

Abstract
Electron bifurcation, or the coupling of exergonic and endergonic oxidation-reduction reactions, was discovered by Peter Mitchell and provides an elegant mechanism to rationalize and understand the logic that underpins the Q cycle of the respiratory chain. Thought to be a unique reaction of respiratory complex III for nearly 40 years, about a decade ago Wolfgang Buckel and Rudolf Thauer discovered that flavin-based electron bifurcation is also an important component of anaerobic microbial metabolism. Their discovery spawned a surge of research activity, providing a basis to understand flavin-based bifurcation, forging fundamental parallels with Mitchell’s Q cycle and leading to the proposal of metal-based bifurcating enzymes. New insights into the mechanism of electron bifurcation provide a foundation to establish the unifying principles and essential elements of this fascinating biochemical phenomenon.
Graphical Abstract

Simple diagram of electron bifurcation illustrating the bifurcation of an electron pair from the electron donor (D) and the subsequent endergonic and exergonic electron transfers acceptors A1 and A2 respectively.
In the simplest terms, electron bifurcation effectively couples exergonic and endergonic oxidation-reduction reactions in an overall thermodynamically spontaneous process [1,2]. Based on Marcus theory, rates of biological electron transfer reactions are governed by the difference in reduction potentials of the donors and acceptors, their mutual distance, and the nature of the physical medium between them, in particular its response to the change in charge – the reorganization energy [3]. Electron transfer reactions are fastest when the thermodynamic driving force matches the reorganization energy and when the separation distance between donor and acceptor is short. Electrons can flow “uphill,” but that comes with a kinetic penalty. Interestingly, when the thermodynamic driving force exceeds the reorganization energy, the reactions slow down; this is known as the “inverted regime” for electron transfer. Electron bifurcation exploits these characteristic properties of electron transfer to direct electrons down spatially separated and energetically distinct landscapes to low and high potential cofactors. Most importantly, the electron flow along the two branches is highly concerted.
Harnessing the free energy of electrochemical potential
Aerobic respiratory chains in biology couple the oxidation of substrates with the reduction of oxygen to generate a transmembrane proton motive force (Figure 1). They are found in the mitochondria of eukaryotes [4], as well as in diverse lineages of bacteria and archaea. Transmembrane electron transfer is a primary mechanism underpinning energy conservation in living systems. The hallmark of electron transfer chain function, and the essence of Mitchell’s “chemiosmotic hypothesis”, is that harnessing the free energy of electron transfer reactions down an electrochemical potential gradient drives proton translocation across a membrane [5]. The transmembrane proton gradient essentially stores free energy in an entropic form (a concentration gradient) as well as in an enthalpic form (if the membrane has a nonzero resting voltage). The redox chains thus convert the electrochemical potential energy of low potential substrates, such as NADH and FADH2, to potential energy in the form of the chemiosmotic potential. In biological energy transduction, the chemiosmotic potential is used to drive ATP synthesis. At the center is an elegant dance of electrons and protons that is of paramount importance to life in general and is driven by electrochemical potential energy.
Figure 1.

The electron transport chain illustrating the electron transfer from the unifying currency of reducing equivalents in biology NADH and FADH2 through three proton translocating respiratory complexes, I, III, and IV from the mitochondrial matrix (M) into the intermembrane space IM resulting in the eventual reduction of oxygen to water and the generation of proton motive force that is harnessed to drive ATP synthesis through an ATPase.
In addition to the chemiosmotic hypothesis, Mitchell is credited with the discovery of “electron bifurcation” in his elucidation of the “Q cycle” that links oxidized quinone (Q) to reduced quinol (QH2) in complex III [6,7] (Figure 2). Complex III is an electron transfer-driven proton pump of the aerobic respiratory chain, oxidizing QH2 equivalents produced by respiratory complexes I and II and reducing cytochrome c, which provides the link to complex IV (cytochrome c oxidase) and oxygen reduction. Mitchell found that complex III functions rather differently from the other respiratory complexes: a fraction of the electron flux and electrochemical potential is diverted to catalyze a seemingly illogical reaction coupling the oxidation of QH2 near the exterior of the membrane to the reduction of Q near the interior of the membrane. The reaction, however, rings of sheer brilliance when considering the mobility of Q/QH2 in the membrane, and its ability to generate a proton gradient through this process.
