PubMed] [Google Scholar]
- 2.Mojica F.J.M., Díez-Villaseñor C., Díez-Villaseñor C., García-Martínez J., García-Martínez J., Soria E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol. 2005;60:174–182. doi: 10.1007/s00239-004-0046-3. [DOI] [PubMed] [Google Scholar]
- 3.Boyaval P., Moineau S., Romero D.A., Horvath P. Against viruses in prokaryotes. Science. 2007;315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 4.Marraffini L.A. CRISPR-Cas immunity in prokaryotes. Nature. 2015;526:55–61. doi: 10.1038/nature15386. [DOI] [PubMed] [Google Scholar]
- 5.Sander J.D., Joung J.K. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 2014;32:347–355. doi: 10.1038/nbt.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Westra E.R., Dowling A.J., Broniewski J.M., van Houte S. Evolution and ecology of CRISPR. Annu. Rev. Ecol. Evol. Syst. 2016;47:307–331. doi: 10.1146/annurev-ecolsys-121415-032428. [DOI] [Google Scholar]
- 7.Cho S.W., Kim S., Kim J.M., Kim J.S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013;31:230–232. doi: 10.1038/nbt.2507. [DOI] [PubMed] [Google Scholar]
- 8.Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M., Cho S.W., Kim S., et al. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fu Y., Foden J.A., Khayter C., Maeder M.L., Reyon D., Joung J.K., Sander J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013;31:822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsai S.Q., Zheng Z., Nguyen N.T., Liebers M., Topkar V.V., Thapar V., Wyvekens N., Khayter C., Iafrate A.J., Le L.P., et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015;33:187–197. doi: 10.1038/nbt.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fu Y., Sander J.D., Reyon D., Cascio V.M., Joung J.K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014;32:279–284. doi: 10.1038/nbt.2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mali P., Aach J., Stranges P.B., Esvelt K.M., Moosburner M., Kosuri S., Yang L., Church G.M. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 2013;31:833–838. doi: 10.1038/nbt.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kleinstiver B.P., Pattanayak V., Prew M.S., Tsai S.Q., Nguyen N.T., Zheng Z., Joung J.K. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529:490–495. doi: 10.1038/nature16526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim D., Kim J., Hur J.K., Been K.W., Yoon S.H., Kim J.S. Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat. Biotechnol. 2016;34:863–868. doi: 10.1038/nbt.3609. [DOI] [PubMed] [Google Scholar]
- 15.Kleinstiver B.P., Tsai S.Q., Prew M.S., Nguyen N.T., Welch M.M., Lopez J.M., McCaw Z.R., Aryee M.J., Joung J.K. Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells. Nat. Biotechnol. 2016;34:869–874. doi: 10.1038/nbt.3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nihongaki Y., Kawano F., Nakajima T., Sato M. Photoactivatable CRISPR-Cas9 for optogenetic genome editing. Nat. Biotechnol. 2015;33:755–760. doi: 10.1038/nbt.3245. [DOI] [PubMed] [Google Scholar]
- 17.Truong D.J.J., Kühner K., Kühn R., Werfel S., Engelhardt S., Wurst W., Ortiz O. Development of an intein-mediated split-Cas9 system for gene therapy. Nucleic Acids Res. 2015;43:6450–6458. doi: 10.1093/nar/gkv601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dow L.E., Fisher J., O’Rourke K.P., Muley A., Kastenhuber E.R., Livshits G., Tschaharganeh D.F., Socci N.D., Lowe S.W. Inducible in vivo genome editing with CRISPR-Cas9. Nat. Biotechnol. 2015;33:390–394. doi: 10.1038/nbt.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oakes B.L., Nadler D.C., Flamholz A., Fellmann C., Staahl B.T., Doudna J.A., Savage D.F. Profiling of engineering hotspots identifies an allosteric CRISPR-Cas9 switch. Nat. Biotechnol. 2016;34:646–651. doi: 10.1038/nbt.3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee J.B., Kim S.H., Lee S.C., Kim H.G., Ahn H.G., Li Z., Yoon K.C. Blue light-induced oxidative stress in human corneal epithelial cells: protective effects of ethanol extracts of various medicinal plant mixtures. Investig. Ophthalmol. Vis. Sci. 2014;55:4119–4127. doi: 10.1167/iovs.13-13441. [DOI] [PubMed] [Google Scholar]
