

SUMMARY
Branching morphogenesis is a fundamental process by which organs in invertebrates and vertebrates form branches to expand their surface areas. The current dogma holds that directional cell migration determines where a new branch forms and thus patterns branching. Here, we asked whether mouse Lgl1, a homolog of the Drosophila tumor suppressor Lgl, regulates epithelial polarity in the mammary gland. Surprisingly, mammary glands lacking Lgl1 have normal epithelial polarity, but they form fewer branches. Moreover, we find that Lgl1 null epithelium is unable to directionally migrate, suggesting that migration is not essential for mammary epithelial branching as expected. We show that LGL1 binds to Integrin β1 and inhibits its downstream signaling, and Integrin β1 overexpression blocks epithelial migration, thus recapitulating the Lgl1 null phenotype. Altogether, we demonstrate that Lgl1 modulation of Integrin β1 signaling is essential for directional migration and that epithelial branching in invertebrates and the mammary gland is fundamentally distinct.
Graphical Abstract
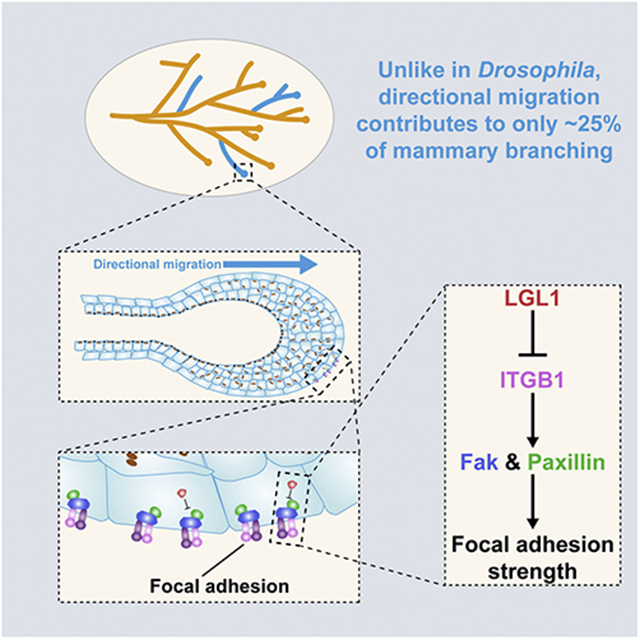
In brief
Ma et al. show that Lgl1 is essential for mammary gland branching morphogenesis but not epithelial polarity. Lgl1 is required for directional migration by regulating Integrin β1 signaling levels and focal adhesion strengths. Finally, branching mechanisms are distinct between mammary gland and Drosophila systems where directional migration is indispensable.
INTRODUCTION
Cell polarity, a basic characteristic of all living cells, refers to the asymmetric distribution of essential cellular components, including fate determinants, organelles, membrane domains, and cytoskeleton (Bryant and Mostov, 2008). At the tissue level, such an asymmetry is often organized into a higher-order polarity. For example, epithelial polarity is the asymmetric arrangement whereby the apical domains of individual epithelial cells face the lumen, i.e., the environment, whereas their basolateral domains face the inside of the tissue. Epithelial polarity is essential for myriad biological processes, such as tissue morphogenesis, cell fate determination, organ physiology, and homeostasis (St Johnston and Ahringer, 2010). By contrast, its loss is an early step during epithelial-mesenchymal transition and a hallmark of cancer (Macara and Mili, 2008). For this reason, epithelial polarity is generally considered a tumor suppressor, and genes involved in this process are thought to be tumor-suppressor genes (TSGs) (Bilder, 2004; Lee and Vasioukhin, 2008; Persa and Niessen, 2019).
Much of what we know about epithelial polarity has been based on studies on invertebrate systems, which show that it is regulated by a similar set of genes that regulate cell polarity (Roignot et al., 2013). In brief, epithelial polarity is controlled by three protein complexes, namely the Par3-atypical protein kinase C (aPKC), the Crumbs, and the Scribble-LGL-DLG complexes (St Johnston and Ahringer, 2010; Tanentzapf and Tepass, 2003). These complexes interact with one another through multiple protein-protein binding and phosphorylation events, which lead to their mutual exclusion and restriction to specific subcellular domains (Goldstein and Macara, 2007; Rolls et al., 2003; Atwood and Prehoda, 2009). Thus, whereas the Crumbs and Par3 complexes localize to the apical surface, the Scribble-LGL-DLG complex is restricted to the basolateral surface and maintains the corresponding subcellular domains (Siller and Doe, 2009; Mellman and Nelson, 2008; Schluter et al., 2009).
Despite the above framework, much has remained unclear regarding the regulation of epithelium polarity, especially in vertebrate organs (Schmidt and Peifer, 2020). This is in part due to the presence of proteins that have similar or redundant functions (Choi et al., 2019). Drosophila Lgl, for example, is a prototypic polarity gene and a TSG. It was discovered because fly larvae lacking Lgl exhibited tumor growth in neuroblasts and imaginal disk epithelium, which became invasive cancers with the forced expression of ras (Albertson and Doe, 2003; Bilder et al., 2000; Justice et al., 2003; Ohshiro et al., 2000; Pagliarini and Xu, 2003). Fly Lgl has two vertebrate homologs, namely Lgl1 and Lgl2 in the mouse (Vasioukhin, 2006; Wirtz-Peitz and Knoblich, 2006). Consistent with the fly data, mammary epithelial cells lacking Lgl1 showed polarity loss in vitro (Russ et al., 2012), while mice lacking Lgl1 suffer from polarity loss in the neuroepithelium, failure of oligodendrocyte differentiation, and development of brain tumors when combined with loss of TSG CDKN2A (Lee and Vasioukhin, 2008; Daynac et al., 2018; Klezovitch et al., 2004). Moreover, Lgl1 is also downregulated in a variety of human cancers, including breast cancer (Grifoni et al., 2004), suggesting it is a candidate TSG. By contrast, mouse embryos lacking Lgl2 developed to term and, despite initial placental defects and runting, eventually developed normally (Sripathy et al., 2011). Surprisingly, Lgl2 is upregulated in breast cancer and its overexpression promotes cancer progression (Saito et al., 2019). It is thus an oncogene in the mammary gland rather than a TSG as expected. These results suggest that Lgl1, rather than Lgl2, is essential for epithelial polarity in the mammary gland. They also underscore the complexity of polarity regulation of vertebrate epithelia and the imperfect parallels with invertebrate epithelial biology.
The mouse mammary gland has emerged in the past decade as a powerful model for studying vertebrate epithelial polarity (Akhtar and Streuli, 2013, Mccaffrey and Macara, 2009b). This is partly due to its postnatal development and to its status as an external appendage, both of which have made the mammary gland very amenable to experimental manipulations (Akhtar and Streuli, 2006; Aggeler et al., 1991; Zhang et al., 2017; Brisken and Ataca, 2015; Watson and Khaled, 2020). Indeed, many epithelial polarity genes, e.g., Par3 and Scribble, are essential for mammary stem cell biology, and their loss promotes breast cancer development (Mccaffrey and Macara, 2009a, Dow et al., 2008; Zhan et al., 2008; Xue et al., 2013). Despite previous in vitro studies (Russ et al., 2012) and correlative studies in human cancers (Grifoni et al., 2004), the in vivo function of Lgl1 in the mouse mammary gland remains unclear. In this study, we hypothesized that Lgl1 regulates mammary gland epithelial polarity and that its loss promotes breast cancer. To test this hypothesis, we examined the mammary glands of mice in which Lgl1 function has been removed.
RESULTS
Lgl1 is downregulated in breast cancer, and its loss promotes cell proliferation in vitro
We first determined Lgl1 mRNA expression in human breast cancers using The Cancer Genome Atlas (TCGA) database to assess whether it is a candidate TSG in the mammary gland, as it is in the brain. We found that Lgl1 is downregulated in the Her2, Luminal A, and Luminal B cancer subtypes, but not the basal subtype, when compared with normal tissue (Figure 1A). Moreover, breast cancer patients with downregulated Lgl1 mRNA expression had a reduced survival rate within ~150 months post diagnosis when compared with patients with high Lgl1 mRNA expression (Figure 1B). These data suggest that unlike Lgl2, Lgl1 is a candidate TSG in the mammary gland.
Figure 1. Lgl1 is downregulated in breast cancer and its loss promotes cell proliferation in vitro.

(A) Lgl1 mRNA expression in human breast cancer subtypes using TCGA database. ****p < 0.0001; N.S., not significant.
(B) Kaplan-Meier survival curve showing that Lgl1 downregulation is correlated with a worse survival.
(C) Lgl1 mRNA expression was measured by qPCR. Lgl1 expression at the 5-week stage was set as the base value against which other stages were compared (n ≥ 3/stage). Graph shows mean ± SD.
(D) MECs were sorted based on their expression of CD24 and CD49f. CD24medCD49fhi cells were basal (ba), whereas CD24hiCD49flow cells and CD24negCD49fneg were luminal (lu) and stromal (st), respectively.
(E) RNA was harvested from the three cell partitions to generate DNA templates for qPCR reactions.
(F) Schematic diagram depicting the experimental procedure of sample preparation, adenoviral infection, FACS, and mammosphere-/acinus-forming assays.
(G) Western blotting analysis of LGL1 expression by Lgl1+/+ and Lgl1Δ/Δ basal cells after 24-h infection by adenovirus-Cre-GFP and enrichment via FACS.