Figure 2.
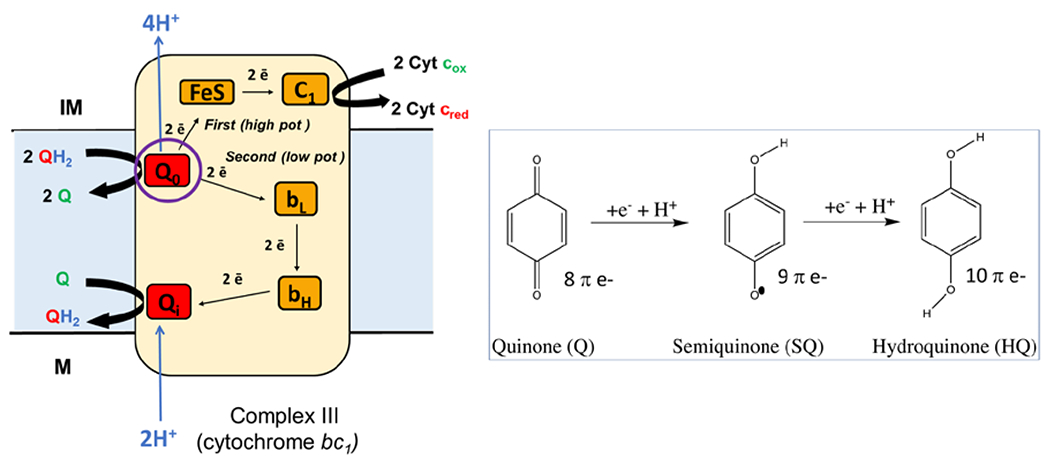
Electron transfer in respiratory complex III and the “Q cycle” illustrating the transfer of electrons from QH2 simultaneously to an FeS cluster and a b-type cytoctrome in an electron bifurcation reaction. The electron transferred to the FeS cluster is on path to the eventual reduction of cytochrome c (the substrate for complex IV). The electron transferred to cytochrome b will be coupled with an additional electron from the subsequent round of the Q cycle to reduce Q to QH2. The link between the Q cycle and the generation of proton motif force is the oxidation of QH2 and the release of protons in the intermembrane space (IM) and the reduction and consumption of protons in the matrix (M). On the right are the structures of the Q, SQ, and QH2 states.
Q cycle and the birth of electron bifurcation
An additional puzzling element of the “Q cycle” reaction is reconciling the thermodynamics of its two-electron chemistry. Complex III-dependent Q oxidation leads to the simultaneous reduction of both cytochrome c (downhill thermodynamically) and the reduction of a b-type cytochrome (uphill thermodynamically), along two different spatially separated redox pathways (Figure 2). “Electron bifurcation” was subsequently proposed to reconcile the two paths within the thermodynamics of the “Q cycle,” and the requirement that all spontaneous reactions at constant pressure require negative values for the Gibbs free energy. Two half reactions were proposed in which the initial one involves the exergonic electron transfer of a single electron to an iron-sulfur cluster, which in turn transfers an electron to cytochrome c. In the second half reaction, the other electron traverses an endergonic path to the reduction of cytochrome b and then Q within complex III. Diffusion of the quinol through the membrane and subsequent oxidation on the outside of the membrane by complex III completes this elegant cycle of redox-driven proton pumping. A key to the mechanism of electron bifurcation is that the exergonic transfer to the iron-sulfur cluster is the production of an energetic intermediate species on Q with a sufficiently low potential to drive the reduction of cytochrome b. The overall energy landscape is thermodynamically favorable ΔG < 0) since the potential difference in the exergonic half-reaction (reduction of FeS and cytochrome c) is larger than the potential difference of endergonic half-reaction.
