

Abstract
Interleukin (IL)-33, a member in the IL-1 family, plays a central role in innate and adaptive immunity; however, how IL-33 mediates cytotoxic T-cell regulation and the downstream signals remain elusive. In this study, we found increased mouse IL-33 expression in CD8+ T cells following cell activation via anti-CD3/CD28 stimulation in vitro or lymphocytic choriomeningitis virus (LCMV) infection in vivo. Our cell adoptive transfer experiment demonstrated that extracellular, but not nuclear, IL-33 contributed to the activation and proliferation of CD8+, but not CD4+ T effector cells in LCMV infection. Importantly, IL-33 induced mTORC1 activation in CD8+ T cells as evidenced by increased phosphorylated S6 ribosomal protein (p-S6) levels both in vitro and in vivo. Meanwhile, this IL-33-induced CD8+ T-cell activation was suppressed by mTORC1 inhibitors. Furthermore, IL-33 elevated glucose uptake and lactate production in CD8+ T cells in both dose-and time-dependent manners. The results of glycolytic rate assay demonstrated the increased glycolytic capacity of IL-33-treated CD8+ T cells compared with that of control cells. Our mechanistic study further revealed the capacity of IL-33 in promoting the expression of glucose transporter 1 (Glut1) and glycolytic enzymes via mTORC1, leading to accelerated aerobic glucose metabolism Warburg effect and increased effector T-cell activation. Together, our data provide new insights into IL-33-mediated regulation of CD8+ T cells, which might be beneficial for therapeutic strategies of inflammatory and infectious diseases in the future.
Keywords: CD8, Glut1, glycolytic metabolism, IL-33, mTORC1, T cells
INTRODUCTION
IL-33 is a damage-associated molecular pattern (DAMP) molecule and a chromatin-associated nuclear factor [1]. Unlike other members in the IL-1 superfamily, IL-33 is rapidly released during cell necrosis or tissue damage, binds to the ST2 receptor and strongly promotes immune responses by inducing immune cell activation and proliferation [2]. The IL-33/ST2 axis contributes to the regulation of T-cell immunity in several diseases, including viral infection, colitis, graft-versus-host disease and cancer [3–8]. IL-33 plays critical roles in both acute and chronic infection. As an alarmin in acute infection, IL-33 synergizes with other immune signals (e.g. IL-12) to boost strong immune cell activity, including Th1, cytotoxic T lymphocyte and type 1 innate lymphoid cell responses, which required for viral clearance and tissue protection [7, 9–11]. The strong IL-33 release may also cause tissue damage due to the induction of severe pro-inflammatory responses. In acute HIV infection, increased numbers and activation of CD8+ T cells were associated with elevated levels of soluble ST2 (sST2), which may be correlated with gut tissue damage [12]. In addition to induction of type 1 immune responses, IL-33 is originally considered a type 2 immune cytokine, which can induce Th2 cells and type 2 innate lymphoid cells as well as regulatory T cells, leading to tissue fibrosis [13, 14]. In chronic HIV infection, the association between CD8+ T-cell activation and sST2 was not appeared, while the alarming IL-33 signal may contribute to the development of lymphatic tissue fibrosis, indicating a phase-dependent and transient role for the IL-33/ST2 axis in viral infection [15]. More recent study showed that IL-33 has been considered the initiator of cytokine storm responses in severe COVID-19 patients by expanding the number of pathogenic granulocyte-macrophage colony-stimulating factor-expressing T cells [16]. IL-33 correlates with CD4+ T-cell activation in PBMCs from convalescent subjects [17] and may amplify pathogenic Th2 cell responses in the lung [18]. However, it is still unclear as to how IL-33 regulates T-cell activity; its associated downstream signalling pathways are not well defined. A better understanding of the underlying mechanisms of IL-33 in T-cell regulation will be beneficial for developing a potential therapeutic approach targeting the IL-33/ST2 axis.
The mammalian target of rapamycin (mTOR) pathway is a key regulator of cell growth and proliferation and is emerging as an attractive target of cancer therapy [19]. In addition to cancer cells, mTOR has been considered crucial for mediating T-cell activation and differentiation [20]. Deletion of the gene encoding tuberous sclerosis complex 2 (TSC2), which is a negative regulator of the mTOR complex 1 (mTORC1), resulted in the generation of highly glycolytic and potent effector CD8+ T cells, while CD8+ T cells with deficient mTORC1 activity failed to differentiate into effector cells [21]. IL-33 has been demonstrated to induce mTOR activation in type 2 innate lymphoid cells, leading to IL-5 and IL-13 production and airway inflammation [22]. While other IL-1 family members such as IL-1β and IL-18 are capable of regulating mTOR activity in NK and regulatory T cells [23, 24], it remains unclear whether IL-33 orchestrates T-cell functions by modulating mTOR signals.
T-cell proliferative and clonal expansion require high levels of energy. This demand is met with dramatic reprogramming of cell metabolism, including specific energetic and biosynthetic pathways to support the unique function needs [25]. Concomitant with effector T-cell activation is the engagement of aerobic glycolysis, which is characteristic of the Warburg effect [26]. In contrast, long-lived memory T cells favour fatty acid oxidation to elevate oxidative phosphorylation to rapidly respond to a reinfection [26]. It is known that mTOR provides a critical link between T-cell metabolism and function [27]. The mTORC1 signal is required for T-cell activation via the promotion of aerobic glycolysis, while the mTORC1 inhibitor rapamycin inhibits TCR-induced upregulation of glucose transporters, glucose uptake and glycolytic enzymes [28]. Although the underlying mechanism of mTORC1-regulated aerobic glycolysis is not totally clear, the transcription factors c-Myc and hypoxia-inducible factor 1-alpha (HIF-1α) have been demonstrated to be the key players in this metabolic programming [29].
In this study, we found that the activation of CD8+ T cells via either TCR stimulation in vitro or LCMV infection in vivo resulted in the upregulation of T cell-derived IL-33 expression. Extracellular, but not nuclear, IL-33 promoted effector CD8+ T-cell activity. Importantly, IL-33 induced mTORC1 activation in CD8+ T cells as evidenced by increased phosphorylated S6 ribosomal protein (p-S6) levels both in vitro and in vivo; this IL-33-induced T-cell activation was diminished by the mTOR inhibitors. IL-33 also facilitated glucose uptake and lactate production in CD8+ T cells in dose- and time-dependent manners. The results of glycolytic rate assay further confirmed the increased glycolytic capacity of IL-33-treated CD8+ T cells compared with that of control cells. Our mechanistic study further revealed that IL-33 could upregulated glucose transporter 1 (Glut1), glycolytic enzymes and key regulators, including c-Myc and HIF-1α. Together, our data provide a new insight into IL-33-mediated regulation of CD8+ T cells and might be beneficial for therapeutic strategies of inflammatory and infectious diseases in the future.
METHODS
Animals and infection
C57BL/6 (B6, #000664) and B6 CD45.1 (#002014) were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). IL-33−/− mice of the B6 background [30] were bred and maintained under specific pathogen-free conditions in the animal care facility at the University of Texas Medical Branch (UTMB, Galveston, TX, USA). All animals were used at 7–12 weeks of age. For LCMV infection, mice were intravenous (i.v.) injected with 2 × 106 focus forming units (FFU) of LCMV Clone 13 and killed at 7 days post-infection. All procedures were approved by UTMB’s Institutional Animal Care and Use Committee and performed according to National Institutes of Health Guidelines.
Propagation and titration of virus
The LCMV stock was prepared and titrated according to the previous study [31]. Briefly, BHK cells were infected with virus for 72 h. The culture supernatant was collected and centrifuged (350 g, 10 min, 4°C) to remove cell debris. Viral stock was stored at −80°C for future use. For titration of virus, Vero cells were infected with a series of 10-fold viral dilutions for 90 min, followed by a methylcellulose overlay. After 4 days of culture, cells were washed and incubated with mouse anti-LCMV polyclonal Ab (Fitzgerald, Acton, MA), followed by incubation with peroxidase (HRP)-conjugated anti-mouse IgG (Southern Biotech, Birmingham, AL). The AEC HRP Substrate Kit (Enzo Life Sciences, Farmingdale, NY) was used for immunocytochemical procedures. Viral titres were calculated by counting the numbers of positive clusters.
