

Abstract
Nitrogen stable-isotope compositions (δ15N) can help track denitrification and N2O production in the environment, as can knowledge of the isotopic discrimination, or isotope effect, inherent to denitrification. However, the isotope effects associated with denitrification as a function of dissolved-oxygen concentration and their influence on the isotopic composition of N2O are not known. We developed a simple steady-state reactor to allow the measurement of denitrification isotope effects in Paracoccus denitrificans. With [dO2] between 0 and 1.2 μM, the N stable-isotope effects of NO3− and N2O reduction were constant at 28.6‰ ± 1.9‰ and 12.9‰ ± 2.6‰, respectively (mean ± standard error, n = 5). This estimate of the isotope effect of N2O reduction is the first in an axenic denitrifying culture and places the δ15N of denitrification-produced N2O midway between those of the nitrogenous oxide substrates and the product N2 in steady-state systems. Application of both isotope effects to N2O cycling studies is discussed.
The importance of denitrification in microbial ecology, N2O production, agricultural N loss and wastewater treatment has prompted a large body of research over the last 25 years. Although the basic pathway is well known (36),
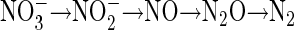 |
1 |
the regulation and distribution of denitrification remain poorly understood. The sensitivity of denitrification to oxygen tension is of particular interest due to (i) the recent demonstration of denitrification under aerobic conditions (40, 41), (ii) increased N2O production under these conditions (12, 20, 28), (iii) the importance of linked nitrification-denitrification in N cycling in natural environments (11, 14), and (iv) development of “single-sludge” wastewater treatment as a low-cost alternative to traditional strategies that employ separate aerobic and anoxic reactors (26, 34, 47).
The natural-abundance 15N ratios of nitrogenous materials have been used to identify or quantify denitrification activity in low-oxygen environments, including the marine water column (24, 53, 54, 55), groundwater (15), sediments (1), and soil (25). These studies exploit the variation in the ratio of 15N to 14N in nitrogenous material that results from the isotopic discrimination of denitrification, in which 14N reacts faster than 15N. Thus, natural-abundance 15N ratios provide a small (≈0.366% 15N) but endogenous in situ tracer of denitrification activity, in contrast to large 15N additions (50 to 99% 15N) traditionally used to trace biological N fixation and other N transformations. However, published estimates of the extent of isotopic discrimination, or isotope effect (ɛ), of denitrification range from 13 to 40‰, reflecting the variety of experimental methods and denitrifying cultures used (5, 6, 10, 13, 27, 50, 52). Because the ɛ of denitrification lies between those of other N transformations, such as N assimilation at 10‰ (9, 19, 30) and nitrification at 13 to 16‰ in situ (21) or 30 to 60‰ in vitro (27, 52), it is desirable that ɛ be better constrained. In addition, possible variation in ɛ as a function of oxygen tension has not been investigated heretofore.
We measured ɛ of denitrification in pure cultures of Paracoccus denitrificans under dissolved-oxygen concentrations between 0 and 1.2 μM. We also measured a unique ɛ for N2O reduction in these cultures. For these experiments, we developed a simple, steady-state reactor which does not require a dedicated mass spectrometer or online sample preparation system, thus offering a flexible approach to investigators who do not routinely use stable-isotope techniques. The reactor was also used to measure oxygen isotope effects, which are reported elsewhere (4). This information expands the utility of stable isotope studies of denitrification in low-oxygen (<10 μM) environments. Here we describe the necessary steady-state fractionation models and the reactor configuration and performance and report the N stable-isotope effects for denitrification.
MATERIALS AND METHODS
Experimental design.
Continuous cultures were used to control the dissolved-oxygen concentration ([dO2]) more easily and to exploit the simplicity of steady-state fractionation models, which relate kinetic ɛ values directly to the isotopic compositions of reactants and products. The isotopic composition of nitrogenous material is commonly expressed as a δ-value relative to atmospheric N2:
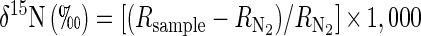 |
2 |
where R = 15N/14N. In a single first-order reaction, in which the substrate pool is infinitely large, ɛ closely approximates the difference between the δ values of the substrate and product (16). The isotopic dynamics of steady-state anoxic denitrification may be idealized as such a one-step process:
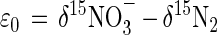 |
3 |
where ɛ0 is the overall ɛ of denitrification. Alternatively, this pseudo-ɛ may be partitioned: ɛ1, δ1ɛ2, δ2 δ15NO3− → δ15N2O → δ15N2 (4)
where ɛ1 and ɛ2 are the ɛ values of NO3− and N2O reduction, respectively, and δ1 and δ2 are the isotopic compositions of the instantaneous products of the two reactions. At steady state, δ15N2O and δ15N2 are constant; thus, δ1 and δ2 both equal δ15N2. Applying the principle of equation 3 to equation 4 yields the following relationships:
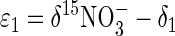 |
5a |
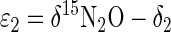 |
5b |
When δ15NO3−, δ15N2O, and δ15N2 are measured experimentally and δ15N2 is substituted for δ1 and δ2, equations 5a and 5b may be solved for ɛ1 and ɛ2, respectively. Note that ɛ1 = ɛ0, which is generally true for unbranched, nonreversible reaction pathways (37).
