

Abstract
Motivation
Building reliable phylogenies from very large collections of sequences with a limited number of phylogenetically informative sites is challenging because sequencing errors and recurrent/backward mutations interfere with the phylogenetic signal, confounding true evolutionary relationships. Massive global efforts of sequencing genomes and reconstructing the phylogeny of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) strains exemplify these difficulties since there are only hundreds of phylogenetically informative sites but millions of genomes. For such datasets, we set out to develop a method for building the phylogenetic tree of genomic haplotypes consisting of positions harboring common variants to improve the signal-to-noise ratio for more accurate and fast phylogenetic inference of resolvable phylogenetic features.
Results
We present the TopHap approach that determines spatiotemporally common haplotypes of common variants and builds their phylogeny at a fraction of the computational time of traditional methods. We develop a bootstrap strategy that resamples genomes spatiotemporally to assess topological robustness. The application of TopHap to build a phylogeny of 68 057 SARS-CoV-2 genomes (68KG) from the first year of the pandemic produced an evolutionary tree of major SARS-CoV-2 haplotypes. This phylogeny is concordant with the mutation tree inferred using the co-occurrence pattern of mutations and recovers key phylogenetic relationships from more traditional analyses. We also evaluated alternative roots of the SARS-CoV-2 phylogeny and found that the earliest sampled genomes in 2019 likely evolved by four mutations of the most recent common ancestor of all SARS-CoV-2 genomes. An application of TopHap to more than 1 million SARS-CoV-2 genomes reconstructed the most comprehensive evolutionary relationships of major variants, which confirmed the 68KG phylogeny and provided evolutionary origins of major and recent variants of concern.
Availability and implementation
TopHap is available at https://github.com/SayakaMiura/TopHap.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
The global health emergency caused by the SARS-CoV-2 coronavirus has catalyzed an unprecedented effort to sequence millions of genomes from all around the world and to analyze them to reveal viral origins and evolutionary patterns (Andersen et al., 2020; Kumar et al., 2021; Rambaut et al., 2020). However, applying classical phylogenetic methods to infer the global SARS-CoV-2 phylogeny has been challenging (Kumar et al., 2021; Morel et al., 2021). This is partly because phylogenetically informative sites are relatively rare due to a low mutation rate and a short evolutionary period of the outbreak. Genome sequences contain random and systematic sequencing errors, which compete with informative phylogenetic variation and mislead phylogenetic inference (Kumar et al., 2021; Morel et al., 2021; Pipes et al., 2021). Consequently, applications of standard phylogenetic methods to the multiple sequence alignments (MSAs) of SARS-CoV-2 genomes have produced many equally plausible phylogenies, particularly when reconstructing early mutational history and the root of the SARS-CoV-2 phylogeny (Nie et al., 2020; Pipes et al., 2021; van Dorp et al., 2020).
Kumar et al. (2021) reconstructed a mutation tree using shared co-occurrence patterns of mutations occurring in >1% of isolates, which they refer to as the mutation order approach (MOA). They applied and advanced a maximum likelihood (ML) method (SCITE) that models false-positive and false-negative variant detections in the absence of recombination (Jahn et al., 2016; Kumar et al., 2021). They reported success deciphering the earliest phases of SARS-CoV-2 evolution and recovered the most recent common ancestor (MRCA) genome, using common variants observed in the early stages of SARS-CoV-2 evolution. Based on the MOA’s success in building the mutation tree using common variants, we hypothesized that it should be possible to build a reliable molecular phylogeny of major SARS-CoV-2 haplotypes by filtering out all genomic positions at which no minor allele rose to a frequency >1%. Such filtering should effectively reduce the noise causing erroneous molecular phylogenetic inferences using standard approaches (e.g. the ML method). If successful, one would prefer a traditional phylogenetic approach because it can better handle multiple substitutions at the same site (homoplasy) and use outgroup sequences more easily than the mutation tree approaches.
However, the approach of excluding alignment sites with only low-frequency variants followed by applying a standard phylogenetic approach on remaining sites did not work. An example in Figure 1 illustrates why. The ancestral genome contains only three polymorphic positions where derived alleles occur at high frequencies (#1, #2 and #3; Fig. 1a). In this case, we expect to see at most four correct haplotypes in the absence of noise: three mutant strains (H1, H2 and H3) and one ancestral haplotype. The addition of a small number of sequencing errors and homoplasy generate additional haplotypes (e.g. H4, H5 and H6) that occur with very low frequency but still misled an ML analysis (Fig. 1b). ML placed two spurious haplotypes (H5 and H6) near the root of the tree (Fig. 1c), albeit without significant statistical support.
Fig. 1.