Antibodies and reagents
Mouse recombinant IL-33 was purchased from Biolegend (San Diego, CA). The mTORC1 inhibitor rapamycin (25 nM), PI3K inhibitor Ly294002 (5 μM), wortmannin (100 nM) and metformin (5 mM) were purchased from Calbiochem (San Diego, CA) and used in cell cultures, according to our previous study [32, 33]. The following antibodies (Abs) were purchased from Thermo Fisher Scientific (San Diego, CA): APC-anti-IFN-γ (XMG1.2), PerCP-efluor 710-anti-TNF-α (MP6-XT22) and Fixable Viability Dye eFluor 506. The following reagents were purchased from BioLegend: Ultra-LEAF purified-anti-CD3ε (145–2C11), Ultra-LEAF purified-anti-CD28 (37·51), PE-Cy7-anti-CD3 (17A2), APC-Cy7-anti-CD8 (53–6·7), Pacific Blue-anti-CD4 (GK1.5), PE-anti-CD44 (IM3), Percp-Cy5.5-anti-CD45.1 (A20), FITC-anti-CD45.2 (104), PE-anti-Ki-67 (SolA15), purified-anti-CD16/32 (2.4G2) and carboxyfluorescein succinimidyl ester (CFSE). Alexa Fluor 647-anti-Glut1 (EPR3915) was purchased from Abcam (Cambridge, United Kingdom). PE-Phospho-S6 Ribosomal Protein (Ser235/236) (D57.2.2E), mouse β-actin and phospho-S6 ribosomal protein (S235/236) antibodies were purchased from Cell Signaling Technology (Danvers, MA).
Lymphocyte isolation, purification and culture
Spleens were gently meshed in the RPMI 1640 medium through a 70-μm cell strainer. Red blood cells were removed by using Red Cell Lysis Buffer (Sigma-Aldrich) and complete RPMI 1640 medium, containing 10% fetal bovine serum, 50 mM β-mercaptoethanol, 50 μg/ml gentamycin, 25 mM HEPES, 100 units/ml penicillin and 50 μg/ml streptomycin. CD4+ and CD8+ T cells were purified from splenocytes using CD4 and CD8 magnetic beads, respectively (Miltenyi Biotec, Auburn, CA). The purities of the target cells were higher than 95%.
Lymphocytes or purified CD4+ and CD8+ T cells were cultured in RPMI 1640 complete medium with 10% fetal bovine serum (FBS) in an anti-CD3 Ab (5 μg/ml)-coated plate with soluble anti-CD28 Ab (1 μg/ml) for 3–4 days. For p-S6 signalling analysis, cultured lymphocytes were rested in RPMI 1640 medium without FBS for 1 h, followed by the stimulation with IL-33 (100 ng/ml) at 37°C. Cells were fixed at the exact time-points, and analysed for p-S6 by flow cytometry.
Glucose and lactate measurement
Cells (5 × 104) were cultured for 2 and 4 days in 96-well plates that were pre-coated with anti-CD3 (5 μg/ml) and soluble anti-CD28 (1 μg/ml), with or without IL-33. Supernatants were collected and analysed for glucose and lactate using Glucose-Glo Assay and Lactate-Glo Assay, respectively (Promega, Madison, WI). Luminescence was read using Synergy HTX Multi-Mode Reader with Gen5 software (BioTek, Winooski, VT).
Adoptive transfer
Splenocytes (1 × 107) from either wild-type (WT) or IL-33−/− mice were adoptively transferred into CD45.1 transgenic recipient mice at −1 days prior to viral infection. In parallel experiments, splenocytes (1 × 107) from naïve CD45.1 transgenic donor mice were adoptively transferred into WT or IL-33−/− recipient mice. All mice were killed at 7 days post-infection (dpi).
Flow cytometry
For surface staining, cells were first incubated with Fc Receptor Blocker (CD16/32), followed by fluorochrome-labelled Abs of surface markers. For intracellular cytokine staining, Brefeldin A (Biolegend) was added for last 6 h of culture. For analysis of virus-specific CD4 and CD8 T-cell responses, cells were incubated with viral peptides GP33 (5 μg/ml) and GP61 (5 μg/ml) for 5 h in the presence of Brefeldin A. After incubation, cells were stained for surface markers first at 4°C for 30 min in the dark, followed by intracellular staining using IC Fixation Buffer (Thermo Fisher Scientific). For Ki-67 staining, the Foxp3/Transcription Factor Staining Buffer Set was used. The phosflow experiments were performed according to the protocol of BD Biosciences. Briefly, cells were stimulated with cytokines for the indicated times, followed by immediate fixation using a pre-warmed Cytofix Fixation Buffer at 37°C for 12 min. The cells were permeabilized using chilled Perm Buffer III for 1 h on ice and then were washed and stained with phosflow Abs. To measure the mitochondrial membrane potential, TMRM assay kit was used (Abcam). Samples were processed on an LSRII FACS Fortessa (BD Bioscience, San Jose, CA) and analysed using FlowJo X software (Tree Star, Ashland, OR).
Quantitative reverse transcriptase-PCR (qRT-PCR)
RNA was extracted using an RNeasy Mini Kit according to the instructions (Qiagen, Valencia, CA). The synthesis of cDNA was proceeded using an iScript Reverse Transcription Kit (Bio-Rad, Hercules, CA). cDNA was amplified in a 10-μl reaction mixture containing 5 μl of iTaq SYBR Green Supermix (Bio-Rad) and 5 μM each of gene-specific forward and reverse primers. The PCR assays were denatured for 30 s at 95°C, followed by 40 cycles of 15 s at 95°C, and 60 s at 60°C, utilizing the CFX96 Touch real-time PCR detection system (Bio-Rad). Relative quantitation of mRNA expression was calculated using the 2−ΔΔCt method. The primers are listed in Table S1.
Western blot analysis
Cell protein was extracted using a RIPA lysis buffer (Cell Signaling Technology) in the presence of the phosphatase inhibitor cocktail (Thermo Fisher Scientific). Protein concentrations were measured using a BCA Protein Assay Kit (Pierce). Protein samples were separated by SDS-PAGE (4–15%) and electro-transferred onto a PVDF membrane, which was then blocked with 5% BSA for 60 min. The membrane was then incubated with primary Abs overnight at 4°C. After incubation, the membrane was washed 3 times with TBST, incubated with secondary Abs for 60 min at room temperature and developed using the ECL Western blotting substrate reagent (Thermo Fisher Scientific). The signal intensity was analysed by ImageJ and normalized to β-actin.
Cell proliferation assay
For the proliferation experiment, carboxyfluorescein succinimidyl ester (CFSE)-labelled T cells from naive mice (1 × 105) were in an anti-CD3 Ab (5 μg/ml)-coated plate with soluble anti-CD28 Ab (1 μg/ml). Metformin (5 mM) was used as a mild mTOCR inhibitor. After 3 days, cell proliferation was evaluated by flow cytometry.
Glycolytic rate assay
T cells were purified from WT mice and cultured in anti CD3/CD28 antibody-coated plate with or without IL-33. At day 3 of culture, cells were harvested and seeded into a 96-well Seahorse plate at a density of 1 × 105/well in Seahorse XF assay medium. Glycolytic rate assay kit (Agilent, Santa Clara, CA) was used to determine extracellular acidification rate (ECAR) and glycolytic proton efflux rate (glycoPER).
Statistical analyses
The data are shown as the mean ± SEM. A two-tailed Student’s t test is used for comparisons between two groups. A one-way ANOVA test was used for statistical analysis of more than two groups. *, **, *** or **** represents p value <0·05, 0·01, 0·001 or 0·0001, respectively. Statistical analyses were performed by the GraphPad Prism software 8.0 (San Diego, CA).
RESULTS
Exogenous, but not nuclear, IL-33 promotes CD8+ T-cell activation
IL-33 is mainly expressed by endothelial and epithelial cells [1]. To determine whether T cells can express IL-33, we assessed IL-33 transcript levels in purified splenic T cells following LCMV infection and found upregulated IL-33 gene expression in T cells of infected mice compared with that of naïve mice (Figure S1a). To confirm this result, we performed an in vitro experiment where we activated T cells via anti-CD3/CD28 cross-linking stimulation. Consistently, we found increased IL-33 transcript levels in activated CD8+ T cells compared with that in naïve controls (Figure S1b). IL-33 levels may also show an increased trend in activated CD4+ T cells, but did not make a significant difference (Figure S1b).