Electron bifurcation thus harnesses the free energy of exergonic electron transfer to drive endergonic electron transfer. Q-based electron bifurcation occurs when QH2 is oxidized by a single electron through an exergonic electron transfer generating a highly energized semiquinone (SQ) intermediate. The SQ intermediate has a low enough reduction potential to favorably drive the endergonic half reaction [8]. The key to high fidelity electron bifurcation is a robust mechanism to direct electron flow along the “orthogonal” pathways, so that electrons are sent down the two pathways in a specific sequence, which is presumably controlled by a combination of distance and free energy effects dictated by electron tunneling and Marcus theory constraints [9]. This electronic choreography is accomplished in complex III through a large scale conformational change that significantly increases the distance of the iron-sulfur cluster acceptor on the exergonic path following electron transfer, making the kinetics for the transfer of a second electron down this pathway much less favorable [10,11]. Electron bifurcation in complex III is an evolutionary innovation that increases the efficiency in the production of a proton motive force by the electron transport chain, and its presence increases the amount of chemiosmotic potential generated in the electron transport chain on the order of 20% without any net change in the overall electron flow.
Flavin-based electron bifurcation
For nearly forty years, electron bifurcation as described by Mitchell was presumed to be unique to complex III. About a decade ago, however, bifurcation re-emerged in the context of anaerobic metabolism [12–14]. Enzymatic activities were described that involved the coupling of the exergonic reduction of NAD+ to an endergonic reaction that allowed the reduction of the low potential electron carrier ferredoxin (Fd) [15]. In anaerobic metabolism, where free energy cannot be squandered without dire consequences to cell viability, electron bifurcation provides a high-fidelity mechanism to promote efficient energy conservation [1,16]. The discovery of electron bifurcation associated with anaerobic metabolism involved the oxidation of NADH coupled to the simultaneous reduction of crotonyl-CoA and Fd in a butyric acid-forming bacterium [13]. The enzyme responsible for this reaction is a member of an electron transferring flavoprotein family (ETF) of enzymes that characteristically possess only flavin redox cofactors, pointing to the fact that electron bifurcation is flavin-based for this class of enzymes [17].
Studies of flavin-based electron bifurcation are emerging rapidly; parallels and new features are being discovered, which compare with a single version of Q-based bifurcation [1]. The best characterized electron bifurcating enzyme is the dimeric NADH-dependent reduced ferredoxin NADP+ oxidoreductase (Nfn) [18,19]. This enzyme has been structurally characterized in multiple states from two different microbial sources (Thermotoga maritima and Pyrococcus furiosus). The dimer consists of a large subunit with one flavin, two [4Fe-4S] clusters, and a small subunit with an additional flavin and a [2Fe-2S] cluster (Figure 3). These five redox cofactors are arranged linearly, with one of the flavins (the bifurcating flavin) located in the large subunit at the site of NADPH oxidation. Exergonic and endergonic electron transfer paths connect the bifurcating flavin with NAD+ and ferredoxin (Fd) reduction sites. The exergonic path contains a [2Fe-2S] cluster about 14 Å from the bifurcating flavin on a pathway to a second flavin located another 9 Å away, at the site of NAD+ reduction. Interestingly, the [2Fe-2S] cluster has a non-canonical coordinating ligand set of three Cys and one Asp, with an unusual reduction potential of +80 mV, estimated by EPR potentiometric titrations [20]. The endergonic branch has two [4Fe-4S] clusters located in the large subunit that connects the bifurcating flavin with the Fd reduction site. The proximal [4Fe-4S] cluster is located less than 8 Å from the bifurcating flavin and, by analogy with the bifurcating flavin proximal [2Fe-2S] cluster, is coordinated with three Cys ligands and an acidic Glu side chain. The distal cluster, which presumably transfers electrons directly to Fd, is approximately 10 Å further removed from the bifurcating flavin.
Figure 3.
