

Abstract
Single-cell image analysis provides a powerful approach for studying cell-to-cell heterogeneity, which is an important attribute of isogenic cell populations, from microbial cultures to individual cells in multicellular organisms. This phenotypic variability must be explained at a mechanistic level if biologists are to fully understand cellular function and address the genotype to phenotype relationship. Variability in single cell phenotypes is obscured by bulk read-outs or averaging of phenotypes from individual cells in a sample, thus single-cell image analysis enables a higher resolution view of cellular function. Here, we consider examples of both small- and large-scale studies carried out with isogenic cell populations assessed by fluorescence microscopy, and we illustrate the advantages, challenges and promise of quantitative single-cell image analysis.
Keywords: single-cell image analysis, phenotypic diversity, phenotypic heterogeneity, high-content screening, fluorescence microscopy
Introduction
Phenotypic diversity is a consistent feature of cell populations, from microbial cultures to individual cells in multicellular organisms. Remarkably, even genetically identical cells grown in the same environment can exhibit different phenotypes depending on their individual physiology and molecular fluctuations in cellular processes and response mechanisms (Geiler-Samerotte et al., 2013). Phenotypic heterogeneity is evolutionarily important as it may represent a bet-hedging strategy that helps microorganisms cope with changing environments, or it may allow the division of labour in a population (Acar et al., 2008; Ackermann, 2015; Levy et al., 2012). In more complex organisms, single-cell heterogeneity is an integral component of development and differentiation, as well as in diseases such as cancer.
Historically, analyses of cell populations have been largely limited to bulk read-outs, producing an average phenotype for cells in a sample, which does not capture information about population heterogeneity. The past two decades have seen an explosion of single-cell technologies, enabling quantitative analysis of the properties and molecular constituents of individual cells (Lee et al., 2020). Many recent studies have analyzed the transcriptome of single cells using RNA-seq, producing key insights into the gene expression changes associated with different cell states. Going a step further, single-cell image analysis has emerged as a powerful technique for exploring the functional consequences of gene expression dynamics, including effects on protein localization and abundance, metabolite concentrations, signaling events and overall cell morphology.
In this review, we focus on microscopy, which is arguably the ‘original’ single-cell technique, with inherent information about single cells present in micrographs. Major advances in single-cell image analysis have been driven by innovations in high through-put microscopy and computational methods, enabling the collection of spatially and temporally resolved data for individual cells, with the potential to characterize diverse phenotypes and states within cell populations. Here, we describe examples of both small- and large-scale studies done with isogenic cell populations assessed by fluorescence microscopy. We illustrate the advantages and challenges associated with single-cell image analysis and the power of this approach to reveal new biological insights.
Looking through the lens: low-throughput studies where single cells matter
Phenotypic heterogeneity within cell populations reflects many factors, including both regulated and stochastic variations in cellular processes and responses, population context and microenvironment, and the consequent divergence of cell behaviours and fates in space and over time (Figure 1). To explore the mechanistic underpinnings of cell-to-cell heterogeneity, biologists have devised powerful single-cell imaging assays that often exploit fluorescent reporters, some examples of which are shown in Table 1. Below, we discuss examples of low throughput imaging studies that query a broad range of bioprocesses and serve to illustrate the potential of single-cell analysis to reveal biological events that are obscured by population-based read-outs.
Figure 1. Sources of phenotypic heterogeneity.
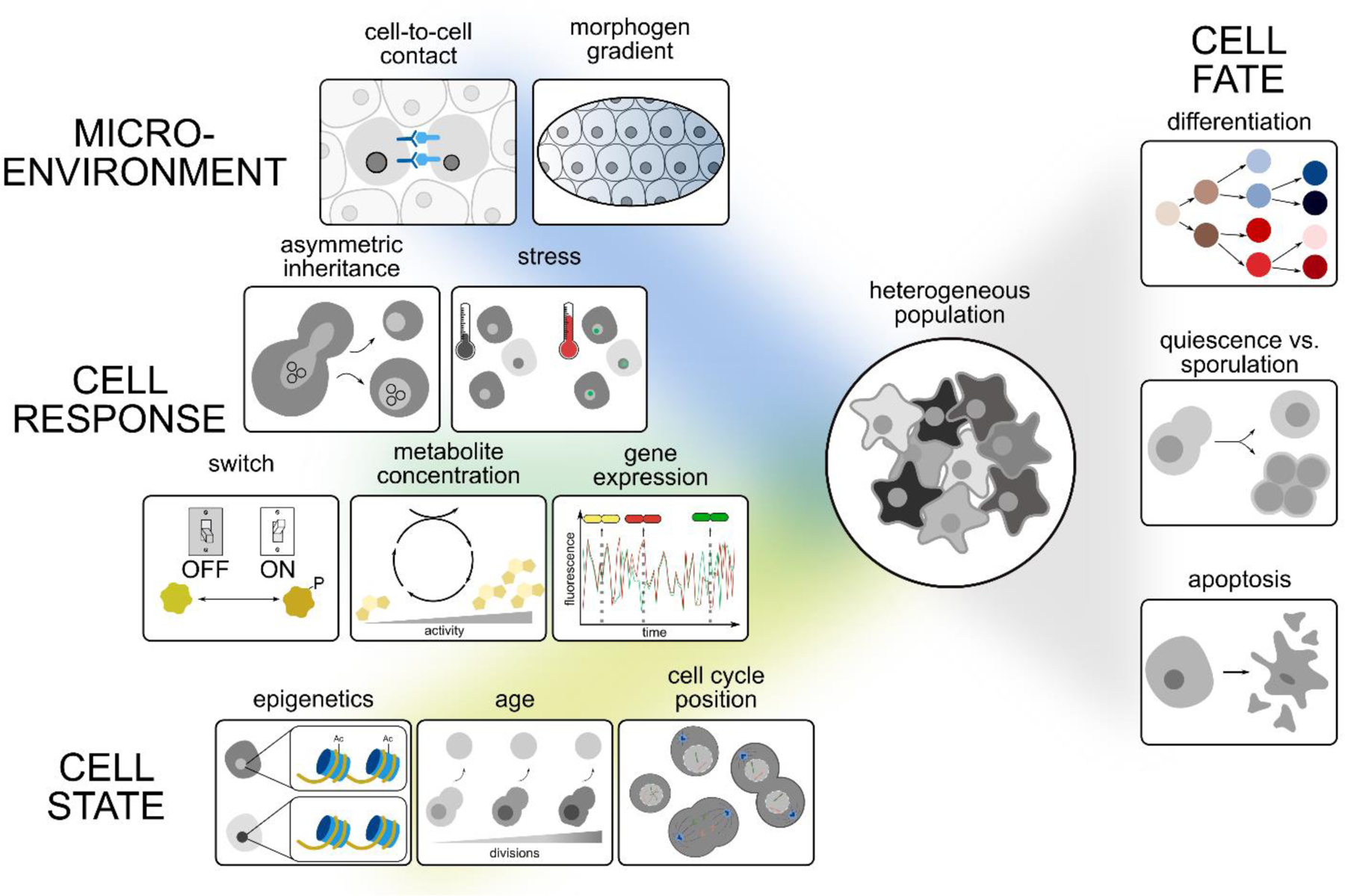
Phenotypic heterogeneity in an isogenic population can arise due to variations in a cell’s microenvironment, and cell-to-cell differences in cell responses and states. These, in turn, can affect a cell’s fate. Shown are some examples from each of these categories, which we discus in more detail in the main text.
Table 1:
Examples of reporters used in single-cell imaging
| Reporter | Information provided | Example [organism] (Reference) |
|---|---|---|
| Molecule-specific | ||
| Fluorescently tagged | Organelle morphology, protein abundance and localization, metabolic state | Gap1-mNeon to assess nitrogen metabolism [Sc] (Arguello-Miranda et al., 2018) |
| tFT linked to protein or organelle localization sequence | Protein or organelle age and stability | tFT-PTS to determine age of peroxisomes [Sc] (Kumar et al., 2018) |
| Antibody system | Organelle morphology, protein abundance and localization | anti-LAMP1 antibody to assess lysosome morphology [Hs] (Liberali et al., 2014) |
| MS2 RNA stem loop + GFP-MS2 coat protein system | RNA localization, abundance | MS2 fused to TFF1 gene to assess RNA abundance [Hs] (Rodriguez et al., 2019) |
| Signalling | ||
| Relocation reporter | Activation of pathway | JNK-KTR to quantify activation of JNK pathway [Hs] (Lane et al., 2019; Regot et al., 2014) |
| TF binding elements or isolated promoter driving reporter expression |
Activation of pathway | HSP70pr-CFP to quantify activation of heat shock pathway [Hs] (Gaglia et al., 2020) |
| Cell State | ||
| Cell cycle reporter | Cell cycle position | FUCCI system to assess G1, G1/S, S/G2, M/early G1 [Hs] (Sakaue-Sawano et al., 2008) |
| pH sensor | Ratiometric readout of intracellular pH | Ratiometric pHluorin to quantify cytosolic pH [Sc] (Mouton et al., 2020) |
| Flux sensor | Metabolic flux readout | CggR-based YFP system to quantify FBP as a measure of metabolic flux [Sc] (Monteiro et al., 2019) |
| Redox sensor | Ratiometric readout of metabolite levels | iNap-cpYFP to quantify intracellular NADPH/NADP+ levels [Hs] (Tao et al., 2017) |
| FRET-based sensor of metabolite | Intracellular concentration of metabolite | ZapCV2 to quantify Zn2+ in cytosol [Hs] (Lo et al., 2020) |
| Live cell stains | Abundance and morphology of organelles, cellular structures, metabolites, proteins | Fluorescently labelled WGA to quantify bud scars (determine replicative age) [Sc] (Mattiazzi Usaj et al., 2020) |
Abbreviations: tFT, tandem fluorescent timer; PTS, peroxisome targeting sequence; JNK-KTR, c-Jun N-terminal kinase-kinase translocation reporter; TF, transcription factor; HSP, Heat Shock Protein; FUCCI, fluorescent ubiquitination-based cell cycle indicator; FBP, fructose-1,6-bisphosphate; iNap-cpYFP, indicator of NADPH fused to circularly permuted YFP; FRET, Förster resonance energy transfer; ZapCV2, Zap1 zinc finger binding domain connecting FRET pair CFP and circularly permuted Venus; WGA, wheat germ agglutinin; Hs, Homo sapiens; Sc, Saccharomyces cerevisiae
Cell-to-cell variability
One prominent source of heterogeneity in single cells is stochastic differences in regulators controlling key molecular processes. The small number of macromolecules involved in some biological processes such as gene expression, means that even modest variation between isogenic cells can have significant phenotypic consequences. A classic study measured the stochasticity of gene expression and distinguished between two mechanisms by which it was generated (Elowitz et al., 2002). The experiment involved constructing bacterial strains expressing two distinguishable versions of GFP with identical promoters regulated by the Lac repressor. Intrinsic variation, quantified as the ratio between the two fluorescent proteins within a single cell, uncovered variation inherent to the biochemical processes associated with gene expression, such as the binding of Lac repressor to each promoter. In contrast, extrinsic variation, measured as the variation in expression of each GFP between cells, was attributed to inter-cellular fluctuations in other cellular components.