To investigate the role of nuclear IL-33 in T cells, we isolated spleen cells from WT and IL-33−/− mice and adoptively transferred of these cells into recipient CD45.1 mice. The recipient mice were then infected with LCMV, and the activation of the donor’s T cells were analysed at 7 dpi. We found comparable numbers of donor CD8+ T cells in the recipient mice between the two groups (Figure 1a,b). Moreover, the activated CD8+ T cells produced similar amounts of IFN-γ, indicating comparable activation levels of CD8+ T cells in these two groups (Figure 1a,b). These results were also observed among CD4+ T cells (Figure S2a). These data suggest that nuclear IL-33 in T cells was not required for cell activation and proliferation in response to viral infection. We speculated therefore that exogenous IL-33 and its signalling pathway were involved in T-cell activation. To test this possibility, we isolated splenocytes from CD45.1 mice and adoptively transferred them into WT and IL-33−/− mice, followed by LCMV infection. Decreased numbers of donor cells were found in IL-33−/− recipient mice (Figure 1c,d). IL-33−/− recipient animals had reduced numbers of transferred CD8+ T cells and decreased IFN-γ production compared with those in the WT recipient mice (Figure 1c,d). However, these differences were not observed in CD4+ T cells (Figure S2b). Together, our data suggest that exogenous, but not nuclear, IL-33 contributed to CD8+ T-cell activation and proliferation during viral infection.
FIGURE 1.
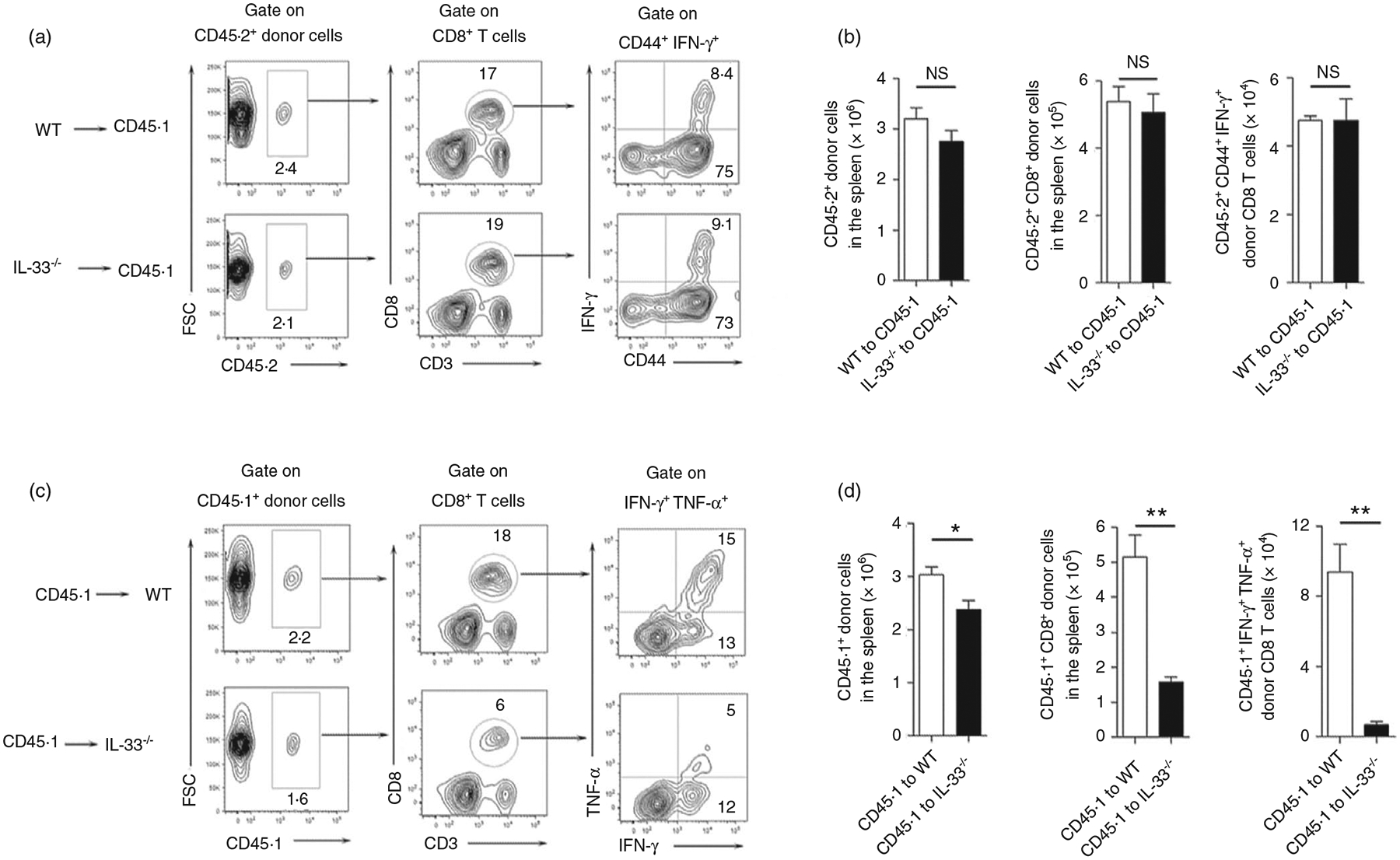
Extracellular, but not nuclear IL-33 contributes to CD8+ T-cell responses. (a and b) Spleen cells were isolated from naïve WT and IL-33−/− mice, followed by adoptively transferring them into CD45.1 recipient mice. (c and d) Spleen cells were isolated from naïve CD45.1 mice, followed by adoptively transferring them into WT and IL-33−/− recipient mice. The number of transferred cells was 1 × 107 for each mouse. Animals were infected with LCMV (2 × 106 FFU/mouse) 1 day after cell adoptive transfer, and killed at 7 days post-infection. Lymphocytes were prepared from spleens and stimulated with viral peptides GP33 (5 μg/ml) in the presence of Brefeldin A for 5 h, followed by surface marker and intracellular cytokine staining. The transferred cells in recipient mice were gated first according to CD45.1 and CD45.2 markers. The data are shown as mean ± SEM of n = 3–5 mice/group from single experiments and are representative of at least three experiments performed. A two-tailed Student’s t test was used for statistical analysis. *p < 0·05; **p < 0·01; NS, no significant difference
IL-33 activates mTORC1 signalling pathway in CD8+ T cells
IL-33 activates the mTOR pathway in Th2 cells and innate lymphoid cells (ILCs), leading to airway inflammation [22]. To investigate whether IL-33 can activate mTOR in CD8+ T cells, we analysed the expression of mTORC1 downstream molecule p-S6 in LCMV-infected WT and IL-33−/− mice. We found a decreased number of cytokine-producing CD8+ T cells accompanied by lower levels of p-S6 expression in IL-33−/− mice compared with that in WT mice (Figure 2a,b). Meanwhile, these differences were not observed in CD4+ T cells (Figure 2a,b). To confirm our in vivo results, T cells were isolated from WT mice and stimulated with IL-33 in vitro, followed by the measurement of p-S6 levels at various time-points. We observed IL-33-stimulated S6 phosphorylation in CD8+ T cells with a peak signal at around 10–15 min of stimulation (Figure 2c). These flow cytometry results were further confirmed by Western blot (Figure 2d). In addition, we found that exogenous IL-33 can also stimulate p-S6 on IL-33-deficient CD8+ T cells, indicating that nuclear IL-33 may not be essential for mTORC1 signalling activation (Figure 2e). In all, we demonstrated both in vitro and in vivo that exogenous IL-33 was capable of activating the mTORC1 signalling pathway in CD8+ T cells.
FIGURE 2.