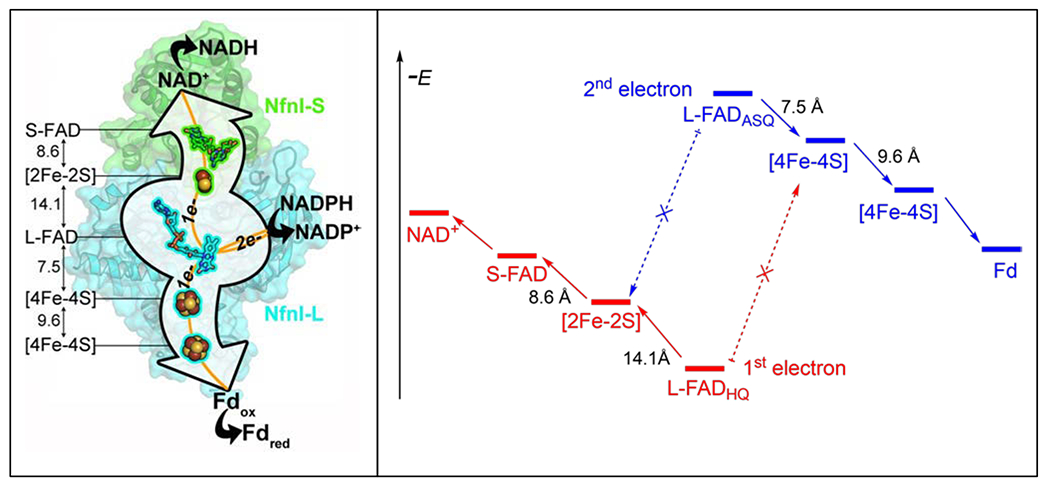
Left: Nfn structure showing cofactor arrangements along pathways and distances between cofactors. Right: Redox potential (E) landscape for bifurcating Nfn. After bifurcation at L-FAD, the first electron flows uphill (red relay) eventually to NAD+, and the second electron flows downhill (blue relay), on a different physical pathway, to ferredoxin (Fd).
A combination of experimental techniques has been employed to provide insights into the mechanism of Nfn-catalyzed electron bifurcation and to understand the requirements for electron bifurcation [19]. Nfn is an excellent model system for probing bifurcating electron transfer due, in part, to the available structures and to accessible interrogation through a variety of biophysical approaches. Electrochemical and spectroscopic methods have been used to estimate the reduction potential of the five redox cofactors, revealing surprising results. The [4Fe-4S] clusters have dramatically different reduction potentials compared to the [2Fe-2S] cluster of the exergonic branch (−718 mV and −513 mV compared to the estimated potential of +80 mV versus the standard hydrogen electrode). This arrangement sets up an unusual energy landscape and indicates that the bifurcating flavin site is likely unique in nature in its ability to generate the driving force needed to reduce the −718 mV cluster at a reasonable rate [21].
Free energy landscape
Key mechanistic insights into Nfn electron bifurcation have been provided through spectroelectrochemistry and transient absorption spectroscopy [19]. The SQ state of the bifurcating flavin cannot be observed in spectroelectrochemical titrations, suggesting that it is very short-lived. The SQ state, however, can be observed using ultrafast transient absorption spectroscopy on a semi-reduced Nfn that is generated by reduction with NADPH in the absence of NAD+ or Fd as electron acceptors which characteristically exists with two electrons residing on the FeS clusters. Laser excitation generates strongly oxidizing flavin sites which are immediately reduced to SQ flavins. Rates of electron transfer back to the FeS clusters can be measured and, conveniently, the S- and L-flavins can be distinguished because they exist in neutral and anionic SQ states, respectively, that have different optical signatures. Electron transfer from the SQ state of the bifurcating flavin to the [4Fe-4S] cluster occurs at a very fast rate (ps) which, when accounting for the reduction potential of the proximal [4Fe-4S], indicates that the reduction potential of SQ to oxidized flavin must be ~ −911 mV, so a HQ to SQ reduction potential would be on the order of +359 mV. This establishes a surprising energy landscape (Figure 3), with the bifurcating flavin having strongly crossed potentials. Although this is not a requirement for bifurcation [21], the potential crossing rationalizes a mechanism to generate the driving force needed to satisfy the energetics required of the endergonic branch, and the overall energy landscape and is in line with the principles of Q-based bifurcation observed in complex III.
