

Abstract
Two 3-hydroxybenzoate-inducible gentisate 1,2-dioxygenases were purified to homogeneity from Pseudomonas alcaligenes NCIB 9867 (P25X) and Pseudomonas putida NCIB 9869 (P35X), respectively. The estimated molecular mass of the purified P25X gentisate 1,2-dioxygenase was 154 kDa, with a subunit mass of 39 kDa. Its structure is deduced to be a tetramer. The pI of this enzyme was established to be 4.8 to 5.0. The subunit mass of P35X gentisate 1,2-dioxygenase was 41 kDa, and this enzyme was deduced to exist as a dimer, with a native molecular mass of about 82 kDa. The pI of P35X gentisate 1,2-dioxygenase was around 4.6 to 4.8. Both of the gentisate 1,2-dioxygenases exhibited typical saturation kinetics and had apparent Kms of 92 and 143 μM for gentisate, respectively. Broad substrate specificities were exhibited towards alkyl and halogenated gentisate analogs. Both enzymes had similar kinetic turnover characteristics for gentisate, with kcat/Km values of 44.08 × 104 s−1 M−1 for the P25X enzyme and 39.34 × 104 s−1 M−1 for the P35X enzyme. Higher kcat/Km values were expressed by both enzymes against the substituted gentisates. Significant differences were observed between the N-terminal sequences of the first 23 amino acid residues of the P25X and P35X gentisate 1,2-dioxygenases. The P25X gentisate 1,2-dioxygenase was stable between pH 5.0 and 7.5, with the optimal pH around 8.0. The P35X enzyme showed a pH stability range between 7.0 and 9.0, and the optimum pH was also 8.0. The optimal temperature for both P25X and P35X gentisate 1,2-dioxygenases was around 50°C, but the P35X enzyme was more heat stable than that from P25X. Both enzymes were strongly stimulated by 0.1 mM Fe2+ but were completely inhibited by the presence of 5 mM Cu2+. Partial inhibition of both enzymes was also observed with 5 mM Mn2+, Zn2+, and EDTA.
The degradative capabilities of microorganisms have contributed significantly to the removal of environmental pollutants. Despite the diversity of chemical structures presented to microorganisms, aromatic hydrocarbons are invariably converted into dihydroxylated aromatic intermediates. The two hydroxyl groups may be placed ortho to each other, as in catechol and protocatechuate, or para to each other, as in gentisate and homogentisate. The formation of these dihydroxy intermediates destabilizes the benzene ring and facilitates their cleavage and subsequent degradation via either the ortho, meta, gentisate, or homogentisate pathway (6, 7).
Gentisate (2,5-dihydroxybenzoic acid) serves as the key intermediate and focal point in the biodegradation of a large number of simple and complex aromatic compounds by microorganisms (8, 16, 17, 19, 21, 22, 25). The gentisate pathway is widely distributed throughout the microbial world (5, 8, 14, 24). Microbial degradation of gentisate is initiated by gentisate 1,2-dioxygenase (EC 1.13.11.4) through extradiol cleavage of the benzene ring (15). The ring fission product, maleylpyruvate, is isomerized to fumarylpyruvate by some microorganisms (2, 15), while in others, it is converted without isomerization to central metabolites, such as citraconic acid (1, 11, 12).
Several enzymes of the gentisate pathway present in Pseudomonas alcaligenes NCIB 9867 and Pseudomonas putida NCIB 9869 were reported to possess broad substrate specificities (11, 12, 18, 19). The presence of 6-hydroxylases, gentisate 1,2-dioxygenases, and maleylpyruvate hydrolases of broad substrate specificity has given these two species the ability to degrade several halogenated xylenols in addition to the unsubstituted xylenols (11, 12, 18, 19). Comparative studies of the properties of different gentisate dioxygenases from different bacterial species will facilitate the identification of the structural determinants of particular functions, such as substrate specificity and catalysis. Such structure-function studies will prove invaluable for designing microorganisms with better enzymes for environmental remediation.
In this paper, we report the purification of two different gentisate 1,2-dioxygenases from P. alcaligenes NCIB 9867 and P. putida NCIB 9869, respectively. The enzymes were characterized with respect to substrate specificities, kinetic properties, and N-terminal amino acid sequences.
MATERIALS AND METHODS
Organisms and growth conditions.
P. alcaligenes NCIB 9867, designated P25X, and P. putida NCIB 9869, designated P35X, were gifts from D. J. Hopper (Wales University, Aberystwyth, United Kingdom). Both strains were isolated from Hull River mud by elective culture enrichment with their respective carbon sources. P25X and P35X cells were grown in 800 ml of minimal medium (10) containing 20 mM sodium lactate at 32°C with shaking at 250 rpm until the optical density reached 0.5 to 0.6 when measured at 580 nm. 3-Hydroxybenzoate (3-HBA) was then introduced to the culture to a final concentration of 2.5 mM, and the culture was allowed to incubate for another 8 h before being harvested.