Traditional phylogenetic approach versus the new TopHap approach for a dataset that contains many sequences with few variants. (a) The true tree shows three simulated mutant haplotypes. In this example, three mutations (α, β and γ) occurred sequentially and gave rise to haplotypes H1, H2 and H3. The size of triangles at each tip is proportional to the number of genomes containing these haplotypes. (b) Phylogenetic approaches use a MSA, simplified here with only three informative variants. Due to sequencing errors, a few spurious haplotypes may be observed (H4–H6) with low frequencies (0.3–1%). The inclusion of these spurious haplotypes misguides standard phylogeny methods (e.g. ML and MP) and produces incorrect evolutionary inference. (c) Result based on a typical ML approach suggests that the spurious haplotypes H6 and H5 were the first to arise. The bootstrap confidence limits for all the branching patterns are low (<50%) because each branch is only one mutation long, a situation where the bootstrap method is known to be powerless (see text). (d) The TopHap approach was able to infer the correct tree because it restricts phylogenetic analysis to haplotypes >1% frequency
However, this behavior is rectified when we removed rare haplotypes (Fig. 1d). This observation prompted us to develop a simple filtering procedure to identify common (top) haplotypes of common variants for molecular phylogenetic analysis. We first present this filtering process and then apply it to infer the early evolutionary history of SARS-CoV-2 by using 68 057 genomes (68KG) previously analyzed by Kumar et al. (2021) for a direct comparison of the TopHap phylogeny with the mutation tree generated by using MOA.
2 Materials and methods
2.1 The TopHap approach
As input, TopHap uses an MSA of genomes (n genomes and m alignment columns). The first step is the selection of common variants by specifying a desired minor allele frequency threshold (e.g., maf > 1%) without using any reference genomes (Fig. 2). All alignment sites containing at least one allele with a frequency greater than maf and another allele with a frequency less than 1-maf are retained (k variant positions). Every genome is then reduced to a haplotype containing k positions. Next, unique haplotype sequences are identified, and their frequencies tallied. TopHap selects the top h haplotypes given a desired hf frequency cutoff. Now, the MSA contains h haplotypes, each k variants long and tagged with its frequency. Outgroup genomes are added into the MSA by converting them into haplotypes containing only k selected positions. TopHap subjects the reduced MSA to the phylogenetic analysis using the Maximum Parsimony (MP) method, which produces the TopHap phylogeny of common haplotypes on common variants.
Fig. 2.

Overview of the TopHap approach. Input to TopHap is an alignment of genome sequences (n sequences, m bases each). TopHap first identifies high-frequency variants (>maf) and produces a restricted alignment with n sequences and k bases. Next, high-frequency haplotypes (>hf) are identified, resulting in a reduced alignment of h haplotypes each with k bases. These haplotypes are subjected to standard phylogenetic inference. To compute bootstrap confidence limits, TopHap resamples n haplotypes with replacement to form a replicate n × k dataset, which is followed by the identification of high-frequency haplotypes (>hf) and the inference of their phylogeny. This process is repeated for the desired number of bootstrap replicates and a consensus phylogeny of haplotypes found in all replicates is produced. Spatiotemporal information can also be used to construct subsets in which variants and haplotypes are identified for each spatiotemporal slice separately (see Supplementary Fig. S1)
When information on sampling location and time of haplotypes is available, TopHap can select variants and haplotypes for each spatiotemporal slice of the dataset that is regionally (e.g., continent, country or city) and temporally (e.g., monthly) partitioned (Supplementary Fig. S1). The same maf and hf thresholds are applied to every spatiotemporal slice, and the final set of variants and haplotypes across all spatiotemporal slices are pooled.
Calculation of the bootstrap support. In the TopHap approach, bootstrap branch support for the inferred phylogeny of common haplotypes is calculated by resampling (with replacement) of haplotypes, which is intended to assess the robustness of the inferred phylogeny to the inclusion/exclusion of haplotypes likely created by sequencing errors and convergent changes that are expected to have relatively low frequencies spatiotemporally. The bootstrap resampling procedure is applied separately to each spatiotemporal slice, and the final set of haplotypes are pooled together. This genome resampling approach is different from Felsenstein’s bootstrap approach of resampling sites to build bootstrap replicate datasets, which needs at least three mutations per branch to achieve a 95% confidence level even without any homoplasy (Felsenstein, 1985). MP method is applied to every bootstrap replicate dataset, and haplotypes that do not appear in all the replicates are pruned from bootstrap phylogenies. Then, a bootstrap consensus tree is generated, which has the bootstrap confidence limits for every clade of haplotypes. Also, one may choose not to prune haplotypes across bootstrap replicates. In this case, phylogenies can be summarized using software that allows for an unequal number of tips across phylogenies (Bouckaert, 2010).
Placement of additional haplotypes into the phylogeny. To place a new genome into the TopHap phylogeny, the first step is to transform it into a haplotype of k positions used to build the TopHap phylogeny. One may use UShER (Turakhia et al., 2021), which is an MP approach, or RAxML-EPA (Berger et al., 2011) and pplacer (Matsen et al., 2010), which are ML approaches. We found RAxML-EPA convenient, so this option is programmed in our TopHap implementation. When the intent is to place a genome with variant(s) in the genomic position that was not used to build the TopHap phylogeny, a TopHap phylogeny needs to be rebuilt by requiring that the position(s) of interest be always included during the TopHap analysis. This step is optional and available in the TopHap analysis.