(H–J) Mammosphere-forming efficiency (MFE) of Lgl1+/+ and Lgl1Δ/Δ basal cells cultured in mammosphere medium. (H and I) Representative photographs of mammospheres. (J) Graph summarizing (H) and (I), mean ± SD. Statistical analysis was performed using unpaired t test (n = 4–5), **p < 0.01.
See also Figure S1.
To examine the in vivo function of Lgl1, we first analyzed its mRNA expression levels during postnatal development of the mouse mammary gland. Using quantitative real-time PCR (qPCR), we found that Lgl1 was expressed at various developmental stages, including 5-week and 10-week virgin, pregnancy day 5 (P5), P12, P17, lactation day 2 (L2), and involution day 2 (I2) stages (Figure 1C). Furthermore, we found that Lgl1 was expressed in all cell subpopulations examined (Figures 1D and 1E).
Next, we examined whether Lgl1 loss affected the self-renewal ability of mammary stem cells (MSCs) using the in vitro mammosphere- and acinus-forming assays. Specifically, we prepared mammary organoids from Lgl1+/+ and Lgl1fl/fl mice and infected them with adenovirus-Cre-GFP to generate control (Lgl1+/+) and mutant (Lgl1Δ/Δ) cells, respectively. Infected mammary epithelial basal cells were enriched for GFP+ expression (Figure 1F). As expected, western blotting analysis showed that LGL1 protein expression was lost in mutant cells but not in control cells (Figure 1G). We then tested the self-renewal ability of control and mutant cells using either the mammosphere- or acinus-forming assay. Interestingly, in both cases we found that mammary epithelial cells (MECs) lacking Lgl1 showed increased efficiency in forming mammospheres (Figures 1H-1J) or acini (Figure S1A).
Thus, these in vitro data show that Lgl1 loss caused increased self-renewal ability in MSCs. They are consistent with our findings showing that Lgl1 is downregulated in subtypes of breast cancers and that Lgl1 downregulation is correlated with a poor survival probability. Together, the results are consistent with the suggestion that Lgl1 may suppress tumorigenesis in the mammary gland.
Lgl1 promotes branching morphogenesis of the mammary gland epithelium
Next, we sought to examine whether loss of Lgl1 causes overgrowths of the mammary gland in vivo. We crossed male mice hemizygous for the MMTV-cre transgene (Wagner et al., 2001) and heterozygous for the Lgl1Δ allele with female mice homozygous for the Lgl1fl/fl allele. All MMTV-cre;Lgl1Δ/fl progeny (Lgl1Mcre-KO) were viable and were used to compare with their control littermates (MMTV-cre;Lgl1fl/+). We then examined mammary gland development in these animals at the 6.5-week stage (Figures 2A and 2B), which is within the 3- to 10-week time frame in which mammary epithelium undergoes active branching in the mouse (Lu et al., 2006). We found that both ductal elongation and branch points were reduced in Lgl1Mcre-KO mice when compared with the control mice (Figures 2C-2E).
Figure 2. Lgl1 promotes branching morphogenesis of the mammary gland epithelium.

(A–E) Branching trees of the #4 mammary glands from control (A, MMTV-cre; Lgl1fl/+) and mutant Lgl1Mcre-KO (B, MMTV-cre; Lgl1Δ/fl) mice at the 6.5-week stage. Arrows indicate the extent of ductal penetration in the fat pad. The dotted white line in (A) and (B) illustrates the epithelial invasion front. Quantitative comparisons of ductal penetration (C), branch points (D), and branch points per millimeter (E) between control and mutant glands (n ≥ 3/genotype).
(F–M) Branching trees of the #4 mammary glands, at the 5-week (F and I), 9-week (G and J), and 13-week (H and K) stages in glands from control (K14-cre; Lgl1fl/+) mice (F–H) and glands from mutant Lgl1Kcre-KO (K14-cre; Lgl1Δ/fl) mice (I–K).
(L and M) Quantitative comparisons of ductal penetration (L) and branching points (M) per millimeter between control and mutant glands.
Plots show mean ± SD (n ≥ 3/genotype); *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; N.S., not significant. Scale bars, 2 mm. See also Figure S2.
The data showing that loss of Lgl1 caused retardation, rather than an acceleration of epithelial branching as we initially predicted, was counter-intuitive. Considering that the MMTV-Cre line has various issues, especially being functional in only some rather than all of the MECs and being hormone dependent (Lu and Werb, 2008), we therefore wanted to determine whether the above unexpected results were restricted to the MMTV-cre-based knockout. To this end, we used an independent cre line, K14-cre, which functions in mammary progenitor cells during the embryonic stages and removes gene function from both the basal and luminal compartments, to conditionally remove Lgl1 function from the mammary epithelium (Dassule et al., 2000).
We crossed male mice hemizygous for the K14-cre transgene and heterozygous for the Lgl1-null allele, Lgl1Δ, with female mice homozygous for the Lgl1 conditional allele, Lgl1fl/fl (Klezovitch et al., 2004). All K14-cre; Lgl1Δ/fl progeny (Lgl1Kcre-KO) were viable and were compared with their morphologically normal “control” (K14-cre; Lgl1fl/+) littermates. We then examined mammary gland development in these animals at several developmental stages, including the 5-week and 9-week stages, during which active branching is ongoing and close to completion, respectively (Lu et al., 2006), and the 13-week stage when branching has already finished in the mouse mammary gland (Figures 2F-2K).
As expected, ductal epithelium progressively elongated and penetrated deeper into the fat pad from the 5-week to the 13-week stages in the control mammary glands (Figures 2F-2H and 2L). Likewise, the branch points also progressively grew with age, and by the 13-week stage each of the three longest epithelial branches had on average 38 branch points (Figure S2A). Despite the increase of both ductal elongation and the total number of branch points, the branch points per millimeter distance remained relatively stable at around 1.8 branch points/mm (Figure 2M).
Both ductal elongation and the total number of branch points also increased with age in Lgl1 null mammary glands (Figures 2I-2K, 2L, and S2A). Compared with the control mammary glands, however, ductal elongation in Lgl1 null mammary glands was ~20% and ~17% decreased at the 5-week and 9-week stages, respectively. No difference in ductal elongation was observed at the 13-week stage, presumably because by then branching had completed in both control and Lgl1 null glands and the slower elongation in the null glands had caught up with that in the control (Figure 2L). Interestingly, the numbers of branch points in Lgl1 null glands remained fewer than those in the control glands at all stages analyzed (Figure S2A). Thus, by the 13-week adult stage, the longest epithelial branch formed 28 branch points in Lgl1 null glands, which was a reduction of ~26% when compared with the control glands (Figure S2A). As in the control glands, the branch points per unit in the null glands remained relatively stable throughout development at around 1.3 branch points/mm (Figure 2M), which was less than in the control glands.
Next, we asked whether the Lgl1 null mammary glands were able to form alveoli and to produce milk. For these experiments, adult females older than 14 weeks of age were crossed with wild-type males. As expected, the control mammary epithelial tree was elaborated with many alveoli at P18.5 (Figure S2B) and the number of alveoli further increased by L3, making the mammary gland appear denser (Figure S2C). We found that Lgl1 null glands formed alveoli at these two stages very similarly to the control glands (Figures S2D and S2E). Moreover, H&E staining of paraffin sections confirmed that the presence and abundance of milk at the P18.5 stage and the L3 stage, respectively, of the control glands (Figures S2F and S2G). Likewise, milk was observed in the Lgl1 null glands very similarly to the control glands at these two stages (Figures S2H and S2I). These data suggest that the adult mutant glands are physiologically normal and can form alveoli and produce milk.
Together, our results show that epithelial branching was retarded in the mutant mice, suggesting that Lgl1 promotes branching morphogenesis of the mammary gland epithelium. They also suggest that loss of a factor that promotes MSC self-renewal or a candidate TSG does not necessarily lead to overgrowth during tissue morphogenesis.
Lgl1 is not essential for epithelial polarity in the mammary gland
It was possible that the retarded branching phenotype was caused by defective epithelial polarity due to Lgl1 loss. To examine this possibility, we used immunofluorescence microscopy to detect the protein expression of several polarity markers and polarity complex components at the 9-week stage. We found that the apical markers ZO-1 and Ezrin were correctly expressed in their normal domains in the Lgl1Kcre-KO epithelium when compared with control (Figures S3A-S3D′). E-Cadherin and Integrin β1, which mark the basolateral domains, were also correctly expressed in Lgl1Kcre-KO mammary glands (Figures S3E-S3H′). Likewise, aPKC and Par3, components of the apical polarity complex, were expressed in the apical domain, and there was no difference in their expression pattern between the Lgl1Kcre-KO and control mammary glands (Figures S3I-S3L′).
These data show that epithelial polarity was normal in mammary glands lacking Lgl1 function. Thus, contrary to our initial hypothesis, Lgl1 is not essential for epithelial polarity in the mammary gland.
Lgl1 is required for collective migration but not ductal elongation of the mammary epithelium
To determine the cause of the epithelial branching defects observed in Lgl1Kcre-KO mammary glands, we turned to two three-dimensional (3D) in vitro cultures that model distinct aspects, i.e., branch/ductal elongation or directional migration of epithelial morphogenesis (Lu et al., 2020; Ewald et al., 2008). Using a fibroblast growth factor 2 (FGF2)-based 3D in vitro culture model, we first assayed the ability of mammary epithelium lacking Lgl1 to undergo ductal elongation (Figures 3A and 3B). To this end, GFP-expressing control (Lgl1+/+) and mutant (Lgl1Δ/Δ) MECs were enriched via fluorescence-activated cell sorting (FACS), and aggregated MECs were subsequently embedded in Matrigel and cultured in basal medium. Under these conditions, control MEC aggregates remained unbranched when cultured in basal medium without FGF2 addition (not shown). Upon adding FGF2, control MECs started to form nascent epithelial branches after 5 days of culture and showed fully branched structures by 7–10 days (Figure 3A). We found that the Lgl1 null MEC aggregates were also able to branch under the same conditions (Figure 3B).