Preparation of crude extracts.
Bacteria were harvested by centrifugation at 10,000 × g for 10 min. The pellet (about 25 g) was washed twice with buffer A (50 mM MOPS [morpholine propane sulfonic acid] buffer containing 0.1 mM ferrous ammonium sulfate, 2 mM l-cysteine, and 10% [wt/vol] glycerol, pH 7.4) and resuspended in 2 vol of the same buffer. The cell suspension was sonicated with a MSE-Soniprep 150 for a total of 20 min, with 15-s cooling intervals between every two 15-s pulses. During sonication, the cell suspension was maintained at about 4°C in an ice-alcohol slurry. Cell debris and unbroken cells were removed by centrifugation at 25,000 × g for 30 min. The supernatant was collected and used as the starting material for purification of gentisate 1,2-dioxygenase.
Purification procedure.
Gentisate 1,2-dioxygenase from P35X was purified by a procedure consisting of the following steps, performed at 4°C unless otherwise stated.
(i) Heat treatment.
Crude extract of P35X was kept in a beaker and heated with constant stirring in a 65°C water bath. When the protein solution reached 60°C, it was removed immediately and then rapidly cooled in an ice bath. The denatured protein was removed by centrifugation at 25,000 × g for 30 min.
(ii) Ammonium sulfate fractionation.
The heat-denatured supernatant was first brought to 40% saturation (242 g/liter) by the addition of ammonium sulfate over a period of 20 min with constant stirring. The suspension was equilibrated for an additional 30 min, followed by centrifugation for 30 min at 25,000 × g. The supernatant was collected and again brought to 55% ammonium sulfate saturation (an additional 96 g/liter) in the same manner as described and then centrifuged. The ammonium sulfate pellet was resuspended in buffer A and dialyzed overnight against buffer A at 4°C.
(iii) DEAE-Sepharose fast-flow chromatography.
The dialyzed sample was loaded onto a column of fast-flow DEAE-Sepharose CL-6B (3 by 10 cm) that had been preequilibrated with buffer A. The column was then washed with 100 ml of buffer A containing 0.1 M NaCl. Gentisate 1,2-dioxygenase activity was eluted with a linear gradient from 0.1 to 0.5 M NaCl in 400 ml of buffer A. The flow rate was maintained at 0.5 ml/min. Fractions containing 40% or greater of the peak fraction were pooled.
(iv) Mini Prep cell.
Pooled fractions from the previous step were concentrated with ultrafiltration membrane cones (Centriflo CF 25; Amicon). The concentrated enzyme was then applied to a cylindrical polyacrylamide gel in a Mini Prep cell (Bio-Rad, Richmond, Calif.). The concentration of the stacking gel was 4%, and that of the separating gel was 5%. After running for about 5 h at 300 V in Tris-glycine running buffer, the gentisate 1,2-dioxygenase was eluted with buffer A at a flow rate of 0.05 ml/min and 0.5-ml fractions were collected. The fractions containing gentisate 1,2-dioxygenase activity were checked for homogeneity by native polyacrylamide gel electrophoresis (PAGE) before they were pooled and concentrated.
The general procedure for the purification of gentisate 1,2-dioxygenase from P25X was similar to that described above, except that the heat treatment step was omitted.
Enzyme assay and protein determination.
Gentisate 1,2-dioxygenase activity was assayed spectrophotometrically at 23°C by measuring the formation of maleylpyruvate (15), which could be monitored by measuring the increase in absorbance at 330 nm with a Shimadzu spectrophotometer model UV 240. Activity was assayed in 3 ml of reaction mixture containing 0.33 mM gentisate in 0.1 M phosphate buffer, pH 7.4. The molar extinction coefficient of 10.8 × 103 M−1 cm−1 was used (5) in the calculation of specific activity. One enzyme unit is defined as the amount of enzyme that produces 1 μmol of maleylpyruvate per min at 23°C. To assay the effect of activation and inhibition by metal ions, enzyme activity was measured after 15 min of incubation with the respective metal ions. Protein concentrations were determined by using the Bradford assay (3) with crystalline bovine serum albumin (Sigma, St. Louis, Mo.) as the protein standard.
PAGE and molecular weight determinations.
Native gel electrophoresis was performed in slab gels containing 10% acrylamide for the separating gel and 5% acrylamide for the stacking gel. Sodium dodecyl sulfate (SDS)-PAGE was carried out in gels containing 10% acrylamide for the separating gel and 5% acrylamide for the stacking gel.
For molecular weight determinations of enzyme subunits by SDS-PAGE, the following molecular weight standards were used: phosphorylase A (Mr, 94,000), bovine serum albumin (Mr, 67,000), ovalbumin (Mr, 43,000), carbonic anhydrase (Mr, 30,000), trypsin inhibitor (Mr, 20,100), and lysozyme (Mr, 14,400).