2.2 Genome data acquisition and assembly
We obtained an MSA containing 68,057 genomes (hereafter, 68KG) of the SARS-CoV-2 coronavirus from human hosts analyzed in Kumar et al. (2021). These genomes were obtained from the GISAID database (https://www.gisaid.org) and covered the period from December 24, 2019 to October 12, 2020. The 68KG alignment was generated after filtering 133,741 SARS-CoV-2 genomes, such that genomes shorter than 28,000 bases and those with many ambiguous bases were removed. Three outgroup coronavirus genomes were added to the alignment: Rhinolophus affinis (RaTG13) and Rhinolophus malayanus (RmYN02) bats and the Manis javanica pangolin (MT040335) (Liu et al., 2020; Zhou et al., 2020). Following the above procedure, we also assembled a bigger dataset containing 1,106,862 genomes (hereafter 1MG) from the GISAID database covering the period from December 24, 2019 to September 11, 2021.
Annotations using Nextstrain and PANGO classifications. To compare TopHap phylogeny with the Nextstrain classification, we annotated all the TopHap haplotypes using the presence and absence of diagnostic Nextstrain mutations (https://nextstrain.org/ncov). We also assigned a PANGO lineage to each genome in the data using the Phylogenetic Assignment of Named Global Outbreak Lineages (PANGOLIN) software (Rambaut et al., 2020). TopHap haplotype ID was also assigned to genomes whose haplotype was identical to the TopHap haplotype. When a TopHap haplotype matched with multiple PANGO lineages, we paired a TopHap haplotype with the major PANGO lineage.
3 Results
We stratified sequence isolates by month of sampling and country to select variants and haplotypes in the TopHap analysis of the 68KG dataset. We used spatial and regional maf and hf cutoffs of 5% to avoid including problematic variants and haplotypes created by recurrent/backward mutations and sequencing error, particularly because of the small number of genomes available for many spatiotemporal slices. When the number of genomes sampled from a country was fewer than 500, we manually pooled them with adjacent countries with fewer than 500 genomes for countries located on the same continent. Also, the numbers of genomes in December 2019 and October 2020 were <500, so we pooled them with January 2020 and September 2020 time slices, respectively. The TopHap’s filtering process (5% threshold for maf and hf) produced an MSA of common haplotypes that consisted of 83 variable sites and 39 unique haplotypes after pruning haplotypes that were not sampled in all bootstrap analyses.
We subjected the final haplotype MSA to an MP analysis in MEGA (Tamura et al., 2021) and an ML analysis in RAxML (Kozlov et al., 2019). The heuristic search was applied with the default option (Subtree-Pruning-Regrafting). For the ML analysis, we used GTR nucleotide substitution model and GAMMA among-site rate heterogeneity (four discrete rate categories) in RAxML (https://raxml-ng.vital-it.ch). We used Lewis’ ascertainment bias correction since the haplotype MSA contains only variable sites (Lewis, 2001). In the ML phylogeny, many branches received low bootstrap support (<52%; Supplementary Fig. S2). Therefore, we disregarded these branches when comparing ML and MP phylogenies and found that the two phylogenies were identical. This result prompted us to implement the MP analysis in the TopHap software.
The TopHap analysis of the 68KG dataset with 100 bootstrap replicates required <1 hour, and all but three groups received >95% bootstrap support (Fig. 3). The remaining groups received >80% bootstrap support. Here, we used Greek symbols for variants designated with symbols in Kumar et al. (2021) (Supplementary Table S2). In this phylogeny, many branches were longer than one mutation, indicating that haplotypes corresponding to intermediate viruses did not rise to high enough frequency in the data or were not sampled. Also, more than two evolutionary lineages originated from the same ancestral lineage in many cases, which is likely to be real because there was no mutational homoplasy around those branches in the phylogeny (see further discussion below).
Fig. 3.