Figure 3. Lgl1 is required for collective migration but not ductal elongation of the mammary epithelium.
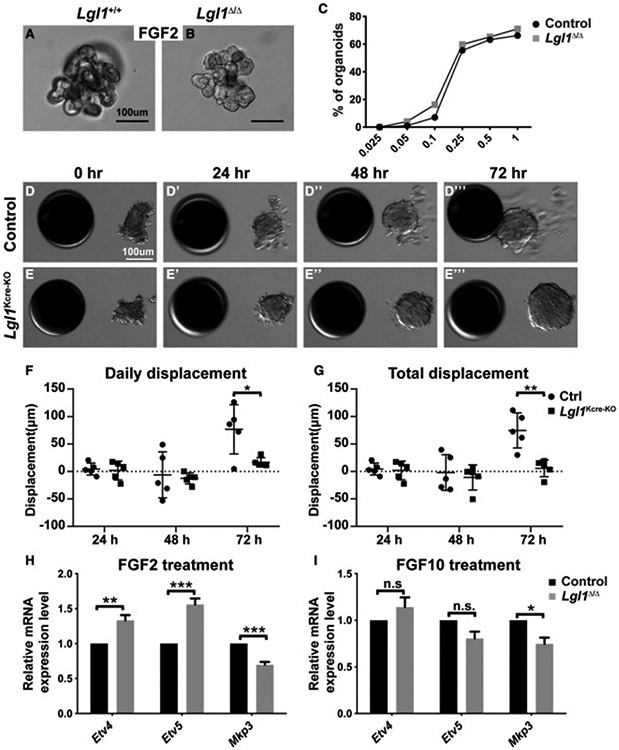
(A–C) In vitro branching assay in which Lgl1+/+ control (A) and Lgl1Δ/Δ mutant MEC aggregates (B) were subjected to cultures in basal medium containing FGF2. When stimulated by FGF2 at progressively higher concentrations from 0.025 nM to 1 nM, a progressively higher percentage of MEC aggregates underwent branching. (C) Quantitative comparisons of control and mutant MECs in their ability to undergo epithelial branching in vitro. Data were from three independent experiments. At least 100–150 organoids were examined for each treatment condition.
(D and E) Time course of control (D–D‴) and mutant Lgl1Kcre-KO (E–E‴) organoid migration toward beads pre-soaked in FGF10.
(F and G) Quantification of the daily (F) and total (G) displacement of control and Lgl1Kcre-KO organoids.
(H and I) Expression of the FGF signaling target genes Etv4, Etv5, and Mkp3 in control and mutant MEC aggregates in response to 24-h treatment of FGF2 (200 ng/mL, H) or FGF10 (200 ng/mL, I). The expression is relative to that of the control samples.
Data are mean ± SD. Statistical analysis was performed using unpaired Student’s t test. *p < 0.05; **p < 0.01; ***p < 0.001; n.s., not significant.
Moreover, we found that control MECs formed branched structures at a progressively higher percentage when FGF2 was used at a progressively higher concentration until a plateau was reached (Figure 3C). The quantitative nature of this assay thus allowed us to examine accurately how Lgl1 loss may affect the branching kinetics of mammary epithelium. Interestingly, we found that the branching kinetics of Lgl1 null epithelium was similar to that of control epithelium (Figure 3C). Together, these in vitro data demonstrate that Lgl1 mutant epithelium did not have defective ductal elongation.
We recently established an FGF10-based 3D in vitro model to study directional migration in the vertebrate epithelium (Lu et al., 2020, 2021; Zhang et al., 2014a). Using this system, we next assessed the effect of Lgl1 loss on the ability of mammary epithelium to undergo directional migration. Thus, mammary organoids from control and Lgl1Kcre-KO glands were juxtaposed with heparin sulfate beads pre-soaked in FGF10, and organoid migration was analyzed for the following 3 days. As expected, we found that control ductal organoids barely moved in the first 2 days, during which time they underwent reconstruction and stratification as we recently reported (Lu et al., 2020) Figures 3D-3D″, 3F, and 3G). They started moving on the second day and by 3 days the control organoids reached the FGF10 beads (Figures 3D‴, 3F, and 3G). Surprisingly, though, Lgl1Kcre-KO organoids completely lost their ability to move and never underwent directional migration (Figures 3E-3E‴, 3F, and 3G). These data show that mutant epithelium was unable to directionally migrate.
It is believed that directional migration is essential for branch initiation and that, in its absence, branching fails as subsequent events including elongation do not take place (Sato and Kornberg, 2002; Cabernard and Affolter, 2005; Cabernard et al., 2004; Davies, 2002; Lu et al., 2006). Our observation that Lgl1Δ/Δ epithelium was unable to undergo directional migration was thus paradoxical because it cannot explain the relatively mild branching defects mentioned above. One possible explanation for the apparent contradiction is that mutant epithelium may have a compromised response to FGF10 signaling as a result of Lgl1 loss and consequently failed to undergo directional migration. We recently found that wild-type MECs respond to FGF2 and FGF10 stimulation by upregulating mRNA expression of several target genes, including Etv4, Etv5, and Mkp3 (Zhang et al., 2014b). Therefore, we next assessed whether the responsiveness of mammary epithelium to FGF stimulation was dampened by Lgl1 loss. First, we analyzed the responsiveness of wild-type and Lgl1Δ/Δ MECs to FGF2 stimulation. We found, when compared with control MECs, that mutant MECs showed a 33% and 56% increase in Etv4 and Etv5 mRNA expression, respectively, and a 30% decrease in Mkp3 mRNA expression after FGF2 stimulation (Figure 3H). By contrast, mutant MECs showed no significant changes in Etv5 or Mkp3 mRNA expression, although there was a 25% drop in Mkp3 mRNA expression, after FGF10 treatment when compared with control MECs (Figure 3I). These data show that MECs lacking Lgl1 function were not desensitized to FGF signaling activities.
Taken together, these results show that Lgl1Δ/Δ epithelium was unable to undergo directional migration. The data thus suggest that, unlike invertebrate systems, directional migration is not essential for mammary gland epithelial branching. Moreover, together with our previous reports showing that the FGF2-based in vitro culture models migration-independent ductal elongation (Lu et al., 2020; Zhang et al., 2014a), the observation that Lgl1Δ/Δ epithelium underwent normal elongation in the FGF2-based model but suffered only a minor elongation defect in vivo suggests that directional migration also plays a role during ductal elongation in vivo (see discussion).
LGL1 binds to and co-localizes with Integrin β1
Next, we wanted to determine the mechanism by which Lgl1 regulates directional migration. To this end, we used the proximity-dependent biotin identification (BioID) labeling method (Sears et al., 2019) to find protein partners of LGL1 that may be required in the migration process. Thus, we created a construct carrying the LGL1-Biotinylase (BirA) fusion protein sequence or BirA alone as an internal control (Figure 4A). The constructs were then used to transfect the mouse neuroblastoma Neuro-2a (N2a) cells, in which the remaining steps of the BioID experiments were performed (Figure 4A).
Figure 4. LGL1 binds to and co-localizes with Integrin β1.

(A) Schematic diagram of BioID design and workflow. Expression plasmids carrying the LGL1-Biotinylase (BirA) fusion protein or BirA was used to transfect the N2a cells to screen for protein partners of LGL1.
(B) Top 50 candidate protein partners of LGL1 from the BioID screen.
(C) Gene ontology analysis of the differentially (p < 0.05) expressed genes to determine the biological processes with which these genes might be involved. Process names are abbreviated. For full names, refer to Figure S4.
(D) Kyoto Encyclopedia of Genes and Genomes analysis of the differentially (p < 0.05) expressed genes to determine the biological processes with which these genes might be involved. Process names are abbreviated. For full names, refer to Figure S4.
(E) Protein binding between ITGB1 and LGL1 as detected by co-IP assays.
(F and G) Time course of localization of ITGB1 and LGL1 as detected by fluorescent microscopy. White arrowheads denote ITGB1 and LGL1 particles over the time course of observation (F). Note that 74% of the ITGB1 particles co-localized with LGL1 particles, as shown in the quantification of the co-localization (G).
Of the top 50 hits from the BioID screen, we found numerous known protein partners of LGL1, including SCRIB and DLG1, which together with LGL1 form the Scribble complex (Figures 4B and S4A). Moreover, both LGL1 and LGL2 were on the list, suggesting that LGL1 can form homophilic and heterophilic interactions with itself and another member of its family. The findings of these known LGL1 partners thus validated the usefulness of the BioID method in finding additional protein partners of LGL1.