The molecular weight of the native protein was further determined by gel filtration with Sephacryl S-200 chromatography (1.6 by 100 cm) with catalase (Mr, 232,000), aldolase (Mr, 158,000), bovine serum albumin (Mr, 67,000), and chymotrypsinogen A (Mr, 25,000) as molecular weight standards. The column was equilibrated and eluted with 0.1 M phosphate buffer (pH 7.4) at a flow rate of 0.2 ml/min.
Determination of amino acid sequence.
Electroblotting of protein bands from a SDS-PAGE minigel to polyvinylidene difluoride membrane (0.2 μm) was carried out with the Bio-Rad blot cell in accordance with the manufacturer’s instructions. The protein band was stained and excised from the membrane, and the NH2-terminal amino acid sequence was determined with an Applied Biosystem model 477A protein sequencer.
RESULTS
Purification of gentisate 1,2-dioxygenases.
The overall schemes for the purification of gentisate 1,2-dioxygenases from P. alcaligenes P25X and P. putida P35X are summarized and presented in Tables 1 and 2, respectively.
TABLE 1.
Purification of gentisate 1,2-dioxygenase from P. alcaligenes P25X
| Purification step | Volume (ml) | Total activity (U) | Total protein (mg) | Sp act (U/mg) | Yield (%) | Purification (fold) |
|---|---|---|---|---|---|---|
| Crude extract | 47 | 1,461 | 3,267 | 0.45 | 100.00 | 1.00 |
| 30–50% (NH4)2SO4 fractionation | 12 | 735 | 1,176 | 0.63 | 58.31 | 1.40 |
| DEAE-Sepharose CL-6B | 33 | 593 | 97 | 6.11 | 40.59 | 13.58 |
| Mini Prep cell | 6 | 174 | 7.5 | 23.20 | 11.91 | 51.56 |
TABLE 2.
Purification of gentisate 1,2-dioxygenase from P. putida P35X
| Purification step | Volume (ml) | Total activity (U) | Total protein (mg) | Sp act (U/mg) | Yield (%) | Purifi-cation (fold) |
|---|---|---|---|---|---|---|
| Crude extract | 50 | 2,473 | 3,435 | 0.72 | 100.00 | 1.00 |
| Heat treatment (60°C) | 41 | 1,562 | 1,838 | 0.85 | 63.20 | 1.18 |
| 40–55% (NH4)2SO4 fractionation | 8.3 | 1,175 | 1,049 | 1.12 | 47.51 | 1.56 |
| DEAE-Sepharose CL-6B | 40 | 847 | 83 | 10.20 | 34.25 | 14.17 |
| Mini Prep cell | 8 | 220 | 5.1 | 43.14 | 8.90 | 59.92 |
Effect of Fe2+ and chemical additives.
The purified P25X and P35X enzymes were found to be highly unstable in 50 mM MOPS buffer with no additives. After 24 h of storage at 4°C in MOPS buffer, the activities of the P25X and P35X enzymes decreased to 22.5 and 33.9% of the original activities, respectively. No activity was detectable for either enzyme after 96 h. Addition of 0.1 mM Fe2+ (ferrous ammonium sulfate) appeared to have greater activating and stabilizing effects on both gentisate 1,2-dioxygenases than that of MOPS buffer containing either 2 mM cysteine or 10% (vol/vol) glycerol alone. Maximum activating and stabilizing effects of both gentisate 1,2-dioxygenases were observed in the presence of all three additives.
The long-term stabilities of both P25X and P35X gentisate 1,2-dioxygenases in MOPS buffer were tested by the addition of all three chemicals mentioned above and storage at −20°C. Under such conditions, 86.5, 70.9, and 42.9% of P25X gentisate 1,2-dioxygenase activity was found to remain after 3, 6, and 12 months of storage, respectively. For the P35X gentisate 1,2-dioxygenase, 42.5, 23.1, and 3.5% of the activity remained after similar periods of storage. However, addition of 0.1 mM freshly prepared Fe2+ could reactivate the activities of both gentisate 1,2-dioxygenases after 12 months of storage in 50 mM MOPS buffer already containing 0.1 mM Fe2+, 2 mM cysteine, and 10% (vol/vol) glycerol. A more pronounced activation effect by Fe2+ was observed for the P35X gentisate 1,2-dioxygenase than for the P25X enzyme, as 82.3% and 77.2%, respectively, of the original activities were regained upon addition of 0.1 mM freshly prepared Fe2+.