The approach described above allows the calculation of ɛ1 and ɛ2 by measuring δ15N of three species at a single steady state. Independent checks of ɛ1 were calculated from isotopic mass balances of the reactor at multiple steady states. Because denitrification intermediates constituted very small fractions of reactor N at steady state, N isotope mass balances were written as follows:
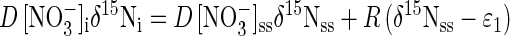 |
6 |
where D is reactor dilution rate (time−1), R is the denitrification rate ([N] time−1), δ15N is the δ value of NO3−, and the subscripts “i” and “ss” refer to initial and steady state, respectively. Equation 6, like equation 3, implies that ɛ1 is equal to the steady-state difference between δ15NO3− and δ15N2. Because R equals the product of D and the concentration of substrate consumed in a continuous culture (chemostat) at steady state, equation 6 can be rewritten to eliminate R:
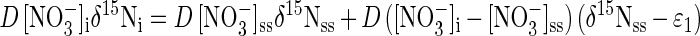 |
7 |
and may be rearranged into linear form:
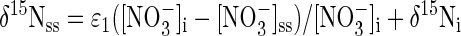 |
8a |
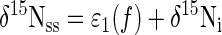 |
8b |
where f is the fraction of NO3− consumed at steady state and ɛ1 equals the slope of steady-state δ15NO3− as a function of f (17). Different values of f were achieved by manipulating the dissolved-oxygen concentration ([dO2]). Experimental [dO2] treatments were 0, 0.1, 0.3, and 1.2 μM.
Reactor configuration.
The reactor system consisted of a medium reservoir, growth chamber, waste carboy, and connecting tubing and flow controls (Fig. 1). The 20-liter Pyrex carboy in which media were sterilized also served as the reservoir. A heavy-gauge aluminum lid and rubber gasket were secured to the reservoir with a collar and screws. The lid contained ports for gas entry, gas and medium exit to the growth chamber, and venting. Gas mixing and flow to the reservoir were controlled by a gas proportioner (Alltech, Deerfield, Ill.). Gas was conducted to the reservoir in 1/8-in. stainless steel tubing, through a 0.5-μm nominal matrix filter (Nupro, Willoughby, Ohio) and a sparging stone. Medium flow from the reservoir to the growth chamber was caused by positive pressure in the reservoir, which was in turn controlled by the sparging rate. Two needle valves (Nupro) were added to enable finer control of medium flow rate to the growth chamber. This mode of medium delivery was chosen over pumping due to the difficulty of maintaining absolutely anoxic connections between steel tubing and peristaltic pump tubing.
FIG. 1.

Reactor configuration.
The growth chamber was a 2-liter Pyrex cylinder equipped with a magnetic stirrer and a stainless steel lid similar to the reservoir lid. In addition to gas and liquid entry ports, it contained a septum port for sampling by syringe, ports to accommodate a pH probe (Orion, Boston, Mass.), and a dO2 probe (Ingold, Wilmington, Mass.), a 3/8-in. port for gas and liquid exit to the waste carboy, and a 3/8-in. port fitted with a shutoff valve for headspace sampling. The pH probe was connected to a pH controller (Cole Parmer, Chicago, Ill.), which activated a peristaltic pump equipped with microbore tubing to introduce HCl into the growth chamber. This tubing entered the growth chamber through the septum port. The waste port was situated to give the growth chamber a working volume of 1.75 liters. Waste liquid and gas were forced by positive pressure into the 20-liter vented waste carboy through 3/8-in. stainless steel tubing.
Organism and media.
P. denitrificans ATCC 17741, a relatively oxygen-sensitive, classic denitrifier, was chosen for the experiments (2). Cultures were purchased from the American Type Culture Collection (Rockville, Md.) and reconstituted in nutrient broth (Difco, Detroit, Mich.) at 30°C. Subcultures were frozen in glycerol and stored at −20°C until needed. The defined medium for continuous-culture experiments contained 30 mM nitrate and 20 mM acetate, which was the sole electron donor, carbon source, and limiting substrate (39). The composition of denitrification medium was as follows (in grams per liter): KNO3, 3.03; CH3COONa · 3H2O, 2.72; K2HPO4, 0.8; KH2PO4, 0.3; NH4Cl, 0.4; MgSO4 · 7H2O, 0.4; trace-elements solution, 2 ml liter−1. The trace-elements solution was modified from that of Vishniac and Santer (48) and contained (in grams per liter) EDTA, 50.0; ZnSO4, 2.2; CaCl2, 5.5; MnCl2 · 4H2O, 5.06; FeSO4 · 7H2O, 5.0; (NH4)6Mo7O24 · 4H2O, 1.1; CuSO4 · 5H2O, 1.57; CoCl2 · 6H2O, 1.61.
Reactor operation and sampling.
Denitrification medium was autoclaved in 16-liter batches at 121°C and 15 lb/in2 for 80 min. Beginning immediately after sterilization, the reservoir was sparged with ultra-high-purity helium or O2 in helium (Med-Tech Gases, Medford, Mass.). The growth chamber, dO2 probe, liquid-sampling needle, waste vessel, and tubing were autoclaved and connected while hot. The pH probe was calibrated with standard buffers (Fisher Scientific, Fair Lawn, N.J.), surface sterilized with 70% ethanol, and inserted into the growth chamber. The chamber was filled to working volume with medium, which was sampled with a syringe when cool. The culture was then inoculated with 30 ml of stationary-phase P. denitrificans and grown in batch mode at 30°C to approximately 108 cells ml−1. Medium was then added at an appropriate dilution rate, and the pH was maintained at 8.0 by automatic addition of 1 M HCl. The [N2O] of the headspace gas was monitored daily until it stabilized within a few ppm (by volume). At this point, the reactor was assumed to have reached steady state (see below).