Of the candidate LGL1 protein partners that were not previously reported, ITGB1 stood out because, as an extracellular matrix (ECM) receptor, it is an essential component of focal adhesions that are known to be important for migration of single cells and fibroblasts (Huttenlocher and Horwitz, 2011) (Figures 4C, 4D, S4B, and S4C). Thus, one possibility was that LGL1 may regulate epithelial migration by binding to ITGB1 by affecting the dynamics of focal adhesions. To test this possibility, we used two independent methods to examine whether LGL1 binds to ITGB1. In the first method, we tagged LGL1 and ITGB1 proteins at their N terminus and C terminus with HA and FLAG peptides, respectively, which were shown to not affect their functions (Zhao et al., 2019; Dahan et al., 2012). These fusion proteins were then subjected to co-immunoprecipitation (co-IP) using HEK293T cells because of their high transfectability to test their potential binding, and we found that ITGB1 was bound to LGL1 (Figure 4E) from this in vitro study.
If LGL1 could bind to ITGB1 protein inside living cells, these proteins should co-localize under live-imaging microscopy. To test this possibility, we fused LGL1 and ITGB1 proteins at their N terminus and C terminus with mCherry and GFP, respectively. Lentiviral constructs expressing these fusion proteins were then used to infect the mammary HC11 line of cells, which were then observed under fluorescence confocal microscopy. We found that LGL1 frequently (~74%) co-localized and co-migrated with the ITGB1 proteins in the cytoplasm of living cells (Figures 4F and 4G; Table S2).
Together, our data show that LGL1 binds to the ECM receptor and focal adhesion component ITGB1.
Modulation of Integrin β1 signaling by Lgl1 is essential for epithelial migration
We predicted that Lgl1 loss resulted in reduced Integrin β1 signaling, consequently leading to a failure in directional migration. To test this prediction, we examined Integrin β1 signaling activities in the control and Lgl1Kcre-KO mammary glands by examining the phosphorylation levels of Fak and Paxillin, two essential components of the signaling pathway (Huttenlocher and Horwitz, 2011). Using western blot analysis, we found that the phospho-Paxillin level had a ~60% increase in Lgl1Kcre-KO glands when compared with control glands (Figures 5A and 5B). However, the phospho-Fak level only showed an insignificant increase in Lgl1Kcre-KO glands (Figures 5A and 5B).
Figure 5. Modulation of Integrin β1 signaling by Lgl1 is essential for epithelial migration.
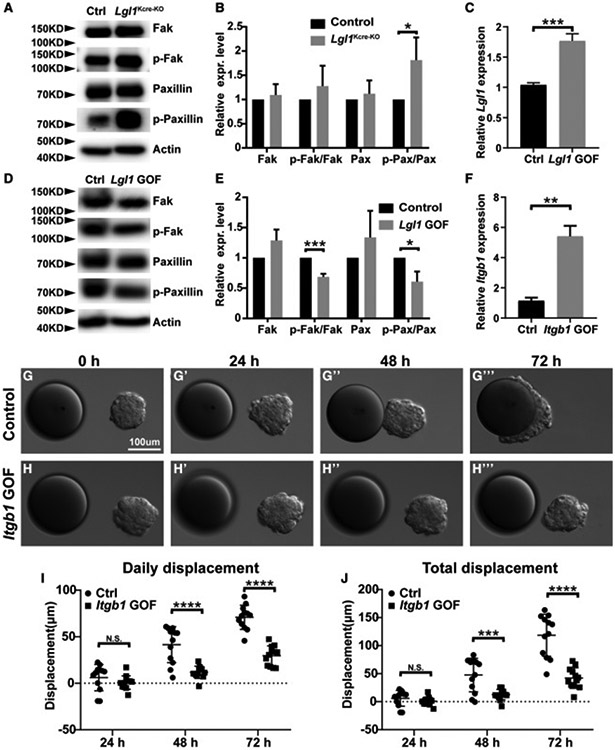
(A and B) Effect of Lgl1 loss on Integrin β1 signaling activation, as measured by the phosphorylation status of Integrin β1 downstream components Fak and Paxillin using western blot analysis. (A) Assays were performed using control and Lgl1 null MECs. (B) Relative levels of active Fak and Paxillin as measured by the ratio of phosphorylated and total forms of Fak and Paxillin.
(C–E) Effect of Lgl1 overexpression on Fak and Paxillin phosphorylation in the HC11 cell line using western blot analysis. (C) Relative levels of Lgl1 overexpression were measured by a qPCR analysis. (D) Assays were performed using control and Lgl1 GOF HC11 cells. (E) Relative levels of active Fak and Paxillin as measured by the ratio of phosphorylated and total forms of Fak and Paxillin.
(F–J) Effect of Itgb1 overexpression on collective migration using the HC11 cell line. (F) Relative levels of Itgb1 overexpression were measured by a qPCR analysis. Time course of control (G–G‴) and Itgb1 GOF (H–H‴) HC11 cell aggregate migration toward beads pre-soaked in FGF10. Quantification of the daily (I) and total (J) displacement of HC11 cells with and without Itgb1 overexpression.
Data are mean ± SD. Statistical analysis was performed using unpaired Student’sttest. *p < 0.05; **p < 0.01; ***p < 0.001; ****p< 0.0001; N.S., not significant. See also Figure S5.
Thus, contrary to our prediction, Lgl1 loss caused an increase rather than a reduction of Integrin β1 signaling activities in the Lgl1Kcre-KO mammary glands. Therefore, we wanted to validate the loss-of-function (LOF) data by performing a gain-of-function (GOF) analysis to examine the effect of Lgl1 overexpression on Integrin β1 signaling activities. To achieve Lgl1 overexpression, we used a modified CRISPR technique in which a deactivated Cas9 protein is fused to the transcriptional activator VP64 (Konermann et al., 2015). Target specificity was achieved by using the single guide RNA sequence from the Lgl1 promoter region (see STAR Methods and Figure S5A). After transfecting HC11 cells, we found that compared with the control samples, HC11 cells infected with the Lgl1 GOF lentivirus had a ~70% increase of Lgl1 mRNA expression (Figure 5C). HC11 cells overexpressing Lgl1 had a 30% and 40% reduction in the phosphorylation levels of Fak and Paxillin, respectively, when compared with control HC11 cells (Figures 5D and 5E).
Together, the results from both the LOF and GOF assays demonstrate that Lgl1 inhibits Integrin β1 signaling activities in the mouse mammary gland. Based on these results, we predicted that an increase in Integrin β1 signaling activities would recapitulate the LOF phenotype of Lgl1 function in the 3D in vitro migration assay. To achieve an increase in Integrin β1 signaling activities, we created an HC11 cell line overexpressing Itgb1 using the above CRISPR-based GOF approach (Figure S5A). Using qPCR analysis, we found that Itgb1 mRNA expression levels had a ~4-fold increase in HC11 cells overexpressing Itgb1 when compared with control cells (Figure 5F).
Next, we aggregated control and Itgb1-GOF HC11 cells overnight and subjected them to the FGF10-based 3D migration assay. We found that control HC11 aggregates migrated toward heparin sulfate beads pre-soaked in FGF10. Interestingly, unlike organoid epithelium, which showed a delayed migration due to epithelial reconstruction and stratification (Figures 3D, 3F, and 3G) (Lu et al., 2020), control HC11 aggregates did not exhibit such a delay and started to directionally migrate by 24 h (Figures 5G-5G″, 5I, and 5J), and reached the FGF10 beads by 48 h (Figures 5G″, 5G‴, 5I, and 5J). Itgb1-GOF HC11 aggregates also migrated toward FGF10 beads. However, they migrated much slower, at only ~30% the speed of the control aggregates, and did not touch the bead even by 72 h (Figures 5H-5J).
Taken together, contrary to our prediction, Lgl1 loss resulted in an increase rather than a decrease in Integrin β1 signaling. Furthermore, we found that overactive Integrin β1 signaling, whether caused by Lgl1 loss or Integrin β1 overexpression, was just as detrimental as loss of Integrin β1 signaling to directional migration of vertebrate epithelium.
DISCUSSION
We initially hypothesized that Lgl1 was a tumor-suppressor gene that regulated epithelial polarity in the mammary gland. Contrary to our hypothesis, however, we found that mammary glands lacking Lgl1 did not have epithelial overgrowth; rather, they had fewer branches, and epithelial polarity was normal. Lgl1 null epithelium was unable to undergo directional migration, thus suggesting that migration is not essential for vertebrate branching. We found that LGL1 was bound to Integrin β1 and inhibited its downstream signaling. Consistent with the Lgl1 null phenotype, Integrin β1 overexpression blocked epithelial migration. Together, our data demonstrate that modulation of Integrin β1 signaling by Lgl1 is essential for directional migration and that epithelial branching in invertebrates and vertebrates is fundamentally different.
Lgl1 modulation of Integrin β1 signaling is essential for epithelial directional migration
Lgl1 null mammary epithelium was unable to undergo directional migration. This, however, was not due to its inability to respond to FGF10 stimulation. We focused on Integrin β1 from the BioID screen because it is well known for its role in migration as a matrix receptor and focal adhesion component. Although we predicted that Lgl1 loss caused downregulation of Integrin β1 signaling, leading to a failure of directional migration, we observed the opposite, with upregulation of Integrin β1 in the mutant epithelium. Consistent with this result, Lgl1 GOF led to reduced Integrin β1 signaling, while Integrin β1 GOF greatly reduced mammary epithelial migration and recapitulated the defect resulted from Lgl1 loss.
It was interesting that Integrin β1 overexpression did not completely block migration as in the Lgl1 LOF phenotype. One possible explanation is that overexpression of Integrin β1 alone was insufficient to bring its downstream signaling to a level comparable with that caused by Lgl1 loss. Alternatively, Lgl1 loss may cause additional defects other than Integrin β1 signaling upregulation that could also be essential for directional migration. Interestingly, a recent study supports our conclusion that Lgl1 loss leads to migration failure as a result of Integrin β1 upregulation. Specifically, Abedrabbo and Ravid (2020) showed that reduction of Lgl1 function by short hairpin RNA knockdown in human breast cancer MDA-MB-231 cells severely reduced collective migration due to defective focal adhesions in mutant cells using the in vitro two-dimensional wound-healing assay.