The TopHap phylogeny of 68KG SARS-CoV-2 major haplotypes. Numbers near nodes are bootstrap confidence limits derived from bootstrap resampling of genomes. Mutations mapped are shown on branches. When the same mutations were included in Kumar et al. (2021), their mutation IDs (Greek symbols) were shown. Their mutations and genomic positions are given in the right side. The Nextstrain clade ID was annotated based on their diagnostic mutations and is provided at the far right. PANGO lineage was annotated for each genome using PANGOLIN software (Rambaut et al., 2020). We also annotated TopHap haplotype for each genome by comparing its haplotype with TopHap haplotypes. When an observed haplotype did not perfectly match any of the TopHap haplotypes, we did not assign any for the genome. Using these genome annotations, we paired each TopHap haplotype with the major PANGO lineage, and the percentage of genomes containing it is presented in the parenthesis
3.1 Temporal trends in variant frequencies
The TopHap approach does not use temporal information from sample isolation dates during the reconstruction of the haplotype phylogeny. Therefore, a TopHap phylogeny can be used to test the concordance between the temporal order of mutation occurrence with the order of their frequency predicted by the phylogeny. For this analysis, we first mapped mutations to every branch in the SARS-CoV-2 phylogeny by reconstructing the most parsimonious ancestral states. All mutations mapped unambiguously (Fig. 3). Frequencies of variants generally decreased from the root to tip on evolutionary lineages (e.g. Fig. 4a). For example, the mutant bases mapping to the earliest diverging branches in the TopHap phylogeny occurred with the highest frequency in the 68KG dataset. Also, the timing of the first sampling date of variants increased on lineages from the root to tips (Fig. 4b). These trends are consistent with the clonal evolution without recombination of SARS-CoV-2 during the early stage of the pandemic.
Fig. 4.

The number of branches from the root to a tip and global mutant nucleotide frequency (a) and the first time the mutation was observed (b). Numbers are the tip identifiers from Figure 3. The same color code was used in (b). Days are counted from the first sample date (December 24, 2019)
3.2 Comparing 68KG TopHap phylogeny with the MOA tree
To directly compare the TopHap phylogeny with the MOA mutation tree reported in Kumar et al. (2021), we also used spatial and regional maf and hf cutoffs of 1% in analyzing the same 68KG dataset. The inferred TopHap phylogeny contained a much larger number of haplotypes (302) and variable sites (570), which included all 83 variants with >1% global maf analyzed in Kumar et al. (2021). The order of these mutations in the TopHap phylogeny was similar to the MOA mutation tree in Kumar et al. (2021), with a few minor differences noted in Supplementary Figure S3. Similarly, TopHap phylogeny agreed well with Nextstrain and PANGO trees (Fig. 5).
Fig. 5.
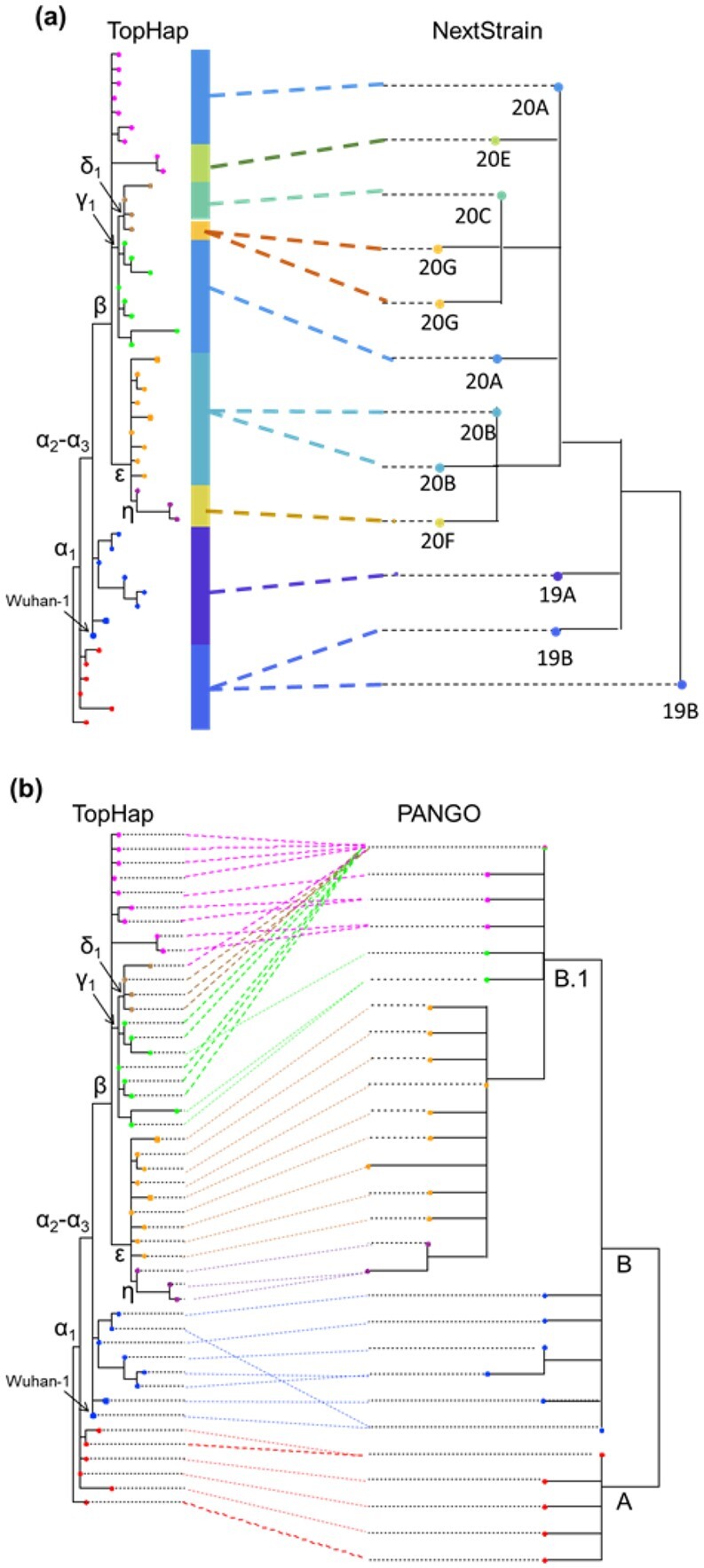
The comparison of TopHap phylogeny with the (a) Nextstrain and (b) PANGO phylogenies. (a) Only clades included in the 68KG data are shown. (b) Only PANGO lineages that were included in the TopHap phylogeny were used. Corresponding PANGO IDs are found in Figure 3
3.3 TopHap analysis of >1 million SARS-CoV-2 genomes
