

Abstract
Aim:
To investigate the effect of nerve growth factor (NGF) neutralization on Na+ channel plasticity of bladder afferent neurons in mice with spinal cord injury (SCI).
Main methods:
Female C57/BL6 mice were randomly divided into spinal intact (SI) group, SCI group and SCI+NGF-Ab group. SCI was induced by spinal cord transection at the Th8/9 level. In SCI+NGF-Ab group, anti-NGF antibodies (10 μg•kg-1 per hour) were continuously administered for 2 weeks using osmotic pumps. Bladder afferent neurons were labelled with Fluoro-gold (FG) injected into the bladder wall. L6-S1 dorsal root ganglion (DRG) neurons were dissociated and whole-cell patch clamp recordings were performed on FG-labelled neurons. Expression of Nav1.7 and Nav1.8 was examined by immunofluorescent staining.
Key findings:
Whole-cell patch clamp recordings showed that TTX only partially inhibited action potentials (AP) and Na+ currents of bladder afferent neuron in SI mice, but it almost completely inhibited them in SCI mice. Total and TTX-sensitive Na+ currents were increased and TTX-resistant currents were decreased in bladder afferent neurons from SCI mice vs. SI mice. These changes in SCI mice were significantly reversed by NGF-antibody treatment. Immunostaining results showed the increased and decreased levels of Nav1.7 and Nav1.8, respectively, in FG-labelled bladder afferent neurons in SCI mice vs. SI mice, which was significantly reversed in SCI+NGF-Ab mice.
Significance:
NGF mediates the Na+ channel plasticity with a shift from TTX-resistant Nav1.8 to TTX-sensitive Nav1.7 in bladder afferent neurons, which could be a possible underlying mechanism of bladder afferent hyperexcitability and detrusor overactivity after SCI.
Keywords: Nerve growth factor, spinal cord injury, Na+ channel, bladder afferent neuron, dorsal root ganglia
Introduction
Lower urinary tract dysfunction (LUTD) is a common complication of spinal cord injury (SCI) and a life-threatening problem in the care of patients[1]. It has been reported that nerve growth factor (NGF) is a crucial mediator involved in the emergence of LUTD after SCI and that the level of NGF in the bladder, dorsal root ganglia (DRG) and the spinal cord are significantly increased after SCI[2, 3]. Our previous studies also demonstrated that anti-NGF antibody treatment that normalizes NGF overexpression in the bladder and the spinal cord can reduce detrusor overactivity detected during cystometry and the hyperexcitability of C-fiber bladder afferent neurons in SCI mice[4, 5]. Furthermore, the improvement of C-fiber bladder afferent neuron excitability after anti-NGF antibody treatment is shown to be induced at least in part by the restoration of A-type K+ channel activity, which was reduced after SCI in mice[5] and rats[6]. However, the role of NGF in the plasticity of another major determinant of afferent neuron excitability; that is, the voltage-gated Na+ (Nav) channel, after SCI has not been examined previously.
Nav channels play an important role in generation and transmission of action potentials in the sensory pathway. There are nine subtypes of Nav channels (Nav1.1–1.9), and TTX-sensitive Nav1.7 channels and TTX-resistant Nav 1.8–1.9 channels are abundantly detected in bladder afferent neurons[7]. Our previous study demonstrated a shift from high-threshold tetrodotoxin (TTX)-resistant Na+ currents to low-threshold TTX-sensitive Na+ currents in bladder afferent neurons in rats with SCI-induced detrusor overactivity[8]. It has also been shown that among TTX-resistant Nav subunits, Nav1.8 & Nav1.9, SCI induced the decreased expression of Nav1.8, but not Nav1.9, in bladder afferent neurons from SCI rats[9]. Furthermore, several studies have reported that NGF can affect DRG neuron excitability by increasing the Na+ channel activity and induce changes in the expression of specific Nav subtypes [10–12]. Thus, we hypothesized that SCI can induce the Na+ channel plasticity due to NGF-dependent changes in the expression of TTX-sensitive Nav1.7 and TTX-resistant NaV1.8 subunits in bladder afferent neurons, resulting in bladder afferent hyperexcitability after SCI.
Therefore, this study was performed to investigate the changes in Na+ channel activity and the expression of Nav1.7 and 1.8 subunits in bladder afferent neurons from SCI mice with or without NGF neutralization using anti-NGF antibody treatment.
Materials and methods
All experiments were performed according to the NIH guidelines and were approved by the Animal Care Committees of University of Pittsburgh and Shanghai Jiao Tong University Affiliated Sixth People’s Hospital.
Animal preparation
A total of thirty-sixth female C57BL/6 mice (8 to 10 weeks old, weighting 18–22g) were randomly divided into three groups; spinally intact (SI) group (n=12), SCI group (n=12) and SCI group treated with anti-NGF antibody (NGF-Ab) (SCI+NGF-Ab group, n=12). The study design was shown in Figure 1.
Figure 1.