Taken together, our data and those from the literature support a model in which the modulation of Integrin β1 signaling levels is essential for vertebrate directional migration. Thus, when Integrin β1 signaling levels are too low, for example when ITGB1 or its downstream components are compromised as reported in the literature, directional epithelial migration is unable to occur (Figures 6A and 6B). On the other hand, too much Integrin β1 signaling can also be detrimental to migration, presumably because too many or too strong focal adhesions, which are large macromolecular assemblies containing Integrin, Fak, Paxillin, and other components through which mechanical force and regulatory signals are transmitted between the ECM and an interacting cell, may prevent cells from disrupting old ones and detaching from the matrix, an important step during forward movement. Thus, upregulation of Integrin β1 signaling levels, for example due to Lgl1 loss, can block epithelial migration as well (Figure 6B).
Figure 6. Model of LGL1 function during vertebrate epithelial migration and branching morphogenesis.

(A) Schematic diagram of the developing mammary gland at the post-pubertal stages during which active branching is occurring. The terminal end buds (TEB) develop at the onset of puberty (3 weeks after birth) at the distal tip of each primary duct. Proximal is to the left and distal to the right.
(B) Modulation of Integrin β1 signaling levels is essential for vertebrate directional migration.
(C) Model of the role of directional migration during invertebrate and vertebrate branching.
Distinct mechanisms of epithelial branching in invertebrate and vertebrate systems
Much of what we know regarding the role of directional migration in branching morphogenesis, including epithelial branching, has been based on studies from invertebrate systems, particularly the fly trachea and air sacs (Lu et al., 2006; Affolter et al., 2003; Affolter and Caussinus, 2008; Ghabrial et al., 2003; Metzger and Krasnow, 1999). In both of these systems, branching does not occur if directional migration of the tracheal or air sac epithelium fails to take place (Figure 6C) (Sato and Kornberg, 2002; Cabernard and Affolter, 2005; Cabernard et al., 2004). Together with their numerous similarities at both the cellular and molecular levels, sometimes referred to as “deep homology,” it has been commonly believed that directional migration determines where a new branch is going to grow out and, thus, the branching pattern not only in invertebrate organs but most likely also in vertebrate organs (Davies, 2002; Lu and Werb, 2008; Lu et al., 2006; Lu and Lu, 2021). This dogma has not been vigorously tested regarding vertebrate epithelial branching because, until now, there has been a lack of a mutant vertebrate branching models in which directional migration does not occur.
Our data show that mammary glands lacking Lgl1 had a 26% decrease in branch-point formation when compared with normal. The data thus cannot be explained by the current dogma, which, given that mammary epithelium lacking Lgl1 was unable to directionally migrate, would have predicted that the null glands formed no branches at all rather than being only partially compromised. Our data thus suggest that the role of directional migration in epithelial branching is different in vertebrates and invertebrates: whereas in invertebrate organs it is required to initiate all of the branch points, in vertebrate organs, at least in the mammary gland, it is only required to initiate a minority of the branch points (Figure 6C).
The generation of branch points in the mammary gland has been traditionally thought to take place in two ways via terminal end bud (TEB) bifurcation, where the epithelial tip is split into two, or side branching, where a secondary branch forms from the side of a main branch (Goodwin and Nelson, 2020; Lu et al., 2006; Affolter et al., 2003). However, Scheele et al. (2017) recently showed that branch points almost exclusively form at the TEBs. Thus, the level of contribution that side branching makes toward mammary gland branching is being debated (Myllymaki and Mikkola, 2019). Together, collective migration plays a relatively minor role in where a branch is going to form in vertebrate branching.
Importantly, our data also show that mammary glands lacking Lgl1 had a 20% reduction in ductal penetration when compared with normal. The data suggest that directional migration also plays a minor role in ductal extension in mammary gland branching (Figure 6C). On the surface, this in vivo phenotype contradicts the observation that Lgl1Δ/Δ epithelium underwent normal elongation in the FGF2-based model. However, this was expected considering that we previously demonstrated that FGF2 does not induce directional migration (Lu et al., 2020; Zhang et al., 2014a). Together, these results reinforce the notion that the FGF2- and FGF10-based models are most well suited for studying migration-independent ductal elongation and directional epithelial migration, respectively.
Limitations of the study
Our current work has yet to address the exact mechanism by which Lgl1 inhibits Integrin β1 signaling (Dahan et al., 2012, 2014). Previous studies have linked Lgl1 to vesicular trafficking and regulation of endocytosis (Daynac et al., 2018). It thus will be interesting to examine in future studies whether Lgl1 regulates the half-life of Integrin β1 or other focal adhesion components via endocytosis. Furthermore, the extent to which directional migration may play a role in ductal extension in invertebrate branching remains elusive. An unequivocal answer to this question will require a temporal mutation in which migration fails only after a branch has been initiated in the fly systems. Finally, it remains unclear whether what we observed in the mammary gland is generally true in other vertebrate branched organs. We await future studies to provide definitive answers to these intriguing questions.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Pengfei Lu (lvpf@shanghaitech.edu.cn).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse strains
Mice carrying the Lgl1fl allele were provided by Dr. Valeri Vasioukhin(Klezovitch et al., 2004). Mice carrying the murine mammary tumor virus (MMTV)-Cre transgene D line (Wagner et al., 2001), the R26RmT/mG reporter allele (JAX Mice, #007576) (Muzumdar et al., 2007) and the K14-cre (Tg(KRT14-cre)1Amc, MGI: 2445832) (Dassule et al., 2000) were purchased from the Jackson Laboratory. Offspring from crosses of the various lines were genotyped according to methods in the publications describing the mouse lines. If it is not specifically mentioned, then 5 to 13 weeks old female mice were used to harvest the mammary glands or organoids. All animals were maintained in a specific pathogen free facility and were kept in a standard light/dark cycle (12h/12h) with free access to food and water. Mice were housed and maintained according to regulations from ShanghaiTech University’s Institutional Animal Care and Use Committee (IACUC# 20200713003).
Cell lines
HEK293T cells were cultured in high glucose DMEM (Gibco, 12430062) supplemented with 10% FBS, and penicillin (5 U/mL), and streptomycin (5μg/mL). HC11 cells were cultured in RPMI 1640 (Gibco, # C11875500CP), supplemented with 10% FBS, insulin (5μg/mL), epidermal growth factor (10 ng/mL), and penicillin (5 U/mL) and streptomycin (5μg/mL). All cell lines were tested for the absence of mycoplasma monthly.
METHOD DETAILS
Mammary gland wholemount preparation, photography, and morphometric analysis
The #4 abdominal mammary glands from estrous cycle-matched female mice were harvested and mounted on glass slides. They were fixed in the Carnoy solution overnight at 4°C, followed by staining in Carmine Red. After clearing, mammary glands were photographed on a Zwiss Axio Zoom V16 stereoscope. ImageJ was used to process images and to measure ductal elongation. Ductal elongation was the mean length of the three longest primary epithelial branches in each mammary gland. This was assessed by measuring the lengths of straight lines from the center of the lymph node to the ends of those three branches. The number of branch points per millimeter of the duct was the mean number of branch points on those three longest primary ducts divided by their mean length.
Preparation of mammary gland epithelial cells and adenovirus infection
Donor mammary glands were harvested, minced, and dissociated in buffer [10 mM HEPES buffer, 5% fetal bovine serum (FBS), DMEM/F12, Penicillin-Streptomycin 100 U/ml] containing collagenase (Sigma C5138-1G, 2 mg/mL) for 1 hr at 37°C. Primary epithelial organoids were purified by five repetitions of washes in the dissociation buffer containing no collagenase and collected using a swing-bucket centrifuge at 400 X g. Purified organoids were resuspended in a growth medium (5 μg/mL insulin, 1 μg/mL hydrocortisone, 10 ng/mL EGF, 10% FBS, Penicillin-Streptomycin 100 U/ml, Gentamicin 50 mg/mL in DMEM/F12) and infected overnight with adenovirus-Cre-GFP (He et al., 1998) at a multiplicity of infection of ~25 particles per cell. The next day, organoids were washed several times with PBS and placed in a fresh growth medium. They were cultured for another 24 hr to allow for recovery from infection before further manipulation.
Fluorescence-activated cell sorting (FACS) and quantitative real-time PCR
MECs or HC11 single cells were dissociated and were fluorescently labeled by infection using Ad-Cre-GFP virus, lentivirus, or antibody staining. Antibodies against CD24 (eFluor 450) and CD49f (APC) were used to sort for luminal and basal populations. Sorting was done using an AriaIII system and analysis using the FACalibur system. Data were processed using FlowJo 10 software.
Trizol reagent (Magen, R4801) or RNA extraction kit (Magen, R4012) was used to prepare total RNA from FACS-based luminal, basal, stromal cell partitions or culture cells. Equal amounts of RNA templates were reverse transcribed into cDNAs using HiScript II Q Select RT SuperMix (Vazyme Biotech co.,ltd, R233). Then cDNAs were used in qPCR reactions using ChamQ Universal SYBR qPCR Master Mix (Vazyme Biotech co.,ltd, Q711) with the BIO-RAD CFX Connect Real-Time Systems according to the manufacturer’s protocol. qPCR was performed using the 7500 Fast Real-Time PCR system (Applied Biosystems) and data were normalized to the expression of at least two of the reference genes, including Actb, and Gapdh. Primer sequences were described in Table S1.