Once steady state was established for a given experimental [dO2] treatment, samples of each type were taken in triplicate. Liquid samples were drawn with a syringe and processed for either dissolved inorganic nitrogen concentrations (DIN) or cell N analysis. Samples (10 ml) for DIN analyses were filtered through 0.2-μm-pore-size cartridges (Gelman, Ann Arbor, Mich.), split into subsamples, and frozen until analysis of DIN or δ15N. Unfiltered liquid samples for cell counts were preserved in 5% formalin and stored at 10°C until analysis. Unfiltered samples for direct cell N measurement were processed immediately after sampling, as described below.
Gas samples were collected in preevacuated, U-shaped Pyrex tubes (34-ml volume) fitted on either end with vacuum stopcocks (Ace Glass, Vineland, N.J.). For N2 collection, the U-tubes were coupled to the gas-sampling port of the growth chamber by using compression fittings with Teflon front ferrules and nylon back ferrules (Swagelok, Solon, Ohio). Each N2 collection tube contained several granules of silica gel for cryogenic absorption of N2 gas (33). With the growth chamber waste vent closed, U-tubes were opened and flushed with outgoing headspace gas (100 ml min−1) for at least 5 min. Each grab sample was then isolated by closing first the stopcock near the sampling port and then the outlet stopcock. This order was necessary to maintain atmospheric pressure and to enable back-calculation of the N2 production rate. Samples were stored in U-tubes for up to 2 days until purified and used for manometric determination of N2.
N2O was quantified by gas chromatography. Samples for gas chromatography were collected by flushing a 30-ml serum bottle with outgoing headspace gas via the gas sampling port. N2O samples for δ15N and δ18O determination were collected by trapping N2O out of the outgoing gas stream. This was necessary because the headspace [N2O] was too low (≈0.1 μM) for grab samples of reasonable volume to yield the 2 to 6 μmol of N required for mass spectrometry. The N2O trap was a U-tube packed with borosilicate glass beads, which increased the trap surface area and dispersed the gas flow enough to trap N2O when chilled in liquid nitrogen (LN2). The efficiency of the N2O trap was verified by measuring zero N2O in the trap effluent. CO2 was removed from N2O samples by a scrubber in line between the growth chamber and the N2O trap. The scrubber consisted of a standard gas purification cartridge (Alltech) packed with a 3-cm layer of Carbosorb granules (Elemental Microanalysis, Manchester, Mass.) between two layers of indicating silica gel.
Cryogenic distillation.
Gas samples were purified by standard cryogenic techniques (7). U-tubes were first immersed in LN2 to freeze the N2 or N2O sample onto silica gel or glass beads, respectively. The large overburden of helium carrier gas was then removed with a vacuum pump. N2 samples were further purified of CO2 and H2O by a LN2-cooled trap; O2 was removed by passing the sample over copper filings at 550°C. The purified N2 was quantified by using a capacitance manometer (MKS Baratron). N2O samples were further purified of H2O by using a glass trap cooled in an ethanol-dry ice slurry. Each N2 or N2O sample was refrozen in a Pyrex ampoule, sealed, and stored until analysis by continuous-flow isotope ratio mass spectrometry.
Analytical methods.
Nitrate [NO3−] and nitrite [NO2−] concentrations were measured by the spongy cadmium reduction method (22). The ammonia [NH3 + NH4+] concentration was determined by the colorimetric method of Strickland and Parsons (45). The δ15N of (NO3− + NO2−) was measured by the ammonia diffusion method as modified by Sigman et al. (43).
The N in bacterial cells was quantified by acridine orange direct counting (18) and a conversion factor for cell N concentration, which was found by performing cell counting and direct cell N measurement on the same samples over a range of cell densities. Direct measurements of cell N were made with a Europa elemental analyzer. To prepare a sample containing 2 to 6 μmol of N, approximately 1.0 ml of cell suspension was filtered onto a precombusted 25-mm-diameter GF/F filter. The filters were dried at 55°C and packed in tin boats before analysis. Cell N was calculated from the following regression (r2 = 0.8835, n = 4):
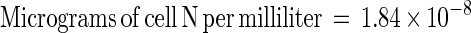 |
9 |
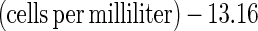 |
N2O production was monitored by using a Hewlett-Packard 5890A gas chromatograph equipped with an electron capture detector (23). A 1/8-in.-diameter stainless steel column packed with Hayesep Q 80/100 mesh was used at 40°C. The injector and detector temperatures were 100 and 350°C, respectively, and the carrier gas was 5% methane in argon at 30 ml min−1. Under these conditions, N2O eluted approximately 1.9 min after sample injection. Calibration curves were prepared daily by using a standard gas mixture of 101 ppm (volume) N2O in N2 (Scott Specialty Gases, Reading, Mass.). Standard injections were performed periodically to check for signal drift.
