

Abstract
Myeloperoxidase (MPO) belongs to the heme peroxidase family, which includes a set of enzymes with potent oxidoreductase activity. MPO is considered an important part of the innate immune system's microbicidal arm and is secreted by neutrophils and macrophages. Interestingly, this enzyme has been implicated in the pathogenesis of several diseases including atherosclerosis. MPO is ubiquitous in atherosclerotic lesions and contributes to the initiation and progression of the disease primarily by oxidizing low-density lipoprotein (LDL) particles. MPO is the only human enzyme with the ability to produce hypochlorous acid (HOCl) at physiological chloride concentrations and HOCl-LDL epitopes were shown to be present inside atheromatous lesions making it a physiologically relevant model for the oxidation of LDL. It has been shown that MPO modified LDL is not able to bind to the native LDL receptor and is recognized instead by scavenger receptors on both endothelial cells and macrophages, which can lead to endothelial dysfunction and foam cell formation, respectively; both of which are instrumental in the progression of the disease. Meanwhile, several studies have proposed MPO as a biomarker for cardiovascular diseases where high levels of this enzyme were linked to an increased risk of developing coronary artery disease. Overall, there is sufficient evidence supporting the value of MPO as a crucial player in health and disease. Thus, future research should be directed towards investigating the still unknown processes associated with this enzyme. This may assist in better understanding the pathophysiological role of MPO, as well in the development of therapeutic strategies for protecting against the deleterious effects of MPO in numerous pathologies such as atherosclerosis.
Keywords: heme peroxidase, myeloperoxidase, inflammation, atherosclerosis, oxidized LDL, myeloperoxidase oxidized LDL, endothelial dysfunction, macrophage activation
1. Introduction
Myeloperoxidase (MPO) belongs to the peroxidase-cyclooxygenase subgroup of the heme peroxidase family of enzymes. It is secreted by neutrophils and monocytes where it plays a crucial role in innate immunity (1,2). In the microbicidal system of phagocytes, MPO catalyzes the formation of hypochlorous acid (HOCl) which is considered one of the strongest oxidant molecules produced in the human body (3). Despite its crucial role in immune defense against pathogens, MPO and its oxidative products react with various lipids, proteins, and nucleic acids causing some detrimental effects in host tissues that are usually associated with ongoing inflammatory states such as atherosclerosis (3). Immunohistochemical analysis reported the presence of MPO in atheroma plaques of human patients and several studies have also shown its co-localization with HOCl-modified low-density lipoproteins (LDLs) in human atherosclerotic lesions where it can be found in both vascular cells and in extracellular spaces (4,5). Thus, MPO and its downstream products, mainly HOCl, are known to be implicated in the pathophysiology of atherosclerotic diseases (6,7). Despite the fact that several processes are reported to be responsible for the oxidation of LDL both in in vitro and in vivo settings, LDL that is oxidized by MPO remains the most physiologically relevant type of oxidized LDL (oxLDL) with considerable evidence confirming this theory. LDL that is modified by the MPO system affects multiple cells that are found in the atheroma plaques; this includes endothelial cells (ECs), macrophages, and smooth muscle cells (7). In this review, we describe the involvement of MPO in health and disease with an emphasis on the role of MPO oxLDL (Mox-LDL) in macrophage and endothelial dysfunction (ED) in the context of atherosclerosis. Overall, this work provides better insights into the crucial role that Mox-LDL plays during atherogenesis, as the ultimate driver of the disease, by presenting an up-to-date review on the different pathways that involve Mox-LDL in both EC and macrophage models of atherosclerosis. This may also pave the way for future research tackling MPO and its oxidation byproducts as important disease biomarkers and targets for novel therapeutic approaches.
2. Heme peroxidase family and MPO
Heme peroxidases
Heme peroxidases are a ubiquitous group of enzymes with robust oxidoreductase activity. They contain a heme group, which acts as a redox cofactor for catalyzing the hydrogen peroxide (H2O2) mediated one- and two-electron oxidation reactions of numerous organic and inorganic molecules. One-electron donors such as organic molecules (AH2) are oxidized into their corresponding radicals (AH) and H2O2 is reduced to water (Reaction 1) (1). Meanwhile, two-electron donors such as halides (X) are oxidized into their respective hypohalous acids (HOX) and H2O2 is also reduced into water (Reaction 2) (1). The heme peroxidase superfamily was previously classified according to the species origin of the enzymes into ‘animal’ and ‘non-animal’ superfamilies; however, this was found to be somewhat misleading since many of these enzymes were reported to be present in all kingdoms (8). Accordingly, the heme peroxidase superfamily is better categorized on the basis of differences in enzymatic activities into four subgroups (8). These four subgroups include the peroxidase-catalase subgroup, the peroxidase-cyclooxygenase subgroup, the peroxidase-chlorite dismutase superfamily, and the peroxidase-peroxygenase superfamily (8). Particularly relevant to this review is the peroxidase-cyclooxygenase subgroup. This subgroup is characteristically unique since its heme group is post-translationally modified, and bound via ester bonds molded by the highly conserved aspartic acid and glutamic acid residues (8). All enzymes of this subgroup catalyze both reactions 1 and 2, with halide oxidation being the more dominant physiological enzymatic activity (Fig. 1A) (8). This subgroup enlists MPO, eosinophil peroxidase (EPO), lactoperoxidase (LPO), and thyroid peroxidase (TPO) as its members (8). MPO, EPO, and LPO are integral mediators of the innate immune system. Conversely, thyroid peroxidase catalyzes the synthesis of the thyroid hormones thyroxine (T4) and triiodothyronine (T3).
Figure 1.
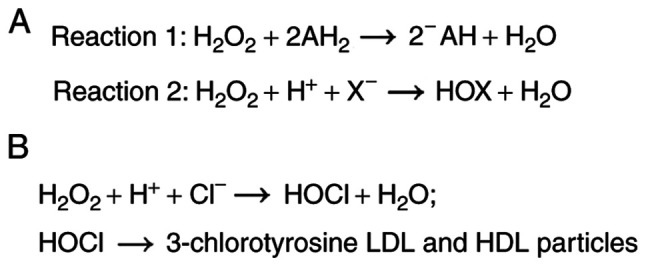
The enzymatic activities of the peroxidase-cyclooxygenase subgroup including MPO. (A) Reactions that are catalyzed by the peroxidase-cyclooxygenase subgroup. (B) The latter group includes MPO, which, though halide oxidation (predominant physiological enzymatic activity) is responsible for the production of modified LDL and HDL particles through the generation of HOCl and the chlorination of tyrosine residues in the protein moieties of those particles. MPO, myeloperoxidase; LDL, low-density lipoproteins; HDL, high-density lipoproteins; HOCl, hypochlorous acid.
MPO is one of the most prominent enzymes of the peroxidase-cyclooxygenase subgroup and is encoded by a gene located on the long arm of chromosome 17 (17q) with a size of 14 kb (9). Synthesis of MPO is exclusive to cells of the myeloid lineage, and it commences during myelopoiesis in the bone marrow in promyelocytes and promonocytes (10). During the differentiation of the myeloid precursors into neutrophils or monocytes, MPO mRNA levels progressively decrease until they reach a point where MPO synthesis is terminated in the fully differentiated monocytes and neutrophils (11). The mature form of MPO is present mainly in the azurophilic granules of neutrophils and to a lesser extent in the lysosomes of monocytes and MPO species isolated from human plasma including both precursor and mature forms of the enzyme (12). The three-dimensional structure of MPO forms a cationic surface (pI >10) at physiological pH, with numerous arginine and lysine residues, and this enables MPO to bind to electronegative molecules such as lipoproteins, proteoglycans, and bacterial surfaces (6).