Controlling electron flow down the exergonic path
Maintaining the 1:1 electron fidelity of bifurcation (moving electrons with an equal flux down the two paths) in complex III is associated with a large conformational change that increases the distance between the outer Q oxidation site and the Rieske FeS site on the exergonic branch [22]. Increasing the distance presumably lowers the electron transfer rate on the exergonic path, in favor of the second electron moving to cytochrome b on the endergonic path. Although this kind of mechanism has been proposed for other members of the bifurcating electron transferring flavoprotein family of enzymes [17,23,24] and suggested to potentially be employed in Nfn, the structural and mass spectrometry results are not consistent with conformational changes on the scale anticipated to support such a mechanism [25]. As an alternative, it has been suggested that the electron transfer is electrochemically controlled and the very large difference between the very low potential bifurcating flavin SQ (−911 mV) and the iron-sulfur cluster on the exergonic branch (+80 mV) places the electron transfer in the Marcus inverted regime: this inverted effect coupled with the nearly 7 Å shorter distance to the acceptor along the endergonic path is predicted to send the second electron down the endergonic path. Exploiting both distances and driving forces of electron transfer reactions between redox cofactors shows how macromolecular machines can exploit the intrinsic biophysical chemistry of electron transfer to direct high value multi-electron redox processes.
New frontiers
The nature of Q based bifurcation revealed decades ago, coupled with the rapidly appearing insights into flavin-based bifurcation, establish the core requirements for electron bifurcation in proteins. The major requirements are 1) a redox site that can perform one- and two-electron chemistry where one redox state is inherently less stable than the other and 2) an effective mechanism to control the 1:1 bifurcation fidelity to affect equal fluxes along exergonic and endergonic pathways. Now that flavin-based bifurcation has been demonstrated, several other bifurcating reactions and associated enzymes have been identified [26] and the structures of these enzymes are emerging [23,24,27,28]. It will be interesting to see if mechanistic features different than those observed for Nfn modulate this unique and elegant electron transfer catalysis. Interestingly, it has been observed that a common multiple redox cofactor protein architecture, termed HydABC, is found to be associated with a canonical Nfn bifurcating architecture as well as bifurcating tungsten-containing formate dehydrogenase [29], [FeFe]-hydrogenase [14], and [NiFe]-hydrogenase sites [30] (Figure 4 [30]). The implication of these observations is that these metal-containing active sites may serve as the site of bifurcation. The role of protons in electron bifurcation is unclear, but a proton-coupled electron transfer process is suspected [31–35]. The growing relevance of electron bifurcation points to a very high level of control that proteins can impose on redox energetics and electron transfer kinetics, pointing to conceivably game changing paradigms to be exploited in future bioinspired energy production and utilization technologies.
Figure 4.
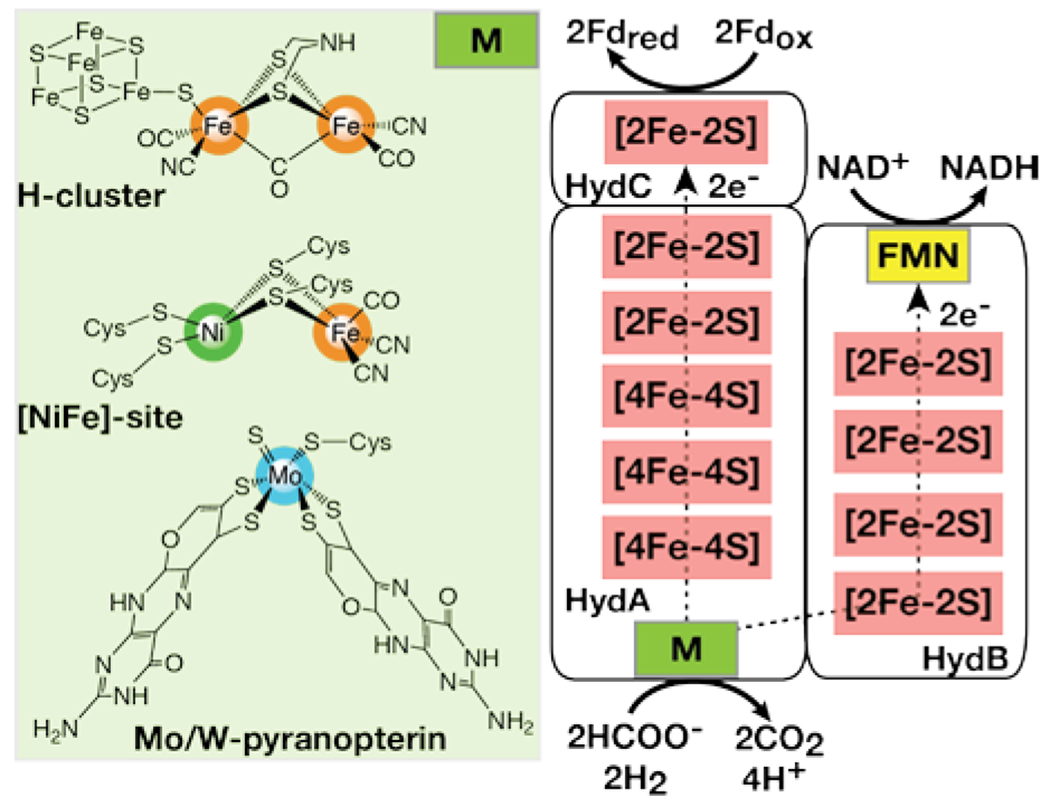
The HydABC scaffold of proposed metal-site bifurcating enzymes (right) where the M site is the H cluster, the [NiFe]-site or the W/Mo-pyranopterin catalytic site (left) of the [FeFe]-hydrogenase, [NiFe]-hydrogenase or formate dehydrogenase, respectively.
