

Abstract
Catalytic methods for the direct introduction of hydrogen isotopes into organic molecules are essential to the development of improved pharmaceuticals and to the alteration of their absorption, distribution, metabolism, and excretion (ADME) properties. However, the development of homogeneous catalysts for selective incorporation of isotopes in the absence of directing groups under practical conditions remains a long-standing challenge. Here, we show that a phosphine-ligated, silver-carbonate complex catalyzes the site-selective deuteration of C–H bonds in five-membered aromatic heterocycles and active pharmaceutical ingredients that have been resistant to catalytic H/D exchange. The reactions occur with CH3OD as a low-cost source of the isotope. The silver catalysts react with five-membered heteroarenes lacking directing groups, tolerate a wide range of functional groups, and react in both polar and nonpolar solvents. Mechanistic experiments, including deuterium kinetic isotope effects, determination of kinetic orders, and identification of the catalyst resting state, support C–H bond cleavage from a phosphine-ligated, silver-carbonate intermediate as the rate-determining step of the catalytic cycle.
Keywords: hydrogen isotope exchange, silver catalyst, C–H activation, heteroarenes, deuteration
Graphical Abstract
INTRODUCTION
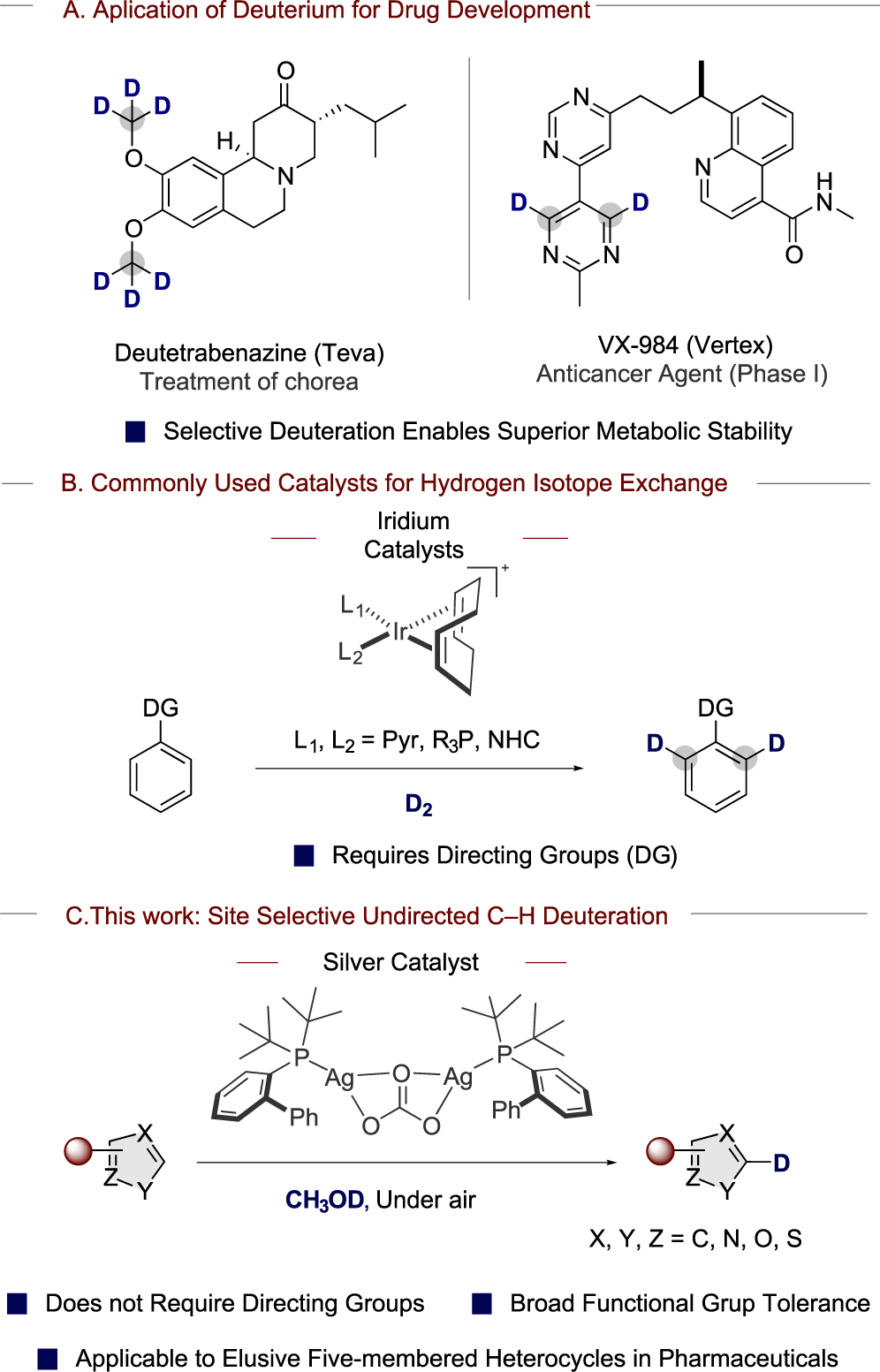
Synthetic methods to incorporate hydrogen isotopes selectively into organic molecules have attracted considerable interest, due to the widespread application of isotopically labeled compounds in chemical and pharmaceutical research. For example, the selective replacement of hydrogen for deuterium at C–H bonds prone to metabolic degradation is a useful approach for the development of new pharmaceuticals with improved pharmacokinetic and pharmacodynamic properties (Scheme 1a).1 Deuterated and tritiated biologically active compounds and pharmaceuticals are also employed in absorption, distribution, metabolism, and excretion (ADME) studies, which are pivotal for the understanding of their of mode of action and toxicology.2 In addition, deuterated molecules are used to investigate reaction mechanisms and are used as standards for mass spectrometry and nuclear magnetic resonance.3