The δ15N of N2O, N2, and (NO3− + NO2−) and the δ18O of N2O were measured on a Finnigan Mat 251 mass spectrometer (55). Samples were conducted by helium carrier flow (50 ml min−1) through Carbosorb and magnesium perchlorate traps to remove trace CO2 and H2O, respectively. The mass 29/28 ratio was measured for N2 samples and expressed as δ15N relative to atmospheric N2. Injections of working standard N2 were made through a septum port in line with the carrier flow. For N2O samples, the mass 45/44 ratio, yielding δ15N values, and the mass 46/44 ratio, yielding δ18O values, were both measured for each sample. The mass spectrometer was calibrated with a standard of pure N2O gas kindly provided by T. Yoshinari, New York State Department of Health. N2O δ values were expressed relative to the δ15N and δ18O of atmospheric N2 and O2, respectively.
Budget calculations.
Dissimilatory (denitrification) and assimilatory N budgets were calculated for the reactor system. Each term in the dissimilatory budget was expressed as a percentage of the NO3− supplied in the medium:
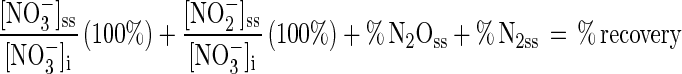 |
10 |
The NO3− and NO2− terms were their respective steady-state concentrations (denoted by the subscript “ss”) expressed as percentages of the initial NO3− concentration. The N2O term (%N2Oss) was the N2O-N production rate expressed as a percentage of the NO3− supply rate:
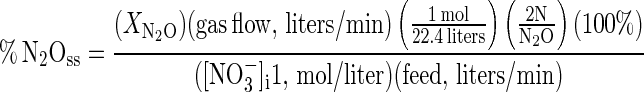 |
11 |
where XN2O is the mole fraction of N2O in growth chamber headspace gas. The analogous N2 term, %N2ss, is shown below. The first factor in this equation was the amount of N contained in a 34-ml grab sample, which was measured manometrically during cryogenic distillation:
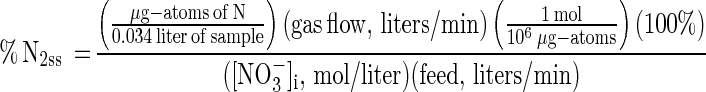 |
(12)
The assimilatory N budget consisted of the sum of cell N and NH3-N present at steady state, expressed as a percentage of NH3 supplied in the medium. Dissolved organic nitrogen compounds which may have been synthesized from NH3 were not included in the budget.
RESULTS
Reactor performance.
Dissimilatory N recovery from the reactor system was near 100% over a range of dilution rates (Fig. 2). Residual [NO3−] increased with increasing dilution rate, but the cultures were not in danger of washout at the rates tested (39). Dissimilatory N recovery varied as a function of the sparging rate (Fig. 3), with particularly high N2 recovery at the lowest sparging rate, as discussed below. Assimilatory N recovery was 80 to 100% over the same range of dilution and sparging rates (data not shown). Dilution and sparging rates of 0.7 day−1 and 100 ml min−1, respectively, were held constant in further experiments, in which variable [dO2] was the sole experimental treatment.
FIG. 2.

Dissimilatory N budget as a function of the dilution rate. Symbols are means and SE (n = 3). NO2− and N2O made up less than 1% of total N.
FIG. 3.

Dissimilatory N budget as a function of the sparge rate. Symbols are means and SE (n = 3).
Experimental results.
Under anoxic steady-state conditions, the reactor system yielded δ15NO3−, δ15N2O, and δ15N2 of 15.5‰ ± 0.3‰, 0.08‰ ± 3.1‰, and −11.0‰ ± 1.2‰, respectively (mean ± standard error [SE], n = 6). According to these data and equations, 5a and 5b, ɛ1 = 26.5‰ ± 1.2‰ and ɛ2 = 11.1‰ ± 3.1‰ under anoxic conditions. The independent estimates of ɛ1 calculated from equation 8b and δ15NO3− and δ15N2O from multiple steady states at different [dO2] are similar although more variable (26.9‰ ± 2.6‰ and 24.3‰ ± 4.0‰, respectively). However, ɛ1 calculated from δ15N2 from multiple steady states was ≈14‰ (Fig. 4). This discrepancy and the high N2 recovery at the low sparging rate (Fig. 3) suggested that significant isotopic signal from atmospheric N2 had biased the δ15N2 values. To quantify this bias, the response of measured δ15N2 to systematic air contamination, or a “handling blank,” was simulated (Fig. 5). The expected δ15N2 was first calculated by subtracting the slope of the NO3− regression in Fig. 4 (i.e., ɛ1) from points on that regression. The resulting straight line represents the δ15N2 expected from constant fractionation and no air contamination. The expected δ15N2 for the highest [dO2] treatment (the least biogenic N2) was compared to the measured value, and the amount of atmospheric N2 necessary to create the difference was calculated by mass balance. Simulation of constant fractionation with constant air contamination was then made by adding the isotopic contribution of this amount (0.5 μmol) of atmospheric N2 to the constant-fractionation values. The resulting curve fits very closely with the measured δ15N2, suggesting that both ɛ1 and the amount of air contamination during sampling were constant for all [dO2] treatments.
FIG. 4.

