

Abstract
Target:
Nighttime agitation is a prevalent symptom in persons with Alzheimer’s disease (AD). Effective treatments are absent due to our limited knowledge of its etiology. We hypothesized that restless legs syndrome (RLS), a common neurologic sensorimotor disorder of uncomfortable leg sensations that appear at night and interfere with sleep, might be a cause for nighttime agitation in persons with AD. RLS is infrequently identified in persons with AD because traditional diagnosis is dependent on patients answering complex questions about their symptoms.
Intervention and Outcomes:
With a validated observational tool for RLS diagnosis, the Behavioral Indicators Test-Restless Legs, we aim to diagnose RLS and determine the effect of an FDA-approved drug for RLS, gabapentin enacarbil (GEn), compared to placebo on nighttime agitation, sleep, antipsychotic medications, and the mechanism for these effects.
Mechanism of Action:
We hypothesize that frequency of RLS behaviors will mediate the relationship between GEn and nighttime agitation.
Design:
This study is an 8-week, double-blind, placebo-controlled, randomized pilot clinical trial, followed by an 8-week open-label trial, that is being conducted in long-term care (LTC) settings and private homes.
Discussion:
The results of this study may shift, personalize, and improve standards of care for treatment of nighttime agitation; reduce aggression and other nighttime agitation behaviors; and improve sleep.
Trial registration:
Clinical Trials.gov Identifier: NCT03082755 (Date of registration 6 March 2017)
Keywords: nighttime agitation, restless legs syndrome, Alzheimer’s disease, gabapentin enacarbil, sundowning, antipsychotics
BACKGROUND
Health care costs in America for Alzheimer’s disease (AD) were 203 billion dollars, and up to 30% of these costs are attributed to the management of neuropsychiatric symptoms (“2014 Alzheimer’s disease facts and figures,” 2014). One prevalent neuropsychiatric symptom is agitation late in the day, also called “sundowning”, defined as emotional distress, loud vocalizations, excessive psychomotor activity, physically aggressive behaviors, disruptive irritability, and disinhibition in the afternoon and/or evening hours (Porsteinsson et al., 2014). Nighttime agitation is often associated with sleep disturbances.
Developing effective and sustainable treatments for nighttime agitation in persons with AD is difficult because knowledge of its etiology is limited. Agitation behaviors may be in response to unmet needs that cannot be communicated any other way. This study focuses on one unmet need, a sleep disorder called restless legs syndrome (RLS) that is infrequently diagnosed or treated in older adults with AD (Richards et al., 2010). Patients with RLS report an urge to move associated with uncomfortable leg sensations that are engendered or worsened by rest or inactivity, relieved by moving the legs, and occur largely in the evening or first part of the night (American Academy of Sleep Medicine., 2014). A relationship between RLS and nighttime agitation is expected, given the nature and etiology of the symptoms (Figure 1), and also supported by our preliminary work. (Rose et al., 2011).
Figure 1.
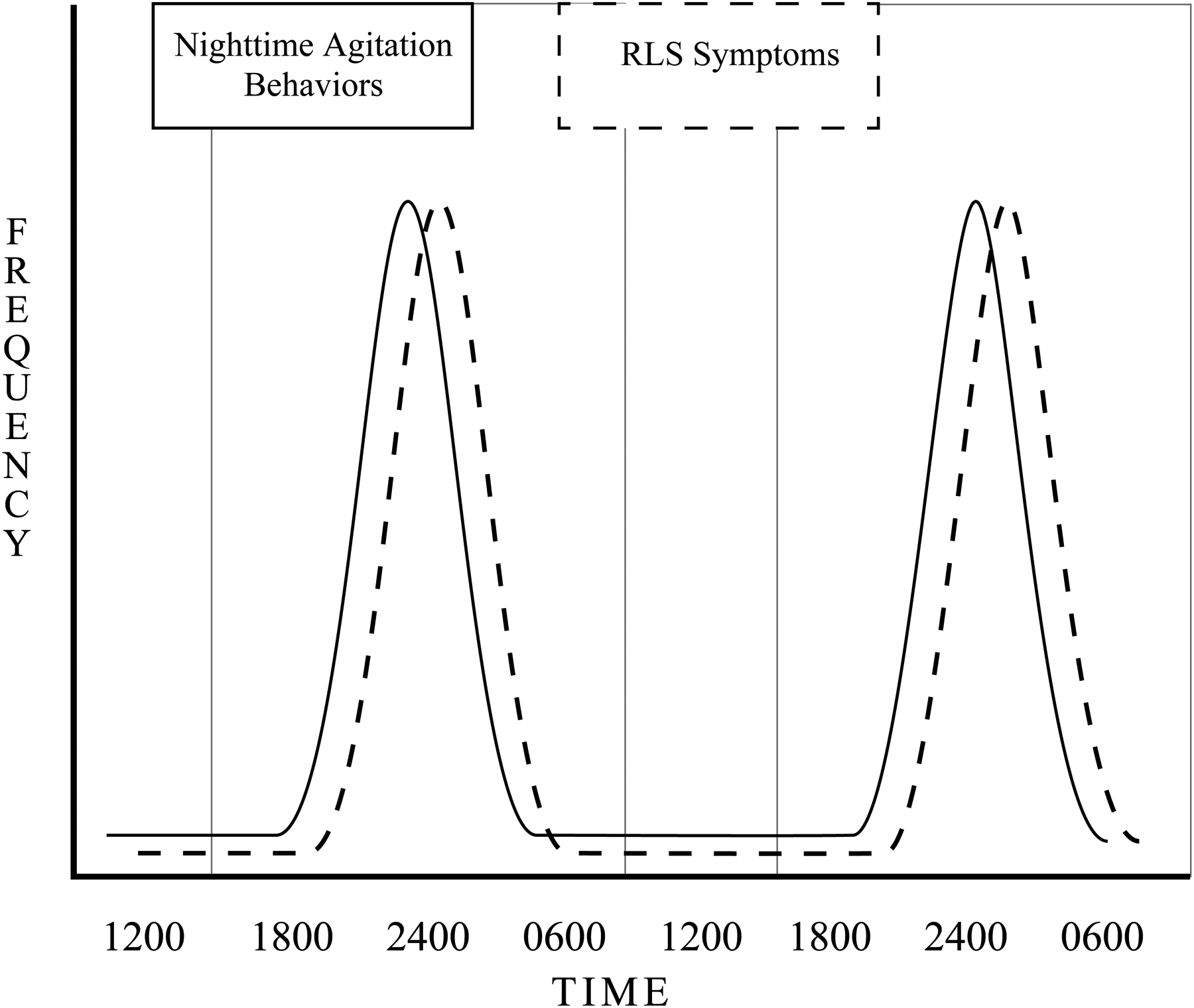
Circadian Rhythms of Nighttime Agitation and Restless Legs Syndrome
Non-pharmacological interventions for treating behavioral symptoms are preferred over medications in older adults because of the potential for adverse medication side effects. Unfortunately, there is insufficient evidence of the efficacy of non-pharmacological interventions for treating RLS. In a survey of 8 geriatric and sleep clinicians, 5 (63%) recommended the alpha 2 delta ligands (α2δ) gabapentin or gabapentin enacarbil (GEn, Horizant®) for treating RLS in frail older adults because of a favorable safety profile and efficacy. The mechanism for the effect of α2δ ligands on RLS is thought to be their effect on pre-synaptic glutamatergic transmission (Gonzalez-Latapi & Malkani, 2019). A growing body of literature links brain iron deficiency, altered glutamatergic transmission, and RLS. While gabapentin is used off-label for treating RLS, it is not approved by the Food and Drug Administration (FDA) for this purpose, thus the study investigative team chose GEn as the intervention for this clinical trial.
Purpose, Aims and Hypotheses.