- 21.Zhang J., Chen L., Zhang J., Wang Y. Drug inducible CRISPR/Cas systems. Comput. Struct. Biotechnol. J. 2019;17:1171–1177. doi: 10.1016/j.csbj.2019.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schumann K., Lin S., Boyer E., Simeonov D.R., Subramaniam M., Gate R.E., Haliburton G.E., Ye C.J., Bluestone J.A., Doudna J.A., Marson A. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proc. Natl. Acad. Sci. U S A. 2015;112:10437–10442. doi: 10.1073/pnas.1512503112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hashimoto M., Takemoto T. Electroporation enables the efficient mRNA delivery into the mouse zygotes and facilitates CRISPR/Cas9-based genome editing. Sci. Rep. 2015;5:11315. doi: 10.1038/srep11315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Horbal L., Luzhetskyy A. Dual control system - a novel scaffolding architecture of an inducible regulatory device for the precise regulation of gene expression. Metab. Eng. 2016;37:11–23. doi: 10.1016/j.ymben.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Das T.A., Tenenbaum L., Berkhout B. Tet-on systems for doxycycline-inducible gene expression. Curr. Gene Ther. 2016;16:156–167. doi: 10.2174/1566523216666160524144041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Senturk S., Shirole N.H., Nowak D.G., Corbo V., Pal D., Vaughan A., Tuveson D.A., Trotman L.C., Kinney J.B., Sordella R. Rapid and tunable method to temporally control gene editing based on conditional Cas9 stabilization. Nat. Commun. 2017;8:14370. doi: 10.1038/ncomms14370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loew R., Heinz N., Hampf M., Bujard H., Gossen M. Improved Tet-responsive promoters with minimized background expression. BMC Biotechnol. 2010;10:13–81. doi: 10.1186/1472-6750-10-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Banaszynski L.A., Chen L.C., Maynard-Smith L.A., Ooi A.G.L., Wandless T.J. A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules. Cell. 2006;126:995–1004. doi: 10.1016/j.cell.2006.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sanjana N.E., Shalem O., Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cao J., Wu L., Zhang S.M., Lu M., Cheung W.K.C., Cai W., Gale M., Xu Q., Yan Q. An easy and efficient inducible CRISPR/Cas9 platform with improved specificity for multiple gene targeting. Nucleic Acids Res. 2016;44:e149. doi: 10.1093/nar/gkw660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim S., Kim D., Cho S.W., Kim J.-S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014;24:1012–1019. doi: 10.1101/gr.171322.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vakulskas C.A., Dever D.P., Rettig G.R., Turk R., Jacobi A.M., Collingwood M.A., Bode N.M., McNeill M.S., Yan S., Camarena J., et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat. Med. 2018;24:1216–1224. doi: 10.1038/s41591-018-0137-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu X., Gao D., Wang P., Chen J., Ruan J., Xu J., Xia X. Efficient homology-directed gene editing by CRISPR/Cas9 in human stem and primary cells using tube electroporation. Sci. Rep. 2018;8:11649. doi: 10.1038/s41598-018-30227-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.González F., Zhu Z., Shi Z.D., Lelli K., Verma N., Li Q.V., Huangfu D. An iCRISPR platform for rapid, multiplexable, and inducible genome editing in human pluripotent stem cells. Cell Stem Cell. 2014;15:215–226. doi: 10.1016/j.stem.2014.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cherry S.R., Biniszkiewicz D., van Parijs L., Baltimore D., Jaenisch R. Retroviral expression in embryonic stem cells and hematopoietic stem cells. Mol. Cell. Biol. 2000;20:7419–7426. doi: 10.1128/mcb.20.20.7419-7426.