Role of MPO in the host defense
MPO is a crucial mediator of the intracellular microbicidal system of phagocytes and the host innate immune system. After synthesis, MPO is stored in the azurophilic granules of resting neutrophils and it is inactivated through inactivated when it is captured by anionic proteoglycans accompanied by the low pH milieu (2). Once a neutrophil becomes activated by ingesting a pathogen, the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase enzyme system is activated and recruited to the internal phagolysosomal membrane surface; concurrently MPO is also released into the phagosome. The majority of cellular MPO is released into the phagosome; however, some MPO is released into the extracellular environment via degranulation or by association with neutrophil extracellular traps (NETs) (6). In circulation, the enzyme is transported by albumin and neutrophil microparticles (9). Inside the phagosome, the activity of the NADPH oxidase results in the formation of a superoxide anion (O2-), which is converted to H2O2 by superoxide dismutase (SOD). MPO can couple H2O2 with halides/pseudohalides to catalyze the formation of powerful reactive oxygen intermediates such as HOCl, hypobromous acid (HOBR), and hypothiocyanous acid (HOSCN). Although chlorine has the lowest reactivity to MPO among the halides, it is considered to be the primary physiological substrate of MPO, due to its high concentration in tissues (10). The unique product of the MPO/H2O2/Cl- system is the potent antimicrobial oxidant hypochlorous acid/hypochlorite. HOCl is a unique product of MPO, since the latter is the only human enzyme with the ability to produce HOCl at physiological chloride concentrations (Fig. 1B) (9). HOCl is able to initiate modification reactions targeting DNA, lipids, and lipoproteins (13). Other notable MPO primary and secondary oxidant products include nitrous radical (NO•) and peroxynitrite (ONOO-), chloramines, hydroxyl radicals (HO·), and ozone (O3). The canonical role of MPO is thought to be the mediation of the host defense against invading pathogens such as fungi and bacteria, although this role of MPO varies among different species (3). In fact, MPO knock-out mice were found to be more susceptible to infections (3). On the other hand, humans with complete and partial MPO-deficiency live normally with rare cases of persistent infections with Candida albicans reported (2).
Role of MPO in inflammation and disease
MPO has been implicated to varying extents in the pathogenesis of several diseases (Table I). These diseases are usually associated with chronic or acute inflammatory states. The detrimental effect of MPO on host tissue is usually mediated by MPO-derived oxidants, which interact with nucleic acids, lipids, and proteins causing deleterious effects. First, MPO is implicated in the pathogenesis of several cancer types such as bladder, breast, colon, larynx, lung, leukemia, and stomach. The carcinogenic role of MPO is mediated by MPO oxidative products which are genotoxic and mutagenic. The role of MPO-downstream oxidants in cancer has been reviewed in depth elsewhere (9,11). MPO also plays a role in neurodegenerative diseases such as Parkinson's disease, Alzheimer's disease, and multiple sclerosis; this role is articulately reviewed in (3). Furthermore, reports have also demonstrated the involvement of MPO in vasculitis, renal illnesses, lung inflammation, rheumatoid arthritis, colitis, pancreatitis, cystic fibrosis, liver diseases, periodontitis, sinusitis, and inflammatory bowel disease (2). MPO is also established to play a role in all stages of atherosclerosis, and this is particularly relevant to the present review.
Table I.
Studies assessing the role of myeloperoxidase in inflammation and disease.
| Disease | (Refs.) |
|---|---|
| Inflammatory diseases | |
| Vasculitis | (2,9,11) |
| Rheumatoid arthritis | (2,3,9,11) |
| Colitis | (2,9) |
| Pancreatitis | (2) |
| Periodontitis | (2) |
| Sinusitis | (2) |
| Inflammatory bowel syndrome | (2,11) |
| Lung inflammation | (2,3,11) |
| Neurodegenerative diseases | |
| Parkinson's disease | (2,3,9,11) |
| Alzheimer's disease | (2,3,9,11) |
| Multiple sclerosis | (2,3,9,11) |
| Cancer | |
| Lung cancer | (9,11) |
| Ovary cancer | (9) |
| Bladder cancer | (9,11) |
| Liver cancer | (9) |
| Stomach cancer | (9) |
| Colon cancer | (11) |
| Lung cancer | (9,11) |
| Larynx cancer | (11) |
| Breast cancer | (11) |
| Pancreatic cancer | (11) |
| Myeloid leukemia | (11) |
| Cardiovascular diseases | |
| Coronary artery disease | (2,3,9,11) |
| Myocardial infarction | (3,11) |
| Heart failure | (11) |
3. MPO and atherosclerosis
Source of MPO in atherosclerosis
MPO is pervasive inside atherosclerotic lesions. MPO has been extensively shown to be present inside human atherosclerotic tissues (4). Inside vascular walls and atherosclerotic lesions, there exist different sources of MPO. For example, during inflammatory states, MPO is secreted by neutrophils into the circulation. Subsequently, MPO binds to ECs through electrostatic interactions which leads to the internalization of MPO into the atherosclerotic lesions in vascular walls by endocytosis (9). Nevertheless, macrophages are thought to be the primary source of MPO inside atherosclerotic lesions. Maturation of monocytes into macrophages is linked to a loss of MPO expression; however, granulocyte-macrophage colony-stimulating factor (GM-CSF) has been found to selectively regulate the ability of macrophages to express MPO in human atherosclerotic lesions (14). Furthermore, it was demonstrated that macrophages present in the earlier stages of atherosclerosis minimally express MPO, whereas macrophages present in the late stages of atherosclerosis highly express it (14). Immunohistochemical evidence showed that antihuman MPO antibodies co-localized with antimacrophage antibodies, and this immunostaining of MPO was particularly prominent in the atherosclerotic lesions of the shoulder region (4). Therefore, both local secretion by macrophages and transcytosis of intraluminal MPO are thought to be the primary sources of MPO in atherosclerotic lesions (15).
Role of MPO in atherosclerosis
Numerous studies including in vitro experiments and pathophysiological observations have implicated MPO and its downstream oxidative products in the pathogenesis of atherosclerosis. MPO contributes to atherosclerosis by oxidizing LDL, impairing high-density lipoprotein (HDL) function, reducing the bioavailability of nitric oxide (NO), causing ED, generating a thrombogenic environment, activating matrix metalloproteinases (MMPs), and inactivating tissue inhibitors of MMPs (TIMPs), and recruiting leukocytes, consequently destabilizing the atherosclerotic plaque (6,15).
High-density lipoprotein is renowned for its athero-protective characteristics including its antioxidant, anti-inflammatory, and antithrombotic properties. A reputed contribution of MPO to atherosclerosis involves targeting and modifying apolipoprotein A-I (apoA-I) of HDL resulting in the attenuation of the athero-protective role of HDL. MPO binds to helix 8 on apoA-I, and converts HDL into a dysfunctional form (16). As previously mentioned, MPO forms the highly reactive HOCl, which will chlorinate electron-rich residues such as lysine and tyrosine, thus forming MPO-specific 3-chlorotyrosine (Fig. 1B) (17). Moreover, MPO oxidative byproducts are capable of producing 3-nitrotyrosine in vitro (18). 3-chlorotyrosine and 3-nitrotyrosine were found to be abundant in apoA-I of HDL recovered from atherosclerotic lesions (19). Furthermore, the levels of 3-chlorotyrosine and 3-nitrotyrosine in serum HDL of individuals with cardiovascular disease (CVD) are markedly higher than in serum HDL of healthy individuals (18-20). In addition, HDL recovered from human aortic atherosclerotic lesions had substantially higher levels of 3-chlorotyrosine in comparison to HDL obtained from plasma (20). Similarly, the mean levels of 3-nitrotyrosine in HDL obtained from human aortic atherosclerotic intima are significantly higher when compared to circulating HDL (18). However, it is important to note that 3-nitrotyrosine is not unique to MPO like 3-chlorotyrosine, since there are other pathways independent of MPO that also produce 3-nitrotyrosine in atherosclerotic lesions (15). Modified HDL is also pro-inflammatory, and it induces the expression of vascular cell adhesion molecule 1 (VCAM1) in ECs (6). Moreover, several studies suggest that MPO modification of HDL attenuates the athero-protective properties of HDL, primarily by hindering the interaction of HDL ATP binding cassette subfamily A member 1 (ABCA1) and ATP binding cassette subfamily G member 1 (ABCG1), consequently impairing the reverse cholesterol efflux process (11,15,21). Interestingly, it was found that HDL-bound MPO retains its enzymatic activity, and binding to HDL may protect MPO from cellular uptake and degradation (15).