The primary study objective [Aim 1] is to determine the effect of an FDA-approved drug for RLS, gabapentin enacarbil (GEn), compared to placebo on nighttime agitation. Secondary objectives are to: [Aim 2] describe the safety of GEn compared to placebo in this population; [Aim 3] estimate the effect size of GEn compared to placebo on nighttime sleep, RLS behaviors, and post-trial antipsychotic use; and [Aim 4] explore whether frequency of RLS behaviors mediates the relationship between GEn and nighttime agitation.
We hypothesize that compared to the placebo control group, the GEn treatment group will have fewer nighttime agitation behaviors, better nighttime sleep, and fewer RLS behaviors and that frequency of RLS behaviors will mediate the effect of GEn on nighttime agitation behaviors.
METHODS
Intervention.
GEn is an actively transported prodrug of gabapentin that provides dose-proportional, sustained exposure to gabapentin (Lal et al., 2009). It is absorbed in the gastrointestinal tract by passive diffusion and active transport. GEn is converted to gabapentin after absorption by nonspecific carboxylesterases which are primarily present in enterocytes. The extended-release formulation allows for sustained delivery of gabapentin to the systemic circulation, thus reducing dosing frequency and producing predictable exposures. The sustained duration of absorption was estimated at 7 hours, with interindividual variability of 22%, and the elimination half-life is approximately 6 hours, similar to the half-life after dosing gabapentin (Lal et al., 2013). GEn is primarily excreted by the kidneys as unchanged drug (Cundy, Annamalai et al., 2004; Cundy, Branch et al., 2004; Cundy et al., 2008).
GEn was approved by the FDA in April 2011 for the treatment of moderate-to-severe primary RLS in adults (NDA 022399-FDA, 2012). Clinical trials have demonstrated a safety profile comparable to traditional gabapentin, improved pharmacokinetics, and significant differences when compared to placebo on RLS symptoms, subjective sleep quality and wake at night (Yaltho & Ondo, 2010). For example, a 2012 review and meta-analysis graded the evidence as high for the effectiveness of GEn for moderate to severe RLS (Aurora et al., 2012). Arbor© Pharmaceuticals LLC markets GEn (Horizant®), and provides GEn 300mg Extended Release tablets free of charge for participants during the research.
Design.
The study is a pilot, 8-week, double-blind, placebo-controlled randomized clinical trial of GEn versus placebo, and an 8-week open-label post-trial follow-up to assess continued RLS treatment and antipsychotic use. The trial control condition is matching placebo because there currently are no FDA-approved medications for nighttime agitation and few nonpharmacological interventions for nighttime agitation have been empirically tested. As described in the Clinical Antipsychotic Trials of Intervention Effectiveness – AD, the proposed trial design encourages prescribing that reflects clinical practice while maintaining the randomized and double-blind treatment assignment (Schneider et al., 2006). We anticipate a sample size of 136; 68 per treatment arm, drawn from approximately 30 sites.
Randomization.
A study statistician, not involved in the study data analysis, developed the randomization schema, by site, by A or B group allocation. Randomization was performed as block randomization with a 1:1 allocation. The statistician sent the schema to the study research pharmacist, who designated whether the A or B group was the GEn or the placebo condition. After the study medical team confirms that all eligibility criteria have been met, the study physician or her designee (MD, nurse practitioner, or physician’s assistant) writes the order, dosage, and time of administration for the study drug: (1) in each participant’s medical record for those residing in LTC facilities or (2) on a standard prescription form. Trained study staff assign participants the next sequential identification number for the LTC facility or community-based setting where they reside, and notify the research pharmacist of the participant’s identification number. The pharmacist consults the randomization schema to determine whether the participant gets GEn or placebo, and dispenses either GEn or placebo based on the schema.
Placebo.
Participants randomized to the control condition receive tablets identical to GEn, but inactive. The tablets match GEn in texture, color, size, thickness, markings, smell, and packaging.
Blinding.
In this double-blind study, the condition is unknown to the participants, the investigators and the entire study team, and those who administer the medication. The research pharmacist, who fills the prescriptions, is unblinded to condition. It is possible that the study RN, the LTC staff, or the home caregivers may notice improvement or side effects in the GEn group, and suspect treatment assignment. The raters of the primary outcome are not involved in assessments for improvement or adverse events and results will not be communicated to them. This will minimize the possibility that rating of the primary outcome will be biased. Unblinding will occur when data collection and entry is complete.
Sample, Setting, and Power Analysis.
Project staff collect data from eligible persons living in LTC facilities and independent living settings (i.e., senior independent living apartments and private homes in the community) over approximately 4 years. Based on our prior research (Richards et al., 2011) and current recruitment efforts, we expect that 450 referrals will be required, about 55% of those referred will consent (n=247), and about 57% of those consented will qualify for randomization. Attrition after randomization due to death or hospitalization and other reasons is expected to be 15% (Richards et al., 2011). Enlisting approximately 25 LTC facilities and 5 independent living settings should provide the required sample of 136, but if needed we will recruit additional facilities and persons living independently.
Power is based on the single primary outcome, nighttime agitation, as measured by the Agitation Behaviors Index (Rose et al., 2011) observed over 8 weeks, and a type I error rate of 0.05. Assuming a mean Agitation Behaviors Index of 1.5 (SD 0.90) in both groups (Rose et al., 2011) at baseline, a within-subject variance of 1.6 and an autocorrelation equal to 0.81, and that the control and treatment groups demonstrate mean values of 1.35 (10% placebo effect) and 1.0, at 8 weeks, respectively; 68 participants per group achieve over 90% power to detect a minimum detectable difference of 0.25 for changes in nighttime agitation over time between the two treatment arms (interaction term in mixed model group × time).
Inclusion and Exclusion Criteria and Measures.
The study inclusion and exclusion criteria and justification are in Table 1. Overall, the inclusion/exclusion criteria, the study design, and the data analysis methods reflect our efforts to reduce measurement “noise” (random fluctuations that hinder detection of true differences, if they exist). The criteria for inclusion/exclusion were also selected to maximize our chances of recruiting and retaining the sample and reduce the potential for risk. For example, inclusion requires a level of observed agitation to avoid a “floor effect”, but avoids including only those who are highly agitated to allow a sufficient recruitment pool. Observations for agitation occur on more than one night and at different times of the night to increase our chances of detecting nighttime agitation, given the circadian and night-to-night variability that often occurs. Persons with dementia frequently cannot reliably report their symptoms, thus in this trial RLS is diagnosed using a validated tool designed for this population. An example of an exclusion criteria to reduce the potential for participant risk is excluding those who consume alcohol because consuming GEn and alcohol together may increase sedation. Fidelity of all measures, including those specifically used for inclusion/exclusion, is addressed by extensive training and testing of study staff, ongoing monitoring of data by the investigators, and assessments of interrater reliability during the study.
Table 1.