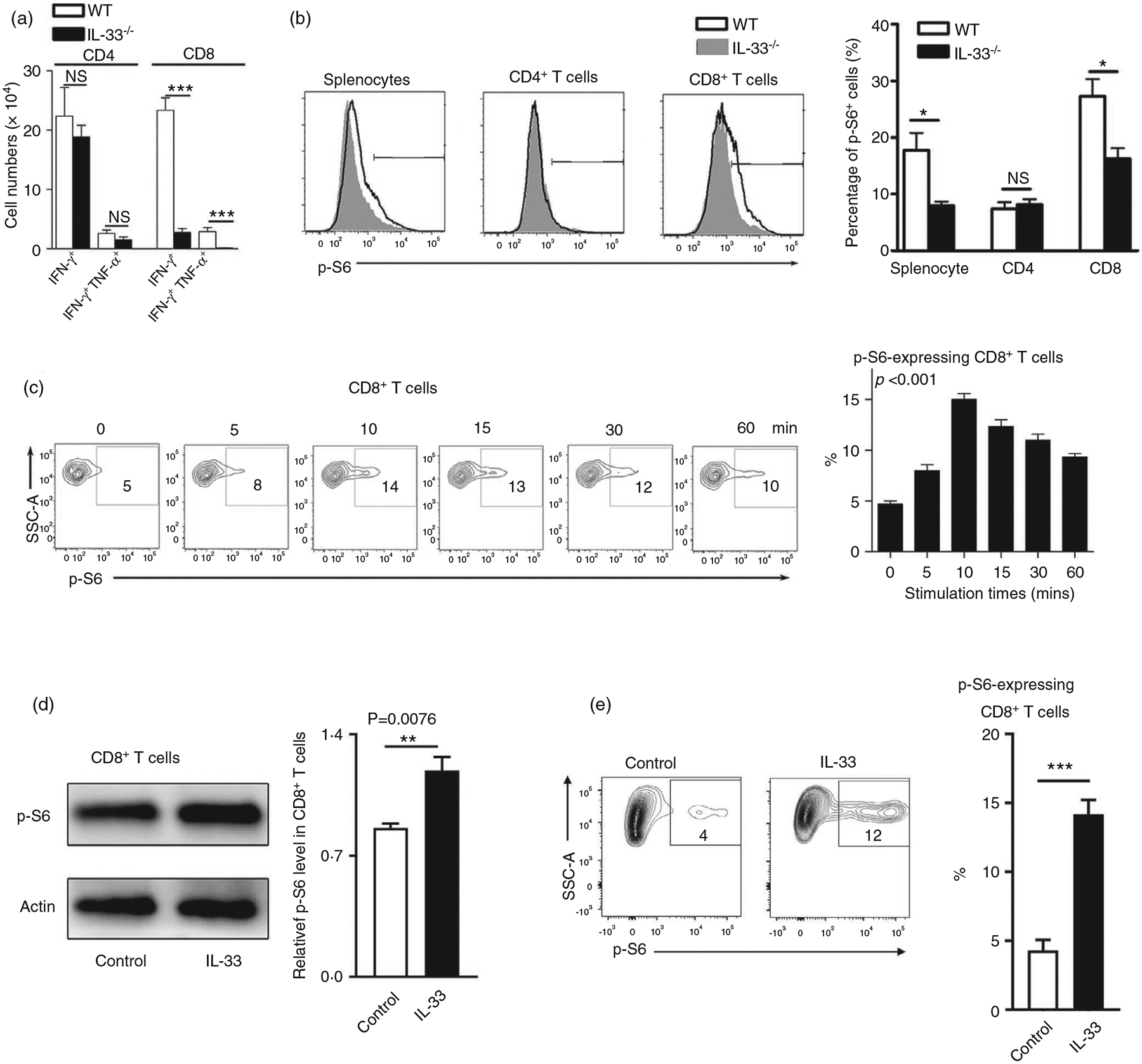
Increased p-S6 expression in CD8+ T cells by IL-33 stimulation. (a) WT and IL-33−/− mice were infected with LCMV (2 × 106 FFU/mouse) and killed at 7 days post-infection. Lymphocytes were prepared from spleens and stimulated with viral peptides GP33 and GP61 in the presence of Brefeldin A for 5 h, followed by surface marker and intracellular cytokine staining. The numbers of IFN-γ+ and IFN-γ+ TNF+ T cells were calculated. (b) Splenocytes isolated from infected mice were fixed and permeabilized for p-S6 staining. The percentages of p-S6+ cells were shown. (c) Splenocytes isolated from naïve mice were cultured in anti-CD3/CD28-coated plates for 3 days. Cells were harvested, rested in RPMI 1640 medium without FBS for 1 h, and incubated with 100 ng/ml IL-33 at 37°C for the indicated times. The p-S6 expression was evaluated according to the BD Phosflow staining protocol, and the percentages of p-S6+ cells are shown. (d) CD8 T cells were purified from WT mouse spleens, followed by the culture in anti-CD3/CD28-coated plates for 3 days. Cells were rested in RPMI 1640 medium without FBS for 1 h, followed by IL-33 treatment (100 ng/ml) at 37°C for the 20 min. Cell protein was extracted using a RIPA buffer with the phosphatase inhibitor cocktail, and the p-S6 expression was evaluated by Western blot. (e) CD8+ T cells were isolated from the spleen of IL-33−/− mice and cultured as in (d). The percentages of p-S6+ cells are shown. The data are shown as mean ± SEM of n = 3–5 mice/group from single experiments and are representative of at least three experiments performed. For in vitro experiments, triplicates were performed for each group. A two-tailed Student’s t test was used for statistical analysis of two groups. One-way ANOVA was used to compare three or more groups. *p < 0·05; ***p < 0·001; NS, no significant difference
mTORC1 signal is required for IL-33-induced CD8+ T-cell activation
To investigate the role of mTORC1 in IL-33-induced CD8+ T-cell activation, we cultured cells with IL-33 in the presence of PI3K/mTOR inhibitors. We found that all inhibitors, including rapamycin, LY294002 and wortmannin, significantly suppressed IL-33-driven p-S6 expression by 3- to 4-fold in CD8+ T cells (Figure 3a). Consistent with our previous report [10], IL-33 strongly induced T-cell activation and proliferation as evidenced by increased IFN-γ production and Ki-67 expression (Figure 3b,c). We further confirmed the T-cell proliferation using CFSE staining and found that IL-33 increased CD8+ T-cell proliferation in vitro (Figure S3). Notably, the CD8+ T cells of IL-33-deficient mice showed lower proliferation compared with that from WT mice (Figure S3). This may suggest that nuclear IL-33, which released from the dead T cells during culture, may promote cell proliferation in vitro. Further analysis showed that IL-33-induced CD8+ T-cell activation was suppressed by PI3K/mTOR inhibitors as demonstrated by significantly decreased IFN-γ and Ki-67 (Figure 3b,c). Interestingly, although activated CD8+ T cells upregulated ST2 expression, metformin treatment did not alter the levels of ST2 (Figure S4). This result suggests that ST2 expression may not be dependent on mTORC1 signals. In all, we concluded that the PI3K/mTORC1 signalling pathway was required for IL-33-induced CD8+ T-cell activation.
FIGURE 3.
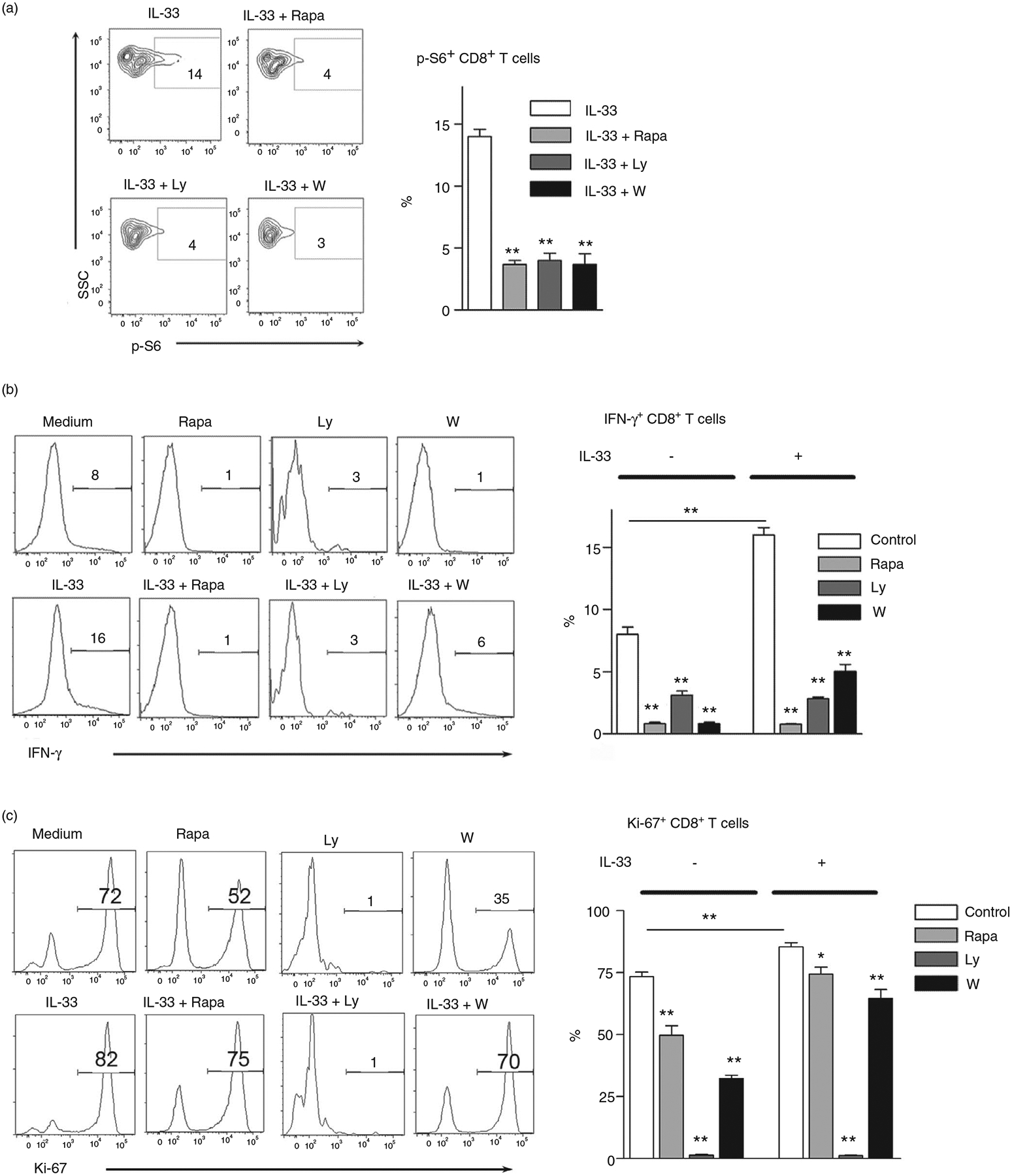
Inhibition of mTORC1 results in impaired CD8 T-cell activation and proliferation by IL-33. (a) Splenocytes isolated from naïve mice were cultured in anti-CD3/CD28-coated plates for 3 days. Cells were treated with rapamycin (25 nM), Ly294002 (5 μM) and wortmannin (100 nM) for 2 h, followed by 100 ng/ml IL-33 stimulation for 10 min at 37°C. DMSO was used as a control. The p-S6 expression was evaluated according to the BD Phosflow staining protocol, and the percentages of p-S6+ cells were shown. The IL-33 group was used as a control for comparison (b and c) Purified CD8+ T cells were cultured in anti-CD3/CD28-coated plates in the presence of IL-33 (100 ng/ml) and inhibitors for 4 days. Brefeldin A was added in the last 6 h of culture. Intracellular IFN-γ and Ki-67 expression were analysed by flow cytometry. The data are shown as mean ± SEM from single experiments and are representative of at least two experiments performed. Triplicates were performed for each group. One-way ANOVA with Dunnett multiple comparisons was used for statistical analysis. *p < 0·05; **p < 0·01
IL-33 promotes glucose uptake and lactate production in CD8+ T cells