Highlights.
Electron bifurcation couples endergonic and exergonic electron transfer reactions.
Electron bifurcation requires redox sites capable of managing single and pairwise electron transfer reactions.
Redox centers in electron bifurcating enzymes exist with an inherently short-lived low-potential redox state.
Crossed potential redox transitions have been observed at electron bifurcating sites, but are not required for electron bifurcation.
Electron bifurcation catalysis can occur at organic and potentially at inorganic cofactors that undergo at least two redox transitions.
Acknowledgements
This work is supported as part of the Biological Electron Transfer and Catalysis (BETCy) EFRC, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science (DE-SC0012518).
References and recommended reading
Papers of particular interest, published within the period of the review, have been highlighted as:
* of special interest
** of outstanding interest
- 1.Buckel W, Thauer RK: Flavin-based electron bifurcation, a new mechanism of biological energy coupling. Chem Rev 2018, 118:3862–3886. [DOI] [PubMed] [Google Scholar]; ** Comprehensive review of the history of research on the biological and biochemical aspects of electron bifrucation emphasizing the new insights into flavin-based bifurcation.
- 2.Peters JW, Miller AF, Jones AK, King PW, Adams MW: Electron bifurcation. Curr Opin Chem Biol 2016, 31:146–152. [DOI] [PubMed] [Google Scholar]
- 3.Marcus RA, Sutin N: Electron Transfers in Chemistry and Biology. Biochimica Et Biophysica Acta 1985, 811:265–322. [Google Scholar]
- 4.Guo RY, Gu JK, Zong S, Wu M, Yang MJ: Structure and mechanism of mitochondrial electron transport chain. Biomedical Journal 2018, 41:9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mitchell P: Possible molecular mechanisms of the protonmotive function of cytochrome systems. J Theor Biol 1976, 62:327–367. [DOI] [PubMed] [Google Scholar]
- 6.Crofts AR: The Q-cycle - A personal perspective. Photosynth Res 2004, 80:223–243. [DOI] [PubMed] [Google Scholar]
- 7.Mitchell P: The protonmotive Q cycle: a general formulation. FEBS Lett 1975, 59:137–139. [DOI] [PubMed] [Google Scholar]
- 8.Bhaduri S, Stadnytskyi V, Zakharov SD, Hasan SS, Bujnowicz L, Sarewicz M,Savikhin S, Osyczka A, Cramer WA: Pathways of transmembrane electron transfer in cytochrome bc complexes: dielectric heterogeneity and interheme coulombic interactions. J Phys Chem B 2017, 121:975–983. [DOI] [PubMed] [Google Scholar]
- 9.Osyczka A, Moser CC, Dutton PL: Fixing the Q cycle. Trends Biochem Sci 2005, 30:176–182. [DOI] [PubMed] [Google Scholar]
- 10.Cooley JW: Protein conformational changes involved in the cytochrome bc1 complex catalytic cycle. Biochim Biophys Acta 2013, 1827:1340–1345. [DOI] [PubMed] [Google Scholar]
- 11.Xiao K, Yu L, Yu CA: Confirmation of the involvement of protein domain movement during the catalytic cycle of the cytochrome bc1 complex by the formation of an intersubunit disulfide bond between cytochrome b and the iron-sulfur protein. J Biol Chem 2000, 275:38597–38604. [DOI] [PubMed] [Google Scholar]