Metal ions, such as K+, Na+, and Ca2+, did not stimulate or inhibit the activities of the purified P25X and P35X gentisate 1,2-dioxygenases. Both enzymes were completely inactivated in the presence of 5 mM Cu2+. Mn2+ and EDTA at 5 mM concentrations were observed to have more drastic reducing effects on the activity of P35X gentisate 1,2-dioxygenase than on that of the P25X enzyme (Table 3).
TABLE 3.
Effects of some metal ions on P25X and P35X gentisate 1,2-dioxygenases
| Metal ion (5 mM) | Relative activity (%)a
|
|
|---|---|---|
| P25X | P35X | |
| None | 100.00 | 100.00 |
| NaCl | 98.33 | 106.02 |
| KCl | 95.70 | 107.76 |
| MgCl2 | 94.62 | 66.00 |
| CaCl2 | 97.63 | 101.00 |
| MnSO4 | 73.96 | 22.36 |
| ZnSO4 | 2.58 | 4.69 |
| CuSO4 | 0.00 | 0.00 |
| EDTA | 60.24 | 4.95 |
Enzymes were preincubated with 5 mM different metal ions for 15 min before the activities were assayed. The activity of the enzyme in the absence of any metal ion was considered as 100% activity.
Effects of temperature and pH on activity and stability.
Optimal activities of both gentisate 1,2-dioxygenases were observed at 50°C. Gentisate 1,2-dioxygenase from P35X was found to retain 73% of its activity when it was exposed to a temperature of 50°C for 5 min, whereas the enzyme from P25X only retained 35% of its activity. Different chemical reagents were tested for their stabilizing effects by exposing the purified gentisate 1,2-dioxygenase to the reagent under test in 50 mM MOPS buffer at 50°C for 20 min. The activities of P25X and P35X gentisate 1,2-dioxygenases could be stabilized and activated by reagents such as 0.1 mM ferrous ammonium sulfate. Twofold- and fivefold-higher activities were observed for the P25X and P35X enzymes, respectively, compared to those when stored in MOPS buffer alone. Higher activities were also detected in the presence of 2 mM l-cysteine and 5 mM 2-mercaptoethanol. Glycerol at 10% (vol/vol) concentration had a greater stabilizing effect on the P25X gentisate 1,2-dioxygenase than on the P35X enzyme. Ammonium sulfate and bovine serum albumin did not stabilize or activate either enzyme (data not shown).
The pH dependence of the purified gentisate 1,2-dioxygenases was investigated by assaying for enzyme activity in 0.1 M phosphate buffers of different pH values. Both P25X and P35X gentisate 1,2-dioxygenases showed maximum activities around pH 8.0. The stability of purified gentisate 1,2-dioxygenase at different pH values was determined by incubating the enzyme in the test buffer for 20 h at 4°C before an assay was performed. Maximum stability of P35X gentisate 1,2-dioxygenase was observed between pH 7.0 and 9.0, while the enzyme from P25X was found to be more stable at lower pHs (5.0 to 7.5).
Molecular properties.
Electrophoresis of the fractions which showed optimal enzyme activities under nondenaturing conditions (Fig. 1A) showed a single band of protein in the gel stained with Coomassie brilliant blue for both P25X and P35X. When analyzed by SDS-PAGE (Fig. 1B), the sample from P25X yielded a single band with an approximate molecular weight of 39,000 ± 1,000 (Fig. 1B, lane 1) while that from P35X exhibited a single band with an approximate molecular weight of 41,000 ± 1,000 (Fig. 1B, lane 2). Molecular weight determination of gentisate 1,2-dioxygenase by gel filtration chromatography with Sephacryl S-200 established a holoenzyme molecular weight of 154,000 ± 6,000 for gentisate 1,2-dioxygenase from P25X and 82,000 ± 4,000 for gentisate 1,2-dioxygenase from P35X. Based on molecular weight estimation of the subunit from the SDS-PAGE gel, the enzyme from P25X appears to be comprised of a tetrameric protein while the enzyme from P35X is deduced to be a dimeric protein. The NH2-terminal amino acid sequences of gentisate 1,2-dioxygenases from both microorganisms were determined by automated Edman degradation (Table 4).
FIG. 1.

Native 10% PAGE (A) and SDS-10% PAGE (B) of purified gentisate 1,2-dioxygenases from P25X and P35X. Lanes M, molecular mass markers (in kilodaltons); lanes 1, 5 μg of purified gentisate 1,2-dioxygenase from P25X; lanes 2, 5 μg of purified gentisate 1,2-dioxygenase from P35X.
TABLE 4.
Alignment of the N-terminal amino acid sequences of P25X, P35X, and P. acidovorans gentisate 1,2-dioxygenases
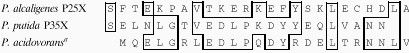 |
Data for P. acidovorans are from reference 9.
When the purified gentisate 1,2-dioxygenase from P25X was analyzed by isoelectric focusing, the pI of the enzyme was found to be 4.8 to 5.0. Similarly, the pI of gentisate 1,2-dioxygenase from P35X was established to be 4.6 to 4.8.