Inclusion and Exclusion Criteria with Rationale
| Inclusion Criteria | Rationale |
|---|---|
| Aged >=55 years | Increase recruitment pool by including those with early onset dementia (aged 55–65) |
| Clinical Dementia Rating (CDR) score of 0.5–3 indicating very mild to severe dementia | Include those with very mild to severe dementia to maximize the sample pool |
| Physician diagnosis Alzheimer’s dementia | Avoid heterogeneity in pathology that may affect manifestation of agitation |
| Nighttime agitation, defined as Cohen-Mansfield Agitation Inventory, Direct Observation total score >=35 | >=35 behaviors is sufficient to avoid a “floor” effect, show differences (if they exist), and maximize the sample pool |
| Opinion of the participant’s physician that medication for agitation is appropriate | Increase generalizability of findings |
| RLS diagnosis by the study medical team, using the Behavioral Indicators Test-Restless Legs | Study design requires that all participants have RLS |
| Medically stable, defined as unchanged medications within 14 days and the absence of fever or other signs and symptoms of acute illness or delirium (e.g. urinary tract infection, pneumonia) | Illness may cause agitation or interfere with the study protocol |
| Able to swallow uncrushed medication | GEn must be swallowed whole |
| Ambulatory, with and without assistance | Inclusion of those who can wander or pace |
| If currently being treated for RLS, may be included if still having RLS symptoms/signs and confirmed as appropriate for inclusion by the medical team | Individuals with RLS may require more than one medication |
| Exclusion Criteria | Rationale |
| Received >=50 morphine milligram equivalents per day during the past 14 days | Morphine and GEn taken together have a higher incidence of sedation and dizziness than either drug alone |
| Currently being treated for RLS with gabapentin or GEn | Potential for overdose |
| Diagnosis of Parkinson’s disease or any other disorder causing tremor | Extrapyramidal symptoms may confound RLS diagnosis and actigraphy |
| Receiving gabapentin | Potential for overdose |
| Severe psychosis | Schizophrenia or other mental illness may cause psychosis; treatment of RLS would not affect this |
| Alcohol consumption | Combining alcohol and GEn may increase sedation and other adverse events |
| Treatment with GEn is contraindicated, such as when a potential participant is receiving multiple antiepileptic drugs, in the opinion of the medical team | GEn may be medically contraindicated |
| Failure of past treatment with gabapentin or GEn | Avoid including those resistant to gabapentin or GEn |
| Compromised renal function as indicated by creatinine clearance <15 or on hemodialysis | GEn is excreted by the kidneys |
| Current participation in a clinical trial or in any study that may affect study outcomes | Avoid confounding caused by other protocols |
| Determined to be at risk for suicide by the study RN, participant’s physician, or medical team | GEn may increase risk for suicide |
| Any condition, that in the opinion of the medical team or the participant’s physician, makes it medically inappropriate for the patient to enroll in the trial | Avoid any condition deemed to increase risk for participants |
| Persons living independently in the community without a live-in caregiver (family or hired) | Caregiver needed to administer study drug and monitor for side effects |
GEn, gabapentin enacarbil; RLS, Restless legs syndrome.
Age, appropriateness of medication for agitation, ability to swallow medication, and other inclusion/exclusion data are collected by the study RN from the medical records, participants’ physicians, caregivers, LTC staff, and assessment. AD diagnosis is determined by either physician diagnosis recorded in the medical record or made by the study physician co-investigator (Fry) or her designee (nurse practitioner or physician’s assistant) (McKhann et al., 2011). Descriptions of the inclusion and exclusion screening measures are in Table 2.
Table 2.
Study Variables and Description of Measures
| Variable | Measure | Level | Description |
|---|---|---|---|
| Restless Legs Syndrome Diagnosis | Behavioral Indicators Test – Restless Legs (BIT-RL) | Screening | The BIT-RL was developed to diagnosis RLS in persons with the dementia stage of AD, and validated in our earlier work. In this study the medical team makes the diagnosis of RLS or no RLS after reviewing the BIT-RL. Only those diagnosed with RLS are included. The BIT-RL (Richards et al., 2015) (range 0–16) consists of two parts: 1) Behavioral Indicators (composite score range 0–10) – 20 minute direct observation by trained RA for RLS behaviors, such as kicking or rubbing legs (8 items), and 2) Clinical Indicators (range 0–6) - medical history or family informant interview (3 items), interviews with caregivers (2 items), and an interview with the participant (1 item). In the diagnostic accuracy study, the BIT-RL had a sensitivity of 78% and specificity of 79%, with 77% correctly classified. Interobserver reliability was >0.90 and intraobserver reliability was >=0 .95 (Richards et al., 2015). The RAs are required to accurately identify RLS behaviors at a high level of agreement (r > .90) with experts and each other prior to data collection and semiannually. |
| Frequency of RLS Behaviors | BIT-RL | Primary Outcomes - Aims 3 and 4 | Frequency of RLS behaviors (range 0–80) is the raw score of the Behavioral Indicators section of the BIT-RL. |
| Compromised Renal Function | Creatinine | Screening | Participants with compromised renal function as indicated by Creatinine Clearance <15 or on hemodialysis are excluded (U.S. Food and Drug Administration, 2012). Project staff collect fasting whole blood samples and LabCorp analyzes the samples. |
| Cognitive Ability | Clinical Dementia Rating (CDR) | Screening | Those with CDR 0.5, 1, 2, and 3 are included. The CDR is a 5-point scale (0, 0.5, 1, 2, 3) for staging severity of dementia. A CDR “0” denotes no cognitive impairment, and “3” severe cognitive impairment. Scale has strong reliability with raters achieving 87% agreement and kappa values 0.66–0.83 (Schafer et al., 2004; Fillenbaum et al., 1996), and criterion validity with other cognitive tests (Morris, 1997). A trained study RA collects the CDR data (Morris, 1993). |
| Nighttime Agitation | Cohen Mansfield Agitation Inventory (CMAI) Caregiver Version | Screening and Secondary Outcome - Aim 1 | Primary caregivers on the evening/night shift use the 14-item CMAI to rate frequency of agitation behaviors, with ratings of 1 = never and 7 = several times per hour. Internal consistency reliabilities ranged from .86 –.91. Interrater reliability values ranged from .88–.92 (Finkel et al., 1992). Validity has been shown in a number of studies (Cohen-Mansfield & Billig, 1986; Cohen-Mansfield et al., 1989). Scores range from 14 to 98, with higher scores indicating more frequent agitation behaviors. |
| CMAI Direct Observation | Screening, Primary Study Outcome - Aims 1 and 4 | The CMAI was modified for direct observation (Chrisman et al., 1991), and later refined for direct nighttime observation by Richards (Rose, et al., 2011). RAs continuously directly observe and record agitation behaviors every 5 minutes. Observations occur on two nights at baseline, 2 weeks, and 8 weeks. Night 1 is 5–10 pm and Night 2 is 10 pm-7am. The RAs note whether the participant is behaviorally awake or asleep, with sleep defined as a quiet state with eyes closed. Nighttime agitation behaviors are scored during wake. After the participant has gone to bed, the RA observes him/her via an infrared camera placed in the bedroom and a remote monitor placed in another location, such as the hallway. The RAs set up and adjust the camera for optimal visualization prior to the participant’s bedtime. RAs are required to identify behaviors at a high level of agreement (r > .90) with the investigator experts prior to and during data collection. If agreement is not acceptable, RAs undergo additional training. The primary outcome is the Agitation Behaviors Index (ABI), defined as the total number of behaviors divided by the total hours of observation. The ABI can range from 0–348, with higher scores indicating more nighttime agitation. | |
| Modified Alzheimer’s Disease Cooperative Study-Clinical Global Impression of Change (mADCS-CGIC) | Secondary Outcome - Aim 1 | The mADCS-CGIC measures clinically meaningful change in the patient’s condition relative to baseline on a 7-point Likert scale (markedly worse to markedly improved) (Schneider et al., 1997; Teri et al., 2000). The scale was modified to assess items specific to agitation, producing a global rating of change in agitation (Porsteinsson et al., 2014). Interrater reliability tests showed strong agreement, with 83% of rater pairs showing either zero to one-point discrepancies (Schneider et al., 1997). Concurrent validity measures showed statistically significant correlations. This scale is completed by the study nurse based on physical examination and interviews with LTC facility caregivers and with the participant (if able). | |
| Safety | Adverse Event Checklist | Primary Outcome - Aim 2 | The adverse event checklist consists of adverse events reported in >1% of participants in previous studies on GEn, and an “other” write-in category, such as falls. Since participants may not be able to verbally report adverse events, the study nurse uses physical examination and systems review, visual observation by study staff, physiological assessment, validated scoring tools, and verbal reports from caregivers, study staff, family members or participants, if able. The main measure is frequency of any adverse event. |