δ15N as a function of NO3− consumption (f). Different fractions of f were caused by dO2 treatments. Symbols are means and SE (n = 3). The slope and SE of the slope of regressions of δ15N versus f are 14.5 and 2.0 (N2), 24.3 and 4.0 (N2O), and 26.9 and 2.6 (NO3−). The asterisk represents the δ15N of the NO3− supplied (−3.6‰).
FIG. 5.

Comparison between measured δ15N2 from dO2 experiments (solid triangles) and dilution rate experiments (open triangles), the expected δ15N2 given constant fractionation (straight line), and the expected value with a constant amount of air contamination (curved line). See the text for details.
To compare ɛ1 and ɛ2 between dO2 treatments, the δ15N2 data were transformed to eliminate the effect of air contamination. This was done by subtracting from each measured δ15N2 value the isotopic bias inherent in 0.5 μmol of atmospheric N2. The corrected δ15N2 values for each treatment were then averaged, and these means were subtracted from the corresponding mean values of δ15NO3− and δ15N2O to yield ɛ1 and ɛ2, respectively (Table 1). Analysis of variance indicates that ɛ1 and ɛ2 varied more between reactor runs than between [dO2] treatments. The 1.2 μM dO2 treatment was excluded from the analysis of variance because low biogenic N2 production made δ15N2 very sensitive to the mass balance correction (Table 1). Given the supporting evidence for systematic air contamination and the good agreement between intratreatment estimates of ɛ1 and regression models of intertreatment δ15N2O and δ15NO3− (Fig. 4), ɛ1 and ɛ2 were assumed to be constant over the range of dO2 treatments and are reported as the grand means 28.6‰ ± 1.9‰ and 12.9‰ ± 2.6‰, respectively (means ± SE, n = 5).
TABLE 1.
Isotope effects for different dO2 treatments, calculated from transformed δ15N2
| ɛ | ɛa at dO2 of:
|
|||||
|---|---|---|---|---|---|---|
| 0 μM | 0 μM | 0.1 μM | 0.1 μM | 0.3 μM | 1.2 μM | |
| ɛ1 | 29.3 ± 0.2a | 26.6 ± 0.8b | 28.8 ± 1.0a | 29.7 ± 0.7a | 28.7 ± 1.1a | 12.4 ± 54b |
| ɛ2 | 16.5 ± 0.8c | 8.6 ± 0.9d | 11.3 ± 1.7e | 13.5 ± 0.9f | 14.7 ± 1.3cf | −2.6 ± 54b |
Data are means ± SE (n = 3). Means sharing a letter are not significantly different (P < 0.05) according to the least-significant-difference test for planned comparisons (44). Columns of data with the same dO2 concentration represent results of different runs.
See the text.
DISCUSSION
The value of ɛ1 reported here falls well within the range in the literature (Table 2). All estimates of biological fractionation are well below 90‰, the maximum theoretical fractionation of N—O bond rupture (50). The precision of ɛ1 as measured in our steady-state system compares favorably with that measured by batch culture experiments (5, 13), in which ɛ1 was calculated by using the classic Rayleigh equation, which is sensitive to error in f (46). The value and precision of ɛ1 are similar to those reported by Mariotti et al. (27), who measured f by the acetylene block technique.
TABLE 2.
Measured values of the overall isotope effect of denitrification
| Experimental system | ɛ (‰) | Reference |
|---|---|---|
| Pseudomonas denitrificans | 13–21 | 13 |
| Pseudomonas stutzeri | 20–30 | 50 |
| Nitrosomonas europaeaa | 35–36 | 52 |
| Soil (amended with glucose) | 14–23 | 5 |
| Soil (unamended) at 20°C | 29.4 ± 2.4 | 27 |
| Soil (unamended) at 30°C | 24.6 ± 0.9 | 27 |
| Eastern tropical North Pacific | 30–40 | 10 |
| Groundwater | 15.9 | 6 |
Assumes that N2O is formed by NO2− reduction.
The results reported here indicate that ɛ1 in P. denitrificans is constant over a range of dO2 concentrations. This constancy supports the validity of using ɛ1 to quantify, identify, or rule out denitrification fluxes in environments containing [dO2] gradients. However, other work indicates that biological kinetic fractionation can vary with environmental conditions. For sulfate reduction, Rees (38) hypothesized that the greater fractionation often measured in situ is due to slower growth in the field than in pure culture, but our dilution rate experiments indicated that the growth rate per se did not cause ɛ1 to vary (Fig. 5). Bryan et al. (8) showed that the overall ɛ of denitrification does vary with the denitrification rate in whole cells and cell extracts of Pseudomonas stutzeri limited by [NO2−], increasing to a maximum value of 25‰ ± 3.2‰ at initial [NO2−] > 2.5 mM. The electron acceptor concentrations in our experiments were well within the asymptotic range reported by Bryan et al. (above 2.5 mM). These authors also found a negative correlation between ɛ and denitrification rate when the rate was increased by higher electron donor concentrations.
The isotopic composition of N2O in our experiments was quite distinct from both δ15N2 and δ15NO3−. The combined effects of ɛ1 and ɛ2 resulted in δ15N2O being about 13‰ heavier and 15‰ lighter than δ15N2 and δ15NO3−, respectively, at steady state. To our knowledge, this is the first report of an isotope effect for nitrous oxide reduction in a denitrifying system. Yoshida et al. (53) cited unpublished data which yielded a value of 27‰ for ɛ2 in P. denitrificans, but they did not specify whether the bacteria were supplied with N2O, NO2−, or NO3−. Yamazaki et al. (51) reported a maximum ɛ of 39‰ for N2O reduction by the N2 fixer Azotobacter vinelandii, but nitrogenase, not nitrous oxide reductase, appeared to be responsible for the observed activity.