Cellular energy needs are met usually by the highly efficient oxidative phosphorylation of pyruvate or less efficient anaerobic glycolysis. The mTOR signalling pathway plays a role in glucose metabolism [34, 35]. Since we had demonstrated that IL-33-driven mTORC1 activation was essential for CD8+ T-cell function (Figures 2 and 3), we speculated that IL-33 can regulate glycolytic metabolism in CD8+ T cells. We first measured the glucose uptake in CD8+ T cells in culture supplemented with IL-33. IL-33 significantly increased glucose uptake in these cells at days 2 and 4 in a dose-and time-dependent manner (Figure 4a). However, IL-33 did not alter glucose uptake in CD4+ T cells (Figure S5a). Supplementing with rapamycin significantly inhibited IL-33-induced glucose uptake by CD8+ T cells (Figure 4b). Consistently, lactate production was significantly increased in CD8+ T cells at days 2 and 4 in the presence of IL-33, and was greatly suppressed by rapamycin (Figure 4c). We further confirmed our results using glycolytic rate assay. Our results demonstrated that IL-33-treated CD8 T cells exhibited higher levels of ECAR and glycoPER compared with the controls (Figure 4d, e). In all, our data demonstrated that IL-33 promoted aerobic glycolysis in CD8+ T cells, reminiscent to the Warburg effect observed in fast growing tumour cells [36]. In addition, although increased mitochondrial membrane potential was found in activated CD8+ T cells compared with naïve ones, IL-33 treatment did not further increase the TMRM levels (Figure S6), indicating that IL-33 may not directly alter the mitochondrial functions in CD8+ T cells.
FIGURE 4.
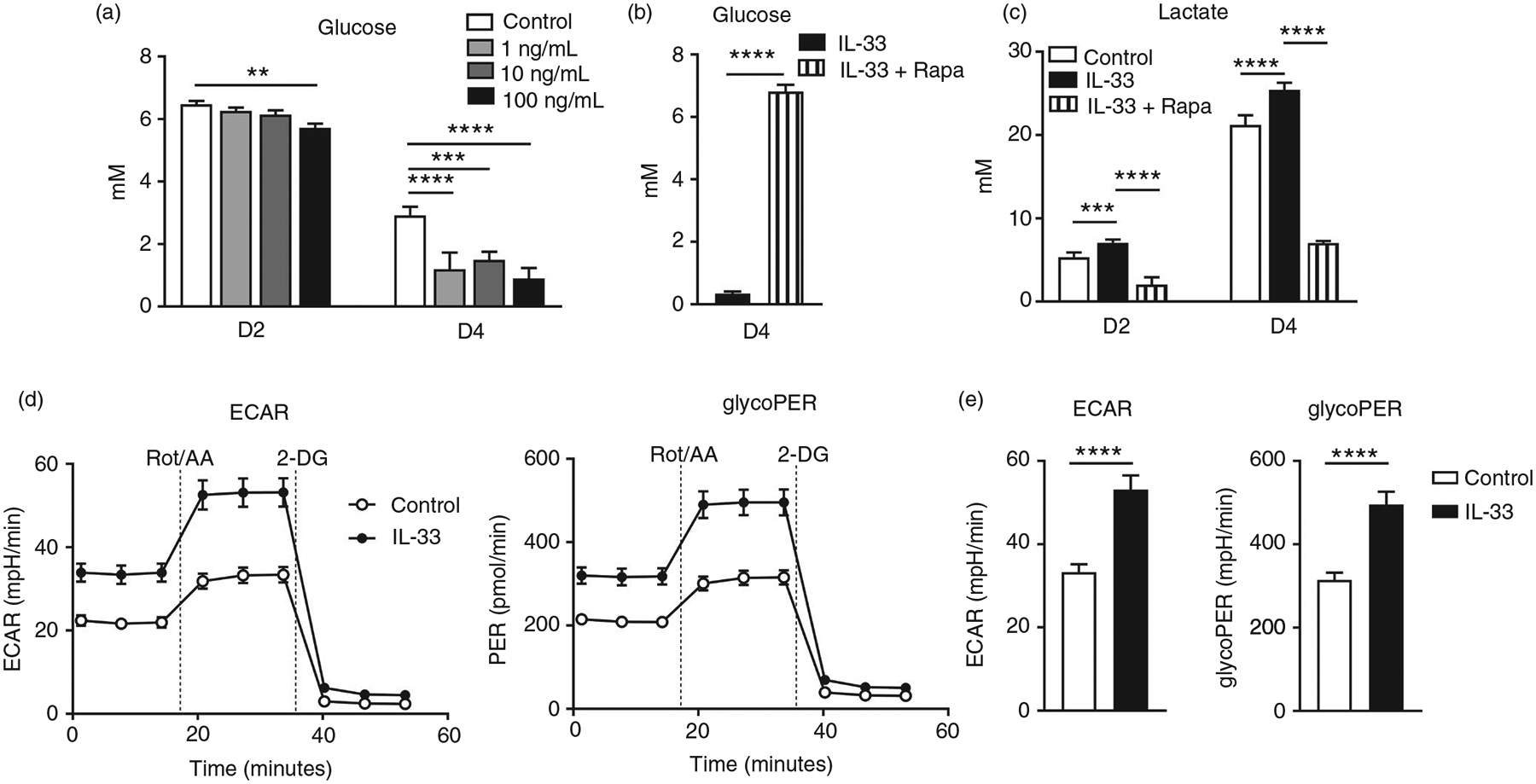
IL-33 promotes glucose uptake and lactate production in CD8 effector cells. CD8+ T cells were purified from naïve mouse spleens and cultured in anti-CD3/CD28-coated plates in the presence of different doses of IL-33. (a) Supernatant was collected at days 2 and 4 for the measurement of glucose. (b and c) CD8+ T cells were cultured in the presence of IL-33 (100 ng/ml) with rapamycin (25 nM) added or omitted. Supernatant was collected for glucose and lactate measurement. (d–e) WT CD8+ T cells were cultured in anti-CD3/CD28-coated plates with or without IL-33 (100 ng/ml) for 3 days. Cells were harvested and seeded into a 96-well Seahorse plate at a density of 1 × 105/well in Seahorse XF assay medium. Glycolytic rate assay kit (Agilent, Santa Clara, CA) was used to determine extracellular acidification rate (ECAR) and glycolytic proton efflux rate (glycoPER) according to the manufacturer’s instruction. The data are shown as mean ± SEM from single experiments and are representative of at least two experiments performed. Triplicates were performed for each group. A two-tailed Student’s t test was used for statistical analysis of two groups. One-way ANOVA with Dunnett multiple comparisons were used for statistical analysis of three or more groups. **p < 0·01; ***p < 0·001; ****p < 0·0001
IL-33 regulates glycolytic metabolism in CD8+ T cells
Rapid induction of aerobic glycolysis after effector T-cell activation is required for cytokine production and cell proliferation [37, 38]. Glut1 facilitates the transport of glucose across the plasma membranes of mammalian cells [39, 40]. Lack of Glut1 results in impaired effector CD4+ T-cell activation in mice [41]. We found that IL-33 upregulated Glut1 expression in purified CD8+ T cells at 3 and 4 days of culture, whereas rapamycin inhibited IL-33-induced Glut1 expression (Figure 5a,b). These results were further confirmed by culturing splenocytes with IL-33. We found that IL-33 increased Glut1 expression in CD8+, but not CD4+ T cells (Figure S5b). The canonical glycolysis pathway comprises several key enzymatic reactions [42]. We further analysed the transcriptional levels of several enzymes in the glycolytic pathway and found that IL-33 stimulation upregulated the gene expression of key genes, including Glut1, HK2, GPI1, TPI, Eno1, PKM2, LDH-α and MCT4 (Figure 5c). Signalling through mTORC1 serves to regulate aerobic glycolysis, at least in part, through the regulation of c-Myc and HIF-1α expression [28]. As we expected, IL-33 increased the transcriptional levels of c-Myc and HIF-1α in CD8+ T cells (Figure 5c). Rapamycin significantly inhibited all IL-33-induced enzymatic gene expression at day 2 (Figure 5c). Therefore, IL-33 promotes glycolytic metabolism and facilitates CD8+ T-cell activation through the mTORC1 signalling pathway (Figure 5d).