Kinetic properties.
Spectrophotometric assays were performed in air-saturated 0.1 M phosphate buffer with the same amount of enzyme for the determination of Km values. The concentrations of gentisate were varied from 2 to 1,000 μM. Both enzymes displayed Michaelis-Menten kinetics, and Lineweaver-Burk plots of enzyme activity yielded apparent Km values of 92 ± 5 and 143 ± 7 μM for the P25X and P35X gentisate 1,2-dioxygenases, respectively. When both enzymes were assayed against gentisate, the P35X enzyme exhibited a slightly higher kinetic turnover of 3,376 min−1 site−1 while the P25X enzyme exhibited a kcat value of 2,433 min−1 site−1.
Substrate specificity.
Both purified gentisate 1,2-dioxygenases exhibited broad substrate specificities towards alkyl and halogenated gentisates. The Km values for gentisates containing substitutions at C-3 position of the ring were generally lower than those towards the unsubstituted gentisate for both enzymes (Table 5). The two enzymes exhibited similar kinetic turnover characteristics for gentisate. The catalytic efficiency as indicated by kcat/Km was established to be 44.08 × 104 s−1 M−1 for the P25X enzyme and 39.34 × 104 s−1 M−1 for the P35X enzyme. Higher kcat/Km values were expressed by both enzymes against the substituted gentisates than against gentisate. However, notable differences were observed between the kcat/Km values of the two enzymes against 3-bromo- and 3-isopropyl-gentisates. The P35X enzyme had a threefold higher kcat/Km value against 3-bromogentisate than against gentisate, while the P25X enzyme showed similar values against both substrates. Instead, a 1.5-fold-higher kcat/Km value was observed against 3-bromogentisate than against gentisate for the P25X enzyme.
TABLE 5.
Substrate specificities of purified gentisate 1,2-dioxygenases from P25X and P35Xa
| Substrate | P25X
|
P35X
|
||||||
|---|---|---|---|---|---|---|---|---|
| % Vmax | Km (μM) | Kcat (min−1 site−1) | Kcat/Km (s−1 M−1) (104) | % Vmax | Km (μM) | Kcat (min−1 site−1) | Kcat/Km (s−1 M−1) (104) | |
| Gentisate | 100 | 92 ± 5 | 2,433 ± 39 | 44.08 ± 3 | 100 | 143 ± 7 | 3,376 ± 41 | 39.34 ± 2 |
| 3-Methylgentisate | 127 ± 2 | 76 ± 3 | 3,098 ± 50 | 67.94 ± 4 | 60 ± 1 | 36 ± 1 | 2,015 ± 32 | 93.28 ± 4 |
| 3-Bromogentisate | 25 ± 0.5 | 15 ± 1 | 613 ± 12 | 68.11 ± 5 | 25 ± 0.5 | 11 ± 0.3 | 833 ± 16 | 126.21 ± 2 |
| 3-Isopropylgentisate | 65 ± 1 | 53 ± 2 | 1,573 ± 25 | 49.47 ± 3 | 3.8 ± 0.1 | 4.3 ± 0.03 | 129 ± 3 | 50.00 ± 1 |
The Vmax values of purified P25X and P35X gentisate 1,2-dioxygenases against gentisate were considered as 100%. This value was 15.8 μmol of product formed min−1 mg−1 for the P25X enzyme and 41.17 μmol of product formed min−1 mg−1 for the P35X enzyme. All other Vmax values were expressed relative to the Vmax against gentisate. Km and kcat were determined at atmospheric O2 saturations. Their values were calibrated from Lineweaver-Burk plots in the presence of 2 to 1,000 μM respective substrate. All values are means ± standard errors from triplicate measurements and are derived from three separate experiments.
DISCUSSION
We have described the purification to homogeneity of gentisate 1,2-dioxygenases from P. alcaligenes P25X and P. putida P35X after ammonium sulfate precipitation of the crude extracts. The data presented here show that P. alcaligenes P25X produced a gentisate dioxygenase which appeared to be similar in both molecular mass and subunit structure to previously reported gentisate 1,2-dioxygenases from Moraxella osloensis (5), Pseudomonas testosteroni (9), Pseudomonas acidovorans (9), Klebsiella pneumoniae (20), and Sphingomonas sp. strain RW5 (23). Gentisate 1,2-dioxygenases from all five bacteria investigated to date have been found to be tetrameric in nature, with identical subunits of around 40 kDa. However, the N-terminal sequence of the P. alcaligenes P25X enzyme showed no homology to those reported for P. testosteroni (9), P. acidovorans (9), and Sphingomonas sp. strain RW5 (23). The P. putida P35X gentisate 1,2-dioxygenase differed both in molecular mass (82 kDa) and subunit structure [(α)2] from all the gentisate 1,2-dioxygenases that have been studied. However, its N-terminal amino acid sequence was 58% identical to that of the gentisate 1,2-dioxygenase from P. acidovorans. Five conserved amino acid residues were shared by P. alcaligenes P25X and P. putida P35X enzymes, while only a single amino acid residue (L) was conserved among the three sequences (Table 4). No homology in the N-terminal amino acid sequences could be established between the gentisate 1,2-dioxygenases of P. putida P35X and those of P. testosteroni (9) and Sphingomonas sp. strain RW5 (23).