More recently, single-cell image analysis of time-lapse images was used to assess reporter gene expression in a cell line model expressing four selected chromatin regulators (CRs) engineered to bind promoters only in the presence of an exogenous inducer (Bintu et al., 2016). When assessing population averages, promoter recruitment of each CR appeared to cause partial inhibition of gene expression. However, assessing individual cells revealed that all four CRs silenced the reporter gene in an all-or-none fashion, each with different dynamics. Likewise, when the CRs were released from binding to chromatin, reactivation of the reporter gene occurred in an all-or-none manner. Thus, single-cell image analysis showed that cells stochastically switch between active and silent states, rather than transitioning gradually through different activity levels. In other studies of transcription, techniques for direct imaging of single RNA molecules in cells have been developed, including multiplexed fluorescence in situ hybridization or insertion of MS2 stem loops, derived from an E. coli phage, in individual RNAs together with the expression of GFP-MS2-binding-protein in live cells (Eng et al., 2019; Femino et al., 1998; Rodriguez et al., 2019). Unlike reporter gene studies, these approaches permit analysis of local neighbourhood relationships such as ligand-receptor interactions, and the spatial organisation and connectivity of neurons (Zhang et al., 2020)(Box 1).
Box 1: Transcriptomics with single molecule FISH.
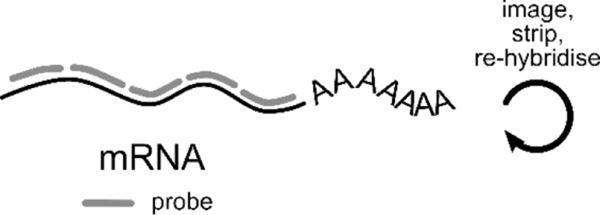
| |
|---|---|
| Fluorescence in situ hybridisation (FISH) of RNA offers spatial information unobtainable from sequencing. It is the gold standard for accurately determining RNA counts and can capture up to 10000 unique transcripts (Torre et al., 2018). Through iterative rounds of imaging, hybridisation, and re-probing (cartoon above), RNA species are identified by different imaging readouts (cartoons below). Some recent single molecule FISH (smFISH) technologies include: | |
readout

|
osmFISH (cyclic-ouroboros
smFISH) The simplest type of FISH, osmFISH, is a non-barcoded strategy that has a high dynamic range. In each cycle, each DNA probe labelled with a unique fluorophore targets a distinct mRNA. The number of targets detected is the product of the number of fluorescent channels and the number of cycles. This technique was used to map the somatosensory cortex in mice (Codeluppi et al., 2018). |
| The remaining approaches we outline here require multiple cycles to identify each RNA. | |
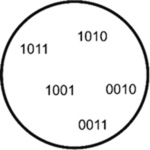
|
MERFISH (Multiplexed
error-robust FISH) MERFISH assigns 2-bit barcodes (0 for absence, 1 for presence of fluorescence) to RNA species through iterative rounds of hybridization and imaging (Moffitt et al., 2016). This way, in 15 cycles, up to 2^15 (~30000) unique barcodes can be assigned. However, to allow for error correction, some barcodes are left intentionally unassigned. MERFISH was used to identify ~1600 cell-cycle dependent genes as well as RNAs enriched in the different compartments in U-2 OS cells (Xia et al., 2019). |
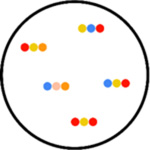
|
STARmap (spatially resolved
transcript amplicon readout mapping) STARmap is useful for 3-D systems. RNA species are paired with DNA probes in order to produce a DNA nanoball using amplification. Here, hydrogel-tissue chemistry is used to preserve the spatial relationships in tissues. Fluorescent-based sequence readout is then used to identify RNA transcripts. STARmap was used in the mapping of 1000 genes of the medial prefrontal cortex of the adult mouse brain (Wang et al., 2018). |
|
seqFISH+ (sequential
fluorescent in situ
hybridisation) seqFISH+ uses super-resolution confocal microscopy to avoid optical crowding, and pseudo-colors to reduce imaging cycles. Each mRNA species has a unique color sequence, so the number of unique barcodes assigned is F^n (F=fluorophores, n=cycles). seqFISH(+) was used to characterize the transcriptomes of tissues such as the cortex, sub-ventricular zone and olfactory bulb in mice and to capture spatial information between ligand-receptor pairs in neighboring cells (Eng et al., 2019; Eng et al., 2017). |
Even when populations are known to be heterogeneous, single-cell image analysis can overturn old ideas about mechanisms of gene regulation based on averaging. HSF1 (heat shock factor 1) is a transcriptional activator of chaperone and other heat stress genes and accumulates in nuclear foci during stress. In bulk cell population analyses, the formation of HSF1 foci correlates with increased heat shock response, leading to the prevailing view that foci formation marks cells that are actively upregulating chaperone genes. Single-cell image analysis of HSF1 in primary human tumour cells revealed heterogeneity, as expected, but surprisingly, the fraction of HSF1 in foci in individual cells was anti-correlated with chaperone protein levels (Gaglia et al., 2020). Inducing proteotoxic stress revealed that dissolution of HSF1 foci - not their formation - correlated with HSF1 activity. Indeed, cells with persistent foci were more prone to undergo apoptosis, indicating that they were not protected from stress.
Another source of phenotypic heterogeneity is replicative aging, which has been well studied in Saccharomyces cerevisiae. Budding yeast, which has a replicative lifespan of less than 30 generations, divides asymmetrically, producing a daughter that is smaller than the mother, and serves as a model for the aging of mitotic cell types, such as stem cells in higher eukaryotes (reviewed by (Janssens and Veenhoff, 2016; Knorre et al., 2018; Steinkraus et al., 2008)). In many cases, due to asymmetric segregation, older more damaged components remain in the mother and the daughter gets the pristine newly synthesized versions. This phenomenon was recently observed with peroxisomes, whose age was determined using a tandem fluorescent timer fused to a peroxisome targeting sequence (Kumar et al., 2018). Tandem fluorescent timers are fusions of two single-color fluorescent proteins that mature with different kinetics; because of the different folding rates, the ratio of the two fluorescent proteins can be used to determine the age of the protein in living cells (Khmelinskii et al., 2012).
The difference in size between yeast mothers and daughters enables the use of microfluidic chambers to retain aging mothers and let daughter cells wash away. Following single cells as they age has revealed that old cells accumulate extrachromosomal DNA circles (Denoth-Lippuner et al., 2014; Hughes and Gottschling, 2012), misassembled nuclear pore complexes (Rempel et al., 2019), aggregated mitochondria (Huberts et al., 2013; Hughes and Gottschling, 2012), oxidatively damaged proteins and protein aggregates, as identified by formation of Hsp104 foci (Aguilaniu et al., 2003), and have higher vacuolar and lower cytosolic pH (Hughes and Gottschling, 2012; Mouton et al., 2020). Because most cells in any population are newly born or young, microfluidics coupled to single-cell image analysis provides a simple way to track the small sub-population of old cells to visualize differential phenotypes associated with aging.
Recently, a modified microfluidics system that retains mother cells throughout their lifespan, and also captures the most recently born daughters, has been used to investigate variability during replicative aging in single S. cerevisiae cells (Jin et al., 2019; Li et al., 2020; Li et al., 2017). Two types of aging were observed with about equal frequency: one characterized by elongated daughter cells with abnormal nucleolar morphology and the other, associated with a shorter lifespan, defined by small round daughter cells with aggregated mitochondria. The largely irreversible decision between the two aging paths occurred within 5–10 divisions after birth. Genetic experiments and computational modeling identified perturbations that increased the fraction of cells undergoing each type of aging and even led to a new longer-lived state.
Cell State
Cell state refers to the overall physiological condition of a cell, which includes its metabolic status, position in the cell cycle, and state of quiescence or growth. In isogenic populations, individual cells may express a spectrum of possible phenotypes, making single-cell image analysis essential for understanding how cell state impacts cell behaviour. Below we describe some of the reporters that have been developed to illuminate cell state, providing insight into cell fate decisions.
Growing cell populations contain cells in different stages of the cell cycle, which represent widely divergent cell states, including S and M phases that require specialized molecular machines to replicate and segregate chromosomes respectively, and growth phases that integrate information about cell state to prepare the cell for these major events. The classic “fluorescent ubiquitination-based cell cycle indicator” (FUCCI) system, offers a method to visually distinguish metazoan cells at different phases of the cell cycle due to the expression of red and green fluorescent proteins that are under the control of two ubiquitin ligases active at different stages of the cell cycle (Sakaue-Sawano et al., 2008). Multiple variants of the FUCCI system that report on cell cycle stages with finer gradations, as well as other cell cycle reporters, have been developed (Sakaue-Sawano et al., 2017). For example, the localization and abundance of fluorescently-tagged endogenous proliferating cell nuclear antigen (PCNA) reports on cell cycle position, as well as transition into quiescence, and uses only a single imaging channel (Zerjatke et al., 2017). At the individual cell level, cell cycle stage can determine the outcome of a perturbation. For example, the cell cycle stage of cells when they were deprived of zinc determined whether they either entered quiescence or began another cell cycle, and then stalled in S-phase (Lo et al., 2020).