| Mini-Mental State Examination (MMSE) | Secondary Outcome - Aim 2 | The MMSE (range 0–30) is a 30-item cognitive screen, with higher scores indicating better cognition (Folstein et al., 1975). Test-retest reliability is .83 (Folstein et al., 1975), and criterion validity is .83 with the Short Portable Mental Status Questionnaire, and .88 with the Cognitive Capacity Screening Examination (Anthony et al., 1982; Foreman, 1987). In this study, we consider a 2 point decrease in MMSE as an adverse event, and trigger for potential dosage adjustment. | |
| Global Rating of Fall Risk (GLORF) | Secondary Outcome - Aim 2 | The GLORF is a single question “how do you judge the risk that Mr. or Mrs. X will fall within 6 months; high or low?” asked of a nurse, aide or caregiver with personal knowledge of the participant. It had a positive likelihood ratio of 2.8 of a future fall in nursing home residents (Nordin et al., 2008). | |
| Physical Mobility Scale (PMS) | Secondary Outcome - Aim 2 | The Physical Mobility Scale (PMS) (range 0–45) is an 8-item performance-based scale to assess mobility (Nitz et al., 2006). Concurrent validity with the Clinical Outcomes Variable Scale was >.7 on each item, and test-retest reliability among raters was >0.9. Pike & Landers (2010) consider a decrease of 4 points “worsened” mobility; thus 4 points is the cut-point for adverse event reporting, and potential dosage adjustment. | |
| Sleep Disturbance | Micro-Mini Motionlogger® Actigraph | Primary Outcome - Aim 3 | The actigraph (Ambulatory Monitoring Inc., Ardsley, NY) is a wrist watch-sized accelerometer, worn by participants who elect to wear the device and do not remove it, on the non-dominant wrist. Sensitivity (actigraphy = sleep when polysomnography = sleep) was 0.97 (Marino et al., 2013). In our previous studies, we used a plastic hospital ID band to hold the actigraph in place that is comfortable to wear, and we use this band in this project. Nighttime total sleep time is the main sleep outcome. Other variables are nighttime wake after sleep onset, sleep efficiency, sleep latency, and awakenings. |
| CMAI Direct Observation | Primary Outcome - Aim 3 | Because specificity of actigraphy for wake is low, direct observation will be the primary wake measure. When the CMAI data are collected, the RA will continuously observe the participant in the evening and night and note every 5 minutes whether the participant is behaviorally awake or asleep. Number and percent of observations the RAs mark as wake will be the main observational sleep outcome. Agreement among raters was .85–.97, and kappa coefficients were .65–.93 in 24 nursing home residents (Bliwise et al., 1990). | |
| Post-Trial Antipsychotic Use | Medication Administration Record or Medication Containers | Primary Outcome - Aim 3 | The study RN collects dosages and times of all medications received by participants, classifies them, and enters the data into REDCap. Changes in antipsychotic therapy over time are assessed by calculating and comparing the antipsychotic dose equivalents for the 8-weeks prior to baseline, for the 8-weeks clinical trial, and for the 8-weeks open label trial. The antipsychotic dose is calculated in REDCap. |
Intervention Dosage, Duration, Titration, Monitoring, and Fidelity.
Prescribing of the study medication, dosage, and monitoring of response is performed by one or more members of the study medical team using an online meeting, desktop sharing, and videoconferencing software package. The medical team consists of the co-investigators [geriatrician (MD); a sleep medicine specialist (MD); a sleep medicine specialist (PhD)]; the study head nurse (BSN, RN); and the study principal investigator (PhD, RN).
The LTC facility staff or home caregivers administers GEn/placebo. The starting dose is one tablet of 300 mg GEn or placebo with food by mouth nightly at 5 PM. The medical team reviews the participant’s response after he/she has been on the study for 2 weeks. They then determine if the patient should continue, or whether or not the dose should remain at 1 tablet or increase to 2 tablets. The dosage increase is based on the creatinine clearance (CC) using GEn prescribing guidelines: CC >= 60 for 600 mg daily and CC 30–59 for 300 mg daily (Administration, 2012) and participant response to the study drug. An example of how the medical team evaluates participant response is review of falls data. All falls during the trial, and their proximity to receiving the study drug and other medications, and their presumed causes are recorded, and compared with any prior history of falls. The falls comparisons, other clinical information such as reports of increased daytime napping, along with their clinical judgment guide the team on whether to increase dosage or discontinue GEn/placebo.
Duration for clinical trials on GEn generally range from 2–64 weeks, with most about 12 weeks (Garcia-Borreguero et al., 2012; VanMeter et al., 2012). A therapeutic response with reduced discomfort and improved sleep occurs in as little as two weeks. An 8-week trial will provide more than adequate duration to achieve a therapeutic response and for assessment of safety outcomes.
At the end of week 9, the study drug is discontinued for all participants. GEn administration guidelines for stopping the drug specify that there is no need to gradually taper the medication if dosage is <=600 mg, but our team takes a more conservative approach at the end of the 8-week clinical trial period by reducing the dose to 300 mg for 1 week (week 9) prior to discontinuing.
The study RN closely monitors for known GEn side effects (i.e., increased dizziness, somnolence/sedation, weight change ± 5%), and other potential effects such as decreased mobility, defined as a > 4 point change on the Pike and Landers (2010) Physical Mobility Scale. In a double-blind, placebo-controlled study in 325 subjects, somnolence and dizziness were the most common adverse events (Lee et al., 2011). Somnolence occurred in 21.7% at the 600 mg dose (2.1% placebo), with a median (range) duration of 35 (1–85) days, which led to discontinuation in 3 subjects. Dizziness occurred in 10.4% at the 600 mg dose (5.2% placebo), with a median duration of 5 days, which led to discontinuation in 2 subjects.
All adverse events and serious adverse events are reported by the study RN and reviewed by the medical team for expectedness, relationship to participation in the study, and the potential that the research may place participants at risk for harm. For a given trial participant, the study intervention may be discontinued by the investigators and protections initiated if harms develop that may reasonably be linked to the study drug: 1) suicidal ideation or behavior - immediately report to participant’s physician, caregiver, nursing home staff to institute suicide precautions and discontinue study drug (no taper required); 2) drug reaction with eosinophilia and systemic symptoms syndrome - immediate evaluation by participant’s physician and discontinue study drug if alternative etiology is not identified (no taper required); and 3) somnolence/sedation - reduce dosage or discontinue study drug (no taper required).
The study RN oversees the study intervention. Because changes in medications during the study may confound results, she requests that providers not change current medications and nonpharmacological interventions during the 8-week clinical trial, and she monitors and records any medication or dosage changes that occur. The study RN also monitors administration of the study drug. She visits the facility and participants living at home 3 times per week for weeks 1 and 2, and weekly for weeks 3 through 8. During these visits she checks the medication administration records and the individual dose packages for any missed doses, documents any missed doses, and resolves any reasons doses were missed.
Open-Label Trial.
When participants complete the clinical trial, the medical team reviews their outcome data, and develops tailored recommendations for treating RLS. Recommendations typically include tapering and discontinuing any medications known to exacerbate RLS, such as selective serotonin reuptake inhibitors; oral iron for those with iron deficiency; and continued treatment with GEn. The study RN shares the treatment plans with each participant’s provider. At the end of the 8-week open-label trial the study RN records adverse events, medications (to determine if providers followed the treatment recommendations), and specifically notes any new or continued use of antipsychotics for management of nighttime agitation.
Measures.
In this study, we will determine if identifying RLS, using a new dementia-specific diagnostic tool that we validated, and treating RLS using an FDA approved medication, reduces or eliminates nighttime agitation, improves sleep, reduces RLS behaviors, and ultimately reduces antipsychotic medications. The study outcome measures were chosen because 1) we expect that participants will show impairment on the measures at baseline; 2) they are less likely than other measures we considered to have floor or ceiling effects; 3) they have been shown to improve after treatment; and 4) when possible, they provide cross verification of the findings because data are collected from multiple sources.
