

Abstract
Motivation
Taxonomic classification of 16S ribosomal RNA gene amplicon is an efficient and economic approach in microbiome analysis. 16S rRNA sequence databases like SILVA, RDP, EzBioCloud and HOMD used in downstream bioinformatic pipelines have limitations on either the sequence redundancy or the delay on new sequence recruitment. To improve the 16S rRNA gene-based taxonomic classification, we merged these widely used databases and a collection of novel sequences systemically into an integrated resource.
Results
MetaSquare version 1.0 is an integrated 16S rRNA sequence database. It is composed of more than 6 million sequences and improves taxonomic classification resolution on both long-read and short-read methods.
Availability and implementation
Accessible at https://hub.docker.com/r/lsbnb/metasquare_db and https://github.com/lsbnb/MetaSquare
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Metagenomics, the collective view of the mass genome of microbes in specified habitats, widely impacts our knowledge about all kinds of biological processes in recent decades. Researchers discover microbes for different purposes and wish to know the composition and contributions of these species. The cost of whole-genome shotgun metagenomics analysis has decreased. Resolving microbiome composition via 16S ribosomal RNA gene amplicon sequencing remains a mainstream strategy for its stable performance and cost efficiency. With the high-throughput next-generation sequencing, an exhausting list of species can be found by bioinformatic pipelines.
Taxonomic classification is a crucial component of microbiome analysis. Bioinformatic pipelines like QIIME 2 (Bolyen et al., 2019) and mothur (Schloss, 2020) rely on 16S rRNA sequence databases for conducting sequence-to-taxon matches. One of the widely used rRNA gene sequence databases, SILVA (Quast et al., 2013), contains ∼9 million ribosomal RNA sequences from bacteria, archaea and some eukarya. Because of the complexity of data sources, sequence duplicates and uneven coverage of clades in these data depository had been argued (Agnihotry et al., 2020). Besides, considerable efforts are required for maintaining the database up to date. Greengenes, another widely used database (DeSantis et al., 2006) with rich taxonomic annotations, was not updated since 2013. The RDP with about 3 million rRNA sequences (Cole et al., 2014) was also stopped updating in 2016. Furthermore, some recent metagenome approaches may reveal new microbe sequences but are delayed on the database due to the curation schedules. For example, the EzBioCloud 16S rRNA gene database, derived from microbe genomic assemblies, contains new bacteria, archaea and eukarya (Yoon et al., 2017). The HOMD is a specified 16S rRNA gene database built for exploring unique taxa in the oral microbiome (Escapa et al., 2020). A database agglomeration work, 16S-UDb, had been presented (Agnihotry et al., 2020). In this work, unified full-length, fully annotated 16S rRNA sequences were collected. This dataset could meet the requirement for conducting 16S rRNA amplicon analyses in various designs, while the recruited taxon number greatly reduced for sequence length constrain.
To improve the resolution of taxonomy analysis, we attempted a data collecting process to build an updated non-redundant 16S rRNA database MetaSquare. This database meets the need for 16S rRNA classification on both long-read and short-read methods.
2 Materials and methods
We adopted the SILVA database (version 138.1) as the starting set for its greatest coverage of sequence entries and its continuing maintenance and agglomerated other entries to form the final dataset. Firstly, we reformatted the sequencing taxonomy assignment of all datasets to comply with Greengenes’ format. Next, we appended the Greengenes (version 13.5) set to the starting set except for those entries that were identical or substrings to an existing entry; RDP (version 11.5), EzBioCloud (visited on 2020.02) and HOMD (version 15.2) were appended in the same criteria. We further recruited 516 sequences of 16S rRNA gene from novel genomes assemblies reported (Pasolli et al., 2019). Sequence duplication was identified using mothur align.seqs on each database appending process. Next, we filtered sequence duplicates from the approximate merged set according to the annotation context, viz. We picked the most detailed taxonomic annotations and preferred entries from the latest renewed database. Finally, the eukaryote sequences were excluded. We collected sequences that met these criteria: (i) 5 or fewer ambiguous bases, (ii) 8 or fewer homopolymers and (iii) longer than 600 bps to ensure the usability for long 16S rRNA amplicon taxonomic classification pipelines. The database construction workflow is in Supplementary Figures S1 and S2.
Two analyses were conducted for database performance: QIIME 2 on a classical short-read/16S rRNA gene amplicon with the V3–V4 amplicon dataset published by NCBI BioProject PRJNA715083 (Kameoka et al., 2021) and Kraken 2 on long-read/16S rRNA gene near-full length amplicon with datasets from PRJDB9744, V1–V9 amplicon (Matsuo et al., 2021) and PRJNA637202, V3–V9 amplicon (Angell et al., 2020). We compared the taxonomic classification output of QIIME 2 with MetaSquare (this study), SILVA, Greengenes and 16-UDb. For 16S rRNA gene V3–V4 region amplicon analyses, the V3–V4 region of 16S rRNA gene sequences were extracted using the V-Xtractor software tool (Hartmann et al., 2010). The benchmarking dataset was listed in Supplementary Table S1 and the workflow for these analyses in Supplementary Figure S1.