A wide range of genetically encoded reporters have been developed that are useful for assessing the metabolic status of a cell, including attributes important for specific cellular processes. For example, Förster resonance energy transfer (FRET)-based ATP probes, referred to as “ATeams”, fused to organelle-targeting sequences, provide a ratiometric readout of ATP levels. Several targeting sequences were used to show that organelles respond differently to glucose depletion: in the mitochondrial matrix, ATP concentrations change rapidly, whereas ATP levels in the cytosol and endoplasmic reticulum are less dynamic (Depaoli et al., 2018). In another example, fluorescent redox sensors have been developed for NADH and NADPH; in these, binding to the reduced form causes a conformational change, resulting in a shift in excitation peak (Tao et al., 2017; Zhao et al., 2015). A proof of principle study monitoring redox dynamics during the cell cycle revealed that levels of cytosolic NADPH rapidly increased just prior to cell division (Zou et al., 2018).
Changes in intracellular pH can have profound effects on many cellular processes, including metabolism, response to growth factors and other external agents, and proper organelle function. Intracellular pH can be tracked in single cells using ratiometric pHluorins, GFP variants whose excitation profiles respond to pH (Miesenbock et al., 1998). For example, a study tracking cytosolic pH in budding yeast revealed modest acidification in early aging and a steep drop in pH during the cells’ final division cycle (Mouton et al., 2020). These observations allowed tests for causality using single-cell image analysis, revealing that acidification was, in fact, a consequence and not a cause of senescence.
Several novel experimental tools have recently been developed to measure levels of metabolites in single cells, many of which make use of metabolite‐binding transcription factors (Hanko et al., 2020). A step beyond quantifying metabolite concentration is measuring flux, the relevant functional output of metabolic activity. Since fructose-1,6-bisphosphate (FBP) levels strictly correlate with glycolytic flux, a modified Bacillus subtilis CggR transcription factor that binds FBP, together with a synthetic promoter, were used to report on flux in budding yeast (Monteiro et al., 2019). Single-cell image analysis revealed that FBP concentration, and thus glycolytic flux, changes during the cell cycle, peaking around cytokinesis and in G1 phase.
Cell Fate
With single-cell image analysis of time-course experiments, markers of cell state can be used to follow the pathways taken by individual cells to their ultimate fate. Below, we illustrate the power of this approach by summarizing examples of studies in which single-cell image analysis has contributed new insights into cell fate trajectories in response to signals such as the presence of bacteria or absence of nutrients, and during differentiation and development.
Protein kinases are broadly involved in cell signaling and function, and many assays have been developed to report on their function. Genetically encoded biosensors for protein kinase activity, kinase translocation reporters (KTRs), are designed to convert phosphorylation of a protein kinase target into a nucleocytoplasmic shuttling event which can be visualized microscopically (Regot et al., 2014). The fate choices made by cells faced with bacterial infection were analyzed by single-cell image analysis using KTRs by simultaneously measuring both fluorescently labeled Salmonella bacteria and host signaling activity (Lane et al., 2019). Remarkably, the dynamics of activation of just two signaling pathways, MAPK and NF-kB, were sufficient to distinguish between soluble bacterial proteins and live bacteria, extracellular and intracellular bacteria, and to define the severity of infection, thus guiding appropriate cell fate decisions.
By tracking multiple readouts in the same cell, single-cell image analysis can be used to deconvolve cell decision-making. In response to starvation, diploid yeast cells divide mitotically 2–3 times, then arrest their cell cycle and either enter meiosis or become quiescent. Six-color live-cell imaging was used to study this decision, with reporters of cell cycle position, entry into the sporulation program, storage carbohydrates, lipid droplets, nitrogen metabolism, and mitochondrial morphology (Arguello-Miranda et al., 2018). Combinations of parameters were identified that had high predictive power, revealing that, for the vast majority of cells, their ultimate fate was determined before the last cell division, far earlier than previously thought. Single-cell image analysis showed that cells process information from multiple sources to select a future course of action and that the metabolic state of the cell, especially the size of the vacuole, determines the meiosis/quiescence cell fate decision.
Single-cell image analysis has also long been crucial for understanding cell fate decisions in multicellular model organisms and their developmental trajectories. Since the 1980s, microscopy has enabled mapping of single-cell fates during development, beginning with pioneering and comprehensive cell lineage studies in Caenorhabditis elegans (Sulston et al., 1983). Current approaches to map the developmental trajectory of single cells typically involve time-lapse imaging to identify lineage relationships. Following imaging, cells are fixed and the cell states of the ‘differentiated’ cells are characterized using RNA FISH, which measures expression of a subset of genes (Hormoz et al., 2016). Alternatively, recent methods make use of engineered genomic target sites that can be mutated using technologies such as CRISPR-Cas9 or serine integrases to enable downstream reconstruction of lineage trees using in situ sequencing of the targeted sites (Askary et al., 2020; Chow et al., 2020; Frieda et al., 2017). This approach bypasses the need for time-lapse imaging, enabling the study of organisms that cannot be easily examined under the microscope.
Time-lapse imaging and single-cell analysis have recently been used to address important questions in a variety of developmental situations, including disease states. For example, single-cell image analysis was used to address the long-standing question of how a key protein regulator of pluripotency, Nanog, is autoregulated. The results of computational simulation of three different regulatory scenarios were compared against single-cell time-lapse data of wild type and cells over-expressing Nanog, supporting a model of weak negative feedback in Nanog dynamics (Feigelman et al., 2016). In another example, the prevailing view that embryonic stem (ES) cells transitioned randomly across different states was challenged. ES cells were imaged over time and endpoint RNA FISH was used to define cell types; this study showed that cell state transitions were stochastic, reversible and structured, occurring in a step-wise fashion (Hormoz et al., 2016). Single cell imaging has also been used to explore the cell fate trajectories of aberrant developmental programs, such as cancer cells. For example, quantitative measurement of overall nuclear and cell morphology revealed distinct and heritable morphological features of breast cancer cells that were predictive of tumorigenic and metastatic potential in mouse models (Wu et al., 2020).
Systematic studies using high-throughput single-cell image analysis
As outlined above, single-cell image analysis has revealed that many different factors, both deterministic and stochastic, can contribute to phenotypic variability. So far, we’ve considered low-throughput approaches that exploit single-cell image analysis to address important hypotheses about cell function that are not otherwise accessible. The availability of microscopes tailored for high-throughput imaging, and associated development of automated imaging methods, has produced high-content screening (HCS) platforms (Boutros et al., 2015; Mattiazzi Usaj et al., 2016; Smith et al., 2018), which enable unbiased assessment of the origins and consequences of cell-to-cell heterogeneity. Below, we summarize systematic HCS approaches, and use selected examples to illustrate the incredible potential of single-cell analysis to advance our understanding of the genotype-to-phenotype relationship.
High-content screening methodology
HCS combines high-throughput imaging of large sample collections with computational image analysis, representing a comprehensive and unbiased approach to investigate diverse biological questions at a global scale. HCS is particularly useful for screens that involve systematic perturbations using genetic methods or bioactive compounds to systematically reveal genes and pathways that influence selected bioprocesses. Comprehensive collections of mutants are available for yeast and other model organisms, such that genome-wide HCS can be carried out in an arrayed format, removing the need for post-processing to identify the underlying genetic mutation. The phenotype of interest is commonly visualized using fluorescent tags or stains to mark compartments or provide functional readouts, with either living or fixed cells. Studies that collect fixed cell end-point measurements use immunostaining or fluorescent dye reporters to detect structures of interest. For example, in yeast, triple staining with fluorescent dyes for cell wall, actin cytoskeleton and nucleus was used for morphological profiling in several studies (Ohya et al., 2015; Ohya et al., 2005). A cognate method called ‘Cell Painting’ has been widely used for morphological profiling of metazoan cells, in which samples are stained with six fluorescent dyes (Bray et al., 2016). More recently, iterative indirect immunofluorescence methods have been used to produce maps of localization and abundance for up to 40 proteins per cell, an approach that can be applied to quantitative single-cell image analysis in various experimental settings (Gut et al., 2018).
Major advances in HCS methods have been propelled by the development of computational image analysis workflows, which are essential for high-throughput and time-lapse studies, where the number of samples and single cells to be analyzed can be in the millions. Developing the image analysis pipeline and setting up the biological assay are best done concurrently, to ensure appropriate fluorescent markers are incorporated for downstream image analysis and that sensible controls for testing the performance of the image analysis method are included. After image acquisition, image analysis includes several key steps, some of which are unique to single cell image analysis, as opposed to other single-cell techniques (reviewed in (Caicedo et al., 2017; Grys et al., 2017; Mattiazzi Usaj et al., 2016)). Importantly, these steps can be used in image analysis of both low-throughput and high-throughput studies.
The first step in image analysis involves locating single cells within images, a crucial task upon which subsequent steps are contingent. Object segmentation is typically done by initially identifying individual fluorescently-labelled nuclei in the image, then defining the cytoplasm and cell boundaries, for example by using a watershed algorithm (Beucher, 1992). Depending on the experimental goal, segmentation of subcellular objects may also be needed and, if time-course data are involved, object tracking to identify the same cell in different images may be required. Second, hundreds of quantitative features that describe each segmented object are extracted. The computer vision field has developed a variety of numeric descriptors of segmented objects, including summaries of the intensity distribution, shape, size, texture, radial distribution, and granularity of the segmented region of interest. Measurements tailored to the problem of interest can be calculated from the extracted quantitative features, such as the ratio of the area of a segmented compartment to the total area of the cell.
Subsequently, dimensionality reduction or feature selection may be required to minimize redundancy of the extracted features, then individual cells are computationally grouped based on the extracted features. Different machine learning approaches for this step abound and are being continuously improved. Two commonly used approaches are: i) unsupervised learning (clustering and outlier detection), which uses unlabeled cells for model training based on commonalities/similarities between vectors comprised of the extracted numeric features; and ii) supervised learning (classification) where a mathematical model is trained using manually labelled representative cells for each predefined phenotype of interest (reviewed in (Caicedo et al., 2017; Grys et al., 2017)). A variety of software tools have been developed for phenotypic image analysis (reviewed in (Smith et al., 2018)).