There is ample evidence showing that MPO and its downstream oxidative products cause ED. First, several peroxidases such as MPO act as a catalytic sink for NO, since NO is a substrate for the one-electron reactions of the peroxidases (9,11). Similarly, downstream oxidative products of MPO can also scavenge NO (15). Furthermore, HOCl chlorinates the nitrogen atoms of arginine the substrate for NO formation, thereby inhibiting nitric oxide synthetase (NOS) (22,23). On a similar note, HOCl oxidizes endothelial NOS (eNOS) and uncouples it, turning eNOS into a superoxide-generating enzyme (24). Scavenging NO and inhibiting its formation ultimately causes ED, which initiates atherosclerosis (25). MPO may also promote EC apoptosis and desquamation in vitro, which provides a clue into its possible role in provoking plaque erosion and thrombus formation in vivo (6). In line with these aforementioned observations, serum MPO levels were found to be associated with ED in humans (26). In addition, MPO contributes to a thrombogenic setting by stimulating ECs to secrete tissue factors and by priming platelet aggregation (6). MPO also promotes atherosclerotic plaque destabilization by activating MMPs and inactivating TIMPs. In fact, HOCl that is produced by MPO has been found to activate MMP-7 in vitro by oxidizing and converting the thiol residue of the cysteine switch to sulfinic acid (27). Moreover, HOCl was shown to oxidize the N-terminal cysteine of TIMP-1, thereby rendering it inactive (28). Atherosclerosis is an inflammatory disorder, and there is increasing evidence revealing a role of MPO-derived products in the perpetuation of inflammation and atherosclerosis by leukocyte recruitment. MPO can contribute to leukocyte recruitment through several mechanisms. First, as previously mentioned, MPO acts as a catalytic sink for NO, and NO has anti-adhesive properties, thus consuming NO promotes the adhesion of leukocytes to the endothelial layer (21). Under physiological conditions, there exists an electrostatic repulsion between the negatively charged endothelium glycocalyx and the negatively charged surfaces of polymorphonuclear neutrophils (PMNs). In inflamed tissues, the cationic MPO is released, and MPO binds to the negatively charged endothelium causing a reduction in the negative surface charge (21). It seems that MPO favors binding to the heparan sulfate glycosaminoglycans, which constitute a major building block of the endothelial glycocalyx (29). This decrease in the negative charge could allow for a new electrostatic attraction between the negatively charged PMN and the now positively charged endothelium, thereby facilitating the binding, rolling, adhesion, and the transmigration of the PMNs into the surrounding tissue and perpetuation of inflammation (21). Furthermore, MPO produces HOSCN, and HOSCN is known to induce the expression of EC surface adhesion molecules (21). MPO also induces EC production of cytokines and chemokines such as IL-6 and IL-8, which enhance inflammation and recruit leukocytes (21).
MPO as a biomarker for CVDs
Several studies have established a relationship between varying levels of MPO and CVD, independent of classical risk factors. For instance, elevated levels of functional MPO per ml of blood and per leukocyte were linked with the risk of coronary artery disease (CAD) (30). Similarly, individuals with the-463 G/A polymorphism of the MPO gene exhibit elevated levels of MPO, and this is associated with an increased risk of CAD (31). On the contrary, cardiovascular problems appear to be very rare among individuals with MPO deficiency (32). Likewise, individuals having a promoter polymorphism linked with a 2-fold reduction in MPO expression appear to be protected from CVD-like problems (33). There is also considerable evidence supporting the role of MPO in the prediction of long-term risk of cardiovascular mortality in patients with cardiovascular problems (6). For example, after a follow-up of 13 years, the top tertile of MPO levels predicted approximately twice the risk of CVD mortality in comparison with patients in the lowest tertile of MPO levels (34).
4. Oxidized LDL (oxLDL) and atherosclerosis
oxLDL
Atherosclerosis is a progressive chronic inflammatory disorder characterized by the interplay of multiple risk factors. High plasma LDL cholesterol levels are one of the most well-established risk factors underlying the development of atherosclerosis and cardiovascular problems. Conversely, low plasma LDL levels are associated with low cardiovascular risk. Indeed, individuals with exceedingly low LDL levels generally do not develop clinically relevant atherosclerosis, irrespective of the presence of other risk factors (35). However, approximately half of the patients with cardiovascular problems have normal levels of cholesterol (36). This might be related to the fact that lipoprotein quality is crucial in mediating atherogenesis; in fact, LDL is post-translationally modified in several processes such as glycation, glycosylation, carbamylation, glycoxidation, acetylation, methylation, ethylation, and oxidation (10,36). Interestingly, one of the most significant LDL modifications is oxidation, which results in the generation of an inflammatory form known as highly oxLDL (7). The oxidation of LDL occurs along an extensive and continuous spectrum. This spectrum ranges from the minimally oxLDL to the highly oxLDL (37). The oxidation of LDL usually begins in the plasma where minimally oxLDL can be detected (38). Measuring the degree of LDL oxidation is usually determined by a fluorometric Thiobarbituric acid reactive substances assay. The degree of oxidation of minimally oxLDL is normally <10 nmol/mg LDL protein; meanwhile, the oxLDL value is usually >30-40 nmol/mg LDL protein (37). These two forms are characteristically very distinct. Minimally oxLDL is oxidized to an extent where it can still be recognized by native LDL receptors and not by scavenger receptors (37). Conversely, oxLDL is oxidized to a level where apolipoprotein B-100 (apoB-100) is modified such that it is no longer able to be recognized by the native LDL receptor (7). Indeed, oxLDL is recognized, bound, and internalized by receptor-mediated endocytosis through multiple scavenger receptors. These scavenger receptors include class A scavenger receptor type 1 and type 2 (SR-A1 & SR-A2), class B scavenger receptor type 1 and class B scavenger receptor type 2 (SR-B1 and SR-B2), macrosialin/CD68, scavenger receptor for phosphatidylserine and oxidized lipoprotein (SRPSOX), and lectin-like oxLDL receptor-1 (LOX-1) (37,39,40). Unlike the native LDL receptor, scavenger receptors are not down-regulated when the cholesterol content of the cell increases (40). Oxidative modification of LDL plays a substantial role in atherosclerosis, and may therefore be a plausible target for interventions to slow the development of atherosclerosis. In fact, several epidemiological studies have found an association between circulating oxLDL levels and clinical cardiovascular events. For example, circulating oxLDL was found to be a sensitive marker for CAD where it correlates with the Framingham risk factors (41). Circulating oxLDL is also associated with other risk factors of atherosclerosis such as diabetes, fatty liver, and obesity (40). A myriad of studies have identified the presence of oxLDL in atherosclerotic lesions (36). Inside atherosclerotic plaques, uptake of oxLDL by receptor-mediated endocytosis through scavenger receptors converts macrophages to foam cells, which constitute the hallmark of atherosclerosis. Besides inducing foam cell formation, oxLDL is pro-inflammatory and it has a wide range of biological effects and properties that contribute to the development of atherosclerosis, which is eloquently summarized in (38,42,43).
Mechanisms of LDL oxidation
There exist several mechanisms for the oxidative modification of LDL. Different cells, enzymes, and cations are involved in the oxidation of LDL. In vitro studies have demonstrated that cells such as fibroblasts, monocytes, smooth muscle cells, macrophages, neutrophils, and ECs can oxidize LDL (44). LDL is oxidized by various enzyme systems such as xanthine oxidase, NADPH oxidase, lipoxygenase, and MPO (10). Moreover, Copper (Cu2+) and Iron (Fe3+) dependent oxidation of LDL has been widely used in experiments; however, these mechanisms require high concentrations of the respective metal ions at the site of oxidation and they should not be considered physiologically relevant (10). Some oxidative mechanisms target the lipid portion of LDL including copper and lipoxygenases, whereas other mechanisms target primarily the protein fraction of LDL such as MPO (10). Particularly relevant to this review is the modification by MPO which forms Mox-LDL.