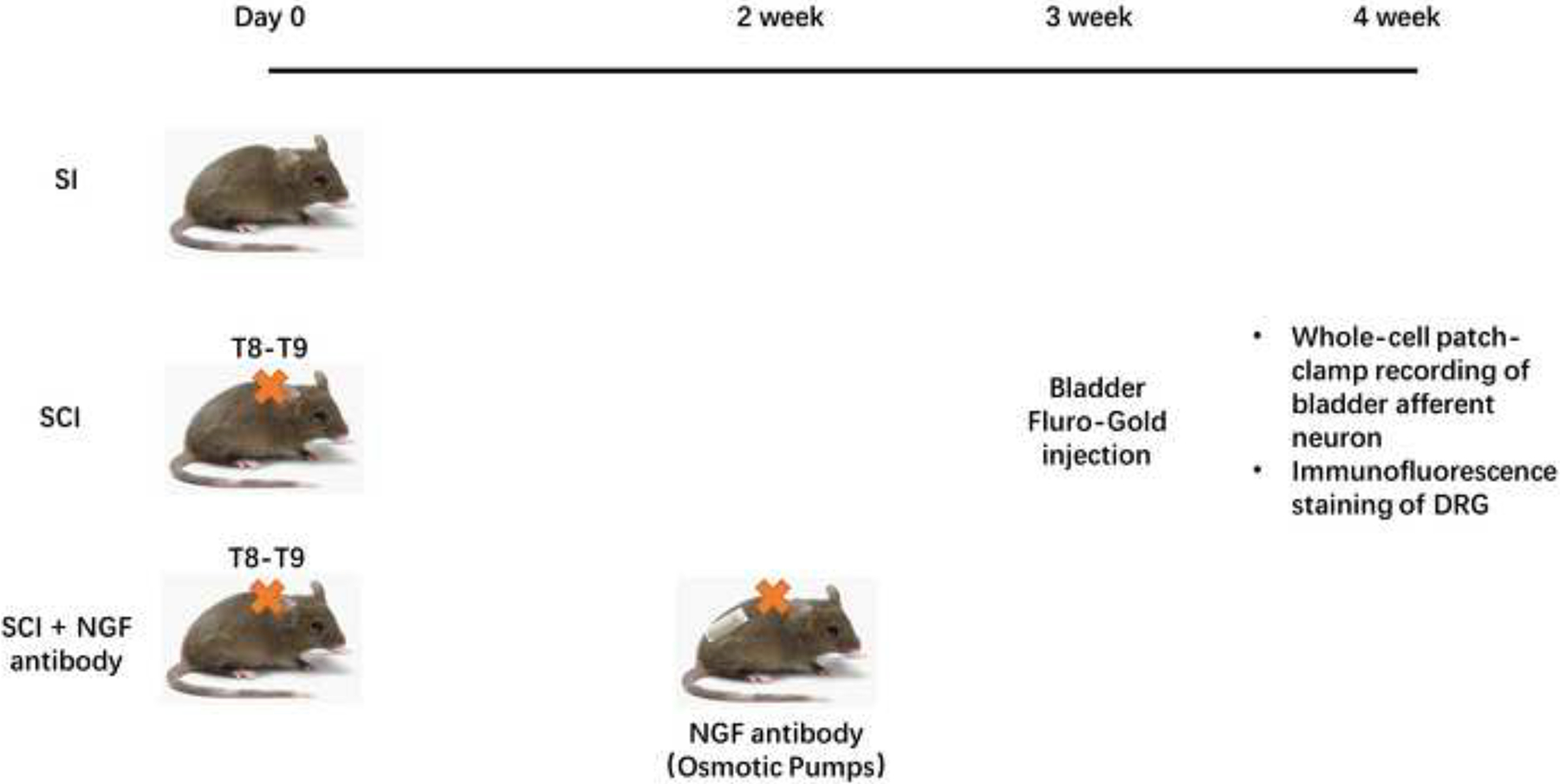
Study design. C57/BL6 mice were randomly divided into three groups: SI group, SCI group, and SCI+NGF Ab group. Two weeks after SCI, an osmotic pump was placed subcutaneously in the back to administer 10 μg/kg/h of anti-NGF antibody (Ab) in mice of SCI+NGF-Ab group. Three weeks after SCI, all mice received bladder injection of Fluoro-gold. Four weeks after SCI, final experiments were performed.
SCI was induced by complete spinal cord transection at the Th8/9 level under isoflurane anesthesia according to the methods described previously[13]. The animals were treated with ampicillin (100 mg/kg, s.c.) for 5 days post-SCI, followed by twice per week to prevent urinary tract infection. The bladder of SCI animals was emptied once a day for 4 weeks post-SCI. In the SCI+NGF-Ab group, at 2 weeks after SCI, an osmotic pump (Alzet Osmotic Pumps, CA, USA) was placed subcutaneously under the back skin to continuously administer 10 μg/kg per hour of anti-NGF antibodies (Exalpha Biologicals Inc., MA, USA) for two weeks. The dosage of the antibody was determined according to previous studies[4, 14]. Three weeks after SCI, 2.5% Fluoro-gold (FG, Fluorochrome, CO, USA) was injected into the bladder wall (4 sites, 10μL/site) using a 30-gauge Hamilton syringe to retrogradely label bladder afferent neurons. Four weeks after SCI, L6-S1 dorsal root ganglia (DRG) were harvested for either whole-cell patch-clamp recordings of dissociated cells or immunofluorescence (IF) staining of DRG sections.
Dissociation of DRG neurons
Neurons were dissociated from L6-S1 DRG by enzymatic and mechanical methods as previously described[5, 15]. Briefly, freshly removed ganglia were digested at 37°C for 15 minutes in DMEM containing 2mg/ml of type 4 collagenase (Sigma C-9891, MO, USA) and 2 mg/ml Trypsin (Sigma T-8253, MO, USA). Trypsin inhibitor (Sigma T-9128, MO, USA) was then added for 5 minutes. The digested ganglia were then dispersed mechanically by pipettes. The cell suspensions were centrifuged at 1000rpm at room temperature for 5 minutes. Cell pellets were re-suspended in DMEM and centrifuged at 1000rpm for 5 minutes. After removing the supernatant, the pellet containing neurons was re-suspended in DMEM. DRG neurons were isolated by trituration, plated on Poly-D-Lysine-coated culture dishes and were kept in a 5% CO2 incubator at 37°C. The patch-clamp recordings were performed within 24 hours after cell dissociation.
Whole-cell patch-clamp recordings
FG-labelled bladder afferent neurons were identified using an inverted phase contrast microscope (Nikon, Tokyo, Japan) with a UV-specific filter. Whole-cell recordings were performed with an Axopatch 200A patch-clamp amplifier (Molecular Devices, Union City, CA, USA), and the data were acquired and analysed with pCLAMP software (Molecular Devices). Previous studies demonstrated that DO after SCI was induced by hyperexcitability of C-fiber bladder afferent pathways[16] and that anti-NGF antibody treatment reduced hyperexcitability of C-fiber bladder afferent neurons, which were sensitive to capsaicin application[5]. Thus, in this study, we selected small-size bladder afferent neurons with their diameter less than 30μm and 35μm in SI and SCI mice, respectively, because the latter study in mice showed that capsaicin-sensitive C-fiber bladder afferent neurons, which underwent cell hypertrophy after SCI, were mostly within these cell diameter ranges[5].
Action potentials (AP) were recorded in the current clamp mode. The internal solution contained (in mM): 140 KCl, 1 CaCl2, 2 MgCl2, 11 EGTA, and 10 HEPES, adjusted to pH 7.4 with CsOH (310 mOsm). The external solutions contained (in mM): 150 NaCl, 5 KCl, 2.5 CaCl2, 1 MgCl2, 10 D-Glucose, and 10 HEPES, adjusted to pH 7.4 with NaOH (340 mOsm). After AP characteristics were evaluated, the effect of TTX on neuronal firing was examined by adding 1μM TTX into the external solution. To evaluate AP firing properties, spike thresholds, at which the initial rise of APs was observed, were measured.
For isolating Na+ currents in the voltage-clamp mode, the external solution contained (in mM): 85 TEA-Cl, 10 MgCl2, 5 4-AP, 50 NaCl, 10 D-Glucose, and 10 HEPES, adjusted to pH 7.2 with TEA-OH (340 mOsm). The internal solution contained (in mM): 10 NaCl, 125 CsF, 2 MgCl2, 10 EGTA, 10 HEPES, 4 ATP-Mg, and 0.4 GTP (Tris salt), adjusted to pH 7.4 with CsOH (310 mOsm). Na+ current–voltage (I-V) relationships were evaluated using a depolarizing pulse protocol (10 mV increments of 100 ms duration) from −90 mV to +30 mV at the holding potential of −100 mV, at which voltage-dependent inactivation of Na+ currents was reportedly minimal in bladder afferent neurons[17]. Thereafter, TTX (1μM) was applied to the bath solution to suppress TTX-sensitive Na+ currents, and the proportion of TTX-sensitive Na+ currents in the total currents was evaluated by off-line subtraction of the remaining TTX-resistant Na+ currents from the total Na+ currents.