More recently, many labs have employed machine learning based on artificial neural networks, also known as deep learning, to segment cells, extract quantitative features from single cells, classify cells into phenotypic classes, and track individual cells (Caicedo et al., 2019; Durr and Sick, 2016; Kraus et al., 2017; Lu et al., 2019; Lugagne et al., 2020; Moen et al., 2019). A key advantage of deep learning classification methods is that they automatically learn suitable features to distinguish between phenotypes directly from image/object pixels. Deep learning is becoming increasingly accessible to experimental labs: collections of deep learning models are enabling users with limited expertise in machine learning to analyze their own data (Chamier et al., 2020), and cloud-based solutions for scaling deep learning workflows are allowing cost effective analyses of large datasets for labs with limited computational resources (Bannon et al., 2021).
Quantitative single-cell data extracted from high-content imaging studies can also be used for mathematical modeling of complex biological systems. Such models interpret and simulate the extracted data in order to obtain a deeper insight into the studied system and predict its behaviour. For example, measurements of protein abundance or status (e.g. phosphorylation) in a biological pathway have contributed to understanding cell state (Miller et al., 2018), cell fate (Min et al., 2020), and drug-resistant mechanisms in cancer cells (Gerosa et al., 2020).
A crucial component of HCS is downstream data analysis, where approaches vary depending on the question being asked. Often, low-throughput studies employ complex data analysis strategies that cannot always be scaled to accommodate larger datasets. Although image-based data have always been available at single-cell resolution, the large amount of quantitative data, inherent variability between single cells, and resultant complexity in the analysis workflow have often led to the use of population averages even when single-cell data is extracted from images. The information content in many imaging screens is thus hugely underutilized. Published imaging datasets represent a tremendous opportunity for re-analysis at the single-cell level to uncover previously hidden information. However, long-term storage, accessibility and distribution of the acquired datasets, particularly raw images, represent a constant challenge. Here, use of open-source bioimage database systems, such as OMERO (Allan et al., 2012), and increased sharing of resources through community-driven public repositories such as IDR (Williams et al., 2017) play a pivotal role. Additionally, pixel data and metadata are frequently stored in different, often proprietary, formats, making reanalysis of the data challenging. The Open Microscopy Environment (OME) consortium, which developed OMERO, has produced Bio-Formats, an open-source software tool that aims to address this issue by enabling the reading and writing of image data using standardized formats (Linkert et al., 2010).
Single-cell HCS in yeast
The most versatile toolbox of genome-scale genetic reagents is available for the budding yeast. The predominant collections used in single-cell image analysis are: the genome-wide gene-deletion collection (Giaever et al., 2002), which enables the analysis of loss-of-function mutations in non-essential genes; the collection of conditional, temperature-sensitive alleles of essential genes (Costanzo et al., 2016; Li et al., 2011), which enables the analysis of the highly-conserved essential gene set and; the GFP collection, where each ORF has been systematically tagged at its C terminus with GFP (Huh et al., 2003). Below, we summarize recent efforts to systematically explore yeast cell biology using single-cell image analysis. We note that to date, most large-scale imaging studies, including those with yeast, have done aggregate analysis rather than single-cell analysis.
Large-scale single-cell image analysis of genetic perturbations.
A major advantage of the yeast system is that well established methods for high-throughput yeast strain construction can be used to introduce markers of a compartment or process of interest into the mutant strain arrays mentioned above. For example, single-cell image analysis has been used to screen yeast mutant collections for specific morphological phenotypes, including defective spindle morphology (Vizeacoumar et al., 2010), increased fraction of cells with DNA-damage foci (Styles et al., 2016), altered fraction of cells with inclusion bodies containing Synphilin-1 (Zhao et al., 2016), defects in asymmetrical segregation of protein aggregates marked by Hsp104 (Hill et al., 2016), and abnormalities in four endocytic compartments (Mattiazzi Usaj et al., 2020). Single-cell image analysis has also been used to screen for non-morphological phenotypes, including a search for mutants that had reduced or improved memory of a previous growth condition, so-called transcription reinduction memory (Bheda et al., 2020). Many of these screens involve phenotypes that are not present in all or even most cells in a mutant population, making single-cell analysis essential for a comprehensive understanding of the genotype-phenotype relationship.
In addition to describing genes and pathways, high-throughput screens can also address global questions about heterogeneity in isogenic populations. For example, our screens of endocytic compartment morphology revealed that incomplete penetrance was reproducible and pervasive, with only ~10% of single mutants showing complete penetrance, where all cells in the population express the measured phenotype (Mattiazzi Usaj et al., 2020). Additionally, ~50% of endocytic morphology mutants exhibited morphological pleiotropy, where a single gene perturbation leads to multiple aberrant phenotypes (Mattiazzi Usaj et al., 2020). Consistent with these results, widespread heterogeneity in cell shape was observed in individual mutants in a genome-wide study in Schizosaccharomyces pombe (Graml et al., 2014). Systematic exploration of penetrance and pleiotropy in isogenic mutant populations is rare and more studies are needed to understand the fundamentals of cell-to-cell heterogeneity.
Single-cell image analysis of the GFP collection.
An advantage of image-based proteome analysis is the ability to study protein localization and protein abundance in single cells, including proteins that localize to multiple compartments, and to observe cell-to-cell differences in protein localization or changes in protein levels. More than 15 years ago, visual inspection of images of the original yeast GFP collection produced the first proteome-scale catalogue of subcellular protein localization (Huh et al., 2003). This pioneering work set the stage for more recent projects that have combined cell images of the GFP collection with various machine learning approaches, including deep convolutional neural networks, for quantitative analysis of the proteome in single cells (Chong et al., 2015; Kraus et al., 2017; Parnamaa and Parts, 2017). These proteome surveys also quantitatively determined that over half of all proteins tested localized to multiple subcellular compartments. Because many bioactive compounds and genetic mutations affect only a subset of a population, single-cell image analysis has also been instrumental in identifying changes in the proteome upon perturbation. By extracting single-cell information, quantitative ‘flux networks’ that describe proteome dynamics in response to environmental factors or mutations, which can include changes that affect only a small fraction of cells, have been constructed (Chong et al., 2015; Kraus et al., 2017). In addition, several groups have used single-cell image analysis to identify localization changes in the presence of chemicals using more broadly defined localization categories or by querying only a subset of localizations (Denervaud et al., 2013; Mazumder et al., 2013). Some of these studies used a single image dataset (Chong et al., 2015) to ask new questions or to develop novel computational methods (Kraus et al., 2017; Lu et al., 2018; Parnamaa and Parts, 2017).
HCS studies with single-cell image analysis beyond yeast
Exploring genetic perturbations.
In general, other model systems lack the extensive molecular toolbox of arrayed collections available in budding yeast. Nonetheless, large-scale RNAi screening has been used extensively to identify mutant phenotypes in the past 15 years, and the availability of arrayed siRNA libraries for some systems has greatly facilitated functional genomics using high content-screening (Heigwer et al., 2018; Mohr et al., 2010). Below, we provide examples of HCS approaches for systematic discovery of phenotypes associated with genetic perturbation in a variety of cell line models.
Due to a number of experimental challenges, large-scale high-content imaging screens with living metazoan cells that use single-cell data for phenotype identification remain sparse. One example is the MitoCheck project, where RNAi of each of the ~21,000 human protein-coding genes was followed by live imaging of fluorescently labelled chromosomes and classification of nuclei into one of 16 morphological classes, identifying hundreds of human genes involved in cell division, migration and survival (Neumann et al., 2010). On the other hand, many informative single-cell imaging screens that rely on end-point imaging of fixed cell samples have been performed, uncovering considerable new cell biology. For example, an RNAi HCS screen in Drosophila cells leveraged single-cell data to explore the heterogeneity of cell morphologies and revealed that most genes regulate the transition between discrete cell shapes rather than generating new morphologies (Yin et al., 2013). An unsupervised outlier detection approach identified HeLa cells with altered Golgi morphology, and after calculating phenotype penetrance for each gene and clustering of outlier cells, ten distinct phenotypic clusters were identified. This phenotypic information was used to build a Golgi phenotypic network that maps similarities between genetic perturbations (Hussain et al., 2017). Single-cell data has also been used to account for population context, such as the influence of neighboring cells, a concept that will also be crucial for informative analysis of more complex cell models (Liberali et al., 2014; Snijder et al., 2009; Snijder et al., 2012).
While very powerful, RNAi screens have several caveats, including off-target effects and variable efficiency of silencing, which confound discovery of true biological heterogeneity and the determination of phenotype penetrance. Recently, several CRISPR-Cas tools for gene editing have been developed with fewer off-target effects than RNAi (Boettcher and McManus, 2015; Pickar-Oliver and Gersbach, 2019). Additionally, with appropriate modifications, CRISPR-Cas-based gene editing appears feasible in almost any organism, including bacteria and yeast. Editing efficiency remains a confounding factor for accurate analysis of penetrance, and thus reporters to distinguish between edited and non-edited cells are needed.
As in yeast, arrayed high-content screens in cell lines enable analysis of multiple subtle or complex phenotypes, arguably at the expense of throughput, and require no post-imaging deconvolution to identify the underlying genetic perturbation. Several groups have developed experimental platforms for arrayed CRISPR screens with human or other metazoan cells (de Groot et al., 2018; Kim et al., 2018; Strezoska et al., 2017). For example, an arrayed library of 2,281 CRISPR‐Cas9 plasmids targeting 1457 human genes was constructed and applied in an image-based screen to identify genes affecting cellular morphology and the subcellular localization of components of the nuclear pore complex (de Groot et al., 2018). Since transient transfection of a targeting plasmid results in a mixed population of wild‐type and genetically perturbed cells, the authors first distinguished between wild‐type cells and cells expressing Cas9 (which may or may not be genetically perturbed). By training classifiers that consider the heterogeneity of specific subsets of features, they were able to identify genes involved in distinct cellular processes.
Nevertheless, large genomes make arrayed screening, and in particular the use of arrays for live-cell imaging, expensive and technically challenging. Several groups have thus proposed pooled CRISPR screening solutions for HCS. For example, targeted in situ sequencing was used to demultiplex a library of genetic perturbations after image-based phenotyping of individual human cells (Feldman et al., 2019). In Visual Cell Sorting, expression of the photoactivatable fluorescent protein Dendra2 allows for selective and high‐throughput labeling of individual cells exhibiting different phenotypes (Hasle et al., 2020). Labeled cell sub-populations can then be sorted using flow cytometry and subjected to different downstream genomics assays. A similar approach that relies on machine learning to identify cells of interest on-the-fly, followed by their photoactivation, sorting and sequencing, was combined with a large-scale genome-wide CRISPR screen to identify genes affecting the nuclear localization of the transcription factor TFEB (Sarraf et al., 2020).