Scheme 1.
Applications of Deuterium and Recent Developments in Catalytic Hydrogen Isotope
Classical methods to synthesize deuterated and tritiated compounds require prefunctionalized starting materials, which are subjected to reductive or dehalogenative processes during the installation of the desired hydrogen isotope.1b,4 However, hydrogen isotope exchange (HIE) catalyzed by homogeneous transition metals has become a widely applicable method for the late-stage introduction of hydrogen isotopes directly into C–H bonds.5 Thus, this method, which can be conducted with commercially available sources of deuterium or tritium, is employed in industry for the isotopic labeling of complex pharmaceuticals.
Ir catalysts, including Crabtree’s catalyst and variants developed by Kerr, are the most used catalyst for H/D exchange because they catalyze the selective isotopic labeling of sp3 and sp2 C–H bonds with the tolerance of many functional groups.6 However, these catalysts require directing groups, and binding of other Lewis basic functional groups to the catalyst reduces the level of isotopic incorporation, presumably because these functional groups bind and poison the catalyst (Scheme 1b).6a,f
Recently, research by Chirik7 and MacMillan8 on base-metal and photoredox catalysts has enabled reactivity and site selectivity that are complementary to those of the Ir-catalyzed methods. As a result, these studies have provided new methods for the selective deuteration and tritiation of sp3 and sp2 C–H bonds in complex pharmaceuticals without the need for directing groups. In spite of these recent advances, common, medicinally relevant molecular scaffolds, such as those based on five-membered ring heteroarenes, remain resistant to HIE under practical conditions with any type of catalyst.