Various data and measures are collected to screen participants for inclusion and describe participants. The study RAs and RN collect a number of descriptive measures, such as age, sex, race, ethnicity; setting; medical history; and mobility requirements. Because RLS is often associated with iron deficiency, study staff collect blood for a complete blood count with differential and an iron panel at baseline.
The study staff work closely with the LTC facility staff and primary home caregivers to train them on the study protocol, specifically on the study measures, using study-specific written materials and one-on-one interactions. Originally, the investigators had planned an orientation session on each shift for the LTC staff, but feedback from staff and administrators was that the orientation session was not needed because staff involvement in data collection is minimal. Table 2 describes the study variables and the measures, and Table 3 lists the assessments, and other study activities, and when the study staff collect the various data.
Table 3.
Study Schema
| Study Steps | Baseline | Randomized, 8-Week Double-Blind, Placebo-Controlled Clinical Trial | 8-Week Open Label | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| WEEK | ||||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 16 | ||
| Referral (facility or medical staff, family) | X | |||||||||
| Informed consent, HIPAA release, MacCAT-CR | X | |||||||||
| Demographic data | X | |||||||||
| Medical history | X | |||||||||
| Laboratory data: Creatinine, CBC w/Diff, Total Iron Binding Capacity, Iron saturation, Ferritin | X | |||||||||
| Clinical Dementia Rating | X | |||||||||
| Mini-Mental State Examination | X | X | X | |||||||
| Behavioral Indicators Test - Restless Legs | X | X | X | |||||||
| Actigraphy | X | X | X | |||||||
| Sleep history | X | X | X | |||||||
| Cohen-Mansfield Agitation Inventory - Caregiver | X | X | X | |||||||
| Cohen-Mansfield Agitation Inventory - Observation | X | X | X | |||||||
| mADCS-CGIC | X | X | X | |||||||
| Global Rating of Fall Risk | X | X | X | |||||||
| Non-pharmacological intervention assessment | X | X | X | |||||||
| Physical Mobility Scale | X | X | X | X | ||||||
| Medication inventory | X | X | X | |||||||
| Study RN: Physical exam, Medication Administration Record review, adverse event assessment | X | x3 | x3 | x1 | x1 | x1 | x1 | x1 | x1 | X |
| Medical team: Restless Legs Syndrome diagnosis, randomization approval, order study drug | X | |||||||||
| Nursing Home Staff or Family caregiver: Administer study drug (Daily ~5pm) | X | X | X | X | X | X | X | X | ||
| Medical team review: Study drug titration, adverse events | X | |||||||||
| Medical team review: Adverse events and Open Label Plan | X | |||||||||
| Participant activity session, certificate, thank you letters | X | |||||||||
MacCat-CR, MacArthur Competence Assessment Tool for Clinical Research; CBC w/Diff, Complete Blood Count with Differential; mADCS-CGIC, Modified Alzheimer’s Disease Cooperative Study-Clinical Global Impression of Change.
Procedures.
Recruitment and Retention.
Project staff recruit eligible persons living in LTC facilities and independent living settings. One of the study co-investigators, a geriatrician and medical director of several long-term care settings, often facilitates the recruitment process by introducing the project team to colleagues and explaining the research to local and corporate LTC facility administrators. Project staff recruit participants living independently by distributing recruitment materials or speaking on topics of interest at non-profit senior organizations, senior-living complexes, and adult daycare programs. Recruitment of minorities is a high priority. Project staff identify LTC facilities, independent living communities, and physician practices with high percentages of minorities and encourage them to support the research. Study staff contact caregivers of potential participants residing in independent living settings who express interest, and nursing or medical staff at the LTC facilities identify potential participants. Strategies to promote retention of study participants include ongoing open communication with participants and their families, and individualized attention for participants (and the home caregivers for those residing in independent living settings) during the study visits.
Informed Consent.
Prior to beginning the research, ethical approval was obtained from the Institutional Review Board at The University of Texas at Austin. Study staff explain the purpose, risks, potential benefits, requirements, and alternatives to potential participants and their legally authorized representatives. They explain all study procedures including administration of the GEn/placebo, cognitive testing, drawing blood, and measuring nighttime agitation and sleep. Also, potential study risks, such as dizziness, are described. Participants, their relatives, and authorized representatives are given opportunity to inquire about details of the study. Project staff administer the MacArthur Competence Assessment Tool for Clinical Research (MacCAT-CR) (Appelbaum & Grisso, 2001) to potential participants and their legally authorized representatives to assess their competency to give consent. If the authorized representative and the potential participant are unable to give consent, they are thanked for their time and no further data is collected. After all explanations are given and all questions answered, the study staff obtain written informed consent from the patient and/or the authorized representative.
Compensation.
Participants are not compensated monetarily for the research. However, those who complete the trial receive a 1-hour social activity session provided by study staff, the resident’s favorite refreshments, and a framed certificate. The caregiver of participants living in the community receive compensation in appreciation for his/her time. The caregiver receives up to four payments totaling $300: $50 after baseline, $75 after 2-week, $125 after 8-week, and $50 after the 16-week assessments.
Modifications to the Study Protocol.
The investigators made several modifications to the study protocol to improve recruitment, reach sample size goals, and maximize participant safety. All modifications are initiated by the study investigators, approved by the Institutional Review Board, and reviewed by the Data Safety Monitoring Board. Table 4 describes each modification and rationale.
Table 4.
Protocol Modifications
| Modification | Rationale |
|---|---|
| Add criterion excluding persons living independently in the community without a live-in caregiver (family or hired). | Persons living independently in the community may not have a live-in caregiver (family or hired) to manage study medication adherence and monitor for potential study medication side effects. |
| Revise the criteria for opioids to exclude those who received > 50 morphine milligram equivalents per day (MME/d) in the 14 days prior to the randomization decision. | The DSMB requested further rationalization and evidence regarding the safety and the decision to enroll participants on opioids. Based on a review of the literature, the medical team proposed excluding those receiving > 50 MME/d in the 14 days prior to the randomization decision. |
| Specify that the exclusion criteria for opioids applies to long-acting not short-acting opioid formulations. | Long-acting opioid formulations may have a synergistic effect with the study drug, GEn, while short-acting opioids do not. Short-acting opioids are commonly prescribed with gabapentin in practice. |
| Change inclusion/exclusion criteria for the CMAI from >=40 to >= 35. | The investigators lowered the CMAI inclusion criteria to increase the pool of potential participants. |
| Change the CDR score to include participants who score 0.5 (Very Mild) to 3 (Severe). | The investigators expanded the CDR inclusion criteria to increase the pool of potential participants. Some persons with mild dementia have severe nighttime agitation and we want to include these individuals in the study. |
| Delete sleep disturbance as an inclusion criterion. | The requirement of <= 6 hours slept by actigraphy excluded participants who otherwise met all inclusion criteria. |
| Expand recruiting sites to include community-based settings (e.g., senior independent living, private homes) in addition to long-term care facilities. | The number of patients who can swallow uncrushed medications in long-term care facilities was lower than we projected, and reduced the pool of potential participants. The study drug, gabapentin enacarbil, must be swallowed whole. |
CDR: Clinical Dementia Rating; CMAI: Cohen Mansfield Agitation Index; DSMB: Data Safety Monitoring Board; GEn: gabapentin enacarbil; MME/d: Morphine milligram equivalents per day; RLS: Restless legs syndrome.
Statistical Analysis.
A 25% improvement in nighttime agitation, the primary study outcome, would represent a clinically significant benefit for patients (Cohen-Mansfield et al., 2007). Estimates are based on computations from 100 simulations for an intent-to-treat mixed effects model of repeated measures over time (2 observations at each of the three time points: baseline, 2 weeks, and 8 weeks). Other outcomes (safety, nighttime sleep, RLS behaviors, and antipsychotic medications) will be considered as exploratory pilot data to inform a larger, more definitive trial. Details of all study outcomes, including variable type and data collection timepoints, are provided in Tables 2 and 3. The comprehensive statistical analysis used for the primary outcome will rely on mixed effects modeling. For specific details of all statistical analyses, please refer to Table 5. Analyses will be performed using SAS V9.4 (SAS Institute Inc., Cary, NC).