A potential pitfall of pooled screens is that the results are dominated by the strongest hits and most common phenotypes. Rare phenotypes or mutations that cause an abnormal phenotype in only a subset of cells (incomplete penetrance) are more difficult to detect with pooled approaches and would require screening of much larger cell numbers. Additionally, as with all classification approaches, a priori knowledge (and classifier training) of the target phenotype is needed.
Exploring the proteome.
To date, by far the largest attempt at mapping the subcellular localization of human proteins has been the Cell Atlas project. Twelve thousand human proteins were localized using immunofluorescence microscopy and in a Herculean effort manually assigned to 30 cellular compartments and substructures. Analysis of single-cell immunofluorescence patterns revealed 1855 proteins with variation in either expression levels or spatial distribution (Thul et al., 2017). Images were subsequently also manually classified with a citizen science approach, where, over the span of one year, more than 320,000 gamers of a mainstream video game annotated the Cell Atlas images as a mini-game, producing more than 33 million classifications of subcellular localization patterns. Besides discovering novel localization patterns, these single-cell annotations were used to improve the classification accuracy of a deep learning model (Sullivan et al., 2018). To identify proteins whose abundance varies in a cell-cycle-dependent manner, 1180 proteins that showed cell-to-cell variability in the Cell Atlas images were reassessed by immunofluorescence in FUCCI U2OS cells. Less than a third of these gave significant cell-cycle variation, confirming that multiple factors contribute to cell-to-cell proteome variation in genetically identical cells (Mahdessian et al., 2021).
CRISPR gene editing technology has also been used for the development of fluorescently tagged cell lines for live-cell imaging of specific compartments in different organisms (Leonetti et al., 2016; Mikuni et al., 2016; Roberts et al., 2017). In one method, electroporation of Cas9 nuclease/single-guide RNA ribonucleoproteins combined with a split-GFP system was used for robust, scarless, and specific tagging of endogenous human genes (Leonetti et al., 2016). A CRISPR-Cas9 genome-editing strategy was also used to systematically tag endogenous proteins with fluorescent tags in human induced pluripotent stem cells, producing cell lines that maintain pluripotency, differentiation potential, and genomic stability (Roberts et al., 2017). These isogenic stem cell lines can be used by the community to explore organelle morphology during differentiation or identify the genetic requirements for organelle function. Even without consideration of splicing variants and tissue-type-specific gene expression, the size of such reagent collections, their maintenance and distribution to the community will no doubt pose a particular challenge. In time, however, these collections of cell lines will allow for live-cell proteome-wide HCS, similar to studies conducted in yeast using the GFP collection.
Challenges and Outlook
We have highlighted selected studies done with yeast and other cells that exemplify the value of unicellular and in vitro cell model systems both to dissect various biological processes and to develop experimental and computational approaches that are transferable to more complex systems. Technological advancements have enabled high-throughput imaging, quantification and profiling of organoids. These 3D model systems better reproduce the micro-environment, cell-cell interactions, homeostasis, and dynamics of tissue and organ development in healthy and diseased states. For example, single nuclei from thousands of organoids have been segmented and quantified to characterize the development of intestinal organoids from single cells (Serra et al., 2019). A platform that combines high-throughput organoid production, whole-organoid immunostaining and tissue clearing, and high-content imaging has enabled the detailed analysis of human midbrain organoids (Renner et al., 2020). Single-cell imaging studies, no matter the experimental system and analysis pipeline, provide information that would otherwise remain hidden when treating all cells as a homogenous population. Analysis of single-cell imaging data highlights the importance of concepts such as penetrance and stochasticity, as well as cell fate decisions.
Both experimental and image analysis methods have advanced considerably in the past two decades; we now have many well-established protocols and advanced know-how to reliably and reproducibly perform HCS, extract single-cell data, and measure an increasing number of cell properties. The number of structures that can be observed by live-cell fluorescence microscopy is still limited by the configuration of the microscope and spectral overlap of fluorophores. Two recent studies (Christiansen et al., 2018; Ounkomol et al., 2018) used convolutional neural networks to predict fluorescence microscopy images from unlabelled transmitted-light z-stacks. With larger training sets covering additional diverse phenotypes, including abnormal subcellular phenotypes, this approach promises to enable multi-marker single-cell phenotyping of live cells by combining in silico labeling of trained phenotypes with fluorescence imaging of novel/untrained/rare phenotypes.
Computationally, we are at the stage where we need to systematically dissect single-cell information available through HTP screening and other imaging studies. The overall goal is to gain an understanding of the events that lead to variability in mutant phenotypes, and to unravel the consequences for the cell. This highly complex data, however, requires a high level of expertise that is not available to most labs, which limits the adoption of single-cell image analysis. With the expansion of data-intensive imaging modalities (e.g. lattice light-sheet) the need for robust and reliable computational approaches for the analysis of multidimensional and information-rich data extracted from single cells is even more urgent. With the bottleneck in analysis shifting downstream, the main question thus becomes ‘how do we take into consideration the multidimensionality, sheer size, and full extent of single-cell data in the analysis pipeline?’ High-content screening is inherently interdisciplinary. In the coming years we expect to see additional interdisciplinary developments at the intersection of fields that will address this challenge, and a concurrent emergence of methods that integrate different single-cell datasets to gain a comprehensive and quantitative view of the cell.
Public data challenges, hackathons, and contributions from citizen scientists have provided state-of-the-art solutions to well defined data analysis problems in the past (Caicedo et al., 2017; Ouyang et al., 2019; Sullivan et al., 2018). A similar engagement will prove beneficial to current and future challenges in the field. Privately funded initiatives have been at the forefront of some of these developments. For example, the non-profit Allen Institute for Cell Science is developing openly available reagents and machine learning image-analysis solutions for live-cell fluorescence microscopy (Chen et al., 2020; Ounkomol et al., 2018; Roberts et al., 2017).
Understanding single-cell biology is also highly relevant for potential future biomedical applications; single-cell heterogeneity is common, for example, both in cancer and in drug efficacy. Broader adoption of single-cell image analysis and integration of different single-cell data types will provide unprecedented insight into cell function and behaviour. Ultimately, this will advance our understanding of the relationship between genotype and phenotype. Instead of ‘hiding’ the messy biological data, it is thus prime time to harness the power of single cells.
Figure 2. Overview of HCS workflows.
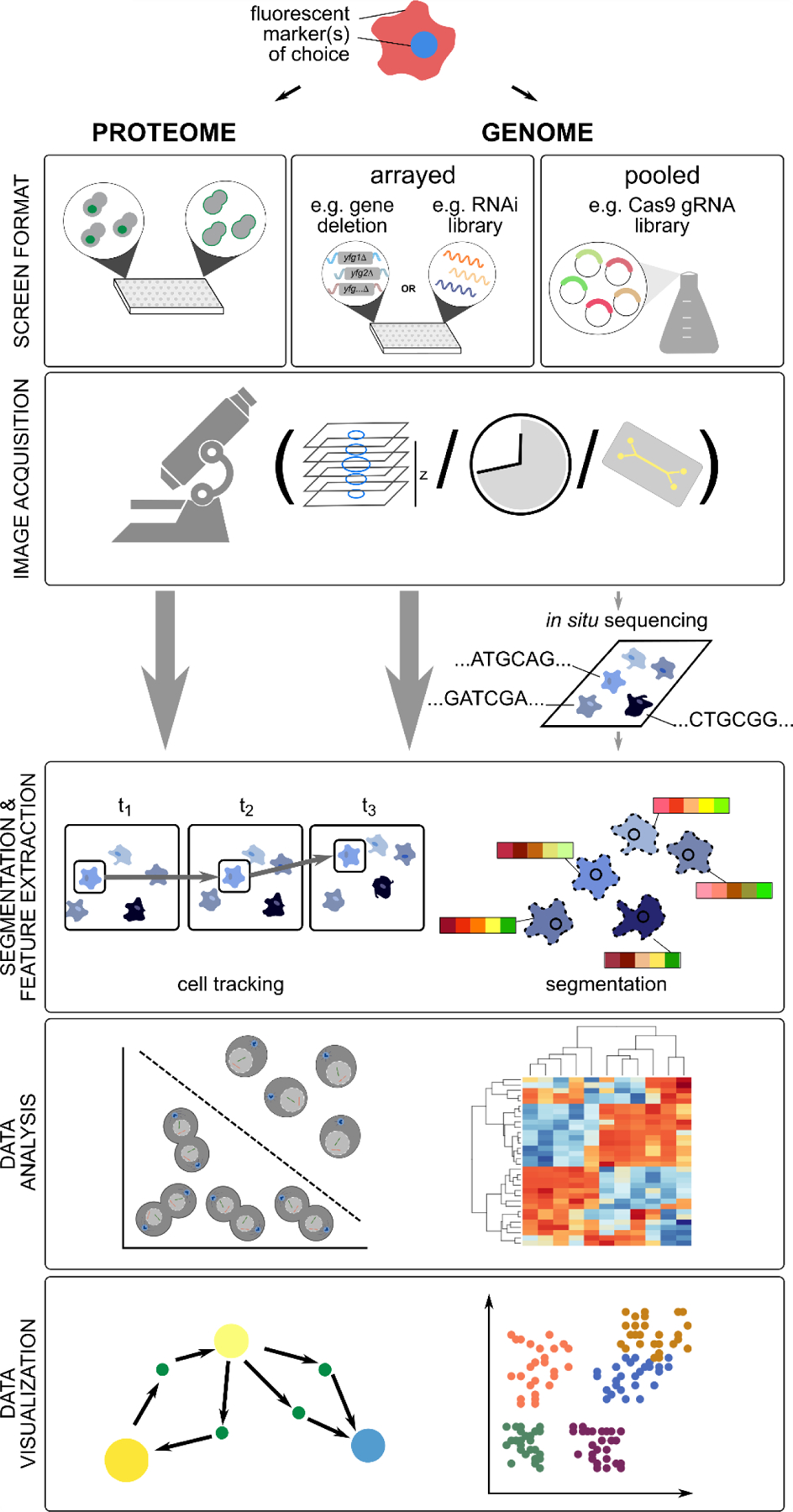
HCS screens, both to monitor proteome-wide changes and large-scale genetic perturbations, are generally performed in arrayed format, but more recently pooled formats have been used. Image acquisition (high-throughput live or fixed-cell imaging of fluorescent markers) may include multiple planes and/or time points. Microfluidics devices may be used to track changes after switching growth conditions or to capture and monitor a subset of cells. Pooled screening approaches require post-imaging deconvolution (e.g. with in situ sequencing) to identify the underlying perturbation. After image acquisition, individual cells are segmented, and in case of time-lapse imaging, tracked, based on positional information. Numeric features describing different cell/phenotype properties are extracted, and the derived feature vectors analyzed with different machine learning approaches, including classification and clustering. Network analysis and feature projection techniques are often used for data visualization. See main text for more details.