Next, we analyzed a recent snapshot of SARS-CoV-2 genome collection acquired 1 year after assembling the 68KG dataset. After filtering out incomplete genome sequences, we constructed an alignment of 1,106,862 genomes (1MG dataset) that is 16 times bigger than the 68KG dataset. Using TopHap with a 5% threshold for maf and hf, we obtained an MSA of 150 haplotypes with 675 variable sites. The number of haplotypes increased only 4-fold between 68KG and 1MG datasets, and the number of variable sites increased by eight times. This greater increase of the number of variable sites than the number of haplotypes is likely due to episodic mutations in the SARS-CoV-2 evolution, where intermediate haplotypes are not found in appreciable frequency. For example, some multi-mutation branches in the TopHap phylogeny correspond well with the unresolved branching order of mutations in Kumar et al. (2021), which was suggested to be due to evolutionary bursts (e.g., three ε mutations). These bursts are also observed in the 1MG phylogeny (Fig. 6a), which shows high concordance with the 68KG phylogeny. Orders of the earliest mutations (α1−α3, β1−β3, ε1−ε3, γ1, δ1 and ν1−ν2) were the same in 1MG and 68KG phylogenies. Therefore, inferences about the early history reported for the 68KG dataset are robust to the expanded sampling of genomes.
Fig. 6.

The 1MG TopHap Phylogeny. (a) Numbers near nodes are bootstrap confidence limits derived from bootstrap resampling of genomes. Early mutations that were predicted in Kumar et al. (2021) are shown on branches using their mutation IDs (Greek symbols). Their mutations and genomic positions are given in Figure 3. The haplotypes with concerning mutations are indicated by using WHO IDs, and 20A EU2 and 20E (EU1) are Nextstrain clade IDs. These haplotypes were identified by annotating PANGO and Nextstrain lineage for each genome. We also annotated TopHap haplotype for each genome by comparing its haplotype with TopHap haplotypes. When an observed haplotype did not perfectly match any of the TopHap haplotypes, we did not assign any for the genome. Using these genome annotations, we paired each TopHap haplotype with the major PANGO and Nextstrain lineage, which contained the WHO annotation. We assigned WHO ID when at least one of the annotations indicated it. Evolutionary relationship of lineages with concerning mutations by (b) Nextstrain and (c) TopHap
The 1MG TopHap phylogeny shows the evolutionary history of key WHO-designated variants of concern (VOC). This includes WHO-ALPHA, WHO-BETA, WHO-DELTA, WHO-ETA, WHO-GAMMA and WHO-LAMBDA variants. We used the WHO-prefix to avoid conflict between Kumar et al. (2021) notations for mutations and WHO’s notation for multi-mutation strains. Notably, Kumar et al. (2021) mutation identifiers were proposed earlier than the WHO designations, so we have retained them.
These VOCs’ placements in TopHap are consistent with those in the Nextstrain taxonomy (Fig. 6b and c). For example, Nextstrain and TopHap infer WHO-ALPHA, WHO-GAMMA and WHO-LAMBDA to be sister lineages. Also, the N501Y Spike recurrent mutation (A23063T) occurred independently in WHO-ALPHA, WHO-GAMMA and WHO-BETA lineages, which are placed correctly by TopHap (Fig. 6a). Since the WHO-OMICRON variant appears to have originated after the last day of sampling the 1MG dataset, the TopHap phylogeny does not contain it. So, we used WHO-OMICRON’s diagnostic mutations listed on the Nextstrain website (https://nextstrain.org/ncov) to place it in the 1MG TopHap phylogeny. WHO-OMICRON is an offspring of the ε lineage, as it contains α, β and ε mutations. This placement agrees with Nextstrains’ inference (Fig. 6b and c).