5. MPO modified LDL
Evidence of the formation of MPO modified LDL in atherosclerosis
The exact processes underlying the formation of oxLDL in vivo are still widely debated among scientists. However, recently, there has considerable evidence confirming the in vivo oxidative modification of apoB-100 of LDL by the MPO/H2O2/Cl- system, and the ability of MPO to bind to LDL and to co-precipitate with it (45). The cationic properties of MPO suggest that MPO binds to the negatively charged amino acids of apoB-100(9). MPO is predicted to strongly bind to and adsorb the NH2-βα1 domain of apoB-100 since this domain is exposed to the outside surface of LDL (10). In fact, the probable site of interaction between apoB-100 and MPO is the amino acid stretch 445-456 (EQIQDDCTGDED) of apoB-100(46). Furthermore, it was found that once MPO adsorbs the surface of LDL, its chlorinating and peroxidase activities are increased by 90% (36). Moreover, MPO kinetic reactivity favors proteins and free amino acids and this will cause MPO to primarily oxidize the amino acid residues of apoB-100(9). Lipid oxidation of LDL by MPO may also occur, albeit to a lesser extent, and may be restricted to acidic conditions with an excess of reactants (10). To study the ability of MPO to oxidize LDL, researchers use either the MPO/H2O2/Cl- system or the MPO/H2O2/Cl- system primary product ‘HOCl’ which is considered the unique product of this system; in fact, MPO is the only human enzyme with the ability to produce HOCl at physiological chloride concentrations (17). However, it is worth noting that HOCl used as a reactant does not 100% mimic the MPO/H2O2/Cl- since as mentioned earlier, MPO adsorbs with a strong interaction with the surface of LDL (9). Nevertheless, in order to facilitate the experimental procedure, HOCl is sometimes used as a reactant instead of the whole MPO/H2O2/Cl- system. Therefore, there exist two different forms when referring to LDL oxidized by MPO: LDL oxidized by the whole MPO/H2O2/Cl- system and LDL oxidized directly by HOCl alone. In this review, we will refer to LDL oxidized by the whole MPO/H2O2/Cl- system as Mox-LDL, and LDL oxidized directly by HOCl as HOCl-LDL. One of the earliest pieces of evidence for LDL oxidation by MPO is the immunohistochemical evidence showing specific monoclonal antibodies cross-reacting with human HOCl-LDL but not with Cu2+-, malondialdehyde-, peroxynitrite-, hemin-, glycated-, acetylated-, or 4-hydroxynonenal-modified LDL (5). These antibodies were specific to epitopes present in LDL after treatment with HOCl added alone as a reagent or generated by the MPO/H2O2/Cl- system (5). It was also reported that the presence of HOCl-LDL epitopes increased in conjunction with the severity of the atherosclerotic lesion (5). Furthermore, HOCl-LDL epitopes were shown to be present inside and around monocytes/macrophages, ECs, as well as in the extracellular matrix (5). Intriguingly, the staining of HOCl-LDL epitopes was co-localized with MPO and CD68-positive cells suggesting that monocytes/macrophages are involved in the production of MPO and MPO-modified epitopes of LDL in advanced human atherosclerotic lesions (5). Another study reported the use of four different monoclonal antibodies specifically raised against LDL oxidized by the MPO (47). These monoclonal antibodies did not cross-react with neither native LDL, very-low-density lipoproteins (VLDL) nor hydrogen peroxide, or copper sulfate oxLDL (Cuox-LDL) (47). Remarkably, these four antibodies demonstrated reactivity with human atherosclerotic aortic and carotid tissues (47). Another line of evidence implicating MPO in the oxidative modification of LDL is mass spectrometry analysis that revealed a total of 97 peptides of apoB-100 modified by either HOCl added as a reagent or by the MPO/H2O2/Cl- system (36). It is important to note that in their study, the authors found differences in the modified residues of LDL oxidized by HOCl or by the MPO/H2O2/Cl- system (36). This same study also demonstrated that many of the modifications of apoB-100 induced by HOCl identified in vitro were also present on LDL isolated from patients who have increased levels of plasma MPO and Mox-LDL (36). HOCl chlorinates electron-rich substrates on apoB-100 such as lysine and tyrosine residues, generating MPO-specific 3-chlorotyrosine. 3-chlorotyrosine is a unique product of MPO-catalyzed oxidation of LDL, and its levels are undetectable in LDL oxidatively modified by traditional oxidizing agents such as cations (copper and iron), enzymes (horseradish peroxidase, lactoperoxidase, and lipoxygenase), and molecules such as hemin, glucose, and peroxynitrite (17). Therefore, 3-chlorotyrosine is used as evidence for the in vivo modification of LDL by MPO (Fig. 1B). Interestingly, gas chromatography-mass spectrometry analysis revealed that the levels of 3-chlorotyrosine recovered from atherosclerotic tissue were 6x greater than those recovered from normal aortic tissue (17). It was also found that the levels of 3-chlorotyrosine were 30x greater in LDL obtained from atherosclerotic tissue compared with circulating LDL (17). LDL oxidation by the MPO/H2O2/Cl- system was presumably restricted to the intima; however, there is recent evidence demonstrating that LDL oxidation by the MPO system may also occur in the plasma (48). This hypothesis was based on the fact that MPO is known to adsorb LDL and the endothelium, and therefore, MPO may oxidize LDL at the surface of EC in the circulation. In vitro experiments were performed using EC, which were treated with native LDL, MPO, and angiotensin II to promote the activity of the NADPH complex (48). The results revealed that Mox-LDL production directly correlated with the LDL and MPO concentrations used in the study (48). These results showed that LDL oxidation may not be restricted to the intima. Furthermore, it was reported that physiological serum concentrations of Mox-LDL range from 10-100 µg/ml (49). LDL can also be oxidized by MPO-generated species other than HOCl. For example, in vitro treatment of LDL with reactive nitrogen species produced by the MPO/H2O2/Nitrite system yields a nitrated form of LDL (NO2-LDL) (50). This nitrated form of LDL does not bind to the native LDL receptor or scavenger-receptor class A, but to CD36 promoting foam cell formation (50). The reactive nitrogen species produced by the MPO/H2O2/Nitrite convert tyrosine into 3-nitrotyrosine, which was found to be enriched in LDL recovered from human atherosclerotic aortic tissue (51). However, it should be noted that the formation of 3-nitrotyrosine is not unique to MPO since other mechanisms may produce 3-nitroyrosine as well (15). In addition, it has been shown that LDL protein carbamylation can be mediated by MPO. MPO/H2O2 oxidizes thiocyanate into cyanate which carbamylates lysine residues on LDL (52). It was found that carbamyl-modified lipoproteins detected in human atheroma plaques co-localized with MPO (52). Moreover, carbamylation of LDL generated an atherogenic molecule that could bind with high affinity to scavenger receptor class A inducing its uptake and foam cell formation (52). Taken together this information demonstrates that MPO is highly implicated in modifying LDL in atherosclerotic lesions. Mox-LDL interacts with a variety of cells such as macrophages, neutrophils, T cells, ECs, platelets, and smooth muscle cells. In this review, we will focus on the interaction of Mox-LDL with macrophages and ECs.
Macrophage interactions with MPO modified LDL
The majority of studies on the effect of oxLDL on monocytes/macrophages have been performed using Cuox-LDL which is not a physiologically relevant model of oxLDL (53-56); meanwhile, Mox- LDL is rarely used. In vitro experiments using murine macrophages (RAW 264.7 cells) showed that HOCl-LDL possesses pro-inflammatory characteristics that activate monocytes, promote monocyte/macrophage differentiation, and induce the expression of CD36 and peroxisome proliferator-activated receptor-γ (PPAR-γ) (57). Furthermore, HOCl-LDL was found to strongly induce the expression of the α-chemokine IL-8 but not the β-chemokine MCP-1 in human monocytes (58). In addition, HOCl-LDL was reported to promote apoptosis in THP-1 monocytes through a caspase-dependent pathway (59). Moreover, HOCl-LDL inhibited lysosomal cysteine protease cathepsin B via a chloramine-dependent mechanism (45). CD36 and SR-B1 were identified as receptors for HOCl-LDL that can take up HOCl-LDL via receptor-mediated endocytosis (60). HOCl-LDL upregulates the expression of CD36 via PPAR-γ (57). This uptake of HOCl-LDL along with the reported protease inhibition causes lipid accumulation in macrophages and consequently foam cell formation. Nevertheless, it should be noted that scavenger receptors that recognize, bind, and take up Mox-LDL on macrophages are yet to be reported. LDL modified by the whole MPO/H2O2/Cl- system also has pro-inflammatory properties that activate macrophages. Treatment of THP-1 monocytes with Mox-LDL at a physiologically relevant concentration of 100 µg/ml for 4 h resulted in a 2-fold increase in the secretion of TNF-α; meanwhile, there was no increase in TNF-α secretion when the cells were treated with native LDL, albumin, or Mox-albumin (61). This shows the specificity of Mox-LDL in the induction of TNF-α release (61). Additionally, incubating RAW264.7 and PBMC-derived macrophages with Mox-LDL resulted in an increase in reactive oxygen species (ROS) production (62). Furthermore, it has been reported that Mox-LDL treatment resulted in overexpression of the antioxidant genes Gclm and Heme oxygenase-1 (HO-1), which are induced by the activation of the transcription factor nuclear factor erythroid 2-related factor 2(62). The effect of Mox-LDL on macrophages can be further elucidated by the ability of Mox-LDL to form a more pronounced foam cell phenotype than is possible via Cuox-LDL in THP-1 cells (63). In this context, it was found that Mox-LDL induced an unfolded protein response (UPR), apoptosis, and autophagy in THP-1 cells (63). Recently, our research group explored the potential role of Mox-LDL in the polarization and repolarization of macrophages by evaluating the in vitro effects of Mox-LDL on the polarization of resting M0-macrophages, as well as on the repolarization of M1- and M2-macrophages using a well-established model of human THP-1-derived macrophages. Our results showed that the surface expression of CD80 was highly significant in M1 macrophages when compared to all other subsets. Similarly, CD209 was significantly expressed in M2 macrophages when compared to the other subtypes. Although IL-6 release was minimal in all macrophage types except in the M1 macrophages where it was upregulated as expected, the same M1 macrophage subtype also showed a surprising and significant increase in IL-10 release (64). In our model, physiological concentrations of Mox-LDL had no significant effects on CD80/CD209 expression or IL-6/IL-10 release in Mox-LDL treated M0 macrophages (64). However, treatment of THP-1 M1 macrophages with Mox-LDL had a significant negative effect on IL-10 cytokine secretion (64). Overall, these results highlighted the inability of Mox-LDL to drive M0 macrophages towards an inflammatory or an alternative phenotype. More importantly, the data revealed an important role of Mox-LDL in increasing the pro-inflammatory state in macrophages by reducing the levels of the anti-inflammatory cytokine IL-10.