FIGURE 5.
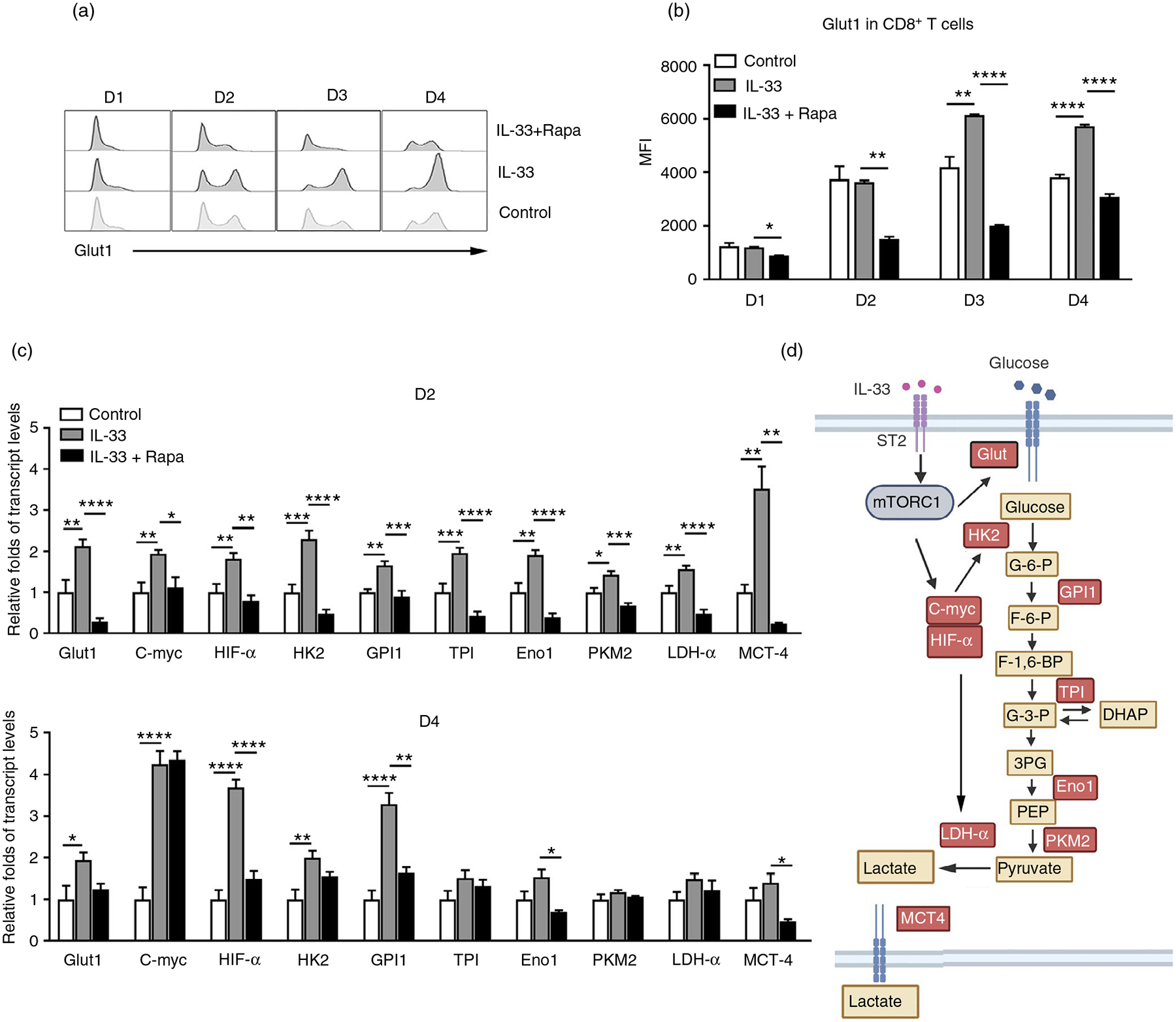
IL-33 modulates glycolytic metabolism in CD8 T cells. CD8+ T cells were purified from naïve WT mouse spleens and cultured in anti-CD3/CD28-coated plates in the presence of IL-33 and rapamycin (25 nM). (a) The expression of Glut1 was analysed by flow cytometry and (b) the mean fluorescent intensity (MFI) of Glut1. (c) Transcript levels of glycolytic pathway associated genes in CD8+ T cells. (d) Schematic figure of IL-33-regulated glycolytic metabolism through the mTORC1 signalling pathway. The data are shown as mean ± SEM from single experiments and are representative of at least three experiments performed. Triplicates were performed for each group. One-way ANOVA with Dunnett multiple comparisons were used for statistical analysis of three or more groups. *p < 0·05; **p < 0·01; ***p < 0·001; ****p < 0·0001
DISCUSSION
The tissue-derived nuclear cytokine IL-33 is mainly produced by epithelial, endothelial and fibroblast cells as well as several hematopoietic cells, such as M2 macrophages and megakaryocytes [43–46]. Upon tissue damage, necrosis or injury, IL-33 is quickly released into the extracellular space where it binds to its cognate receptor, ST2, on the membrane of target cells to potently activate both Th1 and Th2 immune cells [47, 48], as well type 2 innate lymphoid cells and Treg cells [5, 49]. However, whether nuclear IL-33 plays a role intrinsically is still debated [50, 51]. A recent study reported that tumour-infiltrated CD8+ T cells were able to express IL-33 high effective in patients with hepatocellular carcinoma [52]. IL-33-deficient regulatory T cells were found to exhibit attenuated suppressive properties due to epigenetic reprogramming with increased chromatin accessibility of the IFNγ locus [6], suggesting the possible role of nuclear IL-33 in regulating T-cell function. In this study, we observed increased IL-33 expression in activated CD8+ T cells when compared with that in naïve controls (Figure S1). Increased IL-33 was also detected in CD4+ T cells purified from LCMV-infected mice. However, our in vivo data indicated the dispensable role of nuclear IL-33 in T-cell activation following LCMV infection (Figure 1). Consistent with previous findings [8], extracellular IL-33 signalling was critical for virus-specific CTL responses, but not for CD4+ T-cell activation (Figure 1 and Figure S2). It is reported that ST2-deficiency led to impaired antiviral Th1 cell responses, indicating that IL-33/ST2 axis is required for CD4 T-cell activation [53]. The discrepancy might be due to the distinct animal models (IL-33 deficiency vs. ST2 deficiency) we used in current study since IL-33 may have ST2-independent role in promoting inflammation [54]. In addition, our animals were infected with LCMV Cl13 strain, which is not highly pathogenic and can cause persistent infection. However, other researchers used highly pathogenic LCMV WE [53], which can induce liver injury, leading to more IL-33 release [55]. Together, we concluded that exogenous, but not nuclear IL-33 contributes to CD8+ T-cell activation. Since IL-33 may play a different role in acute and chronic infection [15], our study using the acute LCMV infection mouse model also has the limitation. Further investigate is needed to explore the dynamic role of nuclear IL-33 in chronic infection and memory T cells.