Immunofluorescence staining
Mice (n=4 in each group) were transcardially perfused with 4% paraformaldehyde in 0.1 M phosphate buffer. L6-S1 DRG were isolated and post-fixed in the same fixative for additional 4 hours prior to overnight cryopreservation in 20% sucrose. Tissues were mounted in OCT compound, and cryo-sectioned at 20 μm thickness. For immunofluorescence staining, sections were incubated for 1 hours at room temperature in blocking solution, which contained 0.3% Triton X-100, 5% BSA, and 5% goat serum in 1×PBS. Primary antibodies were applied overnight at 4°C, and secondary antibody was thereafter applied for 2 hours at room temperature. Antibodies used for immunohistochemistry include Rabbit Anti-Nav1.7 antibody (ASC-008, ALOMONE LABS, Israel), Rabbit Anti-Nav1.8 antibody (ASC-016, ALOMONE LABS, Israel), and Alexa Fluor 488-labeled Goat Anti-Rabbit IgG (Beyotime, China). Images of sections were obtained using a Leica fluorescence microscope. FG-labelled bladder afferent neurons that were positively stained for Nav1.7 or Nav1.8 were counted in five randomly selected sections per animal by a blinded observer. The staining intensity of Nav1.7 or Nav1.8 immunoreactivity in FG-labelled bladder neurons was measured using ImageJ 1.46 software (NIH, USA).
Statistical analysis
All data were expressed as mean ± SD. Statistical differences were determined using one-way ANOVA, followed by LSD test using SPSS software. P-values less than 0.05 were considered statistically significant. Graphs were produced by GraphPad Prism Software.
Results
Effect of NGF-Ab treatment on excitability of small-sized bladder afferent neurons
The results of whole-cell patch clamp recordings are shown in Figure 2 and Table 1. In the current clamp mode, the resting and peak AP membrane potentials and the duration of APs did not differ among bladder afferent neurons of SI, SCI and SCI+NGF-Ab groups. However, the spike threshold was significantly decreased in bladder afferent neurons from SCI mice compared to those from SI mice (−30.5±5.1mV vs. −22.5±4.8mV, p < 0.05). Also, the number of action potentials during an 800ms membrane depolarization in bladder afferent neurons from SCI mice was significantly increased compared to SI mice (5.2±4.0 spikes vs. 1.2±1.1 spikes, p < 0.05). These results demonstrate that the excitability of bladder afferent neurons was significantly increased in SCI mice as evidenced by lower thresholds of AP and repetitive firing pattern. Then, in the SCI+NGF-Ab group, these SCI-induced changes in firing properties of bladder afferent neurons were significantly restored compared to the SCI group.
Figure 2.
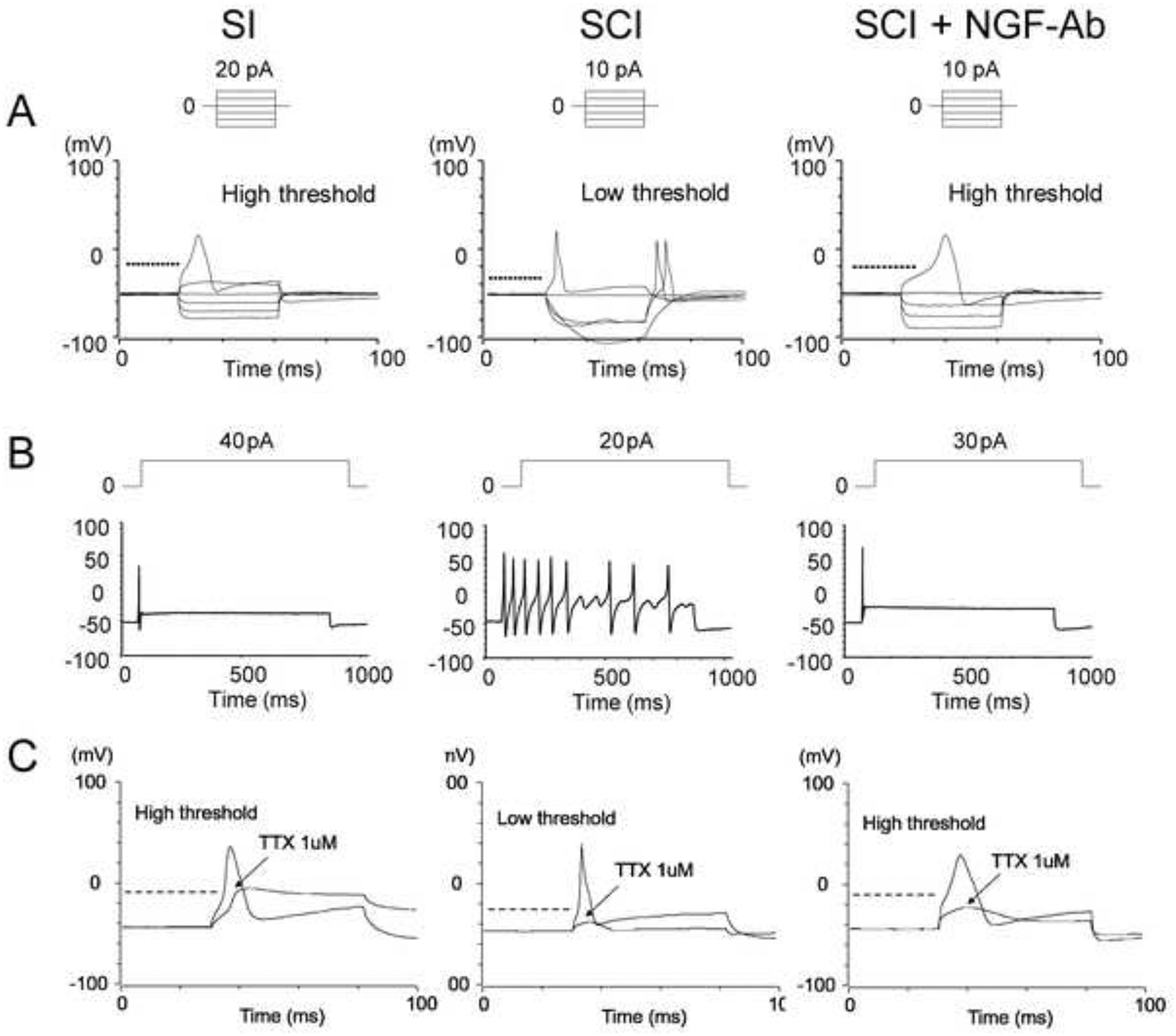
Representative action potential (AP) recordings in bladder afferent neurons from mice. A, B: Excitability of bladder afferent neurons was significantly increased in SCI mice, evident as lower thresholds of APs(A) and multiple firing pattern(B). These SCI-induced changes were significantly reversed in the SCI+NGF-Ab group. C: Bath application of tetrodotoxin (TTX) partly inhibited the AP of bladder afferent neurons in SI mice whereas it almost completely inhibited the AP in SCI mice, but not in SCI+NGF-Ab mice.