The enzymes from both P. alcaligenes P25X and P. putida P35X were found to be activated and stabilized by 0.1 mM Fe2+. Both enzymes were very unstable and could not be purified in the absence of Fe2+. Gentisate 1,2-dioxygenases from both bacteria probably contain Fe2+ in their active sites, as 40 and 95% inhibition of the enzymatic activities by 5 mM EDTA was observed for the P25X and P35X enzymes, respectively. The addition of 2 mM cysteine and 10% glycerol was found to further stabilize the activities of both enzymes. These observations were similar to those reported for gentisate 1,2-dioxygenases from M. osloensis, P. testosteroni, P. acidovorans, and K. pneumoniae.
When investigating the gentisate 1,2-dioxygenase produced by P. acidovorans, Harpel and Lipscomb (9) found that the enzyme routinely migrated in SDS-PAGE gels as multiple closely spaced bands in the molecular weight range of 37,200 to 39,800. They suggested that the multiple bands represented different processing forms or breakdown products of a single enzyme. Unlike the enzyme from P. acidovorans, P. alcaligenes P25X and P. putida P35X enzymes both resolved as single subunit bands of molecular weights 39,000 ± 1,000 and 41,000 ± 1,000, respectively, during SDS-PAGE. The apparent Km of the P25X enzyme was 92 ± 5 μM, and that of the P35X enzyme was 143 ± 7 μM. The Km of the P25X enzyme for gentisate was closer to the Km values reported for P. testosteroni (85 μM) and P. acidovorans (74 μM) but was considerably higher than those reported for M. osloensis (7.1 μM) and K. pneumoniae (52 μM) (Table 6).
TABLE 6.
Properties of gentisate 1,2-dioxygenases from different microorganisms
| Property | Valuea
|
|||||
|---|---|---|---|---|---|---|
| M. osloensis | P. testosteroni | P. acidovorans | K. pneumoniae | P. alcaligenes P25X | P. putida P35X | |
| Holoenzyme (Mr) (±SD) | 154,000 | 158,000 ± 5,000 | 164,000 ± 5,000 | 159,000 ± 6,000 | 154,000 ± 6,000 | 82,000 ± 4,000 |
| Subunit (Mr) (±SD) | 40,000 | 40,800 ± 1,000 | 37,200–39,800 ± 1,000 | 38,000 ± 2,000 | 39,000 ± 1,000 | 41,000 ± 1,000 |
| Deduced structure | (α)4 | (α)4 | (α)4 | (α)4 | (α)4 | (α)2 |
| Optimum pH | 7.0–8.5 | 7.0–9.0 | 7.0–9.0 | 8.0–9.0 | 7.5–9.5 | 7.5–9.5 |
| pH stability range | NA | 6.0–8.0 | 6.0–8.0 | NA | 5.0–7.5 | 7.0–9.0 |
| Optimum temp (°C)b | NA | NA | NA | NA | 50 | 50 |
| Temp stability range (°C)c | NA | NA | NA | NA | Up to 30°C | Up to 40°C |
| Isoelectric point | NA | 5.7–6.0 | 4.6–4.8 | 4.7–5.0 | 4.8–5.0 | 4.6–4.8 |
| Km (μM) | 7.1 | 85 | 74 | 52 | 92 | 143 |
Data for M. osloensis, P. testosteroni, P. acidovorans, and K. pneumoniae are from references 5, 9, and 20, respectively. NA, data not available.
Activity was measured over a temperature range of 0 to 70°C at pH 7.4.
Activity was measured after 15 min of heat treatment over a temperature range of 0 to 70°C.