- 12.Herrmann G, Jayamani E, Mai G, Buckel W: Energy conservation via electron-transferring flavoprotein in anaerobic bacteria. J Bacteriol 2008, 190:784–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li F, Hinderberger J, Seedorf H, Zhang J, Buckel W, Thauer RK: Coupled ferredoxin and crotonyl coenzyme A (CoA) reduction with NADH catalyzed by the butyryl-CoA dehydrogenase/Etf complex from Clostridium kluyveri. J Bacteriol 2008, 190:843–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schut GJ, Adams MW: The iron-hydrogenase of Thermotoga maritima utilizes ferredoxin and NADH synergistically: a new perspective on anaerobic hydrogen production. J Bacteriol 2009, 191:4451–4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buckel W, Thauer RK: Flavin-based electron bifurcation, ferredoxin, flavodoxin, and anaerobic respiration with protons (Ech) or NAD(+) (Rnf) as electron acceptors: a historical review. Front Microbiol 2018, 9:401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Buckel W, Thauer RK: Energy conservation via electron bifurcating ferredoxin reduction and proton/Na(+) translocating ferredoxin oxidation. Biochim Biophys Acta 2013, 1827:94–113. [DOI] [PubMed] [Google Scholar]
- 17.Garcia Costas AM, Poudel S, Miller AF, Schut GJ, Ledbetter RN, Fixen KR, Seefeldt LC, Adams MWW, Harwood CS, Boyd ES, et al. : Defining electron bifurcation in the electron-transferring flavoprotein family. J Bacteriol 2017, 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Demmer JK, Huang H, Wang S, Demmer U, Thauer RK, Ermler U: Insights into flavin-based electron bifurcation via the NADH-dependent reduced ferredoxin:NADP oxidoreductase structure. J Biol Chem 2015, 290:21985–21995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lubner CE, Jennings DP, Mulder DW, Schut GJ, Zadvornyy OA, Hoben JP, Tokmina-Lukaszewska M, Berry L, Nguyen DM, Lipscomb GL, et al. : Mechanistic insights into energy conservation by flavin-based electron bifurcation. Nat Chem Biol 2017, 13:655–659. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** Study describing combined structural, spectrscopic, and electrochemical results providing signficant insights into the mechanism of bifurcating NADH-dependent reduced ferredoxin: NADP+ oxidoreductase I (Nfn).
- 20.Hagen WR, Silva PJ, Amorim MA, Hagedoorn PL, Wassink H, Haaker H, Robb FT: Novel structure and redox chemistry of the prosthetic groups of the iron-sulfur flavoprotein sulfide dehydrogenase from Pyrococcus furiosus; evidence for a [2Fe-2S] cluster with Asp(Cys)3 ligands. J Biol Inorg Chem 2000, 5:527–534. [DOI] [PubMed] [Google Scholar]
- 21.Zhang P, Yuly JL, Lubner CE, Mulder DW, King PW, Peters JW, Beratan DN: Electron bifurcation: thermodynamics and kinetics of two-electron brokering in biological redox chemistry. Acc Chem Res 2017, 50:2410–2417. [DOI] [PubMed] [Google Scholar]; * A comprehensive theoretical analysis of the energy landscape of bifurcating NADH-dependent reduced ferredoxin: NADP+ oxidoreductase I (Nfn) defining the requirements for electron bifurcation.
- 22.Crofts AR, Rose SW, Burton RL, Desai AV, Kenis PJA, Dikanov SA: The Q-cycle mechanism of the bc1 complex: a biologist’s perspective on atomistic studies. J Phys Chem B 2017, 121:3701–3717. [DOI] [PubMed] [Google Scholar]; * Computational study on the mechanism of the Q-cycle of complex III emphasizing the role of comformational change in modulating interactions of the Riske FeS protein with the Q site to gate electron flow.
- 23.Demmer JK, Bertsch J, Oppinger C, Wohlers H, Kayastha K, Demmer U, Ermler U, Muller V: Molecular basis of the flavin-based electron-bifurcating caffeyl-CoA reductase reaction. FEBS Lett 2018, 592:332–342. [DOI] [PubMed] [Google Scholar]; * Structure of a bifurcating electron transfer flavoprotein caffeyl-CoA reductase emaphsizing the role of FeS cluster domain unique to the bifrucating members of the electron trasfer flavoprotein family.