Table 5.
Methods of Analysis
| Aim | Analysis level | Outcome measure | Methods of analysis |
|---|---|---|---|
| Not applicable | Preliminary | All outcome, independent, and control variables | Descriptive statistics and analysis of distributional properties in the overall sample at baseline |
| Preliminary | All outcomes | The mechanism of missing data will be evaluated prior to implementing methodology to minimize bias from missing data. Baseline characteristics will be compared among patients with and without complete follow-up data using two-sample t-tests or nonparametric Wilcoxon statistics, Fisher’s exact tests as appropriate. | |
| Preliminary | All outcomes | Comparison of baseline characteristics and routine medication use between the treatment groups using two-sample t-tests or nonparametric Wilcoxon statistics, Fisher’s exact tests as appropriate; Levine’s test will be used to assess homogeneity of variance. | |
| 1 | Primary | Cohen Mansfield Agitation Inventory (CMAI), Direct Observation - Agitation Behaviors Index (14 items) | Intention-to-treat (ITT) mixed-effects models will be used to compare changes in the primary outcome over time between treatment groups. The most appropriate covariance structure will be selected and used to adjust for within-subject correlation of repeated measures. Predictors will include group, day, and the group × day interaction term (primary effect of interest). Analysis will be adjusted for any variables deemed prognostic in preliminary analyses. |
| Secondary | CMAI, Direct Observation - Agitation Behaviors Index (14 items) | As in the primary analysis, an ITT mixed-effects model will be used. Predictor variables will include group, day, time of night, and their three-way interaction (group × time of night × day). | |
| Secondary | CMAI - Caregiver Version | General linear modeling of 8 week outcomes will be regressed on group and baseline values, along with any variables demonstrating differences at baseline in preliminary analyses. | |
| Secondary | Modified Alzheimer’s Disease Cooperative Study-Clinical Global Impression of Change (mADCS-CGIC) | ||
| 2 | Primary | Adverse Event Checklist | Percentages of participants in the treatment with adverse events will be summarized. The severity, seriousness, likelihood of relationship to GEn, and clinical outcome for each adverse event will be tabulated. Statistical comparisons will rely on Fisher’s exact tests to determine whether participants in the GEn group are at significantly greater risk than the placebo group. |
| Secondary | Global Rating of Fall Risk (GLORF) | As in Aim 1, an ITT mixed-effects model will be used. Predictors will include group, week, and the group × week interaction term (primary effect of interest). | |
| Secondary | Mini-Mental State Examination (MMSE) | ||
| Secondary | Physical Mobility Scale (PMS) | ||
| 3 | Primary | Sleep Disturbance - Direct Observation | General linear modeling of nighttime sleep outcomes and RLS behaviors at 8 weeks will be regressed on group and baseline values, along with any variables demonstrating differences at baseline in preliminary analyses. |
| Primary | Behavioral Indicators Test - Restless Legs (BIT-RL) | ||
| Primary | Sleep Disturbance - Micro-Mini Motionlogger® Actigraph | ||
| Primary | Dose equivalents of antipsychotic medications | Generalized estimating equations modeling specifying a Poisson link will be used to analyze changes in outcomes over time (baseline, 8 weeks, and post-trial week 16) between the treatment groups. Predictors will include group, day, and the group × day interaction term (primary effect of interest). Analysis will be adjusted for any variables deemed prognostic in preliminary analyses. | |
| 4 | Primary | CMAI, Direct Observation - Agitation Behaviors Index (14 items) | Baron and Kenny methodology will be used to explore the mediating effect of the frequency of RLS behaviors on the relationship between the intervention and nighttime agitation. |
| All (Aims 1–4) | Sensitivity | All outcomes | Sensitivity analyses will be performed by excluding patients with medically indicated treatment changes. |
Data Collection and Management.
Data is collected and entered in real-time into a study-specific Research Electronic Data Capture (REDCap) database portal. REDCap is a secure web-based application for building and managing online databases. Data input occurs over a secure web connection with authentication and data logging in a regulation-compliant environment. The project staff uses online study-specific REDCap forms to collect and enter data using tablets and laptop computers. The Data Management Committee led by the study’s database consultant (software systems engineer) developed and designed the study database. Data collection forms were created and organized within 14 REDCap projects based on study design. Project staff initially pilot-tested the data collection system with study staff as the “participants” in the LTC settings and private homes to identify and resolve connectivity, equipment specifications, and optimal camera and research staff placements. Participant data are monitored for completeness and accuracy by study staff, and reviewed by the medical team and research coordinator at baseline, 2, 8, and 16 weeks.
Data Safety Monitoring Board (DSMB).
A DSMB was established to act in an advisory capacity and monitor participant safety, data quality, and evaluate the progress of the study. The DSMB consists of five members, who have no conflicts of interest with the trial and are not collaborators or associates of the investigators, and a safety officer. The study nurse and medical team evaluate a reported event with the standard classifications of severity, outcome and likelihood it was caused by the study drug. Safety assessments determine if the adverse event is serious or non-serious, intensity, severity, duration, episodic nature, and outcome of the adverse event, and the relationship of the adverse event to the drug. During the study recruitment period, adverse events are analyzed by the study unblinded statistician by A and B groups, and reviewed by the DSMB in closed session. Reports containing recommendations, including whether to continue or terminate the study, are sent to the principal investigator and NIH program officer after each meeting.
DISCUSSION
Two prevalent and difficult to manage symptoms in persons with AD are nighttime agitation behaviors, such as wandering and aggression, and sleep disturbances. Multiple interrelated factors, such as frailty, immobility, and cognitive impairment, make the management of these symptoms challenging. There are currently no FDA approved medications for treatment of nighttime agitation. Nighttime agitation and sleep disturbance are associated with more rapid cognitive decline, falls, wandering from home, institutionalization, and caregiver burden. The treatment of comorbidities or deprescribing of medications that exacerbate nighttime agitation and sleep disturbances should always be the first step. Our study is the first randomized controlled trial to focus on treatment of a common comorbid condition, RLS, to determine if RLS diagnosis and treatment affects nighttime agitation and improves sleep in older adults with AD. The trial also will provide evidence on the safety profile of an FDA approved drug for treatment of RLS, GEn, in older adults with AD. This study is highly innovative because it is the first to 1) concurrently address three comorbid chronic conditions in persons with moderate to severe AD: nighttime agitation, sleep disturbances, and RLS; 2) use a precision medicine approach to individually tailor treatment for nighttime agitation to a neglected and unrecognized potential cause – RLS; 3) use a new and innovative standardized diagnostic tool that we developed to identify RLS in persons who cannot verbally report their RLS symptoms; 4) examine whether frequency of RLS behaviors is a mechanism for nighttime agitation, and 5) apply real time observational techniques that we developed to precisely measure nighttime agitation. This project is relevant to public health because the results may provide an alternative to antipsychotics and sedative-hypnotics for treatment of nighttime agitation and sleep disturbances.
Acknowledgements:
Funding support of the National Institute on Aging of the National Institutes of Health (R01AG051588) from April 15, 2018 to March 31, 2021, Arbor Pharmaceuticals for gabapentin enacarbil and matching placebo, long-term care facilities in Austin, Texas, and study participants and their families.
The authors have disclosed no potential conflicts of interest, financial or otherwise. The authors acknowledge funding support from the National Institute on Aging of the National Institutes of Health (R01AG051588); and Arbor Pharmaceuticals for gabapentin enacarbil and matching placebo.
The authors also thank the long-term care facilities in Austin, Texas, and study participants and their families.
Footnotes
Dr. Kovach was not involved in the peer review or decision making of this manuscript.