The TopHap analysis of the 1MB dataset with a 5% threshold for maf and hf was completed in <3 h, including 100 bootstrap replicates. In this phylogeny, 57 out of 72 clusters received 100% bootstrap support, most of which were shallow clusters (close to the tips). This pattern was consistent with the 68KG data analysis.
We explored the impact of using a larger number of bootstrap replicates (1,000), which took 10 times longer, on the estimates of the bootstrap support values. Bootstrap support values from 100 and 1,000 replicates were generally similar (Supplementary Fig. S4). For example, the evolutionary position of WHO-DELTA was 92% and 93% in the two analyses, respectively. Therefore, the use of 100 bootstrap replicates appears to be sufficient.
3.4 TopHap analysis of 1MG dataset with lower maf and hf thresholds
We also reconstructed the 1MG dataset using a 1% cutoff for maf and hf to select regional variants and haplotypes in TopHap. In this phylogeny, the number of variable sites and haplotypes increased to 1,793 with 594, respectively (Supplementary Fig. S5). Restricting the comparison to only haplotypes common in both phylogenies, i.e., 1% and 5% cutoffs, we found a very high concordance, as there were only nine partition differences for some recent strain divergences that were likely caused by the presence of rarer haplotypes containing recurrent/reversal mutations in the 1% cutoff analysis. The evolutionary placements of WHO VOC were also identical between the phylogenies. The inclusion of these haplotypes in the 1% cutoff TopHap phylogeny reduced the bootstrap support in general, except for shallow nodes (close to tips). Therefore, one needs to use high enough maf and hf to avoid haplotypes disrupting phylogenetic inference. When the evolutionary relationship of low-frequency haplotypes needs to be inferred, we suggest using TopHap’s facility to place low-frequency haplotypes of interest into a robust and well-supported phylogeny (Fig. 2).
3.5 Rooting the tree of SARS-CoV-2 genomes
We find that Nextstrain and PANGO phylogeny broadly agree with 68KG and 1MG TopHap phylogenies, except for the root placement (Figs 3, 5 and 6). For example, clade 19A is at the root of the Nextstrain phylogeny, but TopHap phylogenies (using the bat/pangolin outgroups) suggest that Clade 19A is derived. The bootstrap support was modest (>66%) for the root of the TopHap phylogeny, but no bootstrap replicates supported the Nextstrain rooting, and <34% supported the PANGO rooting.
The TopHap rooting is similar to that implied by MOA in Kumar et al. (2021). The TopHap root is also consistent with one of the two preferred roots in Bloom (2021), who analyzed 13 additional partial genomes from the earliest phases of the pandemic in China. Key early mutations analyzed in Bloom (2021) contained an additional variable site (genomic position 29,095), where the minor base occurred with too low a frequency to be included in the TopHap analysis (0.4% in the 68KG dataset). We, therefore, added it to the 68KG MSA and referred to this mutation as x (= 29,095, U is minor and C is major).
We also searched for other rare haplotypes to see if others tend to cluster at or near the root position in the 68KG TopHap phylogeny. We found 936 additional unique haplotypes in the 68KG dataset more than once. We tested their placement one by one in the TopHap phylogeny. Only two were attached at or near the root. One of them had the same haplotype sequence as that of MRCA and was present in 17 isolates. This haplotype is the proCoV2 sequence reported by Kumar et al. (2021); it circulated in early 2020. The other haplotype differed from the proCoV2 sequence in two genomic positions [29,095 (location of x variant) and 18,060 (location of α1 variant)]. It was attached to the trunk of the phylogeny (Fig. 7a). This haplotype is the same as Bloom (2021) suggested to be important in rooting the SARS-CoV-2 phylogeny. Also, Bloom (2021) reported two evolutionary scenarios with this mutation x (Fig. 7b and c), which led us to consider five alternative scenarios based on TopHap, MOA, Bloom (2021), Nextstrain and PANGO (Fig. 7). All these scenarios involved eight positions that experienced early mutations (α1–α3, β1–β3, ν1–ν2 and x) to give rise to seven major haplotypes. Therefore, we inferred phylogenies containing only α1–α3, β1–β3, ν1–ν2 and x mutations using MP, i.e. we attached the haplotype with the x mutation into the phylogenies of TopHap (Fig. 7a and b for two equally parsimonious solutions), MOA (Fig. 7b), Nextstrain (Fig. 7e) and PANGO (Fig. 7d). Our evaluation of these five scenarios is the most detailed comparison to date because of the size of the dataset analyzed and the variants included. For example, ν1 and ν2 variants were absent in Bloom (2021) dataset because the genomes included were only until the end of January 2020, and variant x was missing from Kumar et al. (2021) analysis because its global frequency was <1% in the 68KG dataset.
Fig. 7.