The expression of isotopic fractionation by P. denitrificans was strongly influenced by [dO2]. Within the narrow range between 0 and slightly more than 1.2 μM dO2, the δ15N of NO3− and N2O varied up to 26‰ and δ15N2 probably varied to an equal extent (Fig. 4). However, the usefulness of [dO2] as a predictor of δ15N in denitrifying environments may be limited to the extent to which it controls NO3− consumption. The expression of ɛ in natural and applied systems will also depend on the distribution of denitrifiers. P. denitrificans is very sensitive to dO2 in comparison to some denitrifiers, such as Comamonas sp. (35) and Thiosphaera pantotropha (39), which under aerobic conditions maintain 40 and 25% of their anaerobic denitrifying activity, respectively. However, Pseudomonas fluorescens, which frequently dominates denitrifying environments, denitrifies over approximately the same range of [dO2] as P. denitrificans (28). Chemostat studies of P. halodenitrificans revealed only slightly higher tolerance to dO2, to ∼2 μM (20).
It is hoped that ɛ1 and ɛ2 may be used to help distinguish between denitrification and nitrification as sources of N2O and may serve as in situ tracers of both processes. For example, if the δ15N of the substrate pools for both processes, NO3− and NH4+, respectively, are assumed to be zero, then denitrification- and nitrification-produced δ15N2O in open systems at steady state would be ca. −15‰ and −65‰, respectively (52). However, the isotopic composition of substrate pools in real systems could obscure this distinction. The δ15N of NO3− and NH4+ vary from −23 to +43‰ and −20 to +50‰, respectively, depending upon the source and the combined fractionation effects of redox reactions in the environment (49). Given these ranges, it is possible that denitrification- and nitrification-produced δ15N2O would be indistinguishable. Linked nitrification-denitrification may also confuse δ15N2O signatures by increasing the range of potential substrate molecules, which may in turn may have variable δ15N (56). In environments such as sediments and biofilms, spatial linkage between nitrification and denitrification is on the order of 1 mm or less, and in single microorganisms such as Thiosphaera pantotropha, which both denitrifies and nitrifies in aerobic environments, N2O may be produced from NH4+, NO3−, and NO2− (3). However, in many environments (9, 19, 29, 31), the δ15N of substrate pools can be constrained within a range of 10‰ (see, e.g., references 1, 32, and 42), and the isotopic composition of N2O could provide a simple index to the relative contribution of the two processes producing N2O.
ACKNOWLEDGMENTS
We thank J. Nevins for generous technical assistance and M. Hullar and J.-D. Gu for helpful discussion.
This work was supported in part by NASA NAG 2-843 (awarded to R.M.), NSF OCE-95-30187 and NSF DEB-96-33510 (awarded to J.P.M.), and NSF OCE-95-26356 (awarded to M.A.A.).
REFERENCES
- 1.Altabet M A, Francois R, Murray D W, Prell W L. Climate-related variations in denitrification in the Arabian Sea from sediment 15N/14N ratios. Nature. 1995;373:506–509. [Google Scholar]
- 2.Aragno M, Schlegel H G. The hydrogen-oxidizing bacteria. In: Starr M P, Stolp H, Trüper H G, Balows A, Schlegel H G, editors. The prokaryotes. New York, N.Y: Springer-Verlag; 1981. pp. 865–893. [Google Scholar]
- 3.Arts P A M, Robertson L A, Kuenen J G. Nitrification and denitrification by Thiosphaera pantotropha in aerobic chemostat cultures. FEMS Microbiol Ecol. 1995;18:305–316. doi: 10.1128/aem.54.11.2812-2818.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barford C C. Ph.D. thesis. Cambridge, Mass: Harvard University; 1997. [Google Scholar]
- 5.Blackmer A M, Bremner J M. Nitrogen isotope discrimination in denitrification of nitrate in soils. Soil Biol Biochem. 1977;9:73–77. [Google Scholar]
- 6.Böttcher J, Strebel O, Voerkelius S, Schmidt H-L. Using isotope fractionation of nitrate-nitrogen and nitrate-oxygen for evaluation of microbial denitrification in a sandy aquifer. J Hydrol. 1990;114:413–424. [Google Scholar]
- 7.Boutton T W. Stable carbon isotope ratios of natural materials. I. Sample preparation and mass spectrometric analysis. In: Coleman D C, Fry B, editors. Carbon isotope techniques. London, United Kingdom: Academic Press, Ltd.; 1991. pp. 155–171. [Google Scholar]
- 8.Bryan B A, Shearer G, Skeeters J L, Kohl D H. Variable expression of the nitrogen isotope effect associated with denitrification of nitrite. J Biol Chem. 1983;258:8613–8617. [PubMed] [Google Scholar]