Institutional Review Board: All procedures were approved by the institutional review board at The University of Texas at Austin.
Contributor Information
Kathy Richards, The University of Texas at Austin, School of Nursing, Austin, Texas;.
Janet Morrison, The University of Texas at Austin, School of Nursing, Austin, Texas;.
Yan-yan Wang, The University of Texas at Austin, School of Nursing, Austin, Texas;.
Angelica Rangel, The University of Texas at Austin, School of Nursing, Austin, Texas;.
Ana Loera, The University of Texas at Austin, School of Nursing, Austin, Texas;.
Alexandra Hanlon, Center for Biostatistics and Health Data Science, Virginia Tech, Department of Statistics, Roanoke, Virginia;.
Alicia Lozano, Center for Biostatistics and Health Data Science, Virginia Tech, Department of Statistics, Roanoke, Virginia;.
Christine Kovach, Jewish Home and Care Center, Ovation Communities, Milwaukee, Wisconsin.
Nalaka Gooneratne, The University of Pennsylvania, Philadelphia, Pennsylvania.
Liam Fy, Dell Medical School, The University of Texas at Austin.
Richard Allen, Johns Hopkins University, Baltimore, Maryland.
REFERENCES
- Alzheimer’s Association. (2014). 2014 Alzheimer’s disease facts and figures. Alzheimer’s & Dementia, 10(2), e47–e92. 10.1016/j.jalz.2014.02.001 [DOI] [PubMed] [Google Scholar]
- American Academy of Sleep Medicine. (2014). International classification of sleep disorders. American Academy of Sleep Medicine. https://j2vjt3dnbra3ps7ll1clb4q2-wpengine.netdna-ssl.com/wp-content/uploads/2019/05/ICSD3-TOC.pdf [Google Scholar]
- Anthony JC, LeResche L, Niaz U, Von Korff MR, & Folstein MF (1982). Limits of the ‘Mini-Mental State’as a screening test for dementia and delirium among hospital patients. Psychological medicine, 12(2), 397–408. 10.1017/S0033291700046730 [DOI] [PubMed] [Google Scholar]
- Appelbaum PS, & Grisso T (2001). MacArthur competence assessment tool for clinical research (MacCAT-CR). Professional Resource Press/Professional Resource Exchange. [Google Scholar]
- Aurora RN, Kristo DA, Bista SR, Rowley JA, Zak RS, Casey KR, Lamm CI, Tracy SL, Rosenberg RS & American Academy of Sleep Medicine. (2012). The treatment of restless legs syndrome and periodic limb movement disorder in adults--an update for 2012: practice parameters with an evidence-based systematic review and meta-analyses: an American Academy of Sleep Medicine Clinical Practice Guideline. Sleep, 35(8), 1039–1062. 10.5665/sleep.1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachman D, & Rabins P (2006). “Sundowning” and other temporally associated agitation states in dementia patients. Annu Rev Med, 57:499–511. 10.1146/annurev.med.57.071604.141451 [DOI] [PubMed] [Google Scholar]
- Bliwise DL, Bevier WC, Bliwise NG, Edgar DM, & Dement WC (1990). Systematic 24-hr behavioral observations of sleep and wakefulness in a skilled-care nursing facility. Psychology and Aging, 5(1), 16–24. 10.1037/0882-7974.5.1.16 [DOI] [PubMed] [Google Scholar]
- Chrisman M, Tabar D, Whall AL, & Booth DE (1991). Agitated behavior in the cognitively impaired elderly. J Gerontol Nurs, 17(12), 9–13. 10.3928/0098-9134-19911201-04 [DOI] [PubMed] [Google Scholar]
- Cohen-Mansfield J, & Billig N (1986). Agitated behaviors in the elderly: I. A conceptual review. J Am Geriatr Soc, 34(10), 711–721. 10.1111/j.1532-5415.1986.tb04302.x [DOI] [PubMed] [Google Scholar]
- Cohen-Mansfield J, Libin A, & Marx MS (2007). Nonpharmacological treatment of agitation: a controlled trial of systematic individualized intervention. J Gerontol A Biol Sci Med Sci, 62(8), 908–916. 10.1093/gerona/62.8.908 [DOI] [PubMed] [Google Scholar]
- Cohen-Mansfield J, Werner P, & Marx MS (1989). An observational study of agitation in agitated nursing home residents. Int Psychogeriatr, 1(2), 153–165. 10.1017/S1041610289000165 [DOI] [PubMed] [Google Scholar]
- Cundy KC, Annamalai T, Bu L, De Vera J, Estrela J, Luo W, Shirsat P, Torneros A, Yao F, Zou J, Barrett RW, & Gallop MA (2004). XP13512 [(+/−)-1-([(alpha-isobutanoyloxyethoxy)carbonyl] aminomethyl)-1-cyclohexane acetic acid], a novel gabapentin prodrug: II. Improved oral bioavailability, dose proportionality, and colonic absorption compared with gabapentin in rats and monkeys. J Pharmacol Exp Ther, 311(1):324–333. 10.1124/jpet.104.067959 [DOI] [PubMed] [Google Scholar]
- Cundy KC, Branch R, Chernov-Rogan T, Dias T, Estrada T, Hold K, Koller K, Liu X, Mann A, Panuwat M, Raillard SP, Upadhyay S, Wu QQ, Xiang JN, Yan H, Zerangue N, Zhou CX, Barrett RW, & Gallop MA (2004). XP13512 [(+/−)-1-([(alpha-isobutanoyloxyethoxy)carbonyl] aminomethyl)-1-cyclohexane acetic acid], a novel gabapentin prodrug: I. Design, synthesis, enzymatic conversion to gabapentin, and transport by intestinal solute transporters. J Pharmacol Exp Ther, 311(1):315–323. 10.1124/jpet.104.067934 [DOI] [PubMed] [Google Scholar]
- Cundy KC, Sastry S, Luo W, Zou J, Moors TL, & Canafax DM (2008). Clinical pharmacokinetics of XP13512, a novel transported prodrug of gabapentin. J Clin Pharmacol, 48(12):1378–1388. 10.1177/0091270008322909 [DOI] [PubMed] [Google Scholar]
- Fillenbaum GG, Peterson B, & Morris JC (1996). Estimating the validity of the Clinical Dementia Rating scale: the CERAD experience. Aging Clin Exp Res, 8(6), 379–385. 10.1007/BF03339599 [DOI] [PubMed] [Google Scholar]
- Finkel SI, Lyons JS, & Anderson RL (1992). Reliability and validity of the Cohen–Mansfield agitation inventory in institutionalized elderly. International Journal of Geriatric Psychiatry, 7(7), 487–490. 10.1002/gps.930070706 [DOI] [Google Scholar]
- Folstein MF, Folstein SE, & McHugh PR (1975). “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res, 12(3), 189–198. 10.1016/0022-3956(75)90026-6 [DOI] [PubMed] [Google Scholar]
- Foreman MD (1987). Reliability and validity of mental status questionnaires in elderly hospitalized patients. Nurs Res, 36(4), 216–220. 10.1097/00006199-198707000-00004 [DOI] [PubMed] [Google Scholar]
- Garcia-Borreguero D, Ferini-Strambi L, Kohnen R, O’Keeffe S, Trenkwalder C, Högl B, Benes H, Jennum P, Partinen M, Fer D, Montagna P, Bassetti CL, Iranzo A, Sonka K, Williams AM, European Federation of Neurological Societies, European Neurological Society, & European Sleep Research Society. (2012). European guidelines on management of restless legs syndrome: report of a joint task force by the European Federation of Neurological Societies, the European Neurological Society and the European Sleep Research Society. Eur J Neurol, 19(11), 1385–1396. 10.1111/j.1468-1331.2012.03853.x [DOI] [PubMed] [Google Scholar]
- Gonzalez-Latapi P, & Malkani R (2019). Update on Restless Legs Syndrome: from Mechanisms to Treatment. Curr Neurol Neurosci Rep, 19 (8):54. 10.1007/s11910-019-0965-4 [DOI] [PubMed] [Google Scholar]