The early history of SARS-CoV-2 variants. Five root positions are explored in which the haplotype with mutation x has been added to the TopHap phylogeny in Figure 3 (a and b), Kumar et al. (2021) mutational history (b), Bloom (2021) phylogeny (b and c), PANGO classification (d) and the Nextstrain classification (e). Haplotypes have eight positions that contain variants α1–α3, β1–β3, ν1–ν2 and x. Genomic positions are shown whenever a mutation occurs: green highlighted box with a letter for forward and red highlighted box without a letter for backward mutations. Using the MP criteria, we placed the haplotype with x variant into each phylogeny. TopHap had two equally parsimonious solutions (a and b), where the ML placement predicted scenario A. ML lnL and the number of MP substitutions are shown. WH-1 is the haplotype corresponding to the Wuhan-1 genome. The gray triangle represents all the other SARS-CoV-2 haplotypes of the ongoing infections in the world
We then evaluated these five scenarios (topologies) using MP and ML optimality criteria (Fig. 7). In the MP analysis, scenarios A, B and C were equally parsimonious, and D and E (PANGO and Nextstrain, respectively) were less parsimonious by 1 and 3 mutations. Scenarios D and E were also less likely than A, B and C, where we estimated the log-likelihood (lnL) of all five scenarios (topologies) using a GTR model of nucleotide substitutions in MEGA for the haplotypes shown in Figure 7. While the lnL of scenario A was the highest, it was only slightly higher (difference in lnL < 1.7) than that for B and C that were equally likely. Among scenarios A, B and C, variant x was lost in B, while variant α1 was acquired twice in A and lost once in C.
In all the three equally most parsimonious scenarios (A, B and C), the addition of mutation x pushes back the MRCA of SARS-CoV-2 by one mutation compared to the proCoV2 sequence of Kumar et al. (2021). In these cases, the number of differences between Wuhan-1 and the MRCA is four (Fig. 7). With a mutation rate range of 6.64 × 10−4 to 9.27 × 10−4 substitutions per site per year (Pekar et al., 2021), we can estimate that proCoV2 existed 7.7–10.8 weeks before the December 24, 2019 sampling date of Wuhan-1. This places the progenitor of SARS-CoV-2 to have evolved in mid-September to early-October 2019, many weeks earlier than the mid-November 2019 date proposed by Pekar et al. (2021). For their analysis, Pekar et al. (2021) used the rooting from scenario D in which the lineage containing α2–α3 and β1–β3 (PANGO B) is a sister group of the lineage containing α1 and ν1–ν2 (PANGO A) (Fig. 7d). As noted above, this scenario receives lower bootstrap support than the alternative in which PANGO B arose from the ancestor containing α1. In this sense, Pekar et al. (2021) have likely dated an event that occurred downstream of the MRCA.
4 Conclusions
The ongoing global efforts to monitor the evolution of the SARS-CoV-2 coronavirus have motivated many laboratories worldwide to generate genome sequences continuously. The number of genomes has grown quickly, becoming orders of magnitude greater than the genome size. Rapid growth, low sequence variability and the presence of sequencing error have made the direct use of phylogenetic methods on genome alignments challenging for such data (e.g. Morel et al., 2021).
We have shown that the TopHap phylogeny for common variants and haplotypes in the 68KG SARS-CoV-2 dataset works well and agrees with the mutation tree produced using MOA (Kumar et al., 2021). But, the TopHap approach offers some advantages over MOA. Firstly, MOA assumes the sequencing error rate to be constant throughout the outbreak, which is unlikely to hold for pathogenomic datasets acquired in different laboratories at different times.
Secondly, MOA analysis needs to have mutant bases indicated at the outset, a limitation addressed by Kumar et al. (2021), but at a large computational expense. In contrast, TopHap analyses directly use outgroup in standard phylogenetic analysis. TopHap analysis is certainly more computationally efficient as the analysis of the 68KG dataset took only a few hours. In contrast, MOA took more than a week to compute.
Thirdly, TopHap analysis can use well-established methods to infer phylogeny and ancestral sequences to identify recurrent and backward mutations. In contrast, MOA assumes an infinite site model and, thus, is not suitable for detecting recurrent and backward mutations. Lastly, rarer haplotypes can also be attached to a backbone of a TopHap phylogeny by simply adding the genomic position of interest in constructing the MSA of haplotypes, as demonstrated above.
In conclusion, TopHap is a simple and effective method to build haplotype phylogenies and assess their statistical robustness. TopHap can be applied in any data containing a large number of sequences with a handful of variants, including other pathogens and tumor single-cell sequencing data that is now producing a large number of somatic cell sequences (Navin, 2015).
Supplementary Material
Acknowledgments
We thank Sudip Sharma and reviewers for useful comments. We are thankful to everyone depositing genome data on GISAID (list at http://igem.temple.edu/COVID-19).
Author contributions
S.K. and S.M. developed the original method and designed research; S.M, M.S. and T.D. implemented the technique; M.A.C.-O., S.M., T.D. and Q.T. performed analyses; S.L.K.P. and S.W. assembled sequence alignments; and S.K., S.M., M.A.C.-O., Q.T. and S.L.K.P. wrote the article.