Both P. alcaligenes P25X and P. putida P35X gentisate 1,2-dioxygenases could cleave a wide range of alkyl and halogenated analogs substituted in the 3 position of the ring. The Km values for the substituted gentisates were significantly lower than the Km values for gentisate. Higher affinity towards the substituted gentisates is similarly reported for the enzyme from P. acidovorans. Although both enzymes cleaved the substituted gentisates at much-reduced Vmax and Km values relative to those for gentisate, the catalytic efficiencies as indicated by kcat/Km were higher than for gentisate. The kinetic characteristics of both enzymes are clearly different from those of the gentisate 1,2-dioxygenase expressed by P. testosteroni, which showed higher Km and lower kcat/Km values for 3-methyl-, 3-isopropyl-, and 3-bromogentisates (9). From the lower Km and higher kcat/Km values expressed by both P25X and P35X gentisate 1,2-dioxygenases towards the alkyl- and bromo-substituted gentisates, it is clear that substituent groups at the 3 position were well tolerated and the substrates were efficiently converted. Since the Km values of the P. testosteroni enzyme for substituted gentisates were higher than the Km values of both the P25X and P35X enzymes, the lower catalytic efficiency towards the substituted gentisates expressed by the P. testosteroni enzyme clearly reflects binding or steric differences in the active sites of the three enzymes.
The optimum temperature for both P25X and P35X enzymes was determined to be around 50°C, and this value was higher than the 30°C optimum reported for the K. pneumoniae enzyme (20). The P25X enzyme was relatively stable at temperatures below 30°C, while the P35X enzyme had a higher temperature range for thermal stability. The pH stability range for both P. testosteroni and P. acidovorans gentisate dioxygenases was from 6.0 to 8.0, and this value differed from the pH values established for the P25X and P35X enzymes, which were 5.0 to 7.5 and 7.0 to 9.0, respectively. The pH optimum range was observed to be higher than the pH stability range, being 7.5 to 9.5 for both the P25X and P35X enzymes. These values were quite similar to those previously reported for gentisate 1,2-dioxygenases from P. acidovorans and P. testosteroni, with values between 7.0 and 9.0 (Table 6). Isoelectric focusing of the gentisate 1,2-dioxygenases revealed apparent isoelectric points of 4.6 to 4.8 and 4.8 to 5.0 for the P. putida P35X and P. alcaligenes P25X enzymes, respectively. The pI of the P35X enzyme is similar to the value reported for the enzyme from P. acidovorans, and the pI value of the P25X enzyme is similar to that for the enzyme from K. pneumoniae. The pI of P. testosteroni (5.7 to 6.0) is clearly very different from the pIs of the enzymes present in all the other species reported previously (Table 6). Both enzymes from P. alcaligenes P25X and P. putida P35X were completely inhibited by the presence of 5 mM Cu2+ and Mn2+, while Zn2+ and EDTA were strongly inhibitory. Similar observations were reported for the gentisate 1,2-dioxygenase from K. pneumoniae (20).
The collection of comparative data on the catalytic properties of isofunctional enzymes will enable selection for properties of gentisate 1,2-dioxygenase that will allow it to function as a better biocatalyst. For example, gentisate 1,2-dioxygenases from P. acidovorans, P. alcaligenes P25X, and P. putida clearly have a distinct advantage in the degradation of 3-alkyl and 3-halogenated gentisates over the corresponding isofunctional enzymes from P. testosteroni and M. osloensis, as revealed by the higher catalytic efficiencies (kcat/Km) towards these substrates. Properties such as substrate specificity, temperature-pH optimum, and stability are important determinants to consider when making decisions about the use of microorganisms in remediation of contaminated sites.
ACKNOWLEDGMENT
This work was supported by National University of Singapore academic research grant no. RP95-0383 to C. L. Poh for projects associated with the BioScience Center, NUS.
REFERENCES
- 1.Bayly R C, Chapman P J, Dagley S, DiBerardino D. Purification and some properties of maleylpyruvate hydrolase and fumarylpyruvate hydrolase from Pseudomonas alcaligenes. J Bacteriol. 1980;143:70–77. doi: 10.1128/jb.143.1.70-77.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bayly R C, Barbour M G. The degradation of aromatic compounds by the meta and gentisate pathways. In: Gibson D T, editor. Microbial degradation of organic compounds. Vol. 13. New York, N.Y: Marcel Dekker Inc.; 1984. pp. 253–294. [Google Scholar]