Table 1.
Parameters of Action potential of SI group, SCI group and SCI+NGF-Ab group.
| SI (control) | SCI | SCI+NGF-Ab | |
|---|---|---|---|
| Action potential number of cells/mice | 11(8) | 13(8) | 12(8) |
| Diameter (μm) | 25.1±4.8 | 29.2±3.7* | 29.5±2.9 |
| Input capacitance (pF) | 26.1±28.5 | 36.9±10.5* | 43.2±14.8# |
| Resting membrane potentials (mV) | −50±0.1 | −49.7±0.2 | −49.8±0.5 |
| Spike threshold (mV) | −22.5±4.8 | −30.5±5.1* | −24.5±6.3# |
| Peak membrane potential (mV) | 36.5±11.2 | 37.2±16.1 | 40.5±19.2 |
| Spike duration (ms) | 3.7±1.4 | 3.8±1.4 | 3.4±1.8 |
| Number of spikes (800-ms depolarization) | 1.2±1.1 | 5.2±4.0* | 1.8±1.5# |
p<0.05 compared to SI group.
p<0.05 compared to SCI group.
We also examined the TTX sensitivity of APs in small-sized bladder afferent neurons. In SI mice, TTX application partially inhibit APs whereas it almost completely inhibited APs in bladder afferent neurons from SCI mice. However, APs returned to the TTX- resistant type in the NGF-Ab group of SCI mice. These results indicate that APs of bladder afferent neurons underwent the NGF-dependent transition from a TTX-resistant subtype to a TTX-sensitive subtype after SCI, which was prevented by NGF-Ab treatment.
Effect of NGF-Ab treatment on Na+ currents of small-sized bladder afferent neurons
Next, in bladder afferent neurons obtained from the separate groups of animals, we recorded Na+ currents under the voltage-clamp mode to evaluate their TTX sensitivity and calculate the proportions of TTX-sensitive and TTX-resistant components in the total Na+ currents (Figure 3 and Table 2). TTX application to the bath solution barely influenced Na+ currents evoked from the holding potential of −100mV in bladder afferent neurons from SI mice whereas Na+ currents in bladder afferent neurons from SCI mice were significant reduced after the TTX application. In I-V relationship curves of Na+ currents in each group (Figure 3), total Na+ currents consisted of TTX-resistant and TTX-sensitive current components. The results showed that the total Na+ current density was significantly increased in bladder afferent neurons from SCI compared to those from SI mice and that the TTX-resistant current was predominant in bladder afferent neurons from SI mice whereas the TTX-sensitive current was a major component of Na+ currents in bladder afferent neurons from SCI mice. However, these SCI-induced changes in TTX-sensitive and TTX-resistant Na+ current densities in bladder afferent neurons were significantly reversed by NGF-antibody treatment in SCI mice. (Table 2)
Figure 3.
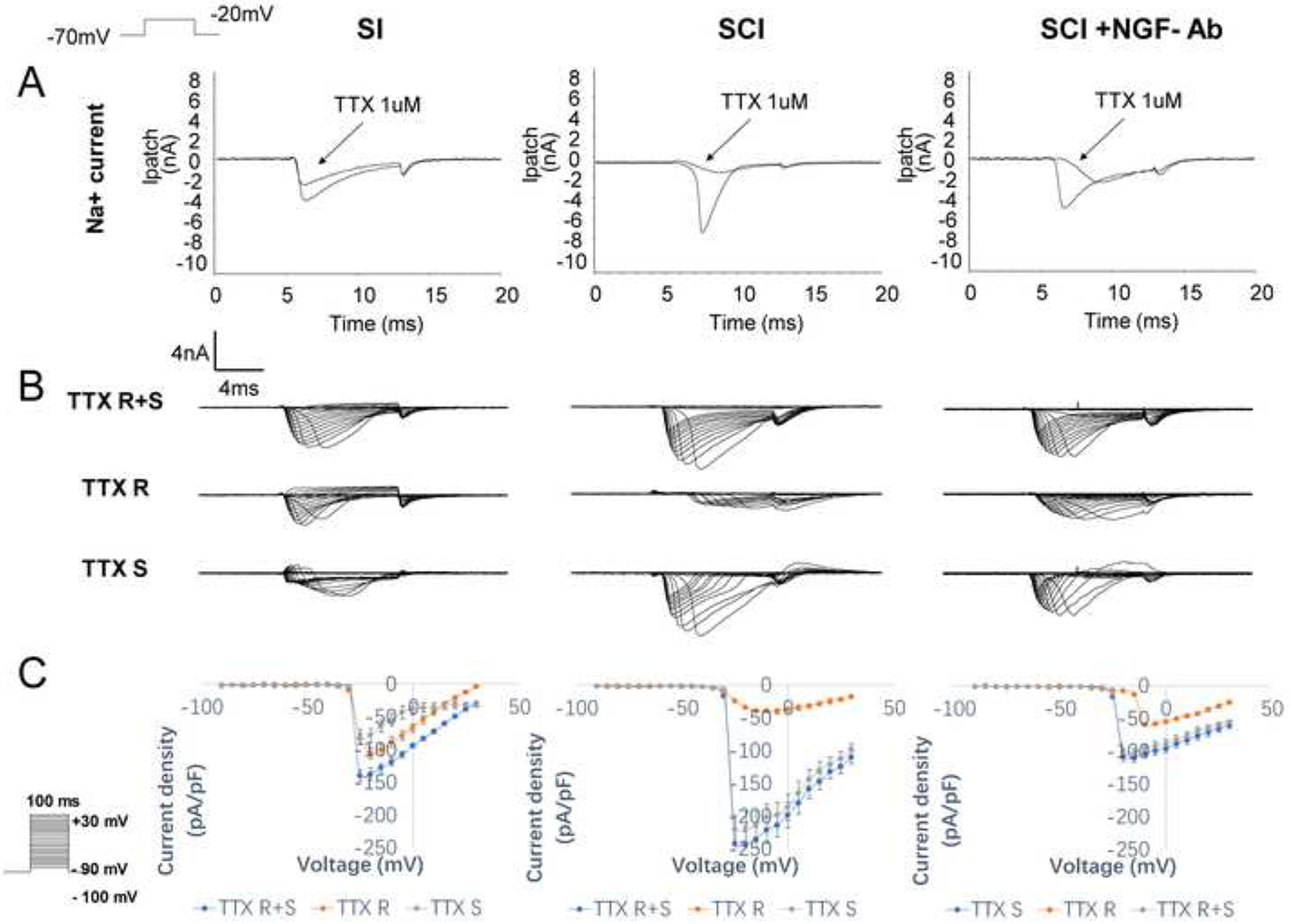
Representative Na+ current recordings with or without TTX application in bladder afferent neurons and I-V curves of total (TTX R+S), TTX-resistant (TTX R) sensitive and TTX-sensitive (TTX S) currents in each of SI, SCI and SCI+ NGF-Ab groups (n=8 each group). A: Bath application of TTX reduced Na+ currents of bladder afferent neurons in SCI mice whereas it only slightly decreased Na+ currents in SI mice. However, this TTX effect on Na+ currents was restored by NGF-Ab treatment in the SCI+NGF-Ab group. B: Representative traces of total (TTX R+S), TTX-resistant (TTX R) sensitive and TTX-sensitive (TTX S) currents induced from the holding potential of −100mV shows reduced TTX R and increased TTX S currents in the SCI group vs. the SI group, which were restored in the SCI+NGF-Ab group. C: I-V relationship curves showed that the amplitudes of TTX-sensitive and TTX-resistant Na+ currents were increased and decreased, respectively, after SCI; however, these changes were restored by NGF-Ab treatment. The depolarizing voltage pulse protocol was shown in an inset.