In summary, LDL modified by the MPO/H2O2/Cl- system or directly by HOCl appears to activate macrophages and induce a stronger reaction in them compared with other forms of oxLDL.
MPO modified LDL and endothelial dysfunction
There is a general consensus among scholars regarding the role of ED in the initiation of atherosclerosis, and oxLDL has the ability to facilitate ED in various manners (7). Meanwhile, the pro-inflammatory nature of LDL oxidized by MPO activates EC at multiple levels (7). In Ea.hy 926 endothelial cells, Mox-LDL treatment induces an increase in the release of IL-8 at concentrations of 100 and 200 mg/ml (61). Similarly, LDL modified by the MPO/H2O2/Cl- system was found to interrupt several EC functions in human umbilical vein endothelial cells (HUVECs) (65). It was reported that pathophysiological concentrations of Mox-LDL decrease tubulogenesis, motility, and wound healing in HUVECs (65). These properties are associated with in vivo angiogenesis. At the molecular level, it was also shown that the observed phenotypic effects can be attributed to miR-22 and the HO-1 gene (Fig. 2) (65). In addition, ECs are known for their role in balancing coagulation and fibrinolysis to form a dynamic equilibrium at their surfaces. Interestingly, Mox-LDL was found to interfere with this role of ECs. In fact, treatment of Ea.hy926 endothelial cells with physiological concentrations of Mox-LDL resulted in an increase in the duration of pericellular fibrinolysis (Fig. 3) (49). Furthermore, this phenotypic change was not related to plasminogen activator inhibitor-1 (PAI-1), tissue plasminogen activator (t-PA), t-PA receptor, or urokinase-type plasminogen activator (uPA) receptor expression, and therefore, it was inferred that Mox-LDL delays pericellular fibrinolysis in a pathway independent of the t-PA- and PAI-1 pathways (49). On a similar note, Mox-LDL treatment was also found to have no transcriptional effect on key fibrinolytic factors such as t-PA, uPA, and their receptors as well the clotting factor XIII in human aortic endothelial cells (HAECs) (66). Fibrin promotes atherosclerosis by disorganizing the normal cobblestone arrangement of EC and causing secretion of IL-8 from ECs (49). Therefore, Mox-LDL may be driving atherosclerotic plaque formation by delaying fibrinolysis, thus prolonging fibrin interaction with ECs that augment endothelial permeability allowing further infiltration and subendothelial accumulation of lipoproteins (Fig. 3). Moreover, Mox-LDL was found to exert differential effects on cell viability between HAECs and bovine aortic endothelial (BAE) cells (66). Indeed, physiological concentrations of Mox-LDL did not exert any significant effect on HAEC viability, whereas the same treatment caused significant death of BAE cells, which is reminiscent of the cytotoxic effect of Cuox-LDL on endothelial cells (66). With regard to ROS production, oxLDL, and particularly the non-physiologically relevant model Cuox-LDL, was reported to increase ROS production in ECs via the receptor LOX-1(67). Interestingly, physiologically relevant concentrations of Mox-LDL did not elevate ROS production in HAECs (66). HOCl-LDL is also reported to exert adverse effects on ECs. Interestingly, HOCl-LDL was reported to exert a dose-dependent anti-proliferative effect on human proximal tubular epithelial cells (HK-2) (68). Additionally, genes associated with inflammation, tissue remodeling, reactive oxygen species metabolism, and cell stress were upregulated in HK-2 cells treated with HOCl-LDL (68). Even though some of the scavenger receptors for HOCl-LDL on macrophages are documented, the receptors that recognize Mox-LDL and enable its uptake by macrophages and ECs are yet to be identified (10). Scavenger receptors known for the uptake of HOCl-LDL in macrophages (CD36 and SR-BI) are also present on the surface of endothelial cells. However, ECs are not capable of forming foam cells when incubated with oxLDL (10). Previously, LOX-1 was reported to bind and take up oxLDL in ECs; however, the notion of whether LOX-1 may function as the receptor for Mox-LDL in EC remains debated. One study reported that LOX-1 is not potentially a receptor for Mox-LDL, since antibodies raised against LOX-1 did not affect IL-8 production induced by Mox-LDL in Ea.hy926(49). Meanwhile, our research group has reported for the first time that the LOX-1 scavenger receptor might regulate the inflammatory response of HAECs to physiological levels of Mox-LDL. It was discovered that Mox-LDL upregulates the expression of its own receptor in HAECs and induces inflammation in the cells via LOX-1 in concert with the induction of this receptor (69). These observations may have important implications with regard to Mox-LDL-driven ED and provide an initial hint to the pathways that are initiated by Mox-LDL during ED and the progression of the atherosclerotic disease (Fig. 3).
Figure 2.
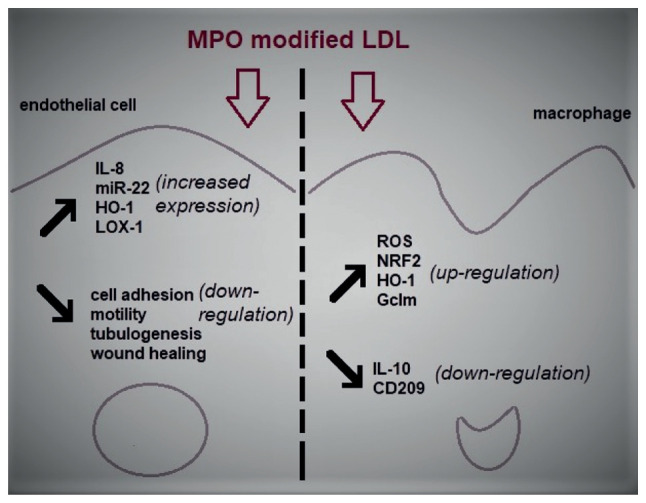
The effects of MPO modified LDL on endothelial cells and macrophage models of atherosclerosis. LDL that has been oxidized by MPO is able to induce ED which is primarily demonstrated by an increase in inflammation and a decrease in the physiological properties of ECs such as cell motility and wound healing. Similarly, Mox-LDL is responsible for the increase in oxidative stress and inflammation in macrophages through the upregulation of the generation of ROS and the downregulation of the major anti-inflammatory cytokine IL-10 in these cells. MPO, myeloperoxidase; LDL, low-density lipoproteins; ED, endothelial dysfunction; EC, endothelial cell; Mox-LDL, MPO oxidized LDL; ROS, reactive oxygen species; miR, microRNA; HO-1, heme oxygenase 1; LOX-1, lectin-like oxLDL receptor-1; NRF2, nuclear factor erythroid 2-related factor 2; Gclm, glutamate-cysteine ligase modifier subunit.
Figure 3.

The primary effects of Mox-LDL during the evolution of atherosclerosis. Mox-LDL is responsible for the activation of ECs through the LOX-1 scavenger receptor. This leads to the recruitment of monocytes to the sub-endothelial layer where they differentiate into macrophages, engulfing modified LDL particles and leading to foam cell formation; at this point, Mox-LDL is capable of driving the polarization of those macrophages towards a more pro-inflammatory phenotype, which may play a major role in the progression of the disease. On the other hand, Mox-LDL can also decrease wound healing and fibrinolysis in ECs leading to more denudation in the endothelium and favoring the formation of blood clots, which will culminate in acute thrombotic events. LDL, low-density lipoproteins; Mox-LDL, myeloperoxidase oxidized LDL; EC, endothelial cell; LOX-1, lectin-like oxLDL receptor-1.
