

Abstract
Summary
Hundreds of gene expression signatures have been developed during the last two decades. However, due to the multitude of development procedures and sometimes a lack of explanation for their implementation, it can become challenging to apply the original method on custom data. Moreover, at present, there is no unified and tidy interface to compute signature scores with different single sample enrichment methods. For these reasons, we developed hacksig, an R package intended as a unified framework to obtain single sample scores with a tidy output as well as a collection of manually curated gene signatures and methods from cancer transcriptomics literature.
Availability and implementation
The hacksig R package is freely available on CRAN (https://CRAN.R-project.org/package=hacksig) under the MIT license. The source code can be found on GitHub at https://github.com/Acare/hacksig.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
A gene signature can be defined as a set of genes sharing a common pattern of expression in relation to a certain phenotype or biological process (Cantini et al., 2018). In years, several enrichment methods have been developed for microarray and RNA-seq data in order to summarize the information coming from gene sets into a single score. This could lead to a more meaningful interpretation of results, both from a biological and clinical point of view, as well as a reduction in the effects of the curse of dimensionality problem affecting genomic studies (i.e. many more variables than samples).
Gene signature scoring methods can follow a number of approaches. For example, by averaging the expression values for genes in a signature (Rooney et al., 2015); or also, by fitting a penalized regression model and then computing single sample scores as a weighted sum between fitted model coefficients and gene expression values (De Cecco et al., 2014). However, it is not always straightforward to directly apply gene signature scoring methods from the literature to custom data. Sometimes details about how a method is implemented are vague and open to interpretation. Other times gene identifiers and eventual model coefficients must be extracted manually from .pdf files or even from images. So, computing signature scores with the original publication method can become a time-consuming procedure even in the best-case scenario.
Gene expression signature scores can be derived using either the original publication procedure or one of five single sample enrichment methods, all of which are collected into two distinct R packages: GSVA, implementing four methods and singscore, enabling to compute enrichment scores with the self-titled procedure (Hänzelmann et al., 2013; Foroutan et al., 2018). The interfaces of these R packages are obviously different and designed to work primarily within the Bioconductor ecosystem (Huber et al., 2015). Hence, none of them do provide a tidy output as intended by Wickham et al. (2019) that is a consistent format for successive data analysis pipelines such as data visualization with ggplot2 and modeling with tidymodels.
Herein, we propose the R package hacksig in order to address the above-mentioned issues and hence to:
compute single sample scores for both custom and manually curated gene expression signatures either with the original publication method or with three alternatives, namely the combined z-score, single sample GSEA (ssGSEA) and singscore (Lee et al., 2008; Barbie et al., 2009; Foroutan et al., 2018);
provide a unified, simple interface and a tidy output.
2 Features and methods
The current release of hacksig includes 23 cancer transcriptomics gene signatures, which were selected mainly due to our interest in the tumor microenvironment biology and its possible influence on response to treatment in head and neck cancer patients (Van den Bossche et al., 2022). The function get_sig_info() can be used to retrieve IDs, associated keywords, DOIs for the original publication and a brief description for each signature (see also Supplementary Table S1).
Most of the package functions require a normalized gene expression matrix as a primary input argument, with genes as rows and samples as columns. Hence, both microarray and RNA-seq normalized data are supported. A list of official HUGO gene symbols composing each implemented signature can be obtained with get_sig_genes().
2.1 The main function
The most important function of the package is hack_sig(), which can be used to easily compute gene expression signature scores in a number of ways. Figure 1A summarizes the syntax and different choices for arguments of the function.
Fig. 1.