Table 2.
Peak amplitudes of total Na+ currents, TTX-resistant (TTX-R) Na+ currents and TTX-sensitive (TTX-S) Na+ currents evoked by depolarization pulses from −70mV to −20mV at the holding potential of −100mV.
| SI (control) | SCI | SCI+NGF-Ab | |
|---|---|---|---|
| number of cells/mice | 10(8) | 11(8) | 12(8) |
| Total Na+ current density(pA/pF) | −138.5±7.1 | −241.5±23.5* | −120.5±7.6# |
| TTX-R Na+ current density(pA/pF) | −102.2±6.4 | −45.1±5.4* | −62.6±6.9# |
| TTX-S Na+ current density(pA/pF) | −79.8±5.6 | −210.4±18.8* | −111.3±7.8# |
p<0.05 compared to SI group.
p<0.05 compared to SCI group.
Immunofluorescence staining
In L6-S1 DRG sections, bladder afferent neurons were identified as FG-labelled cells under a UV filter. Thereafter, the immunofluorescent filter was switched to detect green fluorescence to evaluate the immunostaining intensities of Nav1.7 or Nav1.8 channel subunits in bladder afferent neurons from each group of animals. The results showed that staining intensities of Nav1.7 and Nav1.8 were significantly increased and decreased, respectively, in bladder afferent neurons from SCI mice compared to those from SI mice. (Figure 4) However, these changes of Nav1.7 and Nav1.8 expressions in bladder afferent neurons were reversed by NGF-antibody treatment in SCI mice. In addition, we confirmed that there was no positive staining above background when primary antibody was omitted (data not shown).
Figure 4.
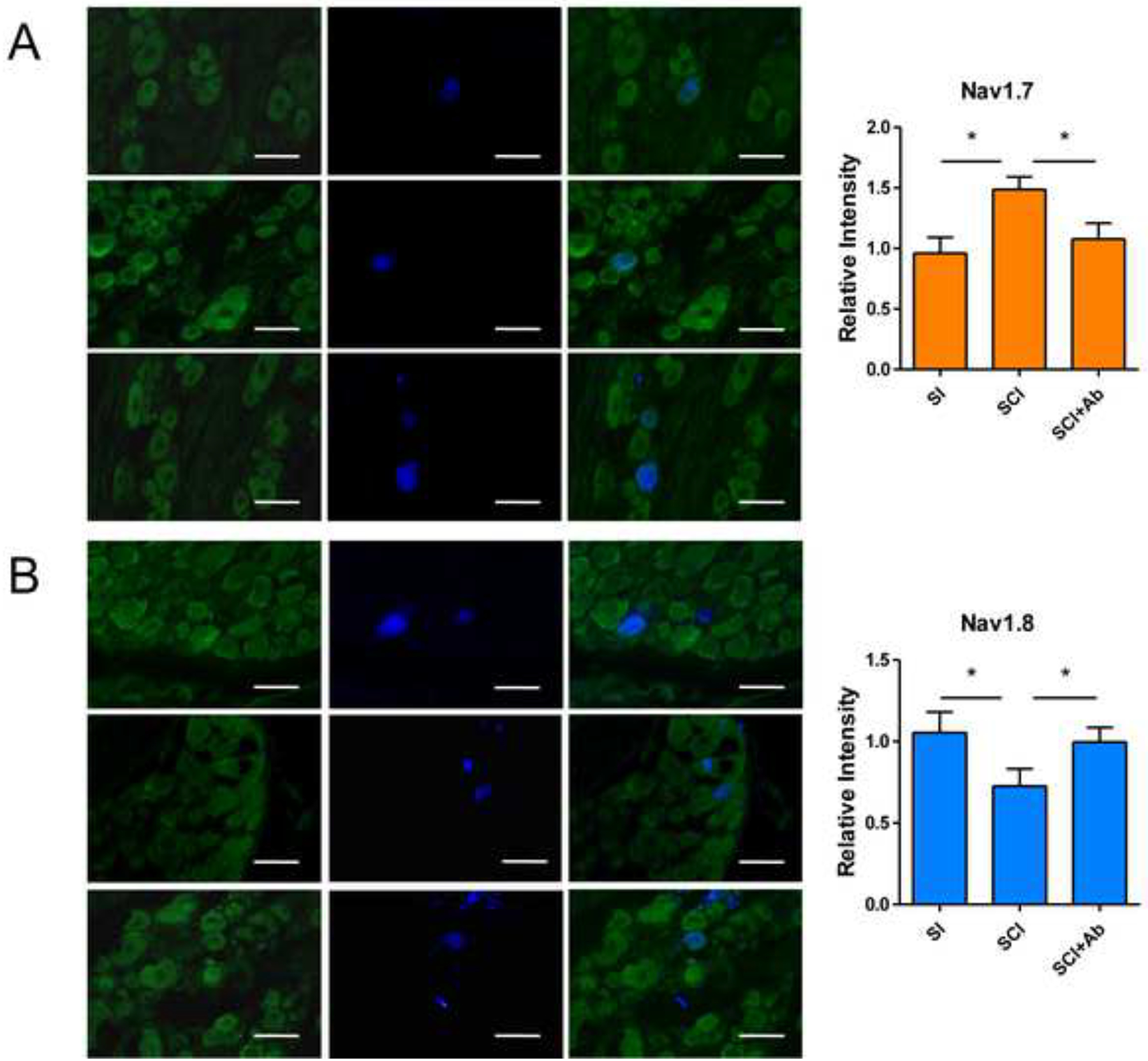
Immunofluorescence (IF) of Nav1.7(A) and Nav1.8(B) subunits in L6-S1 DRG sections. Left panels show the immunoreactivity of Nav1.7 subtype or Nav1.8 subtype (green) in Figure A or B, respectively. Middle panels show Fluoro-gold (FG)-labelled bladder afferent neurons (blue). Right panels show the overlapped images in left and middle panels. IF staining showed that both Nav1.7 and Nav1.8 subunits existed in L6-S1 DRG neurons of SI and SCI mice. In SCI mice, Nav1.7 immunoreactivity was significantly increased and Nav1.8 was significantly decreased in FG-labelled bladder afferent neurons from SCI mice, compared to SI mice. These changes of Nav subunit expression after SCI was reversed by NGF-antibody treatment. Bar scale: 50μm.