- 3.Bradford M M. A rapid and sensitive method for quantitation of microgram quantities of proteins utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 4.Crawford R L. Degradation of 3-hydroxybenzoate by bacteria of the genus Bacillus. Appl Microbiol. 1975;30:439–444. doi: 10.1128/am.30.3.439-444.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crawford R L, Hutton S W, Chapman P J. Purification and properties of gentisate 1,2-dioxygenase from Moraxella osloensis. J Bacteriol. 1975;121:794–799. doi: 10.1128/jb.121.3.794-799.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dagley S. Pathways for the utilization of organic growth substrates. In: Sokatch J R, Ornston L N, editors. The bacteria. Vol. 6. New York, N.Y: Academic Press; 1978. pp. 305–388. [Google Scholar]
- 7.Dagley S. Microbial metabolism of aromatic compounds. In: Murray M Y, editor. Comprehensive biotechnology. Vol. 1. Oxford, England: Pergamon Press; 1985. pp. 483–505. [Google Scholar]
- 8.Goetz F E, Harmuth L J. Gentisate pathway in Salmonella typhimurium: metabolism of m-hydroxybenzoate and gentisate. FEMS Microbiol Lett. 1992;97:45–50. doi: 10.1016/0378-1097(92)90361-q. [DOI] [PubMed] [Google Scholar]
- 9.Harpel M R, Lipscomb J D. Gentisate 1,2-dioxygenase from Pseudomonas: purification, characterization and comparison of the enzymes from Pseudomonas testosteroni and Pseudomonas acidovorans. J Biol Chem. 1990;265:6301–6311. [PubMed] [Google Scholar]
- 10.Hegeman G D. Synthesis of enzymes of the mandelate pathway by Pseudomonas putida. 1. Synthesis of enzymes by the wild type. J Bacteriol. 1966;91:1140–1154. doi: 10.1128/jb.91.3.1140-1154.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hopper D J, Chapman P J. Gentisic acid and its 3- and 4-methyl substituted homologues as intermediates in the bacterial degradation of m-cresol, 3,5-xylenol and 2,5-xylenol. Biochem J. 1971;122:19–28. doi: 10.1042/bj1220019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hopper D J, Chapman P J, Dagley S. The enzymatic degradation of alkyl-substituted gentisates, maleates and malates. Biochem J. 1971;122:29–40. doi: 10.1042/bj1220029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hopper D J, Taylor D G. Pathways for the degradation of m-cresol and p-cresol by Pseudomonas putida: evidence for a plasmid. J Bacteriol. 1975;122:1–6. doi: 10.1128/jb.122.1.1-6.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jones D C, Cooper R A. Catabolism of 3-hydroxybenzoate by the gentisate pathway in Klebsiella pneumoniae M5al. Arch Microbiol. 1990;154:489–495. doi: 10.1007/BF00245233. [DOI] [PubMed] [Google Scholar]
- 15.Lack L. The enzymatic oxidation of gentisic acid. Biochem Biophys Acta. 1959;34:117–123. doi: 10.1016/0006-3002(59)90239-2. [DOI] [PubMed] [Google Scholar]
- 16.Ladd J N. Oxidation of anthranilic acid by a species of Achromobacter isolated from soil. Nature. 1962;194:1099–1100. doi: 10.1038/1941099b0. [DOI] [PubMed] [Google Scholar]
- 17.Ohmoto T, Sakai K, Hamada N, Ohe T. Salicylic acid metabolism through a gentisate pathway by Pseudomonas sp. TA-2. Agric Biol Chem. 1991;55:1733–1737. [Google Scholar]
- 18.Poh C L, Bayly R C. Evidence for isofunctional enzymes used in m-cresol and 2,5-xylenol degradation via the gentisate pathway. J Bacteriol. 1980;143:59–69. doi: 10.1128/jb.143.1.59-69.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Poh C L, Bayly R C. Regulation of isofunctional enzymes in Pseudomonas alcaligenes mutants defective in the gentisate pathway. J Appl Bacteriol. 1988;64:451–458. doi: 10.1111/j.1365-2672.1988.tb05102.x. [DOI] [PubMed] [Google Scholar]
- 20.Suarez M, Ferrer E, Martin M. Purification and biochemical characterization of gentisate 1,2-dioxygenase from Klebsiella pneumoniae M5al. FEMS Lett. 1996;143:89–95. doi: 10.1111/j.1574-6968.1996.tb08466.x. [DOI] [PubMed] [Google Scholar]
- 21.Tomasek P H, Crawford R L. Initial reactions of xanthone biodegradation by an Arthrobacter sp. J Bacteriol. 1986;167:818–827. doi: 10.1128/jb.167.3.818-827.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walker N, Lippert K D. Formation of gentisic acid from 2-naphthol by a Pseudomonas. Biochem J. 1965;95:5c–6c. doi: 10.1042/bj0950005c. [DOI] [PubMed] [Google Scholar]
- 23.Werwath J, Arfmann H-A, Pierer D H, Timmis K N, Wittich R-M. Biochemical and genetic characterization of a gentisate 1,2-dioxygenase from Sphingomonas sp. strain RW5. J Bacteriol. 1998;180:4171–4176. doi: 10.1128/jb.180.16.4171-4176.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wheelis M L, Palleroni N J, Stanier R Y. The metabolism of aromatic acids by Pseudomonas testosteroni and Pseudomonas acidovorans. Arch Microbiol. 1967;59:302–314. doi: 10.1007/BF00406344. [DOI] [PubMed] [Google Scholar]
- 25.Wittich R M, Rast H G, Knackmuss H J. Degradation of naphthalene-2,6- and naphthalene-1,6-disulfonic acid by a Moraxella sp. Appl Environ Microbiol. 1988;54:1842–1847. doi: 10.1128/aem.54.7.1842-1847.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]