Funding
This work was supported by the U.S. National Science Foundation [DEB-2034228 to S.K. and S.M., DBI-2027196 to S.P.]; and U.S. National Institutes of Health [GM-139504 to S.K., AI-134384 to S.P., LM-014005 to S.M.].
Conflict of Interest: none declared.
Contributor Information
Marcos A Caraballo-Ortiz, Institute for Genomics and Evolutionary Medicine, Temple University, Philadelphia, PA 19122, USA; Department of Biology, Temple University, Philadelphia, PA 19122, USA.
Sayaka Miura, Institute for Genomics and Evolutionary Medicine, Temple University, Philadelphia, PA 19122, USA; Department of Biology, Temple University, Philadelphia, PA 19122, USA.
Maxwell Sanderford, Institute for Genomics and Evolutionary Medicine, Temple University, Philadelphia, PA 19122, USA; Department of Biology, Temple University, Philadelphia, PA 19122, USA.
Tenzin Dolker, Institute for Genomics and Evolutionary Medicine, Temple University, Philadelphia, PA 19122, USA; Department of Biology, Temple University, Philadelphia, PA 19122, USA.
Qiqing Tao, Institute for Genomics and Evolutionary Medicine, Temple University, Philadelphia, PA 19122, USA; Department of Biology, Temple University, Philadelphia, PA 19122, USA.
Steven Weaver, Institute for Genomics and Evolutionary Medicine, Temple University, Philadelphia, PA 19122, USA; Department of Biology, Temple University, Philadelphia, PA 19122, USA.
Sergei L K Pond, Institute for Genomics and Evolutionary Medicine, Temple University, Philadelphia, PA 19122, USA; Department of Biology, Temple University, Philadelphia, PA 19122, USA.
Sudhir Kumar, Institute for Genomics and Evolutionary Medicine, Temple University, Philadelphia, PA 19122, USA; Department of Biology, Temple University, Philadelphia, PA 19122, USA; Center of Excellence in Genomic Medicine Research, King Abdulaziz University, Jeddah, Saudi Arabia.
References
- Andersen K.G. et al. (2020) The proximal origin of SARS-CoV-2. Nat. Med., 26, 450–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger S.A. et al. (2011) Performance, accuracy, and web server for evolutionary placement of short sequence reads under maximum likelihood. Syst. Biol., 60, 291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom J.D. (2021) Recovery of deleted deep sequencing data sheds more light on the early Wuhan SARS-CoV-2 epidemic. Mol. Biol. Evol., 38, 5211–5224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouckaert R.R. (2010) DensiTree: making sense of sets of phylogenetic trees. Bioinformatics, 26, 1372–1373. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution, 39, 783–791. [DOI] [PubMed] [Google Scholar]
- Jahn K. et al. (2016) Tree inference for single-cell data. Genome Biol., 17, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlov A.M. et al. (2019) RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics, 35, 4453–4455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S. et al. (2021) An evolutionary portrait of the progenitor SARS-CoV-2 and its dominant offshoots in COVID-19 pandemic. Mol. Biol. Evol., 38, 3046–3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis P.O. (2001) A likelihood approach to estimating phylogeny from discrete morphological character data. Syst. Biol., 50, 913–925. [DOI] [PubMed] [Google Scholar]
- Liu P. et al. (2020) Are pangolins the intermediate host of the 2019 novel coronavirus (SARS-CoV-2)? PLoS Pathog., 16, e1008421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsen F.A. et al. (2010) pplacer: linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinformatics, 11, 538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morel B. et al. (2021) Phylogenetic analysis of SARS-CoV-2 data is difficult. Mol. Biol. Evol., 38, 1777–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navin N.E. (2015) The first five years of single-cell cancer genomics and beyond. Genome Res., 25, 1499–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie Q. et al. (2020) Phylogenetic and phylodynamic analyses of SARS-CoV-2. Virus Res., 287, 198098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekar J. et al. (2021) Evidence against the veracity of SARS-CoV-2 genomes intermediate between lineages A and B. Virological.org (21 March 2022, date last accessed).
- Pipes L. et al. (2021) Assessing uncertainty in the rooting of the SARS-CoV-2 phylogeny. Mol. Biol. Evol., 38, 1537–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A. et al. (2020) A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol., 5, 1403–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K. et al. (2021) MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol., 38, 3022–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turakhia Y. et al. (2021) Ultrafast Sample placement on Existing tRees (UShER) enables real-time phylogenetics for the SARS-CoV-2 pandemic. Nat. Genet., 53, 809–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dorp L. et al. (2020) Emergence of genomic diversity and recurrent mutations in SARS-CoV-2. Infect. Genet. Evol., 83, 104351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P. et al. (2020) A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature, 579, 270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.