Mammosphere and acinar assays
For mammosphere assay, sorted cells were seeded on low-attachment plates [plates treated with poly(2-hydroxyethyl methacrylate) (Sigma)] in phenol red-free DMEM/F12, supplemented with B-27, 100 μg/mL penicillin, 100 U/mL streptomycin, 20 ng/mL EGF, and/ or 20 ng/mL FGF2, and cultured for 7 days. For secondary mammosphere assay, primary mammospheres were collected, trypsinized to obtain a single-cell suspension, and seeded on low-attachment plates at the same density and in the same culture medium as primary mammospheres. For the acinar assay, sorted cells were plated in Matrigel and cultured in DMEM/F12 with 10% (vol/vol) heat-inactivated FBS (Sigma), penicillin, streptomycin, 5 ng/mL EGF, 5 μg/mL insulin, and 1 μg/mL hydrocortisone for 14 days. Mammosphere (or acinus)-forming efficiency was calculated using the following equation: 100 × [number of mammospheres (or acini, respectively) formed/number of cells seeded].
In vitro epithelial branching and migration assays
Either mammary organoids or MEC aggregates were used for branching and migration assays. To aggregate MECs, sorted cells were pelleted, cultured via the “hanging-drop” method whereby a 50μL-drop of growth medium containing single cells was cultured upside-down on the lid of a Petri dish overnight at 37°C. MEC aggregates were then washed in DMEM/F12 to eliminate fetal serum. For branching assay, basal medium (DMEM/F12, ITS, and penicillin-streptomycin) containing growth factors FGF2 (Sigma) were used in the 7–10 day culture (Ewald et al., 2008).
For epithelial migration assay, Heparan sulfate beads (Sigma, #H5263) of ~100–200 μm in sizes were picked and washed in 10μL FGF10 (GenScript, #Z03155-50) solution overnight at 4°C. 15μL (for 24-well plates) or 4μL (for 8-well chamber-slide) of 80% Matrigel was used to coat the plates, which were then heated on a 37°C block for 1 minute. Mammary organoids were put next to beads presoaked in FGF10 at the distance of ~100 μm using a tungsten needle (Figure 1A). The plate was warmed up on a 37°C block for 8 minutes and added 1000μL (for 24 well plates) or 300μL (for 8 well chamber slide) basal organoid culture medium. Samples were cultured in a 37°C incubator with 5% CO2 or transferred to a live imaging microscope for time-lapse imaging.
Immunofluorescence analysis
Mammary glands were harvested and fixed in 4% paraformaldehyde overnight at 4°C. Then they were soaked in 15% sucrose and 30% sucrose prepared in PBS for 12 hours. 10μm frozen sections were cut using a Leica cryostat. Sections were permeabilized with 0.5% Triton X-100 in PBS for 45 minutes. Sections were blocked for 2hr at room temperature (RT) in PBS containing 10% goat serum and 0.2% Tween20, followed by incubation in primary antibodies overnight at 4°C. Primary antibodies used in this study was Zo-1 antibody (Proteintech, #21773-1-AP, 1:100 dilution), Ezrin antibody (Cell Signaling Technology, #3145S, 1:200 dilution), E-cadherin antibody (Innovative Research, #13–1900, 1:200 dilution), Itgb1 antibody (R&D Systems, #MAB2405, 1:200 dilution), aPKC antibody (Abcam, #ab59364, 1:200 dilution), Par3 antibody (Millipore, #07–330, 1:300 dilution). Confocal microscopy was performed on a Zeiss LSM 800 Confocal.
A CRISPRa-based gain of Lgl1 or Itgbl function
Lgl1 or Itgbl gain-of-function was based on a modified CRISPR, in which a deactivated Cas9 protein was used (Konermann et al., 2015). The website (http://www.e-crisp.org/E-CRISP/designcrispr.html) was used to design sgRNA guide sequences (Lgl1, 5′-GGGACTTGTAGTCCGAAGGT-3′; Itgb1, 5′-GTACCGCGCTGAACCACCGA-3′). Corresponding DNA fragments were cloned into lentiSAM v2 (mCherry) (Addgene # 92062).
Viral production was carried out using calcium phosphate–mediated transfection of HEK293T cells. The virus was concentrated by ultracentrifugation and added to cells with polybrene. Stably transduced cells were selected by FACS.
Analysis of Lgl1 mRNA expression in breast cancer and survival probability
Data on Lgl1 mRNA expression in breast cancer subtypes were based on The Cancer Genome Atlas (TCGA) Breast Cancer (BRCA) datasets, available at UCSC Xena (https://xenabrowser.net/). PAM50 was used to analyze 956 cases from “normal” and “cancer” tissues. Survival probability analysis was based on data on 3981 breast cancer patients. Kaplan–Meier Plotter (http://kmplot.com/analysis/index.php?p=service&cancer=breast) was used for the analysis.
Proximity-dependent biotin identification (BioID) assay
Neuro-2a (N2a) cells were used for the BioID screen using a standard protocol (Sears et al., 2019). Mass Spectrometry Data were analyzed using the Perseus (version 1.6). Student t-test was used to detect proteins of significant differences between the control and experimental samples. Those with at least a 1.5 fold difference and p< 0.05 were considered to be “real” partners of LGL1. For KEGG (Kyoto Encyclopedia of Genes and Genomes) and GO (Gene Ontology) analyses, the GSEA website (http://www.gsea-msigdb.org/gsea/msigdb) was used to enrich genes of interests.
Western blotting, co-immuno-precipitation (Co-IP), and co-localization assays
Organoid or HC11 cell preparations were lysed in RIPA buffer (EpiZyme, PC102) with protease inhibitor (Bimake, B14001) and phosphatase inhibitor (Bimake, B15001). Samples were cleared with centrifugation at 15,000 rpm at 4°C for 15 min. Protein was resolved by SDS/PAGE under reducing conditions in Hepes-Tris buffer on a gradient gel 4–20% (Meilunbio, MA0250). Then, Proteins were electrically transferred in a wet-tank to a PVDF membrane. After blocking with 5% BSA (sigma#WXBC3116V), target proteins were visualized using FAK antibody (Cell Signaling Technology, #3285S, 1:1000 dilution), Paxillin antibody (BD Biosciences, #610051, 1:1000 dilution), Phospho-FAK (Tyr397, Invitrogen, #44-624G, 1:1000 dilution), Phospho-Paxillin (Tyr118, Cell Signaling Technology, #2541S, 1:1000 dilution), Actin (Sigma, #A5441, 1:1000 dilution). After the primary antibody was incubated and extensively washed, the membrane was reacted with the HRP-conjugated secondary antibody at room temperature for 2 h. The reactive bands were developed by ECL kit (EpiZyme, SQ202) and tested with Amersham Imager 680. For quantification of protein expression, band density was measured using ImageJ software.
For Co-IP assays, the 293T line of cells expressing ITGB1-Flag and HA-LGL1 were treated with 10μM MG132 (Meilunbio, MB5137) for 6 h before harvest. The cells were lysed in NP-40 buffer (50mM Tris, 150mM NaCl, 1% NP-40, with protease inhibitor). Samples were co-incubated with Anti-Flag magnetic beads (Bimake, B26101) O/N at 4°C. Beads were then washed three times with washing buffer before co-incubated with 40 μL 2 X sample loading buffer to harvest FLAG-tagged proteins before they were subjected to Western Blotting analysis. The antibodies used were anti-Flag (Abcam, #ab49763, 1:1000 dilution), anti-HA (Cell Signaling Technology, #2999S, 1:1000 dilution).
HC11 cells over-expressing both mCherry-LGL1 and EGFP-ITGB1 were cultured in the differentiation medium (DMEM/F12 containing 50U/ml P/S, 5μg/ml Insulin, 50μg/ml gentamycin, 1 μg/ml hydrocortisone, and 3mg/ml prolactin) for four days, and were then visualized on a Zeiss LSM980 confocal. ImageJ was used for analysis and statistical analysis of colocalization coefficients were calculated using the JACoP plugin.