Discussion
In the current study, we revealed that; (1) SCI can induce a transition of Na+ channels from TTX-resistant to TTX-sensitive subtypes, which is at least in part dependent on the increased level of NGF and can be restored by NGF neutralizing treatment, and (2) SCI can induce increased expression of the TTX-sensitive Nav1.7 and decreased expression of the TTX-resistant Nav1.8, which are also restored by NGF neutralizing treatment. Thus, it is assumed that the increased level of NGF after SCI are an important factor to induce Na+ channel plasticity, that is, a shift from the TTX-resistant subtype to the TTX-sensitive subtype, in bladder afferent neurons, leading to bladder afferent hyperexcitability and LUTD such as detrusor overactivity after SCI.
The bladder of various species including rodents and cats is innervated by two types of afferent nerves; Aδ-fiber and C‐ fiber afferents that have different functions. Normal micturition in SI animals is predominantly controlled by Aδ-fiber bladder afferent pathways whereas, in pathological conditions including SCI, increased excitability of bladder afferent pathways, especially the C-fiber population, contributes to the emergence of LUTD such as detrusor overactivity[18, 19]. Among multiple kinds of neurotrophic factors, NGF is widely distributed in the central and autonomic nervous systems[20], and has been reported to increase in bladder afferent pathways via axonal transport from target organs (i.e., bladder) and the spinal cord after SCI[21, 22]. Previous studies using SCI rats showed that immunoneutralization of NGF in the lumbosacral spinal cord reduced the NGF level in bladder afferent pathways and thus improved detrusor overactivity after SCI[22]. Similarly, we reported that, in the same mouse model of SCI as in this study, the similar anti-NGF antibody treatment normalized NGF overexpression in the bladder and the L6-S1 spinal cord, where the major afferent inputs from the bladder terminate, and reduced detrusor overactivity detected during awake cystometry[4, 5], indicating that NGF is a major mediator inducing detrusor overactivity due to afferent hyperexcitability following SCI. In addition, reduced activity of slowly-inactivating A-type K+ channels encoded by the Kv1.4 subunit has been implicated as one of the underlying mechanisms of bladder afferent sensitization after SCI in a rat model[6]. Our recent study further demonstrated that NGF overexpression is significantly involved in this plasticity of A-type K+ channels after SCI because anti-NGF antibody treatment restored the A-type K+ channel function and reduced bladder afferent hyperexcitability in the same mouse model of SCI as used in this study[5].
Moreover, in addition to the changes in A-type K+ channel activity in bladder afferent pathways after SCI, it has also been reported that SCI can induce the plasticity of Na+ channels due to a shift from TTX-resistant to TTX-sensitive Na+ channel phenotypes in bladder afferent pathways after SCI[8] and that the reduction in TTX-resistant channel activity after SCI is associated with the decreased expression of Nav1.8, but not Nav1.9, in bladder afferent neurons using SCI rats[9]. However, the contribution of NGF to this Na+ channel plasticity in bladder afferent neurons after SCI has not been investigated previously. Thus, the present study for the first time provided the direct evidence showing that NGF is also a major mediator inducing the phenotypic changes in Na+ channels in bladder afferent neurons after SCI because NGF neutralization reversed SCI-induced changes in TTX-sensitive and TTX-resistant Na+ channel activity as well as Nav subunits expression in bladder afferent neurons. Taken together, it seems likely that NGF is an important mediator of both Na+ and K+ channel plasticity to induce bladder afferent hyperexcitability, leading to LUTD such as detrusor overactivity. Because TTX-sensitive Na+ channels are activated at lower membrane potentials than TTX-resistant Na+ channels in DRG afferent neurons including bladder-innervating cells[17], the increase of TTX-sensitive channel activity could directly contribute to the lower thresholds for AP activation in bladder afferent neurons from SCI mice compared to those from SI mice (Figure 3, Table 2).
Although this and our previous studies revealed the increased activity of TTX-sensitive Na+ channels in bladder afferent neurons after SCI in rodent models[8], it has not been previously clarified which Nav subunit is responsible for this SCI-induced phenotypic change in Na+ channel activity. The Nav channel family is consisted of 9 isoforms (Nav1.1-Nav1.9) that are divided by their relative sensitivity to TTX as either TTX-sensitive (Nav1.1-Nav1.4, Nav1.6, and Nav1.7) or TTX-resistant (Nav1.5, Nav1.8, and Nav1.9)[23]. A recent study by Grundy et al, demonstrated that, among these Nav isoforms, Nav1.7, 1.8 and Nav1.9 mRNA are abundantly expressed compared to other isoforms in lumbosacral DRG and that Nav1.7, 1.8 and 1.9 isoforms are co-expressed in all bladder afferent neurons in mice[7]. Also, our previous study demonstrated that the reduction of TTX-resistant Na+ currents in bladder afferent neurons was associated with the decreased expression of Nav1.8, but not Nav1.9[9]. Thus, in the present study, we focused on Nav1.7 and Nav1.8 as Nav isoforms encoding TTX-sensitive and TTX-resistant Na+ channels, respectively, and found that NGF-dependent Nav1.7 upregulation was associated with increased activity of TTX-sensitive Na+ channels in bladder afferent neurons from SCI mice. Recent studies in an animal model of neuropathic pain and humans with neuropathy demonstrated that Nav1.7 upregulation in DRG neurons is involved in afferent hyperexcitability and enhanced pain sensation[24]. Moreover, it has been reported that NGF is a significant contributing factor to increased Nav1.7 expression in DRG neurons and enhanced afferent sensitivity in a mouse model of post-surgical pain[12]. Therefore, the present study further elucidated that NGF-dependent Nav1.7 upregulation leading to the increased activity of low-threshold TTX-sensitive channels is also involved in bladder afferent hyperexcitability underlying SCI-induced chronic LUTD (28 days after SCI). Furthermore, recent studies have shown that the treatment with Nav1.7 blocker, protoxin II, reduced burn injury-induced pain in a rat model[25]. Thus, the results of this study suggest that blockade of the upregulated TTX-sensitive Nav1.7 subunit could have a potential for future clinical application as a new therapeutic modality for SCI-induced LUTD such as detrusor overactivity via modulation of bladder afferent activity.