- 9.Cifuentes L A, Fogel M L, Pennock J R, Sharp J H. Biogeochemical factors that influence the stable isotope ratio of dissolved ammonium in the Delaware Estuary. Geochim Cosmochim Acta. 1989;53:2713–2721. [Google Scholar]
- 10.Cline J D, Kaplan I R. Isotopic fractionation of dissolved nitrate during denitrification in the eastern tropical North Pacific Ocean. Marine Chem. 1975;3:271–299. [Google Scholar]
- 11.Codispoti L A, Christensen J P. Nitrification, denitrification and nitrous oxide cycling in the eastern tropical South Pacific Ocean. Marine Chem. 1985;16:277–300. [Google Scholar]
- 12.Davies K J P, Lloyd D, Boddy L. The effect of oxygen on denitrification in Paracoccus denitrificans and Pseudomonas aeruginosa. J Gen Microbiol. 1989;135:2445–2451. doi: 10.1099/00221287-135-9-2445. [DOI] [PubMed] [Google Scholar]
- 13.Delwiche C C, Steyn P L. Nitrogen isotope fractionation in soils and microbial reactions. Environ Sci Technol. 1970;4:929–935. doi: 10.1021/es60047a007. [DOI] [PubMed] [Google Scholar]
- 14.Devol A H, Christensen J P. Benthic fluxes and nitrogen cycling in sediments of the continental margin of the eastern North Pacific. J Marine Res. 1993;51:345–372. [Google Scholar]
- 15.Durka W, Schulze E-D, Gebauer G, Voerkelius S. Effects of forest decline on uptake and leaching of deposited nitrate determined from 15N and 18O measurements. Nature. 1994;372:765–767. [Google Scholar]
- 16.Goericke R, Montoya J P, Fry B. Physiology of isotopic fractionation in algae and cyanobacteria. In: Lajtha K, Michener R H, editors. Stable isotopes in ecology and environmental science. Cambridge, Mass: Blackwell Scientific Publications; 1994. pp. 187–221. [Google Scholar]
- 17.Hayes J M. Fractionation et al.: an introduction to isotopic measurements and terminology. Spectra. 1982;8:3–8. [Google Scholar]
- 18.Hobbie J E, Daley R J, Jasper S. Use of Nuclepore filters for counting bacteria by fluorescence microscopy. Appl Environ Microbiol. 1977;33:1225–1228. doi: 10.1128/aem.33.5.1225-1228.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoch M P, Fogel M L, Kirchman D L. Isotope fractionation during ammonium uptake by marine microbial assemblages. Geomicrobiol J. 1994;12:113–127. [Google Scholar]
- 20.Hochstein L I, Betlach M, Kritikos G. The effect of oxygen on denitrification during steady-state growth of Paracoccus halodenitrificans. Arch Microbiol. 1984;137:74–78. doi: 10.1007/BF00425811. [DOI] [PubMed] [Google Scholar]
- 21.Horrigan S G, Montoya J P, Nevins J L, McCarthy J J. Natural isotopic composition of dissolved inorganic nitrogen in the Chesapeake Bay. Estuarine Coastal Shelf Sci. 1990;30:393–410. [Google Scholar]
- 22.Jones M N. Nitrate reduction by shaking with cadmium. Alternative to cadmium columns. Water Res. 1984;18:643–646. [Google Scholar]
- 23.Kaspar H F, Tiedje J M. Response of electron-capture detector to hydrogen, oxygen, nitrogen, carbon dioxide, nitric oxide and nitrous oxide. J Chromatogr. 1980;193:142–147. [Google Scholar]
- 24.Kim K-R, Craig H. The two-isotope characterization of N2O in the Pacific Ocean and constraints on its origin in deep water. Nature. 1990;347:58–61. [Google Scholar]
- 25.Kim K-R, Craig H. Nitrogen-15 and oxygen-18 characteristics of nitrous oxide: a global perspective. Science. 1993;262:1855–1857. doi: 10.1126/science.262.5141.1855. [DOI] [PubMed] [Google Scholar]
- 26.Kuenen J G, Robertson L A. Combined nitrification-denitrification processes. FEMS Microbiol Rev. 1994;15:109–118. [Google Scholar]
- 27.Mariotti A, Germon J C, Hubert P, Kaiser P, Letolle R, Tardieux A, Tardieux P. Experimental determination of nitrogen kinetic isotope fractionation: some principles; illustration for the denitrification and nitrification processes. Plant Soil. 1981;62:413–430. [Google Scholar]
- 28.McKenney D J, Drury C F, Findlay W I, Mutus B, McDonnell T, Gajda C. Kinetics of denitrification by Pseudomonas fluorescens: oxygen effects. Soil Biol Biochem. 1994;26:901–908. [Google Scholar]
- 29.Montoya J P, Horrigan S G, McCarthy J J. Natural abundance of 15N in particulate nitrogen and zooplankton in the Chesapeake Bay. Mar Ecol Prog Ser. 1990;65:35–61. [Google Scholar]
- 30.Montoya J P, McCarthy J J. Nitrogen isotope fractionation during nitrate uptake by marine phytoplankton in continuous culture. J Plankton Res. 1995;17:439–464. [Google Scholar]
- 31.Nadelhoffer K J, Fry B. Nitrogen isotope studies in forest ecosystems. In: Lajtha K, Michener R H, editors. Stable isotopes in ecology and environmental science. Cambridge, Mass: Blackwell Scientific Publications; 1994. pp. 22–44. [Google Scholar]