3 Results
MetaSquare is composed of a FASTA file and an annotation taxonomy file complied to Greengenes style; 6 449 552 sequences (archaea: 260 555 entries, bacteria: 6 188 997 entries, version 1.0). The composition of MetaSquare by the source is presented in Supplementary Figure S3.
As shown in Figure 1, Supplementary Table S2 and Supplementary Figure S4, MetaSquare outperformed the other three rRNA databases in terms of identified taxon numbers in the 16S rRNA amplicon analysis. Compared with 16-UDb, MetaSquare helps identify much more genera (436 versus 237) on the short-read microbiome dataset (Supplementary Table S1). We also noticed very few unclassified sequences in QIIME 2 + 16-UDb and QIIME 2+Greengenes.
Fig. 1.
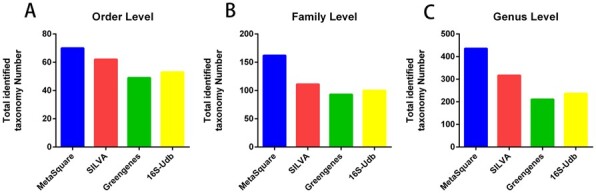
Taxonomic classification result of MetaSquare and competing databases through QIIME 2 pipeline. We counted the non-redundant taxons identified in 16S rRNA gene V3–V4 amplicon (Kameoka et al., 2021) (NCBI PRJNA715083)
Performance of using MetaSquare for long-read 16S rRNA gene amplicon taxonomic classification was accessed by Kraken 2. MetaSquare can help to identify considerably more taxonomic classification genera than the other databases (Supplementary Fig. S5). Details on the results as mentioned above are available in the Supplementary Information.
4 Conclusion
We integrated essential databases to build MetaSquare for microbiome composition profiling based on 16S rRNA gene sequencing data. Overall, MetaSquare included widely used 16S rRNA gene databases with limited data redundancy. Furthermore, it includes novel sequences to increase database coverage. Presently, the update of MetaSquare is scheduled as a biannually semi-automatic process.
Funding
This project was funded by the grants MOST 108-2321-B-037 -001 and 110-2314-B-001 -006 from the Ministry of Science and Technology, Taiwan, and AS-GCS-109-07 from Academia Sinica, Taiwan, to financially support this research and publication.
Conflict of Interest: none declared.
Supplementary Material
Contributor Information
Chun-Chieh Liao, Institute of Information Science, Academia Sinica, 115 Taipei, Taiwan.
Po-Ying Fu, Washington University School of Medicine, St. Louis, MO 63110, USA.
Chih-Wei Huang, Institute of Information Science, Academia Sinica, 115 Taipei, Taiwan.
Chia-Hsien Chuang, Institute of Information Science, Academia Sinica, 115 Taipei, Taiwan.
Yun Yen, TMU Research Center of Cancer Translational Medicine, Taipei Medical University, 110 Taipei, Taiwan.
Chung-Yen Lin, Institute of Information Science, Academia Sinica, 115 Taipei, Taiwan.
Shu-Hwa Chen, TMU Research Center of Cancer Translational Medicine, Taipei Medical University, 110 Taipei, Taiwan.
References
- Agnihotry S. et al. (2020) Construction & assessment of a unified curated reference database for improving the taxonomic classification of bacteria using 16S rRNA sequence data. Indian J. Med. Res., 151, 93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angell I.L. et al. (2020) De novo species identification using 16S rRNA gene nanopore sequencing. PeerJ, 8, e10029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolyen E. et al. (2019) Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol., 37, 852–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole J.R. et al. (2014) Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res., 42, D633–D642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis T.Z. et al. (2006) Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol., 72, 5069–5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escapa I.F. et al. (2020) Construction of habitat-specific training sets to achieve species-level assignment in 16S rRNA gene datasets. Microbiome, 8, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann,M. et al. (2010) V-Xtractor: An open-source, high-throughput software tool to identify and extract hypervariable regions of small subunit (16S/18S) ribosomal RNA gene sequences. J. Microbiol. Methods, 83, 250–253. [DOI] [PubMed] [Google Scholar]
- Kameoka S. et al. (2021) Benchmark of 16S rRNA gene amplicon sequencing using Japanese gut microbiome data from the V1-V2 and V3-V4 primer sets. BMC Genomics, 22, 527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo Y. et al. (2021) Full-length 16S rRNA gene amplicon analysis of human gut microbiota using MinION nanopore sequencing confers species-level resolution. BMC Microbiol., 21, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasolli E. et al. (2019) Extensive unexplored human microbiome diversity revealed by over 150,000 genomes from metagenomes spanning age, geography, and lifestyle. Cell, 176, 649–662 e620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss P.D. (2020) Reintroducing mothur: 10 years later. Appl. Environ. Microbiol., 86, e02343-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon S.H. et al. (2017) Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol., 67, 1613–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.