- 24.Demmer JK, Pal Chowdhury N, Selmer T, Ermler U, Buckel W: The semiquinone swing in the bifurcating electron transferring flavoprotein/butyryl-CoA dehydrogenase complex from Clostridium difficile. Nat Commun 2017,8: 1577. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** The structure of bifurcating electron transferring flavoprotein butyrl-CoA dehydogenase complex is described, and a semiquinone swing mechanism is proposed for gating electron flow.
- 25.Berry L, Poudel S, Tokmina-Lukaszewska M, Colman DR, Nguyen DMN, Schut GJ, Adams MWW, Peters JW, Boyd ES, Bothner B: H/D exchange mass spectrometry and statistical coupling analysis reveal a role for allostery in a ferredoxin-dependent bifurcating transhydrogenase catalytic cycle. Biochim Biophys Acta 2018, 1862:9–17. [DOI] [PubMed] [Google Scholar]
- 26.Ledbetter RN, Garcia Costas AM, Lubner CE, Mulder DW, Tokmina-Lukaszewska M, Artz JH, Patterson A, Magnuson TS, Jay ZJ, Duan HD, et al. : The electron bifurcating fixABCX protein complex from Azotobacter vinelandii: generation of low-potential reducing equivalents for nitrogenase catalysis.Biochemistry 2017, 56:4177–4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chowdhury NP, Mowafy AM, Demmer JK, Upadhyay V, Koelzer S, Jayamani E, Kahnt J, Hornung M, Demmer U, Ermler U, et al. : Studies on the mechanism of electron bifurcation catalyzed by electron transferring flavoprotein (Etf) and butyryl-CoA dehydrogenase (Bcd) of Acidaminococcus fermentans. J Biol Chem 2014, 289:5145–5157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wagner T, Koch J, Ermler U, Shima S: Methanogenic heterodisulfide reductase (HdrABC-MvhAGD) uses two noncubane [4Fe-4S] clusters for reduction. Science 2017, 357:699–703. [DOI] [PubMed] [Google Scholar]; * Structure of the methanogen electron bifurcating heterodisuflide reductase-[NiFe]-hydrogenase complex revealing an ellaborate array of FeS clusters, some having unique structures, for trafficking electrons.
- 29.Redecke L, Nass K, DePonte DP, White TA, Rehders D, Barty A, Stellato F, Liang M, Barends TR, Boutet S, et al. : Natively inhibited Trypanosoma brucei cathepsin B structure determined by using an X-ray laser. Science 2013, 339:227–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peters JW, Beratan DN, Schut GJ, Adams MWW: On the nature of organic and inorganic centers that bifurcate electrons, coupling exergonic and endergonic oxidation-reduction reactions. Chem Commun (Camb) 2018, 54:4091–4099. [DOI] [PubMed] [Google Scholar]; ** Description of the current state of research on electron bifrucating enzymes presenting a new proposal of specific metal sites that might act as bifurcating center, implicating bifurcation could occur at inorganic sites in addition to organic quinone and flavin sites where bifurcation has been demonstrated.
- 31.Dempsey JL, Winkler JR, Gray HB: Proton-coupled electron flow in protein redox machines. Chem Rev 2010, 110:7024–7039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huynh MH, Meyer TJ: Proton-coupled electron transfer. Chem Rev 2007, 107:5004–5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mayer JM, Rhile IJ: Thermodynamics and kinetics of proton-coupled electron transfer: stepwise vs. concerted pathways. Biochim Biophys Acta 2004, 1655:51–58. [DOI] [PubMed] [Google Scholar]
- 34.Reece SY, Nocera DG: Proton-coupled electron transfer in biology: results from synergistic studies in natural and model systems. Annu Rev Biochem 2009, 78:673–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weinberg DR, Gagliardi CJ, Hull JF, Murphy CF, Kent CA, Westlake BC, Paul A, Ess DH, McCafferty DG, Meyer TJ: Proton-coupled electron transfer. Chem Rev 2012, 112:4016–4093. [DOI] [PubMed] [Google Scholar]