- 32.Naqvi S W A, Yoshinari T, Jayakumar D A, Altabet M A, Narvekar P V, Devol A H, Brandes J A, Codispoti L A. Budgetary and biogeochemical implications of N2O isotope signatures in the Arabian Sea. Nature. 1998;394:462–464. [Google Scholar]
- 33.Nevins J L, Altabet M A, McCarthy J J. Nitrogen isotope ratio analysis of small samples: sample preparation and calibration. Anal Chem. 1985;57:2143–2145. [Google Scholar]
- 34.O’Neill M, Horan N J. Achieving simultaneous nitrification and denitrification of wastewaters at reduced cost. Water Sci Technol. 1995;32:303–312. [Google Scholar]
- 35.Patreau D, Bernet N, Moletta R. Effect of oxygen on denitrification in continuous chemostat culture with Comamonas sp SGLY2. J Ind Microbiol. 1996;16:124–128. [Google Scholar]
- 36.Payne W J. Reduction of nitrogenous oxides by microorganisms. Microbiol Rev. 1973;37:409–452. doi: 10.1128/br.37.4.409-452.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peterson B J, Fry B. Stable isotopes in ecosystem studies. Annu Rev Ecol Syst. 1987;18:293–320. [Google Scholar]
- 38.Rees C E. A steady-state model for sulphur isotope fractionation in bacterial reduction processes. Geochim Cosmochim Acta. 1973;37:1141–1162. [Google Scholar]
- 39.Robertson L A. Aerobic denitrification and heterotrophic nitrification in Thiosphaera pantotropha and other bacteria. Ph.D. thesis. Delft, The Netherlands: Technical University of Delft; 1988. [Google Scholar]
- 40.Robertson L A, Kuenen J G. Aerobic denitrification: a controversy revived. Arch Microbiol. 1984;139:351–354. [Google Scholar]
- 41.Robertson L A, van Neil E W J, Torremans R A M, Kuenen J G. Simultaneous nitrification and denitrification in aerobic chemostat cultures of Thiosphaera pantotropha. Appl Environ Microbiol. 1988;54:2812–2818. doi: 10.1128/aem.54.11.2812-2818.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schäfer P, Ittekkot V. Seasonal variability of δ15N in settling particles in the Arabian Sea and its palaeogeochemical significance. Naturwissenschaften. 1993;80:511–513. [Google Scholar]
- 43.Sigman D M, Altabet M A, Michener R, McCorkle D C, Fry B, Holmes R M. Natural abundance-level measurement of the nitrogen isotopic composition of oceanic nitrate: an adaptation of the ammonia diffusion method. Marine Chem. 1997;57:227–242. [Google Scholar]
- 44.Sokal R R, Rohlf F J. Biometry. W. H. New York, N.Y: Freeman & Co.; 1995. [Google Scholar]
- 45.Strickland J D H, Parsons T R. A practical handbook of seawater analysis. Ottawa, Canada: Fisheries Research Board of Canada; 1968. [Google Scholar]
- 46.Tong J Y, Yankwich P E. Calculation of experimental isotope effects for pseudo first-order irreversible reactions. J Phys Chem. 1957;61:540–543. [Google Scholar]
- 47.U.S. Environmental Protection Agency. Nitrogen control. Publication EPA/625/R-93/010. U.S. Washington, D.C: Environmental Protection Agency; 1993. [Google Scholar]
- 48.Vishniac W, Santer M. The thiobacilli. Bacteriol Rev. 1957;21:195–213. doi: 10.1128/br.21.3.195-213.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wada E, Hattori A. Nitrogen in the sea: forms, abundances, and rate processes. Boca Raton, Fla: CRC Press, Inc.; 1991. [Google Scholar]
- 50.Wellman R P, Cook F D, Krouse H R. Nitrogen-15: microbiological alteration of abundance. Science. 1968;161:269–270. doi: 10.1126/science.161.3838.269. [DOI] [PubMed] [Google Scholar]
- 51.Yamazaki T, Yoshida N, Wada E, Matsuo S. N2O reduction by Azotobacter vinelandii with emphasis on kinetic nitrogen isotope effects. Plant Cell Physiol. 1987;28:263–271. [Google Scholar]
- 52.Yoshida N. 15N-depleted N2O as a product of nitrification. Nature. 1988;335:528–529. [Google Scholar]
- 53.Yoshida N, Hattori A, Saino T, Matsuo S, Wada E. 15N/14N ratio of dissolved N2O in the eastern tropical Pacific Ocean. Nature. 1984;307:442–444. [Google Scholar]
- 54.Yoshida N, Morimoto H, Hirano M, Koike I, Matsuo S, Wada E, Saino T, Hattori A. Nitrification rates and 15N abundances of N2O and NO3− in the western North Pacific. Nature. 1989;342:895–897. [Google Scholar]
- 55.Yoshinari T, Altabet M A, Naqvi S W A, Codispoti L, Jayakumar A, Kuhland M, Devol A. Nitrogen and oxygen isotopic composition of N2O from suboxic waters of the eastern tropical North Pacific and the Arabian Sea - measurement by continuous-flow isotope-ratio monitoring. Marine Chem. 1997;56:253–264. [Google Scholar]
- 56.Yoshinari T, Koike I. The use of stable isotopes for the study of gaseous nitrogen species in marine environments. In: Lajtha K, Michener R H, editors. Stable isotopes in ecology and environmental science. Cambridge, Mass: Blackwell Scientific Publications; 1994. pp. 114–137. [Google Scholar]