There are some limitations in this study. First, we studied SCI-induced changes in only two Na+ channel subunits, Nav1.7 and Nav1.8, based on the findings in previous studies including ours. However, further studies are needed to examine the change of other Nav isoforms to fully investigate the NGF-mediated mechanism of Na+ channel plasticity after SCI. Second, the downstream molecular mechanisms underlying NGF-mediated changes in Nav expression in bladder afferent neurons were not examined in this study. We previously reported that the p38 MAP kinase pathway is involved in the emergence of SCI-induced detrusor overactivity in the same mouse model as used in this study [26]. Also, a recent study has shown that the NGF-dependent increase in Nav1.7 expression in DRG neurons is possibly mediated via a TrkA-SGK1-Nedd4-2 pathway, in a mouse model of post-surgical pain[12]. Thus, further studies are warranted to examine the downstream signalling pathways inducing Nav1.7 upregulation or Nav1.8 downregulation in bladder afferent pathways after SCI. Finally, we did not include another control group of spinal intact mice with NGF antibody treatment because the aim of this study was to identify the role of NGF overexpression in SCI-induced bladder afferent hyperexcitability, and reduced NGF expression below the control level reportedly induces opposite pathological conditions of afferent hypo-activity and bladder underactivity, as shown in our previous study using rats with diabetes mellitus [27]. Because the role of bladder NGF downregulation is out of focus of this study, we will plan to examine its effects on bladder function and afferent activity in future studies.
Conclusion
Our results indicate that SCI can induce the NGF-dependent voltage-gated Na+ channel plasticity, that is, a shift from a TTX-resistant subtype to a TTX-sensitive subtype, leading to bladder afferent hyperexcitability, which is likely to be an underlying mechanism of LUTD after SCI (Figure 5). Also, this SCI-induced Na+ channel plasticity was associated with TTX-resistant Nav1.8 downregulation and TTX-sensitive Nav1.7 upregulation in bladder afferent neurons, suggesting that these Na+ channel subunits could be potential therapeutic targets for the future development of new treatments SCI-induced LUTD.
Figure 5.
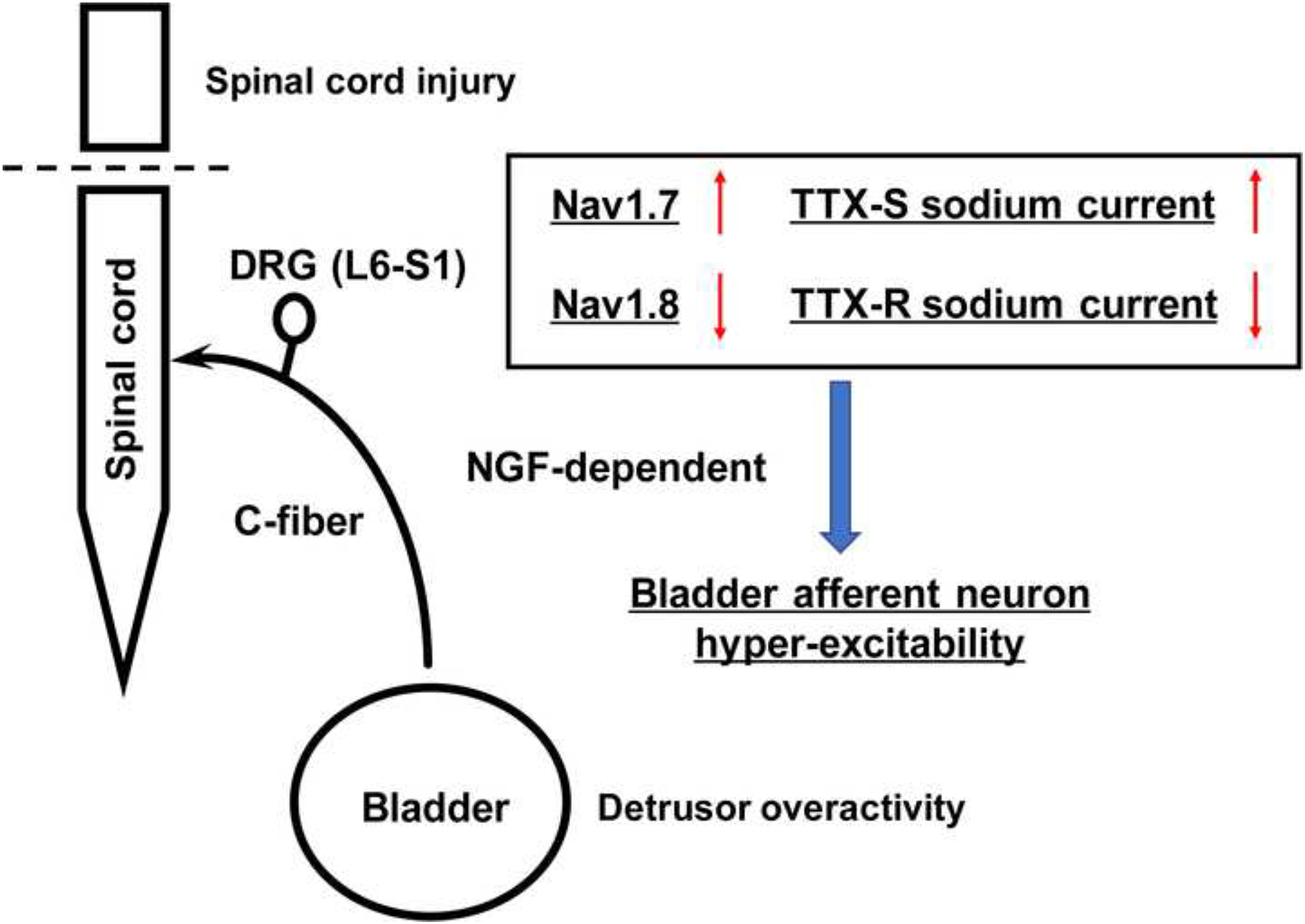
Schematic diagram of nerve growth factor (NGF)-mediated Na+ channel plasticity of bladder afferent neurons in mice with spinal cord injury.
Acknowledgement
This work was supported by the National Institutes of Health (No. R01DK129194), the National Natural Science Foundation of China (No. 81870521) and the Shanghai Sailing Program (No. 21YF1423400).
Footnotes
Declaration of competing interest
The authors declare no conflict of interest.