6. Conclusions and future perspectives
In summary, there is a substantial body of evidence suggesting that MPO is implicated in health and in disease. Physiologically, MPO plays a role in innate microbial defenses by catalyzing the formation of powerful reactive oxygen intermediates, which are potent antimicrobial tools against phagocytosed pathogens. Beyond the role of MPO in antimicrobial defenses, there exist other uncharted important physiological roles of MPO (9). Future research should focus on exploring these still unknown or incompletely understood roles in order to fully comprehend the pathophysiological functions of MPO. In addition, it could be argued that considerably more is known regarding the pathological role of MPO than its physiological role. MPO is reported to be implicated in a range of diseases characterized by an ongoing inflammatory state. In prolonged inflammatory states, MPO can cause tissue damage through the chemical modification of molecules as a result of MPO enzymatic activities and through the action of MPO-generated oxidants. In such circumstances, targeting and inhibiting MPO may induce a therapeutic effect. Thus, in the future, it would be an intriguing prospect to determine whether it is possible to design MPO inhibitors that are efficient in vivo, and these may be able to protect against the deleterious effects of MPO while preserving its physiological role. Additionally, there exist species-specific differences in the role of MPO in atherosclerosis and therefore, researchers should be vigilant when translating and interpreting the role of MPO in atherosclerosis.
On that same note, the ubiquitous presence of MPO in atherosclerotic lesions along with the newly reported effects of MPO-modified LDL in this particular context supports the notion that LDL that is oxidized by MPO should now be considered as the more pathophysiological model for LDL oxidation and that future research involving oxLDL should now use this form of modified LDL as a preferred model to study atherosclerosis. However, despite recent advancements in the field and the increase in our understanding of the effects of Mox-LDL in the development of atherosclerosis, there remain several knowledge gaps pertaining to the mechanisms regulating the role of Mox-LDL in macrophage and EC pathobiology during the progression of the disease. In the latter model, as already mentioned, our recent results provide an updated knowledge on the signaling pathways that are promoted by Mox-LDL by providing initial insights into its corresponding scavenger receptor LOX-1(69). In light of the latter finding, conceiving anti-atherogenic strategies that target the Mox-LDL-LOX-1 signaling axis as a therapeutic target may be extremely valuable in the context of treating atherosclerotic diseases and alleviating its symptoms. Moreover, the same strategies could be of an important benefit in the management of other MPO-related pathologies, which include cancer, and neurodegenerative and inflammatory diseases.
Nevertheless, these results are still insufficient and thus, should pave the way for ongoing research to further elucidate additional signaling pathways and novel functions of Mox-LDL; this would ultimately direct scientists towards a more complete understanding of the complex paradigm of ED in atherosclerosis. Moreover, regarding the effect of Mox-LDL on macrophage biology, we recently showed that Mox-LDL might play a potential role in the repolarization of THP-1 macrophages by increasing their pro-inflammatory state through the downregulation of the major anti-inflammatory cytokine IL-10(64). Hopefully, these results will also lead the way to future investigations that will give the scientific community better insights into the immunomodulatory effects of Mox-LDL and the instrumental role that this particular form of modified LDL may play in the progression of atherosclerosis. Additional data that could be generated at this level are highly anticipated since they would certainly be crucial in introducing research strategies that aim to develop therapeutics that target the major pro-inflammatory immunological processes underlying Mox-LDL-driven atherogenesis.
Acknowledgements
Not applicable.
Funding Statement
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
CF drafted the manuscript. JD conceived the review topic, and wrote and edited the manuscript. Both authors have read and approved the final manuscript. Data authentication is not applicable.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Zamocky M, Jakopitsch C, Furtmüller PG, Dunand C, Obinger C. The peroxidase-cyclooxygenase superfamily: Reconstructed evolution of critical enzymes of the innate immune system. Proteins. 2008;72:589–605. doi: 10.1002/prot.21950. [DOI] [PubMed] [Google Scholar]
- 2.Arnhold J. The dual role of myeloperoxidase in immune response. Int J Mol Sci. 2020;21(8057) doi: 10.3390/ijms21218057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aratani Y. Myeloperoxidase: Its role for host defense, inflammation, and neutrophil function. Arch Biochem Biophys. 2018;640:47–52. doi: 10.1016/j.abb.2018.01.004. [DOI] [PubMed] [Google Scholar]
- 4.Daugherty A, Dunn JL, Rateri DL, Heinecke JW. Myeloperoxidase, a catalyst for lipoprotein oxidation, is expressed in human atherosclerotic lesions. J Clin Invest. 1994;94:437–444. doi: 10.1172/JCI117342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Malle E, Waeg G, Schreiber R, Gröne EF, Sattler W, Gröne HJ. Immunohistochemical evidence for the myeloperoxidase/H2O2/halide system in human atherosclerotic lesions: Colocalization of myeloperoxidase and hypochlorite-modified proteins. Eur J Biochem. 2000;267:4495–4503. doi: 10.1046/j.1432-1327.2000.01498.x. [DOI] [PubMed] [Google Scholar]
- 6.Teng N, Maghzal GJ, Talib J, Rashid I, Lau AK, Stocker R. The roles of myeloperoxidase in coronary artery disease and its potential implication in plaque rupture. Redox Rep. 2017;22:51–73. doi: 10.1080/13510002.2016.1256119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Daher J. Other forms of oxidized LDL: Emerging functions (Review) World Acad Sci J. 2020;2(4) [Google Scholar]
- 8.Zámocký M, Hofbauer S, Schaffner I, Gasselhuber B, Nicolussi A, Soudi M, Pirker KF, Furtmüller PG, Obinger C. Independent evolution of four heme peroxidase superfamilies. Arch Biochem Biophys. 2015;574:108–119. doi: 10.1016/j.abb.2014.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vanhamme L, Zouaoui Boudjeltia K, Van Antwerpen P, Delporte C. The other myeloperoxidase: Emerging functions. Arch Biochem Biophys. 2018;649:1–14. doi: 10.1016/j.abb.2018.03.037. [DOI] [PubMed] [Google Scholar]
- 10.Delporte C, Van Antwerpen P, Vanhamme L, Roumeguère T, Zouaoui Boudjeltia K. Low-density lipoprotein modified by myeloperoxidase in inflammatory pathways and clinical studies. Mediators Inflamm. 2013;2013(971579) doi: 10.1155/2013/971579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van der Veen BS, de Winther MP, Heeringa P. Myeloperoxidase: Molecular mechanisms of action and their relevance to human health and disease. Antioxid Redox Signal. 2009;11:2899–2937. doi: 10.1089/ars.2009.2538. [DOI] [PubMed] [Google Scholar]
- 12.Hansson M, Olsson I, Nauseef WM. Biosynthesis, processing, and sorting of human myeloperoxidase. Arch Biochem Biophys. 2006;445:214–224. doi: 10.1016/j.abb.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 13.Malle E, Furtmüller PG, Sattler W, Obinger C. Myeloperoxidase: A target for new drug development? Br J Pharmacol. 2007;152:838–854. doi: 10.1038/sj.bjp.0707358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sugiyama S, Okada Y, Sukhova GK, Virmani R, Heinecke JW, Libby P. Macrophage myeloperoxidase regulation by granulocyte macrophage colony-stimulating factor in human atherosclerosis and implications in acute coronary syndromes. Am J Pathol. 2001;158:879–891. doi: 10.1016/S0002-9440(10)64036-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schindhelm RK, van der Zwan LP, Teerlink T, Scheffer PG. Myeloperoxidase: A useful biomarker for cardiovascular disease risk stratification? Clin Chem. 2009;55:1462–1470. doi: 10.1373/clinchem.2009.126029. [DOI] [PubMed] [Google Scholar]