QUANTIFICATION AND STATISTICAL ANALYSIS
Sample size for each figure is denoted in the figure legends. The statistical significance between groups were analyzed by GraphPad Prism. Statistical significance between conditions was assessed by two-tailed unpaired Student’s t tests. All error bars represent SD, and significance is denoted as *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001. n.s. denotes not significant.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| ZO-1 Polyclonal antibody | Proteintech | Cat#21773-1-AP; RRID: AB_10733242 |
| Ezrin Antibody | Cell Signaling Technology | Cat#3145S; RRID: AB_2100309 |
| Anti-PKC zeta antibody | Abcam | Cat#ab59364; RRID: AB_944858 |
| FAK Antibody | Cell Signaling Technology | Cat#3285S; RRID: AB_2269034 |
| Purified Mouse Anti-Paxillin Clone 349/Paxillin (RUO) | BD Biosciences | Cat#610051; RRID: AB_397463 |
| Phospho-FAK (Tyr397) Polyclonal Antibody | Invitrogen | Cat#44-624G; RRID: AB_2533701 |
| Phospho-Paxillin (Tyr118) Antibody | Cell Signaling Technology | Cat#2541S; RRID: AB_2174466 |
| Anti-E-CADHERIN Monoclonal Antibody, Unconjugated, Clone ECCD-2 | Innovative Research | Cat#13-1900; RRID: AB_86571 |
| Mouse Integrin beta 1/CD29 Antibody | R&D Systems | Cat#MAB2405; RRID: AB_2249264 |
| Anti-Partitioning-defective 3 Antibody | Millipore | Cat#07-330; RRID: AB_2101325 |
| HA-Tag (6E2) Mouse mAb (HRP Conjugate) | Cell Signaling Technology | Cat#2999S; RRID: AB_1264166 |
| HRP Anti-DDDDK tag (Binds to FLAG® tag sequence) antibody [M2] | Abcam | Cat#ab49763; RRID: AB_869428 |
| Bacterial and virus strains | ||
| Adeno-Cre-GFP | Obio Technology (Shanghai) Corp., Ltd. | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Collagenase from Clostridium histolyticum | Sigma-Aldrich | Cat#C5138 |
| EGF | novoprotein | Cat#C029 |
| EZ-LINK NHS-BIOTIN | Thermo Fisher Scientific | Cat#20217 |
| FGF2 | GenScript | Cat#Z03116 |
| FGF10 | GenScript | Cat#Z03155 |
| ITS Liquid Media Supplement (100×) | Sigma-Aldrich | Cat#I3146 |
| DAPI | Sigma-Aldrich | Cat#D9542; CAS#28718-90-3 |
| Deoxyribonuclease I | Sigma-Aldrich | Cat#D4527 |
| Growth Factor Reduced (GFR) Basement Membrane Matrix | Corning | Cat#354230 |
| Experimental models: Cell lines | ||
| HC11 Mammary Epithelium | ATCC | Cat#CRL-3062; RRID: CVCL_0288 |
| HEK293T | Kindly provided by Dr. Jianlong Sun | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: Tg(MMTV-cre)4Mam/J | Jackson Laboratory | JAX: 003553; RRID: IMSR_JAX:003553 |
| Mouse: B6.129(Cg)-Gt(ROSA) 26Sortm4(ACTB-tdTomato,-EGFP)Luo/J | Jackson Laboratory | JAX: 007676; RRID: IMSR_JAX:007676 |
| Mouse: Tg(KRT14-cre)1Amc/J | Jackson Laboratory | JAX: 004782; RRID: IMSR_JAX:004782 |
| Mouse: Lgl1fl/fl | Kindly provided by Dr. Valeri Vasioukhin | N/A |
| Oligonucleotides | ||
| qRT-PCR primers | This paper | Table S1 |
| Recombinant DNA | ||
| pcDNA3.1-Itgb1-Flag | This paper | N/A |
| pcDNA3.1-HA-Lgl1 | This paper | N/A |
| pCDH-Itgb1-EGFP | This paper | N/A |
| pCDH-Lgl1-mCherry | This paper | N/A |
| lentiSAM v2 (mCherry)-Lgl1 | This paper | N/A |
| lentiSAM v2 (mCherry)-Itgb1 | This paper | N/A |
| lentiMPH v2 | Addgene | Cat#89308 |
| Software and algorithms | ||
| ImageJ | https://imagej.net/Welcome | RRID: SCR_003070 |
| GraphPad Prism | https://www.graphpad.com/ | RRID: SCR_002798 |
| Perseus | https://maxquant.net/perseus/ | RRID: SCR_015753 |
Highlights.
Lgl1 promotes mammary gland branching morphogenesis
Lgl1 is required for collective migration but not epithelial polarity
LGL1 binds to Integrin β1 and inhibits its downstream signaling
Branching mechanisms are distinct between mammary gland and invertebrate epithelia
ACKNOWLEDGMENTS
We thank Dr. Xiaohong Zhang for her assistance in the in vitro branching, mammosphere, migration, and qPCR assays; Tiezhu Shi for conducting the BioID experiment; Zichao Zhang and Ruolan Deng for providing ITGB1-tagged HC11 cells. We thank the Mouse Core Facility at the National Institute for Protein Science Center in Shanghai. We are greatly indebted for the invaluable and timely technical support from the Molecular Imaging Core Facility, the Molecular and Cell Biology Core Facility, and the Bio-Mass Spectrometry Core Facility in the School of Life Science and Technology at ShanghaiTech University. This work was supported by grants from the Ministry of Science and Technology of China (2017YFA0103502) and the National Natural Science Foundation of China (31671494) and a startup fund from ShanghaiTech University (to P.L.); and National Institutes of Health grants R35-DE026602 (to O.D.K.) and R01CA164746 (to C.K.P.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110375.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abedrabbo M, and Ravid S (2020). Scribble, Lgl1, and myosin II form a complex in vivo to promote directed cell migration. Mol. Biol. Cell 31, 2234–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Affolter M, Bellusci S, Itoh N, Shilo B, Thiery JP, and Werb Z (2003). Tube or not tube: remodeling epithelial tissues by branching morphogenesis. Dev. Cell 4, 11–18. [DOI] [PubMed] [Google Scholar]
- Affolter M, and Caussinus E (2008). Tracheal branching morphogenesis in Drosophila: new insights into cell behaviour and organ architecture. Development 135, 2055–2064. [DOI] [PubMed] [Google Scholar]
- Aggeler J, Ward J, Blackie LM, Barcellos-Hoff MH, Streuli CH, and Bissell MJ (1991). Cytodifferentiation of mouse mammary epithelial cells cultured on a reconstituted basement membrane reveals striking similarities to development in vivo. J. Cell Sci 99, 407–417. [DOI] [PubMed] [Google Scholar]
- Akhtar N, and Streuli CH (2006). Rac1 links integrin-mediated adhesion to the control of lactational differentiation in mammary epithelia. J. Cell Biol 173, 781–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhtar N, and Streuli CH (2013). An integrin-ILK-microtubule network orients cell polarity and lumen formation in glandular epithelium. Nat. Cell Biol 15, 17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albertson R, and Doe CQ (2003). Dlg, Scrib and Lgl regulate neuroblast cell size and mitotic spindle asymmetry. Nat. Cell Biol 5, 166–170. [DOI] [PubMed] [Google Scholar]
- Atwood SX, and Prehoda KE (2009). aPKC phosphorylates Miranda to polarize fate determinants during neuroblast asymmetric cell division. Curr. Biol 19, 723–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilder D (2004). Epithelial polarity and proliferation control: links from the Drosophila neoplastic tumor suppressors. Genes Dev. 18, 1909–1925. [DOI] [PubMed] [Google Scholar]
- Bilder D, Li M, and Perrimon N (2000). Cooperative regulation of cell polarity and growth by Drosophila tumor suppressors. Science 289, 113–116. [DOI] [PubMed] [Google Scholar]
- Brisken C, and Ataca D (2015). Endocrine hormones and local signals during the development of the mouse mammary gland. Wiley Interdiscip. Rev. Dev. Biol 4, 181–195. [DOI] [PubMed] [Google Scholar]
- Bryant DM, and Mostov KE (2008). From cells to organs: building polarized tissue. Nat. Rev. Mol. Cell Biol 9, 887–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabernard C, and Affolter M (2005). Distinct roles for two receptor tyrosine kinases in epithelial branching morphogenesis in Drosophila. Dev. Cell 9, 831–842. [DOI] [PubMed] [Google Scholar]
- Cabernard C, Neumann M, and Affolter M (2004). Cellular and molecular mechanisms involved in branching morphogenesis of the Drosophila tracheal system. J. Appl. Physiol 97, 2347–2353. [DOI] [PubMed] [Google Scholar]
- Choi J, Troyanovsky RB, Indra I, Mitchell BJ, and Troyanovsky SM (2019). Scribble, Erbin, and Lano redundantly regulate epithelial polarity and apical adhesion complex. J. Cell Biol 218, 2277–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahan I, Petrov D, Cohen-Kfir E, and Ravid S (2014). The tumor suppressor Lgl1 forms discrete complexes with NMII-A and Par6alpha-aPKCzeta that are affected by Lgl1 phosphorylation. J. Cell Sci 127, 295–304. [DOI] [PubMed] [Google Scholar]
- Dahan I, Yearim A, Touboul Y, and Ravid S (2012). The tumor suppressor Lgl1 regulates NMII-A cellular distribution and focal adhesion morphology to optimize cell migration. Mol. Biol. Cell 23, 591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dassule HR, Lewis P, Bei M, Maas R, and Mcmahon AP (2000). Sonic hedgehog regulates growth and morphogenesis of the tooth. Development 127, 4775–4785. [DOI] [PubMed] [Google Scholar]
- Davies JA (2002). Do different branching epithelia use a conserved developmental mechanism? Bioessays 24, 937–948. [DOI] [PubMed] [Google Scholar]
- Daynac M, Chouchane M, Collins HY, Murphy NE, Andor N, Niu J, Fancy SPJ, Stallcup WB, and Petritsch CK (2018). Lgl1 controls NG2 endocytic pathway to regulate oligodendrocyte differentiation and asymmetric cell division and gliomagenesis. Nat. Commun 9, 2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dow LE, Elsum IA, King CL, Kinross KM, Richardson HE, and Humbert PO (2008). Loss of human Scribble cooperates with H-Ras to promote cell invasion through deregulation of MAPK signalling. Oncogene 27, 5988–6001. [DOI] [PubMed] [Google Scholar]
- Ewald AJ, Brenot A, Duong M, Chan BS, and Werb Z (2008). Collective epithelial migration and cell rearrangements drive mammary branching morphogenesis. Dev. Cell 14, 570–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghabrial A, Luschnig S, Metzstein MM, and Krasnow MA (2003). Branching morphogenesis of the Drosophila tracheal system. Annu. Rev. Cell Dev. Biol 19, 623–647. [DOI] [PubMed] [Google Scholar]