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Herbst F., Ball C.R., Tuorto F., Nowrouzi A., Wang W., Zavidij O., Dieter S.M., Fessler S., Van Der Hoeven F., Kloz U., et al. Extensive methylation of promoter sequences silences lentiviral transgene expression during stem cell differentiation in vivo. Mol. Ther. 2012;20:1014–1021. doi: 10.1038/mt.2012.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chavez A., Scheiman J., Vora S., Pruitt B.W., Tuttle M., P R Iyer E., Lin S., Kiani S., Guzman C.D., Wiegand D.J., et al. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods. 2015;12:326–328. doi: 10.1038/nmeth.3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cho S.W., Kim S., Kim Y., Kweon J., Kim H.S., Bae S., Kim J.S. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014;24:132–141. doi: 10.1101/gr.162339.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim D., Bae S., Park J., Kim E., Kim S., Yu H.R., Hwang J., Kim J. Il, Kim J.S. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat. Methods. 2015;12:237–243. doi: 10.1038/nmeth.3284. [DOI] [PubMed] [Google Scholar]
- 40.Ellis J. Silencing and variegation of gammaretrovirus and lentivirus vectors. Hum. Gene Ther. 2005;16:1241–1246. doi: 10.1089/hum.2005.16.ft-126. [DOI] [PubMed] [Google Scholar]
- 41.Hamaguchi I., Woods N.-B., Panagopoulos I., Andersson E., Mikkola H., Fahlman C., Zufferey R., Carlsson L., Trono D., Karlsson S. Lentivirus vector gene expression during ES cell-derived hematopoietic development in vitro. J. Virol. 2000;74:10778–10784. doi: 10.1128/jvi.74.22.10778-10784.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.MacArthur C.C., Xue H., Van Hoof D., Lieu P.T., Dudas M., Fontes A., Swistowski A., Touboul T., Seerke R., Laurent L.C., et al. Chromatin insulator elements block transgene silencing in engineered human embryonic stem cell lines at a defined chromosome 13 locus. Stem Cells Dev. 2012;21:191–205. doi: 10.1089/scd.2011.0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vieyra D.S., Goodell M.A. Pluripotentiality and conditional transgene regulation in human embryonic stem cells expressing insulated tetracycline-ON transactivator. Stem Cells. 2007;25:2559–2566. doi: 10.1634/stemcells.2007-0248. [DOI] [PubMed] [Google Scholar]
- 44.Behr M., Zhou J., Xu B., Zhang H. In vivo delivery of CRISPR-Cas9 therapeutics: progress and challenges. Acta Pharm. Sin. B. 2021;11:2150–2171. doi: 10.1016/j.apsb.2021.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim D.Y., Lee J.M., Moon S. Bin, Chin H.J., Park S., Lim Y., Kim D., Koo T., Ko J.H., Kim Y.S. Efficient CRISPR editing with a hypercompact Cas12f1 and engineered guide RNAs delivered by adeno-associated virus. Nat. Biotechnol. 2022;40:94–102. doi: 10.1038/s41587-021-01009-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carpentier A., Tesfaye A., Chu V., Nimgaonkar I., Zhang F., Lee S.B., Thorgeirsson S.S., Feinstone S.M., Liang T.J. Engrafted human stem cell-derived hepatocytes establish an infectious HCV murine model. J. Clin. Invest. 2014;124:4953–4964. doi: 10.1172/jci75456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carpentier A., Nimgaonkar I., Chu V., Xia Y., Hu Z., Liang T.J. Hepatic differentiation of human pluripotent stem cells in miniaturized format suitable for high-throughput screen. Stem Cell Res. 2016;16:640–650. doi: 10.1016/j.scr.2016.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alasoo K., Martinez F.O., Hale C., Gordon S., Powrie F., Dougan G., Mukhopadhyay S., Gaffney D.J. Transcriptional profiling of macrophages derived from monocytes and iPS cells identifies a conserved response to LPS and novel alternative transcription. Sci. Rep. 2015;5:12524. doi: 10.1038/srep12524. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.