- 16.Smith JD. Dysfunctional HDL as a diagnostic and therapeutic target. Arterioscler Thromb Vasc Biol. 2010;30:151–155. doi: 10.1161/ATVBAHA.108.179226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hazen SL, Heinecke JW. 3-Chlorotyrosine, a specific marker of myeloperoxidase-catalyzed oxidation, is markedly elevated in low density lipoprotein isolated from human atherosclerotic intima. J Clin Invest. 1997;99:2075–2081. doi: 10.1172/JCI119379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pennathur S, Bergt C, Shao B, Byun J, Kassim SY, Singh P, Green PS, McDonald TO, Brunzell J, Chait A, et al. Human atherosclerotic intima and blood of patients with established coronary artery disease contain high density lipoprotein damaged by reactive nitrogen species. J Biol Chem. 2004;279:42977–42983. doi: 10.1074/jbc.M406762200. [DOI] [PubMed] [Google Scholar]
- 19.Zheng L, Nukuna B, Brennan ML, Sun M, Goormastic M, Settle M, Schmitt D, Fu X, Thomson L, Fox PL, et al. Apolipoprotein A-I is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. J Clin Invest. 2004;114:529–541. doi: 10.1172/JCI21109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bergt C, Pennathur S, Fu X, Byun J, O'Brien K, McDonald TO, Singh P, Anantharamaiah GM, Chait A, Brunzell J, et al. The myeloperoxidase product hypochlorous acid oxidizes HDL in the human artery wall and impairs ABCA1-dependent cholesterol transport. Proc Natl Acad Sci USA. 2004;101:13032–13037. doi: 10.1073/pnas.0405292101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nussbaum C, Klinke A, Adam M, Baldus S, Sperandio M. Myeloperoxidase: A leukocyte-derived protagonist of inflammation and cardiovascular disease. Antioxid Redox Signal. 2013;18:692–713. doi: 10.1089/ars.2012.4783. [DOI] [PubMed] [Google Scholar]
- 22.Yang J, Ji R, Cheng Y, Sun JZ, Jennings LK, Zhang C. L-arginine chlorination results in the formation of a nonselective nitric-oxide synthase inhibitor. J Pharmacol Exp Ther. 2006;318:1044–1049. doi: 10.1124/jpet.106.104422. [DOI] [PubMed] [Google Scholar]
- 23.Zhang C, Reiter C, Eiserich JP, Boersma B, Parks DA, Beckman JS, Barnes S, Kirk M, Baldus S, Darley-Usmar VM, White CR. L-arginine chlorination products inhibit endothelial nitric oxide production. J Biol Chem. 2001;276:27159–27165. doi: 10.1074/jbc.M100191200. [DOI] [PubMed] [Google Scholar]
- 24.Stocker R, Huang A, Jeranian E, Hou JY, Wu TT, Thomas SR, Keaney JF Jr. Hypochlorous acid impairs endothelium-derived nitric oxide bioactivity through a superoxide-dependent mechanism. Arterioscler Thromb Vasc Biol. 2004;24:2028–2033. doi: 10.1161/01.ATV.0000143388.20994.fa. [DOI] [PubMed] [Google Scholar]
- 25.Gimbrone MA Jr, García-Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. 2006;118:620–636. doi: 10.1161/CIRCRESAHA.115.306301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vita JA, Brennan ML, Gokce N, Mann SA, Goormastic M, Shishehbor MH, Penn MS, Keaney JF Jr, Hazen SL. Serum myeloperoxidase levels independently predict endothelial dysfunction in humans. Circulation. 2004;110:1134–1139. doi: 10.1161/01.CIR.0000140262.20831.8F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fu X, Kassim SY, Parks WC, Heinecke JW. Hypochlorous acid oxygenates the cysteine switch domain of pro-matrilysin (MMP-7). A mechanism for matrix metalloproteinase activation and atherosclerotic plaque rupture by myeloperoxidase. J Biol Chem. 2001;276:41279–41287. doi: 10.1074/jbc.M106958200. [DOI] [PubMed] [Google Scholar]
- 28.Wang Y, Rosen H, Madtes DK, Shao B, Martin TR, Heinecke JW, Fu X. Myeloperoxidase inactivates TIMP-1 by oxidizing its N-terminal cysteine residue: An oxidative mechanism for regulating proteolysis during inflammation. J Biol Chem. 2007;282:31826–31834. doi: 10.1074/jbc.M704894200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klebanoff SJ, Kinsella MG, Wight TN. Degradation of endothelial cell matrix heparan sulfate proteoglycan by elastase and the myeloperoxidase-H2O2-chloride system. Am J Pathol. 1993;143:907–917. [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang R, Brennan ML, Fu X, Aviles RJ, Pearce GL, Penn MS, Topol EJ, Sprecher DL, Hazen SL. Association between myeloperoxidase levels and risk of coronary artery disease. JAMA. 2001;286:2136–2142. doi: 10.1001/jama.286.17.2136. [DOI] [PubMed] [Google Scholar]
- 31.Nikpoor B, Turecki G, Fournier C, Théroux P, Rouleau GA. A functional myeloperoxidase polymorphic variant is associated with coronary artery disease in French-Canadians. Am Heart J. 2001;142:336–339. doi: 10.1067/mhj.2001.116769. [DOI] [PubMed] [Google Scholar]
- 32.Kutter D, Devaquet P, Vanderstocken G, Paulus JM, Marchal V, Gothot A. Consequences of total and subtotal myeloperoxidase deficiency: Risk or benefit? Acta Haematol. 2000;104:10–15. doi: 10.1159/000041062. [DOI] [PubMed] [Google Scholar]
- 33.Nicholls SJ, Hazen SL. Myeloperoxidase and cardiovascular disease. Arterioscler Thromb Vasc Biol. 2005;25:1102–1111. doi: 10.1161/01.ATV.0000163262.83456.6d. [DOI] [PubMed] [Google Scholar]
- 34.Heslop CL, Frohlich JJ, Hill JS. Myeloperoxidase and C-reactive protein have combined utility for long-term prediction of cardiovascular mortality after coronary angiography. J Am Coll Cardiol. 2010;55:1102–1109. doi: 10.1016/j.jacc.2009.11.050. [DOI] [PubMed] [Google Scholar]
- 35.Bentzon JF, Otsuka F, Virmani R, Falk E. Mechanisms of plaque formation and rupture. Circ Res. 2014;114:1852–1866. doi: 10.1161/CIRCRESAHA.114.302721. [DOI] [PubMed] [Google Scholar]
- 36.Delporte C, Boudjeltia KZ, Noyon C, Furtmüller PG, Nuyens V, Slomianny MC, Madhoun P, Desmet JM, Raynal P, Dufour D, et al. Impact of myeloperoxidase-LDL interactions on enzyme activity and subsequent posttranslational oxidative modifications of apoB-100. J Lipid Res. 2014;55:747–757. doi: 10.1194/jlr.M047449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yoshida H, Quehenberger O, Kondratenko N, Green S, Steinberg D. Minimally oxidized low-density lipoprotein increases expression of scavenger receptor A, CD36, and macrosialin in resident mouse peritoneal macrophages. Arterioscler Thromb Vasc Biol. 1998;18:794–802. doi: 10.1161/01.atv.18.5.794. [DOI] [PubMed] [Google Scholar]
- 38.Steinberg D. Low density lipoprotein oxidation and its pathobiological significance. J Biol Chem. 1997;272:20963–20966. doi: 10.1074/jbc.272.34.20963. [DOI] [PubMed] [Google Scholar]
- 39.Mehta JL, Chen J, Hermonat PL, Romeo F, Novelli G. Lectin-like, oxidized low-density lipoprotein receptor-1 (LOX-1): A critical player in the development of atherosclerosis and related disorders. Cardiovasc Res. 2006;69:36–45. doi: 10.1016/j.cardiores.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 40.Gao S, Zhao D, Wang M, Zhao F, Han X, Qi Y, Liu J. GW28-e0393 Association between circulating oxidized low-density lipoprotein and atherosclerotic cardiovascular disease: A meta-analysis of prospective observational studies. J Am Coll Cardiol. 2017;70 (16 Suppl):C79–C80. doi: 10.1016/j.cjca.2017.07.015. [DOI] [PubMed] [Google Scholar]
- 41.Holvoet P, Mertens A, Verhamme P, Bogaerts K, Beyens G, Verhaeghe R, Collen D, Muls E, Van de Werf F. Circulating oxidized LDL is a useful marker for identifying patients with coronary artery disease. Arterioscler Thromb Vasc Biol. 2001;21:844–848. doi: 10.1161/01.atv.21.5.844. [DOI] [PubMed] [Google Scholar]
- 42.Kita T, Kume N, Minami M, Hayashida K, Murayama T, Sano H, Moriwaki H, Kataoka H, Nishi E, Horiuchi H, et al. Role of oxidized LDL in atherosclerosis. Ann N Y Acad Sci. 2001;947:199–206. doi: 10.1111/j.1749-6632.2001.tb03941.x. [DOI] [PubMed] [Google Scholar]