Acknowledgments
Work in the Andrews and Boone labs is supported by grants from the Canadian Institutes of Health Research (FDN-143264, FDN-143265), the National Institutes of Health (R01HG005853), the Ontario Research Fund (RE09–11) and Genome Canada (OGI-163). Equipment for high content screening was supported by grants from the Canadian Foundation for Innovation and the Ontario Research Fund. C.B. is a fellow of the Canadian Institute for Advanced Research.
References
- Acar M, Mettetal JT, and van Oudenaarden A (2008). Stochastic switching as a survival strategy in fluctuating environments. Nat Genet 40, 471–475. [DOI] [PubMed] [Google Scholar]
- Ackermann M (2015). A functional perspective on phenotypic heterogeneity in microorganisms. Nat Rev Microbiol 13, 497–508. [DOI] [PubMed] [Google Scholar]
- Aguilaniu H, Gustafsson L, Rigoulet M, and Nystrom T (2003). Asymmetric inheritance of oxidatively damaged proteins during cytokinesis. Science 299, 1751–1753. [DOI] [PubMed] [Google Scholar]
- Allan C, Burel JM, Moore J, Blackburn C, Linkert M, Loynton S, Macdonald D, Moore WJ, Neves C, Patterson A, et al. (2012). OMERO: flexible, model-driven data management for experimental biology. Nat Methods 9, 245–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arguello-Miranda O, Liu Y, Wood NE, Kositangool P, and Doncic A (2018). Integration of Multiple Metabolic Signals Determines Cell Fate Prior to Commitment. Mol Cell 71, 733–744 e711. [DOI] [PubMed] [Google Scholar]
- Askary A, Sanchez-Guardado L, Linton JM, Chadly DM, Budde MW, Cai L, Lois C, and Elowitz MB (2020). In situ readout of DNA barcodes and single base edits facilitated by in vitro transcription. Nat Biotechnol 38, 66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannon D, Moen E, Schwartz M, Borba E, Kudo T, Greenwald N, Vijayakumar V, Chang B, Pao E, Osterman E, et al. (2021). DeepCell Kiosk: scaling deep learning-enabled cellular image analysis with Kubernetes. Nat Methods 18, 43–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beucher S (1992). The watershed transformation applied to image segmentation. Scanning microscopy-supplement-, 299–299. [Google Scholar]
- Bheda P, Aguilar-Gomez D, Becker NB, Becker J, Stavrou E, Kukhtevich I, Hofer T, Maerkl S, Charvin G, Marr C, et al. (2020). Single-Cell Tracing Dissects Regulation of Maintenance and Inheritance of Transcriptional Reinduction Memory. Mol Cell 78, 915–925 e917. [DOI] [PubMed] [Google Scholar]
- Bintu L, Yong J, Antebi YE, McCue K, Kazuki Y, Uno N, Oshimura M, and Elowitz MB (2016). Dynamics of epigenetic regulation at the single-cell level. Science 351, 720–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boettcher M, and McManus MT (2015). Choosing the Right Tool for the Job: RNAi, TALEN, or CRISPR. Mol Cell 58, 575–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutros M, Heigwer F, and Laufer C (2015). Microscopy-Based High-Content Screening. Cell 163, 1314–1325. [DOI] [PubMed] [Google Scholar]
- Bray MA, Singh S, Han H, Davis CT, Borgeson B, Hartland C, Kost-Alimova M, Gustafsdottir SM, Gibson CC, and Carpenter AE (2016). Cell Painting, a high-content image-based assay for morphological profiling using multiplexed fluorescent dyes. Nat Protoc 11, 1757–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caicedo JC, Cooper S, Heigwer F, Warchal S, Qiu P, Molnar C, Vasilevich AS, Barry JD, Bansal HS, Kraus O, et al. (2017). Data-analysis strategies for image-based cell profiling. Nat Methods 14, 849–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caicedo JC, Roth J, Goodman A, Becker T, Karhohs KW, Broisin M, Molnar C, McQuin C, Singh S, Theis FJ, et al. (2019). Evaluation of Deep Learning Strategies for Nucleus Segmentation in Fluorescence Images. Cytometry A 95, 952–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamier L.v., Laine RF, Jukkala J, Spahn C, Krentzel D, Nehme E, Lerche M, Hernández-Pérez S, Mattila PK, Karinou E, et al. (2020). ZeroCostDL4Mic: an open platform to use Deep-Learning in Microscopy. bioRxiv [Google Scholar]
- Chen J, Ding L, Viana MP, Lee H, Sluezwski MF, Morris B, Hendershott MC, Yang R, Mueller IA, and Rafelski SM (2020). The Allen Cell and Structure Segmenter: a new open source toolkit for segmenting 3D intracellular structures in fluorescence microscopy images. bioRxiv [Google Scholar]
- Chong YT, Koh JL, Friesen H, Duffy SK, Cox MJ, Moses A, Moffat J, Boone C, and Andrews BJ (2015). Yeast Proteome Dynamics from Single Cell Imaging and Automated Analysis. Cell 161, 1413–1424. [DOI] [PubMed] [Google Scholar]
- Chow K-HK, Budde MW, Granados AA, Cabrera M, Yoon S, Cho S, Huang T. h., Koulena N, Frieda KL, Cai L, et al. (2020). Imaging cell lineage with a synthetic digital recording system. bioRxiv [DOI] [PubMed] [Google Scholar]
- Christiansen EM, Yang SJ, Ando DM, Javaherian A, Skibinski G, Lipnick S, Mount E, O’Neil A, Shah K, Lee AK, et al. (2018). In Silico Labeling: Predicting Fluorescent Labels in Unlabeled Images. Cell 173, 792–803 e719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codeluppi S, Borm LE, Zeisel A, La Manno G, van Lunteren JA, Svensson CI, and Linnarsson S (2018). Spatial organization of the somatosensory cortex revealed by osmFISH. Nat Methods 15, 932–935. [DOI] [PubMed] [Google Scholar]
- Costanzo M, VanderSluis B, Koch EN, Baryshnikova A, Pons C, Tan G, Wang W, Usaj M, Hanchard J, Lee SD, et al. (2016). A global genetic interaction network maps a wiring diagram of cellular function. Science 353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groot R, Luthi J, Lindsay H, Holtackers R, and Pelkmans L (2018). Large-scale image-based profiling of single-cell phenotypes in arrayed CRISPR-Cas9 gene perturbation screens. Mol Syst Biol 14, e8064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denervaud N, Becker J, Delgado-Gonzalo R, Damay P, Rajkumar AS, Unser M, Shore D, Naef F, and Maerkl SJ (2013). A chemostat array enables the spatio-temporal analysis of the yeast proteome. Proc Natl Acad Sci U S A 110, 15842–15847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denoth-Lippuner A, Krzyzanowski MK, Stober C, and Barral Y (2014). Role of SAGA in the asymmetric segregation of DNA circles during yeast ageing. Elife 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depaoli MR, Karsten F, Madreiter-Sokolowski CT, Klec C, Gottschalk B, Bischof H, Eroglu E, Waldeck-Weiermair M, Simmen T, Graier WF, et al. (2018). Real-Time Imaging of Mitochondrial ATP Dynamics Reveals the Metabolic Setting of Single Cells. Cell Rep 25, 501–512 e503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durr O, and Sick B (2016). Single-Cell Phenotype Classification Using Deep Convolutional Neural Networks. J Biomol Screen 21, 998–1003. [DOI] [PubMed] [Google Scholar]
- Elowitz MB, Levine AJ, Siggia ED, and Swain PS (2002). Stochastic gene expression in a single cell. Science 297, 1183–1186. [DOI] [PubMed] [Google Scholar]
- Eng CL, Lawson M, Zhu Q, Dries R, Koulena N, Takei Y, Yun J, Cronin C, Karp C, Yuan GC, et al. (2019). Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature 568, 235–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng CL, Shah S, Thomassie J, and Cai L (2017). Profiling the transcriptome with RNA SPOTs. Nat Methods 14, 1153–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feigelman J, Ganscha S, Hastreiter S, Schwarzfischer M, Filipczyk A, Schroeder T, Theis FJ, Marr C, and Claassen M (2016). Analysis of Cell Lineage Trees by Exact Bayesian Inference Identifies Negative Autoregulation of Nanog in Mouse Embryonic Stem Cells. Cell Syst 3, 480–490 e413. [DOI] [PubMed] [Google Scholar]
- Feldman D, Singh A, Schmid-Burgk JL, Carlson RJ, Mezger A, Garrity AJ, Zhang F, and Blainey PC (2019). Optical Pooled Screens in Human Cells. Cell 179, 787–799 e717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Femino AM, Fay FS, Fogarty K, and Singer RH (1998). Visualization of single RNA transcripts in situ. Science 280, 585–590. [DOI] [PubMed] [Google Scholar]
- Frieda KL, Linton JM, Hormoz S, Choi J, Chow KK, Singer ZS, Budde MW, Elowitz MB, and Cai L (2017). Synthetic recording and in situ readout of lineage information in single cells. Nature 541, 107–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaglia G, Rashid R, Yapp C, Joshi GN, Li CG, Lindquist SL, Sarosiek KA, Whitesell L, Sorger PK, and Santagata S (2020). HSF1 phase transition mediates stress adaptation and cell fate decisions. Nat Cell Biol 22, 151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiler-Samerotte KA, Bauer CR, Li S, Ziv N, Gresham D, and Siegal ML (2013). The details in the distributions: why and how to study phenotypic variability. Curr Opin Biotechnol 24, 752–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerosa L, Chidley C, Frohlich F, Sanchez G, Lim SK, Muhlich J, Chen JY, Vallabhaneni S, Baker GJ, Schapiro D, et al. (2020). Receptor-Driven ERK Pulses Reconfigure MAPK Signaling and Enable Persistence of Drug-Adapted BRAF-Mutant Melanoma Cells. Cell Syst 11, 478–494 e479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaever G, Chu AM, Ni L, Connelly C, Riles L, Veronneau S, Dow S, Lucau-Danila A, Anderson K, Andre B, et al. (2002). Functional profiling of the Saccharomyces cerevisiae genome. Nature 418, 387–391. [DOI] [PubMed] [Google Scholar]
- Graml V, Studera X, Lawson JLD, Chessel A, Geymonat M, Bortfeld-Miller M, Walter T, Wagstaff L, Piddini E, and Carazo Salas RE (2014). A genomic Multiprocess survey of machineries that control and link cell shape, microtubule organization, and cell-cycle progression. Dev Cell 31, 227–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grys BT, Lo DS, Sahin N, Kraus OZ, Morris Q, Boone C, and Andrews BJ (2017). Machine learning and computer vision approaches for phenotypic profiling. J Cell Biol 216, 65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gut G, Herrmann MD, and Pelkmans L (2018). Multiplexed protein maps link subcellular organization to cellular states. Science 361. [DOI] [PubMed] [Google Scholar]