The downstream of IL-33 signalling in T cells is not well understood and has mostly been investigated in Th2 cells [56]. IL-33 activates NF-κB and MAP kinases by binding ST2 and drives production of Th2-associated cytokines [1]. The p38 MAP kinase pathway is also found as a central downstream target of the IL-33/ST2 axis in memory Th2 cells [57]. Recently, it was reported that the activation of mTORC1 is required for type 2 cytokine production in ILC2 [22, 58], indicating that the mTOR signalling pathway is a key downstream target of IL-33. Our data demonstrated that IL-33 induced mTORC1 activation in CD8+ T cells (Figure 2), suggesting the critical role of mTORC1 in IL-33-amplified CTL responses. We previously reported that IL-33 promotes CTL responses in viral hepatitis [7]. Using CFSE staining, we demonstrated that exogenous IL-33 also increased CD8+ T-cell proliferation in vitro. Interestingly, CD8+ T cells from IL-33-deficient mice showed lower level of proliferation compared with that from WT mice (Figure S3). This may suggest that endogenous IL-33 may play a role for cell proliferation when they are directly released from the dead T cells in vitro culture. Importantly, inhibition of mTORC1 activity by the inhibitors decreased IL-33-mediated IFN-γ production and proliferation marker Ki-67 expression in CD8+ T cells (Figure 3). In addition, the levels of ST2 on CD8+ T cells was not altered by metformin treatment (Figure S4), indicating that ST2 regulation in T cells might be independent of mTOR signalling pathway.
T-cell activation, differentiation and function are marked by striking changes in cellular metabolism. Accumulating evidence has elucidated that T-cell effector and memory responses are regulated by the Warburg effect, which is commonly observed in fast growing cancer cells [36]. This phenomenon is characterized by high rates of glucose uptake and lactate secretion, even in the presence of oxygen [28]. Aerobic glycolysis drives T-cell effector responses, whereas low levels of glycolysis and lipid metabolism is associated with memory and regulatory T-cell differentiation [26, 37]. It has been reported that mTORC1 signalling drives aerobic glycolysis and promotes T effector function via upregulation of glucose transporters and metabolic enzymes [59, 60], indicating the key role of mTORC1 in T-cell metabolism. In this study, we found that IL-33 can increase glucose uptake and lactate production in CD8 effector T cells, whereas this effect of IL-33 was abolished by the mTORC1 inhibitor rapamycin (Figure 4). Our mechanistic studies showed that IL-33 treatment elevated expression of the glucose transporter Glut1 and increased the transcript levels of several glycolytic enzymes (Figure 5 and Figure S5). Furthermore, rapamycin inhibited IL-33-induced aerobic glycolysis as evidenced by downregulating the expression of Glut1 and glucose enzymes (Figure 5 and Figure S5). These data suggest that IL-33 may facilitate CD8 effector T-cell responses through activating mTORC1 and promoting aerobic glycolysis. However, we found that IL-33 may not be able to alter the glycolytic metabolism in CD4+ T cells in vitro (Figure S5). Further study is needed to explore the different metabolic mechanism between CD4 and CD8 T cells by IL-33.
Together, this study indicates that extracellular but not nuclear IL-33 promotes CD8 effector T-cell activation through the mTORC1 signalling pathway. Our mechanistic study revealed that IL-33 can orchestrate glycolytic metabolism by regulating glucose transporters and glycolytic enzymes, leading to vigorous CD8 effector responses. Our study delineates the key downstream signalling pathway of IL-33 and provides new insight into IL-33-mediated cell metabolic reprogramming.
Supplementary Material
ACKNOWLEDGMENT
We thank Dr. Sherry Haller for her assistance with the manuscript preparation.
Funding information
This work was supported in part by grants from the NIH (EY028773 to JC and JS, AI132674 and AI156536 to LS, and AI153586 to YL), as well as the UTMB Institute of Human Infections & Immunity Pilot grant (to LS and YL).
Abbreviations:
- CFSE
carboxyfluorescein succinimidyl ester
- DAMP
damage-associated molecular pattern
- dpi
days post-infection
- FBS
fetal bovine serum
- FFU
focus forming units
- Glut1
glucose transporter 1
- HIF-1 α
hypoxia-inducible factor 1-alpha
- ILCs
innate lymphoid cells
- mTOR
mammalian target of rapamycin
- mTORC1
mTOR complex 1
- p-S6
phosphorylated S6 Ribosomal Protein
- sST2
soluble ST2
- TSC2
tuberous sclerosis complex 2
- WT
wild-type
Footnotes
CONFLICT OF INTEREST
The authors have no financial conflicts of interest.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section.
REFERENCES
- 1.Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–90. [DOI] [PubMed] [Google Scholar]
- 2.Chan BCL, Lam CWK, Tam LS, Wong CK. IL33: roles in allergic inflammation and therapeutic perspectives. Front Immunol. 2019;10:364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gao X, Wang X, Yang Q, Zhao X, Wen W, Li G, et al. Tumoral expression of IL-33 inhibits tumor growth and modifies the tumor microenvironment through CD8+ T and NK cells. J Immunol. 2015;194:438–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reichenbach DK, Schwarze V, Matta BM, Tkachev V, Lieberknecht E, Liu Q, et al. The IL-33/ST2 axis augments effector T-cell responses during acute GVHD. Blood. 2015;125:3183–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schiering C, Krausgruber T, Chomka A, Frohlich A, Adelmann K, Wohlfert EA, et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature. 2014;513:564–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hatzioannou A, Banos A, Sakelaropoulos T, Fedonidis C, Vidali MS, Kohne M, et al. An intrinsic role of IL-33 in Treg cell-mediated tumor immunoevasion. Nat Immunol. 2020;21:75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liang Y, Jie Z, Hou L, Aguilar-Valenzuela R, Vu D, Soong L, et al. IL-33 induces nuocytes and modulates liver injury in viral hepatitis. J Immunol. 2013;190:5666–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonilla WV, Frohlich A, Senn K, Kallert S, Fernandez M, Johnson S, et al. The alarmin interleukin-33 drives protective antiviral CD8(+) T cell responses. Science. 2012;335:984–9. [DOI] [PubMed] [Google Scholar]
- 9.Clark JT, Christian DA, Gullicksrud JA, Perry JA, Park J, Jacquet M, et al. IL-33 promotes innate lymphoid cell-dependent IFN-gamma production required for innate immunity to Toxoplasma gondii. Elife. 2021;10:e65614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liang Y, Jie Z, Hou L, Yi P, Wang W, Kwota Z, et al. IL-33 promotes innate IFN-gamma production and modulates dendritic cell response in LCMV-induced hepatitis in mice. Eur J Immunol. 2015;45:3052–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang Q, Li G, Zhu Y, Liu L, Chen E, Turnquist H, et al. IL-33 synergizes with TCR and IL-12 signaling to promote the effector function of CD8+ T cells. Eur J Immunol. 2011;41:3351–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mehraj V, Jenabian MA, Ponte R, Lebouche B, Costiniuk C, Thomas R, et al. The plasma levels of soluble ST2 as a marker of gut mucosal damage in early HIV infection. AIDS. 2016;30:1617–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McHedlidze T, Waldner M, Zopf S, Walker J, Rankin AL, Schuchmann M, et al. Interleukin-33-dependent innate lymphoid cells mediate hepatic fibrosis. Immunity. 2013;39:357–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li D, Guabiraba R, Besnard AG, Komai-Koma M, Jabir MS, Zhang L, et al. IL-33 promotes ST2-dependent lung fibrosis by the induction of alternatively activated macrophages and innate lymphoid cells in mice. J Allergy Clin Immunol. 2014;134:1422–32 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mehraj V, Ponte R, Routy JP. The dynamic role of the IL-33/ST2 axis in chronic viral-infections: alarming and adjuvanting the immune response. EBioMedicine. 2016;9:37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zizzo G, Cohen PL. Imperfect storm: is interleukin-33 the Achilles heel of COVID-19? Lancet Rheumatol. 2020;2(12):e779–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stanczak MA, Sanin DE, Apostolova P, Nerz G, Lampaki D, Hofmann M, et al. IL-33 expression in response to SARS-CoV-2 correlates with seropositivity in COVID-19 convalescent individuals. Nat Commun. 2021;12:2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Calise J, Garabatos N, Bajzik V, Farrington M, Robinson D, Jeong D, et al. Optimal human pathogenic TH2 cell effector function requires local epithelial cytokine signaling. J Allergy Clin Immunol. 