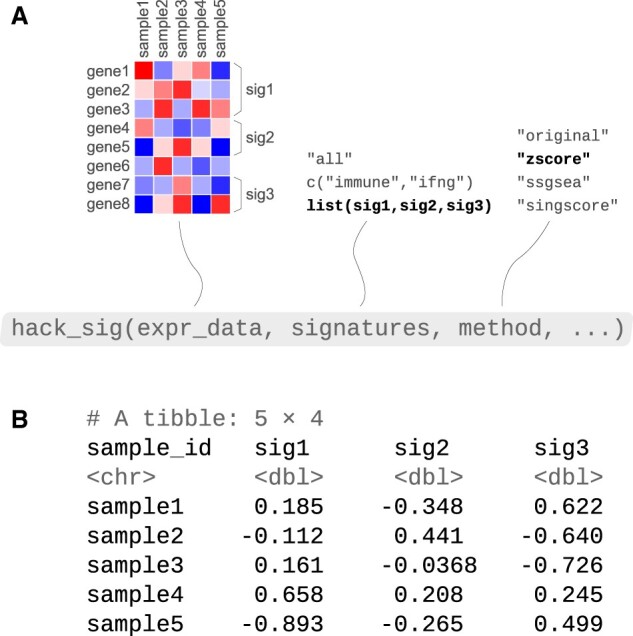
Syntax and output for the main function hack_sig(). (A) Possible choices for its three main arguments expr_data, signatures and method are shown. (B) An example output resulting from choosing arguments in bold is shown (i.e. a custom list of gene sets and the combined z-score method). The ellipsis represents additional arguments controlling options for the enrichment methods
If only an expression matrix is given in input, then hack_sig() will compute scores with the original procedure for all the implemented signatures, except those related to CINSARC (Chibon et al., 2010), ESTIMATE (Yoshihara et al., 2013) and Immunophenoscore (Charoentong et al., 2017), for which dedicated functions exist (see next section). Anyway, signature scores can also be derived with one of the three possible single sample enrichment methods by setting the argument method to one of ‘zscore’, ‘ssgsea’ or ‘singscore’, corresponding to the combined z-score, ssGSEA and singscore, respectively. This will cause hack_sig() to compute enrichment scores with that particular procedure for all the implemented signatures. In addition, other optional arguments regarding single sample methods can be modified, such as the exponent in the running sum statistic of ssGSEA or its type of normalization.
It is possible to select just a particular group of signatures or to compute scores for a custom list of gene sets by means of the argument signatures, which is set to ‘all’ (i.e. all the implemented signatures) by default. If signatures is a character vector (e.g. c(‘immune’,‘ifng’)) with one or more valid keywords, hack_sig() will compute scores only for signatures matching those strings, either with the original procedure or with one of the three single sample alternatives depending on the choice of method. If signatures is a custom list of gene sets, then hack_sig() will compute scores with the procedure specified in the method argument, which cannot be set to ‘original’ in this case. If method is not specified, raw ssGSEA scores will be obtained by default for custom gene sets.
In general, the result of calling hack_sig() will be a tibble (i.e. a modern redefining of the data.frame R class) with one row per sample, a column indicating sample IDs and one column for each considered gene signature giving the corresponding scores (Fig. 1B).
2.2 Other features
Although hack_sig() can be used to compute scores for most of the gene signatures included in the package, there are three particular methods which for us deserve their own function implementation. These are hack_cinsarc(), which implements the CINSARC classification (Chibon et al., 2010; Lesluyes and Chibon, 2020); hack_estimate(), which computes the immune, stroma, ESTIMATE and tumor purity scores as in Yoshihara et al. (2013); hack_immunophenoscore(), giving immune marker scores together with the Immunophenoscore (Charoentong et al., 2017).
Before computing enrichment scores, it should be considered good practice to always check if genes composing a signature are well represented in the expression matrix. For this reason, we developed check_sig(), a function that returns counts and proportions of how many genes are present in a gene expression matrix for every input signature as well as possible missing genes.
Finally, the package supports the future framework to parallelize and speed-up computations either on a local machine or a computer cluster (Bengtsson, 2021).
More details about the usage of hacksig are reported in the package vignette, either on CRAN or running vignette(‘hacksig’) in R.