- Goldstein B, and Macara IG (2007). The PAR proteins: fundamental players in animal cell polarization. Dev. Cell 13, 609–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin K, and Nelson CM (2020). Branching morphogenesis. Development 147, dev184499. [DOI] [PubMed] [Google Scholar]
- Grifoni D, Garoia F, Schimanski CC, Schmitz G, Laurenti E, Galle PR, Pession A, Cavicchi S, and Strand D (2004). The human protein Hugl-1 substitutes for Drosophila lethal giant larvae tumour suppressor function in vivo. Oncogene 23, 8688–8694. [DOI] [PubMed] [Google Scholar]
- He TC, Zhou S, Da Costa LT, Yu J, Kinzler KW, and Vogelstein B (1998). A simplified system for generating recombinant adenoviruses. Proc. Natl. Acad. Sci. USA 95, 2509–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenlocher A, and Horwitz AR (2011). Integrins in cell migration. Cold Spring Harb. Perspect. Biol 3, a005074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justice N, Roegiers F, Jan LY, and Jan YN (2003). Lethal giant larvae acts together with numb in notch inhibition and cell fate specification in the Drosophila adult sensory organ precursor lineage. Curr. Biol 13, 778–783. [DOI] [PubMed] [Google Scholar]
- Klezovitch O, Fernandez TE, Tapscott SJ, and Vasioukhin V (2004). Loss of cell polarity causes severe brain dysplasia in Lgl1 knockout mice. Genes Dev. 18, 559–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, et al. (2015). Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517, 583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, and Vasioukhin V (2008). Cell polarity and cancer—cell and tissue polarity as a non-canonical tumor suppressor. J. Cell Sci 121, 1141–1150. [DOI] [PubMed] [Google Scholar]
- Lu P, and Lu Y (2021). Born to run? Diverse modes of epithelial migration. Front. Cell Dev. Biol 9, 704939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P, Sternlicht MD, and Werb Z (2006). Comparative mechanisms of branching morphogenesis in diverse systems. J. Mammary Gland Biol. Neoplasia 11,213–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P, and Werb Z (2008). Patterning mechanisms of branched organs. Science 322,1506–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Deng R, You H, and Lu P (2021). 3D in vitro culture system to study collective migration in mammary organoid epithelium. STAR Protoc. 2, 100778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Deng R, You H, Xu Y, Antos C, Sun J, Klein OD, and Lu P (2020). Asymmetric stratification-induced polarity loss and coordinated individual cell movements drive directional migration of vertebrate epithelium. Cell Rep. 33, 108246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macara IG, and Mili S (2008). Polarity and differential inheritance—universal attributes of life? Cell 135, 801–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mccaffrey LM, and Macara IG (2009a). The Par3/aPKC interaction is essential for end bud remodeling and progenitor differentiation during mammary gland morphogenesis. Genes Dev. 23, 1450–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mccaffrey LM, and Macara IG (2009b). Widely conserved signaling pathways in the establishment of cell polarity. Cold Spring Harb. Perspect. Biol 1, a001370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellman I, and Nelson WJ (2008). Coordinated protein sorting, targeting and distribution in polarized cells. Nat. Rev. Mol. Cell Biol 9, 833–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger RJ, and Krasnow MA (1999). Genetic control of branching morphogenesis. Science 284, 1635–1639. [DOI] [PubMed] [Google Scholar]
- Muzumdar M, Tasic B, Miyamichi K, Li L, and Luo LJG (2007).A global double-fluorescent Cre reporter mouse. Genesis 45, 593–605. [DOI] [PubMed] [Google Scholar]
- Myllymaki SM, and Mikkola ML (2019). Inductive signals in branching morphogenesis—lessons from mammary and salivary glands. Curr. Opin. Cell Biol 61,72–78. [DOI] [PubMed] [Google Scholar]
- Ohshiro T, Yagami T, Zhang C, and Matsuzaki F (2000). Role of cortical tumour-suppressor proteins in asymmetric division of Drosophila neuroblast. Nature 408, 593–596. [DOI] [PubMed] [Google Scholar]
- Pagliarini RA, and Xu T (2003).A genetic screen in Drosophila for metastatic behavior. Science 302, 1227–1231. [DOI] [PubMed] [Google Scholar]
- Persa OD, and Niessen CM (2019). Epithelial polarity limits EMT. Nat. Cell Biol 21, 299–300. [DOI] [PubMed] [Google Scholar]
- Roignot J, Peng X, and Mostov K (2013). Polarity in mammalian epithelial morphogenesis. Cold Spring Harb. Perspect. Biol 5, a013789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolls MM, Albertson R, Shih HP, Lee CY, and Doe CQ (2003). Drosophila aPKC regulates cell polarity and cell proliferation in neuroblasts and epithelia. J. Cell Biol 163, 1089–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russ A, Louderbough JM, Zarnescu D, and Schroeder JA (2012). Hugl1 and Hugl2 in mammary epithelial cells: polarity, proliferation, and differentiation. PLoS One 7, e47734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito Y, Li L, Coyaud E, Luna A, Sander C, Raught B, Asara JM, Brown M, and Muthuswamy SK (2019). LLGL2 rescues nutrient stress by promoting leucine uptake in ER(+) breast cancer. Nature 569, 275–279. [DOI] [PubMed] [Google Scholar]
- Sato M, and Kornberg TB (2002). FGF is an essential mitogen and chemoattractant for the air sacs of the Drosophila tracheal system. Dev. Cell 3, 195–207. [DOI] [PubMed] [Google Scholar]
- Scheele CL, Hannezo E, Muraro MJ, Zomer A, Langedijk NS, Van Oudenaarden A, Simons BD, and Van Rheenen J (2017). Identity and dynamics of mammary stem cells during branching morphogenesis. Nature 542, 313–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schluter MA, Pfarr CS, Pieczynski J, Whiteman EL, Hurd TW, Fan S, Liu CJ, and Margolis B (2009). Trafficking of Crumbs3 during cytokinesis is crucial for lumen formation. Mol. Biol. Cell 20, 4652–4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A, and Peifer M (2020). Scribble and Dlg organize a protection racket to ensure apical-basal polarity. Proc Natl Acad Sci U S A 117, 13188–13190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears RM, May DG, and Roux KJ (2019). BioID as a tool for protein-proximity labeling in living cells. Methods Mol. Biol 2012, 299–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siller KH, and Doe CQ (2009). Spindle orientation during asymmetric cell division. Nat. Cell Biol 11, 365–374. [DOI] [PubMed] [Google Scholar]
- Sripathy S, Lee M, and Vasioukhin V (2011). Mammalian Llgl2 is necessary for proper branching morphogenesis during placental development. Mol. Cell Biol 31,2920–2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Johnston D, and Ahringer J (2010). Cell polarity in eggs and epithelia: parallels and diversity. Cell 141, 757–774. [DOI] [PubMed] [Google Scholar]
- Tanentzapf G, and Tepass U (2003). Interactions between the crumbs, lethal giant larvae and bazooka pathways in epithelial polarization. Nat. Cell Biol 5, 46–52. [DOI] [PubMed] [Google Scholar]
- Vasioukhin V (2006). Lethal giant puzzle of Lgl. Dev. Neurosci 28, 13–24. [DOI] [PubMed] [Google Scholar]
- Wagner KU, Mcallister K, Ward T, Davis B, Wiseman R, and Hennighausen L (2001). Spatial and temporal expression of the Cre gene under the control of the MMTV-LTR in different lines of transgenic mice. Transgenic Res. 10, 545–553. [DOI] [PubMed] [Google Scholar]
- Watson CJ, and Khaled WT (2020). Mammary development in the embryo and adult: new insights into the journey of morphogenesis and commitment. Development 147, dev169862. [DOI] [PubMed] [Google Scholar]
- Wirtz-Peitz F, and Knoblich JA (2006). Lethal giant larvae take on a life of their own. Trends Cell Biol. 16, 234–241. [DOI] [PubMed] [Google Scholar]
- Xue B, Krishnamurthy K, Allred DC, and Muthuswamy SK (2013). Loss of Par3 promotes breast cancer metastasis by compromising cell-cell cohesion. Nat. Cell Biol 15, 189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan L, Rosenberg A, Bergami KC, Yu M, Xuan Z, Jaffe AB, Allred C, and Muthuswamy SK (2008). Deregulation of scribble promotes mammary tumorigenesis and reveals a role for cell polarity in carcinoma. Cell 135, 865–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Lee AV, and Rosen JM (2017). The cellular origin and evolution of breast cancer. Cold Spring Harb. Perspect. Med 7, a027128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Martinez D, Koledova Z, Qiao G, Streuli CH, and Lu P (2014a). FGF ligands of the postnatal mammary stroma regulate distinct aspects of epithelial morphogenesis. Development 141, 3352–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Qiao G, and Lu P (2014b). Modulation of fibroblast growth factor signaling is essential for mammary epithelial morphogenesis. PLoS One 9, e92735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao G, Gong L, Su D, Jin Y, Guo C, Yue M, Yao S, Qin Z, Ye Y, Tang Y, et al. (2019). Cullin5 deficiency promotes small-cell lung cancer metastasis by stabilizing integrin beta1. J. Clin. Invest 129, 972–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.