2021;S0091–6749:282–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hua H, Kong Q, Zhang H, Wang J, Luo T, Jiang Y. Targeting mTOR for cancer therapy. J Hematol Oncol. 2019;12:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chi H. Regulation and function of mTOR signalling in T cell fate decisions. Nat Rev Immunol. 2012;12:325–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pollizzi KN, Patel CH, Sun IH, Oh MH, Waickman AT, Wen J, et al. mTORC1 and mTORC2 selectively regulate CD8(+) T cell differentiation. J Clin Invest. 2015;125:2090–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Salmond RJ, Mirchandani AS, Besnard AG, Bain CC, Thomson NC, Liew FY. IL-33 induces innate lymphoid cell-mediated airway inflammation by activating mammalian target of rapamycin. J Allergy Clin Immunol. 2012;130:1159–66.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marcais A, Walzer T. mTOR: a gate to NK cell maturation and activation. Cell Cycle. 2014;13:3315–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ikeda S, Saijo S, Murayama MA, Shimizu K, Akitsu A, Iwakura Y. Excess IL-1 signaling enhances the development of Th17 cells by downregulating TGF-beta-induced Foxp3 expression. J Immunol. 2014;192:1449–58. [DOI] [PubMed] [Google Scholar]
- 25.MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol. 2013;31:259–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Buck MD, O’Sullivan D, Klein Geltink RI, Curtis JD, Chang CH, Sanin DE, et al. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell. 2016;166:63–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Powell JD, Delgoffe GM. The mammalian target of rapamycin: linking T cell differentiation, function, and metabolism. Immunity. 2010;33:301–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salmond RJ. mTOR regulation of glycolytic metabolism in T cells. Front Cell Dev Biol. 2018;6:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35:871–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Louten J, Rankin AL, Li Y, Murphy EE, Beaumont M, Moon C, et al. Endogenous IL-33 enhances Th2 cytokine production and T-cell responses during allergic airway inflammation. Int Immunol. 2011;23:307–15. [DOI] [PubMed] [Google Scholar]
- 31.Liang Y, Yi P, Wang X, Zhang B, Jie Z, Soong L, et al. Retinoic acid modulates hyperactive T cell responses and protects vitamin A-deficient mice against persistent lymphocytic choriomeningitis virus infection. J Immunol. 2020;204:2984–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yi P, Liang Y, Yuan DMK, Jie Z, Kwota Z, Chen Y, et al. A tightly regulated IL-22 response maintains immune functions and homeostasis in systemic viral infection. Sci Rep. 2017;7:3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu L, Wang X, Chen Y, Soong L, Chen Y, Cai J, et al. Metformin modulates T cell function and alleviates liver injury through bioenergetic regulation in viral hepatitis. Front Immunol. 2021;12:638575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sanguesa G, Roglans N, Baena M, Velazquez AM, Laguna JC, Alegret M. mTOR is a key protein involved in the metabolic effects of simple sugars. Int J Mol Sci. 2019;20:1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mao Z, Zhang W. Role of mTOR in glucose and lipid metabolism. Int J Mol Sci. 2018;19:2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Damaghi M, West J, Robertson-Tessi M, Xu L, Ferrall-Fairbanks MC, Stewart PA, et al. The harsh microenvironment in early breast cancer selects for a Warburg phenotype. Proc Natl Acad Sci USA. 2021;118:e2011342118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Menk AV, Scharping NE, Moreci RS, Zeng X, Guy C, Salvatore S, et al. Early TCR signaling induces rapid aerobic glycolysis enabling distinct acute T cell effector functions. Cell Rep. 2018;22:1509–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cao Y, Rathmell JC, Macintyre AN. Metabolic reprogramming towards aerobic glycolysis correlates with greater proliferative ability and resistance to metabolic inhibition in CD8 versus CD4 T cells. PLoS ONE. 2014;9:e104104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Olson AL, Pessin JE. Structure, function, and regulation of the mammalian facilitative glucose transporter gene family. Annu Rev Nutr. 1996;16:235–56. [DOI] [PubMed] [Google Scholar]
- 40.Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity. 2002;16:769–77. [DOI] [PubMed] [Google Scholar]
- 41.Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 2014;20:61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hay N. Reprogramming glucose metabolism in cancer: can it be exploited for cancer therapy? Nat Rev Cancer. 2016;16:635–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cayrol C, Girard JP. Interleukin-33 (IL-33): a nuclear cytokine from the IL-1 family. Immunol Rev. 2018;281:154–68. [DOI] [PubMed] [Google Scholar]
- 44.Furukawa S, Moriyama M, Miyake K, Nakashima H, Tanaka A, Maehara T, et al. Interleukin-33 produced by M2 macrophages and other immune cells contributes to Th2 immune reaction of IgG4-related disease. Sci Rep. 2017;7:42413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mirchandani AS, Salmond RJ, Liew FY. Interleukin-33 and the function of innate lymphoid cells. Trends Immunol. 2012;33:389–96. [DOI] [PubMed] [Google Scholar]
- 46.Pariser DN, Hilt ZT, Ture SK, Blick-Nitko SK, Looney MR, Cleary SJ, et al. Lung megakaryocytes are immune modulatory cells. J Clin Invest. 2021;131:e137377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liew FY, Girard JP, Turnquist HR. Interleukin-33 in health and disease. Nat Rev Immunol. 2016;16:676–89. [DOI] [PubMed] [Google Scholar]
- 48.Smithgall MD, Comeau MR, Yoon BR, Kaufman D, Armitage R, Smith DE. IL-33 amplifies both Th1-and Th2-type responses through its activity on human basophils, allergen-reactive Th2 cells, iNKT and NK cells. Int Immunol. 2008;20:1019–30. [DOI] [PubMed] [Google Scholar]
- 49.Xia N, Lu Y, Gu M, Li N, Liu M, Jiao J, et al. A unique population of regulatory T cells in heart potentiates cardiac protection from myocardial infarction. Circulation. 2020;142:1956–73. [DOI] [PubMed] [Google Scholar]
- 50.Gautier V, Cayrol C, Farache D, Roga S, Monsarrat B, Burlet-Schiltz O, et al. Extracellular IL-33 cytokine, but not endogenous nuclear IL-33, regulates protein expression in endothelial cells. Sci Rep. 2016;6:34255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Choi YS, Park JA, Kim J, Rho SS, Park H, Kim YM, et al. Nuclear IL-33 is a transcriptional regulator of NF-kappaB p65 and induces endothelial cell activation. Biochem Biophys Res Commun. 2012;421:305–11. [DOI] [PubMed] [Google Scholar]
- 52.Brunner SM, Rubner C, Kesselring R, Martin M, Griesshammer E, Ruemmele P, et al. Tumor-infiltrating, interleukin-33-producing effector-memory CD8(+) T cells in resected hepatocellular carcinoma prolong patient survival. Hepatology. 2015;61:1957–67. [DOI] [PubMed] [Google Scholar]
- 53.Baumann C, Bonilla WV, Frohlich A, Helmstetter C, Peine M, Hegazy AN, et al. T-bet-and STAT4-dependent IL-33 receptor expression directly promotes antiviral Th1 cell responses. Proc Natl Acad Sci USA. 2015;112:4056–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Luzina IG, Pickering EM, Kopach P, Kang PH, Lockatell V, Todd NW, et al. Full-length IL-33 promotes inflammation but not Th2 response in vivo in an ST2-independent fashion. J Immunol. 2012;189:403–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lukashevich IS, Rodas JD, Tikhonov II, Zapata JC, Yang Y, Djavani M, et al. LCMV-mediated hepatitis in rhesus macaques: WE but not ARM strain activates hepatocytes and induces liver regeneration. Arch Virol. 2004;149:2319–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alvarez F, Fritz JH, Piccirillo CA. Pleiotropic effects of IL-33 on CD4(+) T cell differentiation and effector functions. Front Immunol. 2019;10:522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Endo Y, Hirahara K, Iinuma T, Shinoda K, Tumes DJ, Asou HK, et al. The interleukin-33-p38 kinase axis confers memory T helper 2 cell pathogenicity in the airway. Immunity. 2015;42:294–308. [DOI] [PubMed] [Google Scholar]
- 58.Petrova T, Pesic J, Pardali K, Gaestel M, Arthur JSC. p38 MAPK signalling regulates cytokine production in IL-33 stimulated Type 2 Innate Lymphoid cells. Sci Rep. 2020;10:3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. 2011;208:1367–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shyer JA, Flavell RA, Bailis W. Metabolic signaling in T cells. Cell Res. 2020;30:649–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.