Five-membered aromatic heterocycles are prevalent in pharmaceuticals, agrochemicals, and natural products.9 The introduction of these heterocycles into biologically active agents is a widely used approach for the optimization of their toxicology properties10 because they can alter lipophilicity, polarity, and hydrogen-bonding capacity.10a Five-membered aromatic heterocycles are also used in drug discovery as bioisosteres of benzene rings, carboxylic acids, and esters.10b,11 Despite the value of such heteroarenes, homogeneous catalysts for the selective incorporation of hydrogen isotopes in a broad range of five-membered aromatic heterocycles embedded in complex pharmaceuticals without the assistance of directing groups have been elusive. Base-induced H/D exchange under stoichiometric conditions has been employed for the isotopic labeling of a handful of five-membered heterocycles at multiple positions with limited tolerance of functional groups; however, these methods require high reaction temperatures (over 120 °C) and in some cases supercritical conditions (high temperatures combined with high pressures).12
Our group13 recently reported the activation of C–H bonds in arenes by silver complexes as part of a study of the mechanism of the direct allylation of arenes catalyzed by palladium complexes with silver additives. The groups of Larrosa,14 Sanford,15 and Zhu16 have reported that related silver complexes cleave the C–H bonds of arenes bound to Cr(CO)3, activated arenes containing multiple fluorine substituents, thiophene, and benzothiophene. The reactions of less activated arenes and heteroarenes have not occurred.
These previous studies suggested that silver salts and phosphine-ligated silver complexes bearing carboxylate ligands were the species in palladium-catalyzed couplings that cleaved C–H bonds, presumably by a carboxylate-assisted concerted metalation deprotonation step.14–16 However, characterization of a silver carboxylate species that effects C–H bond cleavage, particularly cleavage of the C–H bonds in heteroarenes, has not been conducted,14a and ligands that accelerate the rate of the C–H activation step were not identified. Thus, high concentrations of silver catalysts and stoichiometric amounts of bases were needed to activate the C–H bonds, even of these more reactive arenes, and moderate degrees of isotopic incorporation were observed during mechanistic experiments.
We considered that this C–H activation chemistry could be applied to unsolved problems in H/D exchange by creating silver complexes that cleave the C–H bonds of medicinally important five-membered heteroarenes and enable H/D exchange under practical conditions. This goal would require the development of discrete silver catalysts that cleave C–H bonds at faster reaction rates and with greater tolerance of functional groups than prior silver systems that were part of catalytic C–C bond-forming processes.
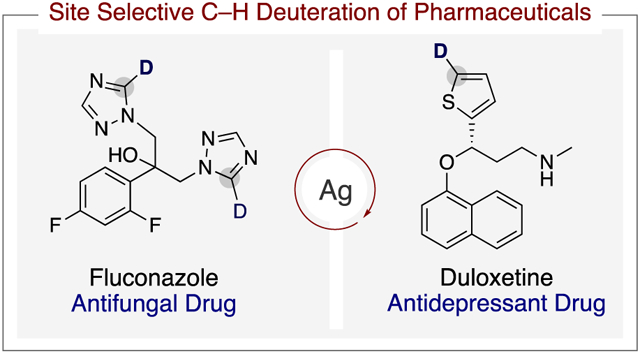
Herein, we describe the identification of a catalytic system based on silver for such site-selective deuteration of C–H bonds in medicinally relevant five-membered aromatic heterocycles, including imidazole, thiazole, triazole, thiophene, and indole. The developed method proceeds in the absence of directing groups, and it uses commercially available silver catalysts, phosphine ligands, and CH3OD as a low-cost source of deuterium (Scheme 1c). Moreover, we demonstrate that the functional group compatibility of this silver-catalyzed method is broad, and this property enables the isotopic labeling of complex pharmaceutical ingredients to produce new isotopologues.
RESULTS AND DISCUSSION
Development of Silver-Catalyzed C–H Deuteration.