3 Conclusions and future perspectives
The R package hacksig offers a tidy and unified framework aimed at simplifying the computation of gene signature scores following both the original methods or three single sample alternatives. We acknowledge that our implementations of enrichment methods using ranks (i.e. ssGSEA and singscore) are slower than those in the GSVA and singscore packages (see Supplementary Material). Parallelization through the future R package is supported and can definitely decrease computation time. Nonetheless, the code for some functions might be rewritten in order to improve performance even more. More features are planned to be added, and we want to encourage future users of the package to open an issue on GitHub for every signature or method they would wish to be implemented.
Supplementary Material
Acknowledgements
The authors are grateful to Drs Stefano Cavalieri and Silvana Canevari for the critical revision of this manuscript.
Funding
This work was supported by the Associazione Italiana per la Ricerca sul Cancro (AIRC) [IG23573 to L.D.C.].
Conflict of Interest: none declared.
Contributor Information
Andrea Carenzo, Molecular Mechanisms Unit, Department of Research, Fondazione IRCCS Istituto Nazionale dei Tumori, Milan 20133, Italy.
Federico Pistore, Head and Neck Cancer Medical Oncology 3 Department, Fondazione IRCCS Istituto Nazionale dei Tumori, Milan 20133, Italy.
Mara S Serafini, Molecular Mechanisms Unit, Department of Research, Fondazione IRCCS Istituto Nazionale dei Tumori, Milan 20133, Italy.
Deborah Lenoci, Molecular Mechanisms Unit, Department of Research, Fondazione IRCCS Istituto Nazionale dei Tumori, Milan 20133, Italy.
Armando G Licata, Molecular Mechanisms Unit, Department of Research, Fondazione IRCCS Istituto Nazionale dei Tumori, Milan 20133, Italy.
Loris De Cecco, Molecular Mechanisms Unit, Department of Research, Fondazione IRCCS Istituto Nazionale dei Tumori, Milan 20133, Italy.
References
- Barbie D.A. et al. (2009) Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature, 462, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtsson H. (2021) A unifying framework for parallel and distributed processing in R using futures. R J., 13, 208. [Google Scholar]
- Cantini L. et al. (2018) Classification of gene signatures for their information value and functional redundancy. NPJ Syst. Biol. Appl., 4, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charoentong P. et al. (2017) Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep., 18, 248–262. [DOI] [PubMed] [Google Scholar]
- Chibon F. et al. (2010) Validated prediction of clinical outcome in sarcomas and multiple types of cancer on the basis of a gene expression signature related to genome complexity. Nat. Med., 16, 781–787. [DOI] [PubMed] [Google Scholar]
- De Cecco L. et al. (2014) Comprehensive gene expression meta-analysis of head and neck squamous cell carcinoma microarray data defines a robust survival predictor. Ann. Oncol., 25, 1628–1635. [DOI] [PubMed] [Google Scholar]
- Foroutan M. et al. (2018) Single sample scoring of molecular phenotypes. BMC Bioinformatics, 19, 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hänzelmann S. et al. (2013) GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics, 14, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber W. et al. (2015) Orchestrating high-throughput genomic analysis with bioconductor. Nature Methods, 12, 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E. et al. (2008) Inferring pathway activity toward precise disease classification. PLoS Comput. Biol., 4, e1000217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesluyes T., Chibon F. (2020) A global and integrated analysis of CINSARC-associated genetic defects. Cancer Res., 80, 5282–5290. [DOI] [PubMed] [Google Scholar]
- Rooney M.S. et al. (2015) Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell, 160, 48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Bossche V. et al. (2022) Microenvironment-driven intratumoral heterogeneity in head and neck cancers: clinical challenges and opportunities for precision medicine. Drug Resist. Updat., 60, 100806. [DOI] [PubMed] [Google Scholar]
- Wickham H. et al. (2019) Welcome to the tidyverse. J. Open Source Softw., 4, 1686. [Google Scholar]
- Yoshihara K. et al. (2013) Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun., 4, 2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.