- Hanko EKR, Paiva AC, Jonczyk M, Abbott M, Minton NP, and Malys N (2020). A genome-wide approach for identification and characterisation of metabolite-inducible systems. Nat Commun 11, 1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasle N, Cooke A, Srivatsan S, Huang H, Stephany JJ, Krieger Z, Jackson D, Tang W, Pendyala S, Monnat RJ Jr., et al. (2020). High-throughput, microscope-based sorting to dissect cellular heterogeneity. Mol Syst Biol 16, e9442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heigwer F, Port F, and Boutros M (2018). RNA Interference (RNAi) Screening in Drosophila. Genetics 208, 853–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill SM, Hao X, Gronvall J, Spikings-Nordby S, Widlund PO, Amen T, Jorhov A, Josefson R, Kaganovich D, Liu B, et al. (2016). Asymmetric Inheritance of Aggregated Proteins and Age Reset in Yeast Are Regulated by Vac17-Dependent Vacuolar Functions. Cell Rep 16, 826–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hormoz S, Singer ZS, Linton JM, Antebi YE, Shraiman BI, and Elowitz MB (2016). Inferring Cell-State Transition Dynamics from Lineage Trees and Endpoint Single-Cell Measurements. Cell Syst 3, 419–433 e418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huberts DH, Janssens GE, Lee SS, Vizcarra IA, and Heinemann M (2013). Continuous high-resolution microscopic observation of replicative aging in budding yeast. J Vis Exp, e50143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL, and Gottschling DE (2012). An early age increase in vacuolar pH limits mitochondrial function and lifespan in yeast. Nature 492, 261–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, and O’Shea EK (2003). Global analysis of protein localization in budding yeast. Nature 425, 686–691. [DOI] [PubMed] [Google Scholar]
- Hussain S, Le Guezennec X, Yi W, Dong H, Chia J, Yiping K, Khoon LK, and Bard F (2017). Digging deep into Golgi phenotypic diversity with unsupervised machine learning. Mol Biol Cell 28, 3686–3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens GE, and Veenhoff LM (2016). Evidence for the hallmarks of human aging in replicatively aging yeast. Microb Cell 3, 263–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M, Li Y, O’Laughlin R, Bittihn P, Pillus L, Tsimring LS, Hasty J, and Hao N (2019). Divergent Aging of Isogenic Yeast Cells Revealed through Single-Cell Phenotypic Dynamics. Cell Syst 8, 242–253 e243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khmelinskii A, Keller PJ, Bartosik A, Meurer M, Barry JD, Mardin BR, Kaufmann A, Trautmann S, Wachsmuth M, Pereira G, et al. (2012). Tandem fluorescent protein timers for in vivo analysis of protein dynamics. Nat Biotechnol 30, 708–714. [DOI] [PubMed] [Google Scholar]
- Kim HS, Lee K, Kim SJ, Cho S, Shin HJ, Kim C, and Kim JS (2018). Arrayed CRISPR screen with image-based assay reliably uncovers host genes required for coxsackievirus infection. Genome Res 28, 859–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knorre DA, Azbarova AV, Galkina KV, Feniouk BA, and Severin FF (2018). Replicative aging as a source of cell heterogeneity in budding yeast. Mech Ageing Dev 176, 24–31. [DOI] [PubMed] [Google Scholar]
- Kraus OZ, Grys BT, Ba J, Chong Y, Frey BJ, Boone C, and Andrews BJ (2017). Automated analysis of high-content microscopy data with deep learning. Mol Syst Biol 13, 924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, de Boer R, and van der Klei IJ (2018). Yeast cells contain a heterogeneous population of peroxisomes that segregate asymmetrically during cell division. J Cell Sci 131. [DOI] [PubMed] [Google Scholar]
- Lane K, Andres-Terre M, Kudo T, Monack DM, and Covert MW (2019). Escalating Threat Levels of Bacterial Infection Can Be Discriminated by Distinct MAPK and NF-kappaB Signaling Dynamics in Single Host Cells. Cell Syst 8, 183–196 e184. [DOI] [PubMed] [Google Scholar]
- Lee J, Hyeon DY, and Hwang D (2020). Single-cell multiomics: technologies and data analysis methods. Exp Mol Med 52, 1428–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonetti MD, Sekine S, Kamiyama D, Weissman JS, and Huang B (2016). A scalable strategy for high-throughput GFP tagging of endogenous human proteins. Proc Natl Acad Sci U S A 113, E3501–3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy SF, Ziv N, and Siegal ML (2012). Bet hedging in yeast by heterogeneous, age-correlated expression of a stress protectant. PLoS Biol 10, e1001325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Jiang Y, Paxman J, O’Laughlin R, Klepin S, Zhu Y, Pillus L, Tsimring LS, Hasty J, and Hao N (2020). A programmable fate decision landscape underlies single-cell aging in yeast. Science 369, 325–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Jin M, O’Laughlin R, Bittihn P, Tsimring LS, Pillus L, Hasty J, and Hao N (2017). Multigenerational silencing dynamics control cell aging. Proc Natl Acad Sci U S A 114, 11253–11258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Vizeacoumar FJ, Bahr S, Li J, Warringer J, Vizeacoumar FS, Min R, Vandersluis B, Bellay J, Devit M, et al. (2011). Systematic exploration of essential yeast gene function with temperature-sensitive mutants. Nat Biotechnol 29, 361–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberali P, Snijder B, and Pelkmans L (2014). A hierarchical map of regulatory genetic interactions in membrane trafficking. Cell 157, 1473–1487. [DOI] [PubMed] [Google Scholar]
- Linkert M, Rueden CT, Allan C, Burel JM, Moore W, Patterson A, Loranger B, Moore J, Neves C, Macdonald D, et al. (2010). Metadata matters: access to image data in the real world. J Cell Biol 189, 777–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo MN, Damon LJ, Wei Tay J, Jia S, and Palmer AE (2020). Single cell analysis reveals multiple requirements for zinc in the mammalian cell cycle. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu AX, Chong YT, Hsu IS, Strome B, Handfield LF, Kraus O, Andrews BJ, and Moses AM (2018). Integrating images from multiple microscopy screens reveals diverse patterns of change in the subcellular localization of proteins. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu AX, Kraus OZ, Cooper S, and Moses AM (2019). Learning unsupervised feature representations for single cell microscopy images with paired cell inpainting. PLoS Comput Biol 15, e1007348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugagne JB, Lin H, and Dunlop MJ (2020). DeLTA: Automated cell segmentation, tracking, and lineage reconstruction using deep learning. PLoS Comput Biol 16, e1007673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahdessian D, Cesnik AJ, Gnann C, Danielsson F, Stenstrom L, Arif M, Zhang C, Le T, Johansson F, Shutten R, et al. (2021). Spatiotemporal dissection of the cell cycle with single-cell proteogenomics. Nature 590, 649–654. [DOI] [PubMed] [Google Scholar]
- Mattiazzi Usaj M, Sahin N, Friesen H, Pons C, Usaj M, Masinas MPD, Shuteriqi E, Shkurin A, Aloy P, Morris Q, et al. (2020). Systematic genetics and single-cell imaging reveal widespread morphological pleiotropy and cell-to-cell variability. Mol Syst Biol 16, e9243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattiazzi Usaj M, Styles EB, Verster AJ, Friesen H, Boone C, and Andrews BJ (2016). High-Content Screening for Quantitative Cell Biology. Trends Cell Biol 26, 598–611. [DOI] [PubMed] [Google Scholar]
- Mazumder A, Pesudo LQ, McRee S, Bathe M, and Samson LD (2013). Genome-wide single-cell-level screen for protein abundance and localization changes in response to DNA damage in S. cerevisiae. Nucleic Acids Res 41, 9310–9324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miesenbock G, De Angelis DA, and Rothman JE (1998). Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature 394, 192–195. [DOI] [PubMed] [Google Scholar]
- Mikuni T, Nishiyama J, Sun Y, Kamasawa N, and Yasuda R (2016). High-Throughput, High-Resolution Mapping of Protein Localization in Mammalian Brain by In Vivo Genome Editing. Cell 165, 1803–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller I, Min M, Yang C, Tian C, Gookin S, Carter D, and Spencer SL (2018). Ki67 is a Graded Rather than a Binary Marker of Proliferation versus Quiescence. Cell Rep 24, 1105–1112 e1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min M, Rong Y, Tian C, and Spencer SL (2020). Temporal integration of mitogen history in mother cells controls proliferation of daughter cells. Science 368, 1261–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moen E, Bannon D, Kudo T, Graf W, Covert M, and Van Valen D (2019). Deep learning for cellular image analysis. Nat Methods 16, 1233–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffitt JR, Hao J, Wang G, Chen KH, Babcock HP, and Zhuang X (2016). High-throughput single-cell gene-expression profiling with multiplexed error-robust fluorescence in situ hybridization. Proc Natl Acad Sci U S A 113, 11046–11051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr S, Bakal C, and Perrimon N (2010). Genomic screening with RNAi: results and challenges. Annu Rev Biochem 79, 37–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro F, Hubmann G, Takhaveev V, Vedelaar SR, Norder J, Hekelaar J, Saldida J, Litsios A, Wijma HJ, Schmidt A, et al. (2019). Measuring glycolytic flux in single yeast cells with an orthogonal synthetic biosensor. Mol Syst Biol 15, e9071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouton SN, Thaller DJ, Crane MM, Rempel IL, Terpstra OT, Steen A, Kaeberlein M, Lusk CP, Boersma AJ, and Veenhoff LM (2020). A physicochemical perspective of aging from single-cell analysis of pH, macromolecular and organellar crowding in yeast. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann B, Walter T, Heriche JK, Bulkescher J, Erfle H, Conrad C, Rogers P, Poser I, Held M, Liebel U, et al. (2010). Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature 464, 721–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohya Y, Kimori Y, Okada H, and Ohnuki S (2015). Single-cell phenomics in budding yeast. Mol Biol Cell 26, 3920–3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohya Y, Sese J, Yukawa M, Sano F, Nakatani Y, Saito TL, Saka A, Fukuda T, Ishihara S, Oka S, et al. (2005). High-dimensional and large-scale phenotyping of yeast mutants. Proc Natl Acad Sci U S A 102, 19015–19020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ounkomol C, Seshamani S, Maleckar MM, Collman F, and Johnson GR (2018). Label-free prediction of three-dimensional fluorescence images from transmitted-light microscopy. Nat Methods 15, 917–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang W, Winsnes CF, Hjelmare M, Cesnik AJ, Akesson L, Xu H, Sullivan DP, Dai S, Lan J, Jinmo P, et al. (2019). Analysis of the Human Protein Atlas Image Classification competition. Nat Methods 16, 1254–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnamaa T, and Parts L (2017). Accurate Classification of Protein Subcellular Localization from High-Throughput Microscopy Images Using Deep Learning. G3 (Bethesda) 7, 1385–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickar-Oliver A, and Gersbach CA (2019). The next generation of CRISPR-Cas technologies and applications. Nat Rev Mol Cell Biol 20, 490–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regot S, Hughey JJ, Bajar BT, Carrasco S, and Covert MW (2014). High-sensitivity measurements of multiple kinase activities in live single cells. Cell 157, 1724–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rempel IL, Crane MM, Thaller DJ, Mishra A, Jansen DP, Janssens G, Popken P, Aksit A, Kaeberlein M, van der Giessen E, et al. (2019). Age-dependent deterioration of nuclear pore assembly in mitotic cells decreases transport dynamics. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner H, Grabos M, Becker KJ, Kagermeier TE, Wu J, Otto M, Peischard S, Zeuschner D, TsyTsyura Y, Disse P, et al. (2020). A fully automated high-throughput workflow for 3D-based chemical screening in human midbrain organoids. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts B, Haupt A, Tucker A, Grancharova T, Arakaki J, Fuqua MA, Nelson A, Hookway C, Ludmann SA, Mueller IA, et al. (2017). Systematic gene tagging using CRISPR/Cas9 in human stem cells to illuminate cell organization. Mol Biol Cell 28, 2854–2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez J, Ren G, Day CR, Zhao K, Chow CC, and Larson DR (2019). Intrinsic Dynamics of a Human Gene Reveal the Basis of Expression Heterogeneity. Cell 176, 213–226 e218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, et al. (2008). Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 132, 487–498. [DOI] [PubMed] [Google Scholar]
- Sakaue-Sawano A, Yo M, Komatsu N, Hiratsuka T, Kogure T, Hoshida T, Goshima N, Matsuda M, Miyoshi H, and Miyawaki A (2017). Genetically Encoded Tools for Optical Dissection of the Mammalian Cell Cycle. Mol Cell 68, 626–640 e625. [DOI] [PubMed] [Google Scholar]
- Sarraf SA, Maman Y, Baldwin H, Johnson KR, Ward ME, Kampmann M, Lippincott-Schwartz J, and Youle RJ (2020). Image-based pooled whole genome CRISPR screening for Parkin and TFEB subcellular localization. bioRxiv [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra D, Mayr U, Boni A, Lukonin I, Rempfler M, Challet Meylan L, Stadler MB, Strnad P, Papasaikas P, Vischi D, et al. (2019). Self-organization and symmetry breaking in intestinal organoid development. Nature 569, 66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith K, Piccinini F, Balassa T, Koos K, Danka T, Azizpour H, and Horvath P (2018). Phenotypic Image Analysis Software Tools for Exploring and Understanding Big Image Data from Cell-Based Assays. Cell Syst 6, 636–653. [DOI] [PubMed] [Google Scholar]
- Snijder B, Sacher R, Ramo P, Damm EM, Liberali P, and Pelkmans L (2009). Population context determines cell-to-cell variability in endocytosis and virus infection. Nature 461, 520–523. [DOI] [PubMed] [Google Scholar]
- Snijder B, Sacher R, Ramo P, Liberali P, Mench K, Wolfrum N, Burleigh L, Scott CC, Verheije MH, Mercer J, et al. (2012). Single-cell analysis of population context advances RNAi screening at multiple levels. Mol Syst Biol 8, 579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinkraus KA, Kaeberlein M, and Kennedy BK (2008). Replicative aging in yeast: the means to the end. Annu Rev Cell Dev Biol 24, 29–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strezoska Z, Perkett MR, Chou ET, Maksimova E, Anderson EM, McClelland S, Kelley ML, Vermeulen A, and Smith AVB (2017). High-content analysis screening for cell cycle regulators using arrayed synthetic crRNA libraries. J Biotechnol 251, 189–200. [DOI] [PubMed] [Google Scholar]
- Styles EB, Founk KJ, Zamparo LA, Sing TL, Altintas D, Ribeyre C, Ribaud V, Rougemont J, Mayhew D, Costanzo M, et al. (2016). Exploring Quantitative Yeast Phenomics with Single-Cell Analysis of DNA Damage Foci. Cell Syst 3, 264–277 e210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan DP, Winsnes CF, Akesson L, Hjelmare M, Wiking M, Schutten R, Campbell L, Leifsson H, Rhodes S, Nordgren A, et al. (2018). Deep learning is combined with massive-scale citizen science to improve large-scale image classification. Nat Biotechnol 36, 820–828. [DOI] [PubMed] [Google Scholar]
- Sulston JE, Schierenberg E, White JG, and Thomson JN (1983). The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev Biol 100, 64–119. [DOI] [PubMed] [Google Scholar]
- Tao R, Zhao Y, Chu H, Wang A, Zhu J, Chen X, Zou Y, Shi M, Liu R, Su N, et al. (2017). Genetically encoded fluorescent sensors reveal dynamic regulation of NADPH metabolism. Nat Methods 14, 720–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thul PJ, Akesson L, Wiking M, Mahdessian D, Geladaki A, Ait Blal H, Alm T, Asplund A, Bjork L, Breckels LM, et al. (2017). A subcellular map of the human proteome. Science 356. [DOI] [PubMed] [Google Scholar]
- Torre E, Dueck H, Shaffer S, Gospocic J, Gupte R, Bonasio R, Kim J, Murray J, and Raj A (2018). Rare Cell Detection by Single-Cell RNA Sequencing as Guided by Single-Molecule RNA FISH. Cell Syst 6, 171–179 e175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizeacoumar FJ, van Dyk N, F, S.V., Cheung V, Li J, Sydorskyy Y, Case N, Li Z, Datti A, Nislow C, et al. (2010). Integrating high-throughput genetic interaction mapping and high-content screening to explore yeast spindle morphogenesis. J Cell Biol 188, 69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Allen WE, Wright MA, Sylwestrak EL, Samusik N, Vesuna S, Evans K, Liu C, Ramakrishnan C, Liu J, et al. (2018). Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams E, Moore J, Li SW, Rustici G, Tarkowska A, Chessel A, Leo S, Antal B, Ferguson RK, Sarkans U, et al. (2017). The Image Data Resource: A Bioimage Data Integration and Publication Platform. Nat Methods 14, 775–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu PH, Gilkes DM, Phillip JM, Narkar A, Cheng TW, Marchand J, Lee MH, Li R, and Wirtz D (2020). Single-cell morphology encodes metastatic potential. Sci Adv 6, eaaw6938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia C, Fan J, Emanuel G, Hao J, and Zhuang X (2019). Spatial transcriptome profiling by MERFISH reveals subcellular RNA compartmentalization and cell cycle-dependent gene expression. Proc Natl Acad Sci U S A 116, 19490–19499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Z, Sadok A, Sailem H, McCarthy A, Xia X, Li F, Garcia MA, Evans L, Barr AR, Perrimon N, et al. (2013). A screen for morphological complexity identifies regulators of switch-like transitions between discrete cell shapes. Nat Cell Biol 15, 860–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerjatke T, Gak IA, Kirova D, Fuhrmann M, Daniel K, Gonciarz M, Muller D, Glauche I, and Mansfeld J (2017). Quantitative Cell Cycle Analysis Based on an Endogenous All-in-One Reporter for Cell Tracking and Classification. Cell Rep 19, 1953–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Eichhorn SW, Zingg B, Yao Z, Zeng H, Dong H, and Zhuang X (2020). Molecular, spatial and projection diversity of neurons in primary motor cortex revealed by in situ single-cell transcriptomics. bioRxiv [Google Scholar]
- Zhao L, Yang Q, Zheng J, Zhu X, Hao X, Song J, Lebacq T, Franssens V, Winderickx J, Nystrom T, et al. (2016). A genome-wide imaging-based screening to identify genes involved in synphilin-1 inclusion formation in Saccharomyces cerevisiae. Sci Rep 6, 30134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Hu Q, Cheng F, Su N, Wang A, Zou Y, Hu H, Chen X, Zhou HM, Huang X, et al. (2015). SoNar, a Highly Responsive NAD+/NADH Sensor, Allows High-Throughput Metabolic Screening of Anti-tumor Agents. Cell Metab 21, 777–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y, Wang A, Shi M, Chen X, Liu R, Li T, Zhang C, Zhang Z, Zhu L, Ju Z, et al. (2018). Analysis of redox landscapes and dynamics in living cells and in vivo using genetically encoded fluorescent sensors. Nat Protoc 13, 2362–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]