- Lal R, Sukbuntherng J, Luo W, Chen D, Vu A, Tovera J, & Cundy KC (2009). Pharmacokinetics and tolerability of single escalating doses of gabapentin enacarbil: a randomized-sequence, double-blind, placebo-controlled crossover study in healthy volunteers. Clin Ther, 31(8):1776–1786. 10.1016/j.clinthera.2009.07.026 [DOI] [PubMed] [Google Scholar]
- Lal R, Sukbuntherng, Luo W, Tovera J, Lassauzet ML, & Cundy KC (2013). Population pharmacokinetics and pharmacodynamics of gabapentin after administration of gabapentin enacarbil. J Clin Pharmacol, 53(1):29–40. 10.1177/0091270012439209 [DOI] [PubMed] [Google Scholar]
- Lee DO, Ziman RB, Perkins AT, Poceta JS, Walters AS, Barrett RW, & XP053 Study Group. (2011). A randomized, double-blind, placebo-controlled study to assess the efficacy and tolerability of gabapentin enacarbil in subjects with restless legs syndrome. J Clin Sleep Med, 7(3), 282–292. 10.5664/JCSM.1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino M, Li Y, Rueschman MN, Winkelman JW, Ellenbogen JM, Solet JM, Dulin H, Berkman LF, & Buxton OM (2013). Measuring sleep: accuracy, sensitivity, and specificity of wrist actigraphy compared to polysomnography. Sleep, 36(11):1747–1755. 10.5665/sleep.3142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr, Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, Mohs RC, Morris JC, Rossor MN, Scheltens P, Carrillo MC, Thies B, Weintraub S, &Phelps CH (2011). The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement, 7(3):263–269. 10.1016/j.jalz.2011.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JC (1993). The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology, 43(11):2412–2414. 10.1212/WNL.43.11.2412-a [DOI] [PubMed] [Google Scholar]
- Morris JC (1997). Clinical dementia rating: a reliable and valid diagnostic and staging measure for dementia of the Alzheimer type. Int psychoger, 9(S1), 173–176. 10.1017/S1041610297004870 [DOI] [PubMed] [Google Scholar]
- Nitz JC, Hourigan SR, & Brown A (2006). Measuring mobility in frail older people: reliability and validity of the Physical Mobility Scale. Australasian Journal on Ageing, 25(1), 31–35. 10.1111/j.1741-6612.2006.00137.x [DOI] [Google Scholar]
- Nordin E, Lindelöf N, Rosendahl E, Jensen J, & Lundin-olsson L (2008). Prognostic validity of the Timed Up-and-Go test, a modified Get-Up-and-Go test, staff’s global judgement and fall history in evaluating fall risk in residential care facilities. Age Ageing, 37(4), 442–448. 10.1093/ageing/afn101 [DOI] [PubMed] [Google Scholar]
- Pike E, & Landers MR (2010). Responsiveness of the physical mobility scale in LTC facility residents. J Geriatr Phys Ther, 33(2), 92–98. doi: 10.1097/JPT.0b013e3181df019f. [DOI] [PubMed] [Google Scholar]
- Porsteinsson AP, Drye LT, Pollock BG, Devanand DP, Frangakis C, Ismail Z, Marano C, Meinert CL, Mintzer JE, Munro CA, Pelton G, Rabins PV, Rosenberg PB, Schneider LS, Shade DM, Weintraub D, Yesavage J, Lyketsos CG, & CitAD Research Group. (2014). Effect of citalopram on agitation in Alzheimer disease: the CitAD randomized clinical trial. JAMA, 311(7), 682–691. 10.1001/jama.2014.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards KC, Bost JE, Rogers VE, Hutchison LC, Beck CK, Bliwise DL, Kovach CR, Cuellar N, & Allen RP (2015). Diagnostic accuracy of behavioral, activity, ferritin, and clinical indicators of restless legs syndrome. Sleep, 38(3), 371–380. 10.5665/sleep.4492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards KC, Lambert C, Beck CK, Bliwise DL, Evans WJ, Kalra GK, Kleban MH, Lorenz R, Rose K, Gooneratne NS, & Sullivan DH (2011). Strength training and walking exercise and social activity improve sleep in nursing home and assisted living residents: Randomized controlled trial. J Amer Geri Soc, 59 (2): 214–223. 10.1111/j.1532-5415.2010.03246.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards KC, Shue VM, Beck CK, Lambert CW, & Bliwise DL (2010). Restless legs syndrome risk factors, behaviors, and diagnoses in persons with early to moderate dementia and sleep disturbance. Behavioral Sleep Medicine, 8 (1):48–61. 10.1080/15402000903425769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose KM, Beck C, Tsai PF, Liem PH, Davila DG, Kleban M, Gooneratne NS, Kalra G, & Richards KC (2011). Sleep disturbances and nocturnal agitation behaviors in older adults with dementia. Sleep, 34(6), 779–786. 10.5665/SLEEP.1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer KA, Tractenberg RE, Sano M, Mackell JA, Thomas RG, Gamst A, Thal LJ, Morris JC, & Alzheimer’s Disease Cooperative Study. (2004). Reliability of monitoring the clinical dementia rating in multicenter clinical trials. Alzheimer Dis Assoc Disord, 18(4), 219–222. http://ovidsp.ovid.com/ovidweb.cgi?T=JS&PAGE=reference&D=ovftg&NEWS=N&AN=00002093-200410000-00011 [PMC free article] [PubMed] [Google Scholar]
- Schneider LS, Dagerman K, & Insel PS (2006). Efficacy and adverse effects of atypical antipsychotics for dementia: meta-analysis of randomized, placebo-controlled trials. Am J Geriatr Psychiatry, 14(3), 191–210. 10.1097/01.JGP.0000200589.01396.6d [DOI] [PubMed] [Google Scholar]
- Schneider LS, Olin JT, Doody RS, Clark CM, Morris JC, Reisberg B, Schmitt FA, Grundman M, Thomas RG, & Ferris SH (1997). Validity and reliability of the Alzheimer’s Disease Cooperative Study-Clinical Global Impression of Change. The Alzheimer’s Disease Cooperative Study. Alzheimer Dis Assoc Disord., 11 Suppl 2: S22–32. 10.1007/978-1-4612-4116-4_64 [DOI] [PubMed] [Google Scholar]
- Teri L, Logsdon RG, Peskind E, Raskind M, Weiner MF, Tractenberg RE, Foster NL, Schneider LS, Sano M, Whitehouse P, Tariot P, Mellow AM, Auchus AP, Grundman M, Thomas RG, Schafer K, Thal LJ & Alzheimer’s Disease Cooperative Study. (2000). Treatment of agitation in AD: a randomized, placebo-controlled clinical trial. Neurology, 55(9), 1271–1278. 10.1212/WNL.55.9.1271 [DOI] [PubMed] [Google Scholar]
- U.S. Food and Drug Administration. (2012, December). NDA 022399 -- FDA Approved Labeling Text dated December 2012: Highlights of prescribing information. http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/022399s006,s007lbl.pdf
- VanMeter SA, Kavanagh ST, Warren S, & Barrett RW (2012). Dose response of Gabapentin Enacarbil versus placebo in subjects with moderate-to-severe primary restless legs syndrome: an integrated analysis of three 12-week studies. CNS Drugs, 26(9), 773–780. 10.2165/11634870-000000000-00000 [DOI] [PubMed] [Google Scholar]
- Yaltho TC, & Ondo WG (2010). The use of gabapentin enacarbil in the treatment of restless legs syndrome. Ther Adv Neurol Disord, 3(5), 269–275. 10.1177/175628561037805 [DOI] [PMC free article] [PubMed] [Google Scholar]