References
- [1].de Groat WC, Griffiths D, Yoshimura N. Neural control of the lower urinary tract. Comprehensive Physiology. 2015;5:327–96. 10.1002/cphy.c130056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Vizzard MA. Neurochemical plasticity and the role of neurotrophic factors in bladder reflex pathways after spinal cord injury. Progress in brain research. 2006;152:97–115. 10.1016/s0079-6123(05)52007-7. [DOI] [PubMed] [Google Scholar]
- [3].Ochodnicky P, Cruz CD, Yoshimura N, Cruz F. Neurotrophins as regulators of urinary bladder function. Nature reviews Urology. 2012;9:628–37. 10.1038/nrurol.2012.178. [DOI] [PubMed] [Google Scholar]
- [4].Wada N, Shimizu T, Shimizu N, de Groat WC, Kanai AJ, Tyagi P, et al. The effect of neutralization of nerve growth factor (NGF) on bladder and urethral dysfunction in mice with spinal cord injury. Neurourology and urodynamics. 2018;37:1889–96. 10.1002/nau.23539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Shimizu T, Majima T, Suzuki T, Shimizu N, Wada N, Kadekawa K, et al. Nerve growth factor-dependent hyperexcitability of capsaicin-sensitive bladder afferent neurones in mice with spinal cord injury. Experimental physiology. 2018;103:896–904. 10.1113/ep086951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Takahashi R, Yoshizawa T, Yunoki T, Tyagi P, Naito S, de Groat WC, et al. Hyperexcitability of bladder afferent neurons associated with reduction of Kv1.4 α-subunit in rats with spinal cord injury. The Journal of urology. 2013;190:2296–304. 10.1016/j.juro.2013.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Grundy L, Erickson A, Caldwell A, Garcia-Caraballo S, Rychkov G, Harrington A, et al. Tetrodotoxin-sensitive voltage-gated sodium channels regulate bladder afferent responses to distension. Pain. 2018;159:2573–84. 10.1097/j.pain.0000000000001368. [DOI] [PubMed] [Google Scholar]
- [8].Yoshimura N, de Groat WC. Plasticity of Na+ channels in afferent neurones innervating rat urinary bladder following spinal cord injury. The Journal of physiology. 1997;503 (Pt 2):269–76. 10.1111/j.1469-7793.1997.269bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Black JA, Cummins TR, Yoshimura N, de Groat WC, Waxman SG. Tetrodotoxin-resistant sodium channels Na(v)1.8/SNS and Na(v)1.9/NaN in afferent neurons innervating urinary bladder in control and spinal cord injured rats. Brain research. 2003;963:132–8. 10.1016/s0006-8993(02)03957-4. [DOI] [PubMed] [Google Scholar]
- [10].Jonas R, Klusch A, Schmelz M, Petersen M, Carr RW. Assessment of TTX-s and TTX-r Action Potential Conduction along Neurites of NGF and GDNF Cultured Porcine DRG Somata. PloS one. 2015;10:e0139107. 10.1371/journal.pone.0139107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Rudy B, Kirschenbaum B, Rukenstein A, Greene LA. Nerve growth factor increases the number of functional Na channels and induces TTX-resistant Na channels in PC12 pheochromocytoma cells. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1987;7:1613–25. 10.1523/jneurosci.07-06-01613.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Liu BW, Zhang J, Hong YS, Li NB, Liu Y, Zhang M, et al. NGF-Induced Nav1.7 Upregulation Contributes to Chronic Post-surgical Pain by Activating SGK1-Dependent Nedd4-2 Phosphorylation. Molecular neurobiology. 2020. 10.1007/s12035-020-02156-1. [DOI] [PubMed] [Google Scholar]
- [13].Ryu JC, Tooke K, Malley SE, Soulas A, Weiss T, Ganesh N, et al. Role of proNGF/p75 signaling in bladder dysfunction after spinal cord injury. The Journal of clinical investigation. 2018;128:1772–86. 10.1172/jci97837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Shimizu N, Wada N, Shimizu T, Suzuki T, Takaoka EI, Kanai AJ, et al. Effects of nerve growth factor neutralization on TRP channel expression in laser-captured bladder afferent neurons in mice with spinal cord injury. Neuroscience letters. 2018;683:100–3. 10.1016/j.neulet.2018.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yoshimura N, Bennett NE, Hayashi Y, Ogawa T, Nishizawa O, Chancellor MB, et al. Bladder overactivity and hyperexcitability of bladder afferent neurons after intrathecal delivery of nerve growth factor in rats. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2006;26:10847–55. 10.1523/jneurosci.3023-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kadekawa K, Majima T, Shimizu T, Wada N, de Groat WC, Kanai AJ, et al. The role of capsaicin-sensitive C-fiber afferent pathways in the control of micturition in spinal-intact and spinal cord-injured mice. American journal of physiology Renal physiology. 2017;313:F796–f804. 10.1152/ajprenal.00097.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Yoshimura N, White G, Weight FF, de Groat WC. Different types of Na+ and A-type K+ currents in dorsal root ganglion neurones innervating the rat urinary bladder. The Journal of physiology. 1996;494 (Pt 1):1–16. 10.1113/jphysiol.1996.sp021471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].de Groat WC, Yoshimura N. Plasticity in reflex pathways to the lower urinary tract following spinal cord injury. Experimental neurology. 2012;235:123–32. 10.1016/j.expneurol.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].de Groat WC, Yoshimura N. Changes in afferent activity after spinal cord injury. Neurourology and urodynamics. 2010;29:63–76. 10.1002/nau.20761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Jiang L, Ye B, Wang Y, Yu T, Xu H. Effect and mechanisms of sacral nerve stimulation on visceral hypersensitivity mediated by nerve growth factor. J Cell Mol Med. 2019;23:8019–24. 10.1111/jcmm.14660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Vizzard MA. Changes in urinary bladder neurotrophic factor mRNA and NGF protein following urinary bladder dysfunction. Experimental neurology. 2000;161:273–84. 10.1006/exnr.1999.7254. [DOI] [PubMed] [Google Scholar]
- [22].Seki S, Sasaki K, Fraser MO, Igawa Y, Nishizawa O, Chancellor MB, et al. Immunoneutralization of nerve growth factor in lumbosacral spinal cord reduces bladder hyperreflexia in spinal cord injured rats. The Journal of urology. 2002;168:2269–74. 10.1097/01.ju.0000025338.65642.09. [DOI] [PubMed] [Google Scholar]
- [23].Catterall WA, Goldin AL, Waxman SG. International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacological reviews. 2005;57:397–409. 10.1124/pr.57.4.4. [DOI] [PubMed] [Google Scholar]
- [24].Li Y, North RY, Rhines LD, Tatsui CE, Rao G, Edwards DD, et al. DRG Voltage-Gated Sodium Channel 1.7 Is Upregulated in Paclitaxel-Induced Neuropathy in Rats and in Humans with Neuropathic Pain. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2018;38:1124–36. 10.1523/jneurosci.0899-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Torres-Pérez JV, Adamek P, Palecek J, Vizcaychipi M, Nagy I, Varga A. The NA(v)1.7 blocker protoxin II reduces burn injury-induced spinal nociceptive processing. Journal of molecular medicine (Berlin, Germany). 2018;96:75–84. 10.1007/s00109-017-1599-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Shimizu N, Wada N, Shimizu T, Suzuki T, Kurobe M, Kanai AJ, et al. Role of p38 MAP kinase signaling pathways in storage and voiding dysfunction in mice with spinal cord injury. Neurourology and urodynamics. 2020;39:108–15. 10.1002/nau.24170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sasaki K, Chancellor MB, Goins WF, Phelan MW, Glorioso JC, de Groat WC, et al. Gene therapy using replication-defective herpes simplex virus vectors expressing nerve growth factor in a rat model of diabetic cystopathy. Diabetes. 2004;53:2723–30. 10.2337/diabetes.53.10.2723. [DOI] [PubMed] [Google Scholar]