- 43.Mitra S, Goyal T, Mehta JL. Oxidized LDL, LOX-1 and atherosclerosis. Cardiovasc Drugs Ther. 2011;25:419–429. doi: 10.1007/s10557-011-6341-5. [DOI] [PubMed] [Google Scholar]
- 44.Yoshida H, Kisugi R. Mechanisms of LDL oxidation. Clin Chim Acta. 2010;411:1875–1882. doi: 10.1016/j.cca.2010.08.038. [DOI] [PubMed] [Google Scholar]
- 45.Carr AC. Hypochlorous acid-modified low-density lipoprotein inactivates the lysosomal protease cathepsin B: Protection by ascorbic and lipoic acids. Redox Rep. 2001;6:343–349. doi: 10.1179/135100001101536526. [DOI] [PubMed] [Google Scholar]
- 46.Sokolov AV, Chekanov AV, Kostevich VA, Aksenov DV, Vasilyev VB, Panasenko OM. Revealing binding sites for myeloperoxidase on the surface of human low density lipoproteins. Chem Phys Lipids. 2011;164:49–53. doi: 10.1016/j.chemphyslip.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 47.Moguilevsky N, Zouaoui Boudjeltia K, Babar S, Delrée P, Legssyer I, Carpentier Y, Vanhaeverbeek M, Ducobu J. Monoclonal antibodies against LDL progressively oxidized by myeloperoxidase react with ApoB-100 protein moiety and human atherosclerotic lesions. Biochem Biophys Res Commun. 2004;323:1223–1228. doi: 10.1016/j.bbrc.2004.08.220. [DOI] [PubMed] [Google Scholar]
- 48.Vaes M, Zouaoui Boudjeltia KZ, Van Antwerpen P, Babar S, Deger F, Neve J, Vanhaeverbeek M, Ducobu J. ThP15:146 Low-density lipoprotein oxidation by myeloperoxidase occurs in the blood circulation during hemodialysis. Atherosclerosis Suppl. 2006;7(525) [Google Scholar]
- 49.Zouaoui Boudjeltia K, Daher J, Van Antwerpen P, Moguilevsky N, Delree P, Ducobu J, Raes M, Badran B, Vanhaeverbeek M, Brohee D, et al. Exposure of endothelial cells to physiological levels of myeloperoxidase-modified LDL delays pericellular fibrinolysis. PLoS One. 2012;7(e38810) doi: 10.1371/journal.pone.0038810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Podrez EA, Schmitt D, Hoff HF, Hazen SL. Myeloperoxidase-generated reactive nitrogen species convert LDL into an atherogenic form in vitro. J Clin Invest. 1999;103:1547–1560. doi: 10.1172/JCI5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leeuwenburgh C, Hardy MM, Hazen SL, Wagner P, Oh-ishi S, Steinbrecher UP, Heinecke JW. Reactive nitrogen intermediates promote low density lipoprotein oxidation in human atherosclerotic intima. J Biol Chem. 1997;272:1433–1436. doi: 10.1074/jbc.272.3.1433. [DOI] [PubMed] [Google Scholar]
- 52.Wang Z, Nicholls SJ, Rodriguez ER, Kummu O, Hörkkö S, Barnard J, Reynolds WF, Topol EJ, DiDonato JA, Hazen SL. Protein carbamylation links inflammation, smoking, uremia and atherogenesis. Nat Med. 2007;13:1176–1184. doi: 10.1038/nm1637. [DOI] [PubMed] [Google Scholar]
- 53.Nicholson AC, Frieda S, Pearce A, Silverstein RL. Oxidized LDL binds to CD36 on human monocyte-derived macrophages and transfected cell lines. Evidence implicating the lipid moiety of the lipoprotein as the binding site. Arterioscler Thromb Vasc Biol. 1995;15:269–275. doi: 10.1161/01.atv.15.2.269. [DOI] [PubMed] [Google Scholar]
- 54.Isa SA, Ruffino JS, Ahluwalia M, Thomas AW, Morris K, Webb R. M2 macrophages exhibit higher sensitivity to oxLDL-induced lipotoxicity than other monocyte/macrophage subtypes. Lipids Health Dis. 2011;10(229) doi: 10.1186/1476-511X-10-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Montano EN, Boullier A, Almazan F, Binder CJ, Witztum JL, Hartvigsen K. Development and application of a nonradioactive binding assay of oxidized low-density lipoprotein to macrophage scavenger receptors. J Lipid Res. 2013;54:3206–3214. doi: 10.1194/jlr.D040923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Houben T, Oligschlaeger Y, Bitorina AV, Hendrikx T, Walenbergh SMA, Lenders MH, Gijbels MJJ, Verheyen F, Lütjohann D, Hofker MH, et al. Blood-derived macrophages prone to accumulate lysosomal lipids trigger oxLDL-dependent murine hepatic inflammation. Sci Rep. 2017;7(12550) doi: 10.1038/s41598-017-13058-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Westendorf T, Graessler J, Kopprasch S. Hypochlorite-oxidized low-density lipoprotein upregulates CD36 and PPARgamma mRNA expression and modulates SR-BI gene expression in murine macrophages. Mol Cell Biochem. 2005;277:143–152. doi: 10.1007/s11010-005-5873-z. [DOI] [PubMed] [Google Scholar]
- 58.Woenckhaus C, Kaufmann A, Bussfeld D, Gemsa D, Sprenger H, Gröne HJ. Hypochlorite-modified LDL: Chemotactic potential and chemokine induction in human monocytes. Clin Immunol Immunopathol. 1998;86:27–33. doi: 10.1006/clin.1997.4453. [DOI] [PubMed] [Google Scholar]
- 59.Vicca S, Hennequin C, Nguyen-Khoa T, Massy ZA, Descamps-Latscha B, Drüeke TB, Lacour B. Caspase-dependent apoptosis in THP-1 cells exposed to oxidized low-density lipoproteins. Biochem Biophys Res Commun. 2000;273:948–954. doi: 10.1006/bbrc.2000.3017. [DOI] [PubMed] [Google Scholar]
- 60.Marsche G, Zimmermann R, Horiuchi S, Tandon NN, Sattler W, Malle E. Class B scavenger receptors CD36 and SR-BI are receptors for hypochlorite-modified low density lipoprotein. J Biol Chem. 2003;278:47562–47570. doi: 10.1074/jbc.M308428200. [DOI] [PubMed] [Google Scholar]
- 61.Boudjeltia KZ, Legssyer I, Van Antwerpen P, Kisoka RL, Babar S, Moguilevsky N, Delree P, Ducobu J, Remacle C, Vanhaeverbeek M, Brohee D. Triggering of inflammatory response by myeloperoxidase-oxidized LDL. Biochem Cell Biol. 2006;84:805–812. doi: 10.1139/o06-061. [DOI] [PubMed] [Google Scholar]
- 62.Calay D, Rousseau A, Mattart L, Nuyens V, Delporte C, Van Antwerpen P, Moguilevsky N, Arnould T, Boudjeltia KZ, Raes M. Copper and myeloperoxidase-modified LDLs activate Nrf2 through different pathways of ROS production in macrophages. Antioxid Redox Signal. 2010;13:1491–1502. doi: 10.1089/ars.2009.2971. [DOI] [PubMed] [Google Scholar]
- 63.Vlaminck B, Calay D, Genin M, Sauvage A, Ninane N, Zouaoui Boudjeltia K, Raes M, Michiels C. Effects of copper sulfate-oxidized or myeloperoxidase-modified LDL on lipid loading and programmed cell death in macrophages under hypoxia. Hypoxia (Auckl) 2014;2:153–169. doi: 10.2147/HP.S65242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bazzi S, Frangie C, Azar E, Daher J. The effect of myeloperoxidase-oxidized LDL on THP-1 macrophage polarization and repolarization. Innate Immun. 2022;28(2):91–103. doi: 10.1177/17534259221090679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Daher J, Martin M, Rousseau A, Nuyens V, Fayyad-Kazan H, Van Antwerpen P, Courbebaisse G, Martiat P, Badran B, Dequiedt F, et al. Myeloperoxidase oxidized LDL interferes with endothelial cell motility through miR-22 and heme oxygenase 1 induction: Possible involvement in reendothelialization of vascular injuries. Mediators Inflamm. 2014;2014(134635) doi: 10.1155/2014/134635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.El Samad G, Bazzi S, Karam M, Boudjeltia KZ, Vanhamme L, Daher J. Effect of myeloperoxidase modified LDL on bovine and human aortic endothelial cells. Exp Ther Med. 2019;18:4567–4574. doi: 10.3892/etm.2019.8109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Neri Serneri GG, Coppo M, Bandinelli M, Paoletti P, Toscano T, Micalizzi E, Chiostri M, Boddi M. Exaggerated myocardial oxLDL amount and LOX-1 receptor over-expression associated with coronary microvessel inflammation in unstable angina. Atherosclerosis. 2013;226:476–482. doi: 10.1016/j.atherosclerosis.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 68.Porubsky S, Schmid H, Bonrouhi M, Kretzler M, Malle E, Nelson PJ, Gröne HJ. Influence of native and hypochlorite-modified low-density lipoprotein on gene expression in human proximal tubular epithelium. Am J Pathol. 2004;164:2175–2187. doi: 10.1016/S0002-9440(10)63775-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.El-Hajjar L, Hindieh J, Andraos R, El-Sabban M, Daher J. Myeloperoxidase-oxidized LDL activates human aortic endothelial cells through the LOX-1 scavenger receptor. Int J Mol Sci. 2002;23(2837) doi: 10.3390/ijms23052837. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.