We began our studies to achieve mild, silver-catalyzed HIE with 2-methylthiophene (1) as a model substrate for an electron-rich, five-membered heterocycle (Table 1). Initial experiments showed that the deuterated thiophene [2H]1 formed with moderate isotopic incorporation and perfect site selectivity toward the most acidic sp2 C–H bond in the presence of Ag2CO3 (2.5 mol %) and PPh3 (5 mol %) in CH3OD solvent after 8 h at 40 °C (Table 1, entry 1). A series of experiments with a range of commercially available phosphines demonstrated that [2H]1 formed in high isotopic incorporation (up to 95%) when the silver catalyst contained the electron-rich 2-(biphenyl)di-tert-butylphosphine (JohnPhos) ligand (Table 1, entry 5). Catalysts containing related electron-rich dialkylbiaryl phosphines, namely, 2-dicyclohexylphosphino-2′,6′-dimethoxybiphenyl (SPhos) and 2-(dicyclohexylphosphino)biphenyl (Cy-JohnPhos), led to lower degrees of isotope exchange. A notable feature of the silver catalyst ligated by JohnPhos is its high activity in the presence of atmospheric oxygen, which allowed the synthesis of [2H]1 in air with high isotopic composition (Table 1, entry 8) and with rates that were identical to those measured under nitrogen (see SI for details). Silver catalysts containing other anionic ligands were also tested. The analogues containing acetate and pivalate ligands were not active toward the deuteration of 1 (Table 1, entries 9–10). Control experiments in the absence of JohnPhos or Ag2CO3 showed the necessity of both components for the C–H bond deuteration reaction to proceed (Table 1, entries 11–12). Other commercially available sources of deuterium, including CH3CH2OD and D2O, in different organic solvents were also investigated, but reactions with these sources led to lower degrees of deuterium incorporation into 1 than did reactions conducted in CH3OD (see SI for further details).
Table 1.
Evaluation of Reaction Conditions for the Site Selective C–H Deuteration of 2-methylthiophene (1)

| |||
|---|---|---|---|
| entry | silver catalyst | ligand | %Da |
| 1 | Ag2CO3 | Ph3P | 29 |
| 2 | Ag2CO3 | Cy3P | 39 |
| 3 | Ag2CO3 | tBu3P | 52 |
| 4 | Ag2CO3 | tBu2PPh | 77 |
| 5 | Ag2CO3 | JohnPhos | 95 |
| 6 | Ag2CO3 | CyJohnPhos | 92 |
| 7 | Ag2CO3 | SPhos | 90 |
| 8b | Ag2CO3 | JohnPhos | 95 |
| 9b,c | AgOAc | JohnPhos | 0 |
| 10b,d | AgOPiv | JohnPhos | 0 |
| 11b | JohnPhos | 0 | |
| 12b | Ag2CO3 | 0 | |
Determined by 1H NMR in CDCl3.
Under air.
With 5 mol % of AgOAc.
With 5 mol % of AgOAc.
Scope of the Silver-Catalyzed C–H Deuteration.
The functional group compatibility of the silver-catalyzed C–H deuteration was first examined with mono- and disubstituted thiophenes.17 During the course of these experiments, we observed that a high degree of isotopic incorporation (up to 99%) was obtained after two cycles of C–H deuteration in air after 10–48 h at temperatures between 50 and 80 °C (see SI for details). After the first reaction cycle, purification of the intermediate deuterated product was not required, although addition of further silver catalyst was necessary in a few cases to obtain high isotopic incorporation (see SI for complete experimental details).
As shown in Scheme 2, thiophene 2, containing the electron-donating 3,4-ethylendioxyl moiety, underwent high deuterium incorporation at the position α to the sulfur atom. The thiophene in 3 was more reactive toward C–H bond deuteration than the phenyl unit, giving [2H]3 selectively in high yield. Reactions of thiophenes containing ester, benzoyl, carboxamide, and carbamate moieties showed that these coordinating functionalities did not direct C–H activation; selective deuteration at the C5-position of the thiophenes provided [2H]4–[2H]7 in high yield. The lack of directing effect of the pyridine in 2-(2-pyridyl)thiophene 8 is particularly noteworthy. The major site of incorporation of deuterium was the C–H bond distal to, rather than adjacent to, the basic nitrogen.
Scheme 2. Scope of Thiophenes That Undergo Selective C–H Deuterationa.
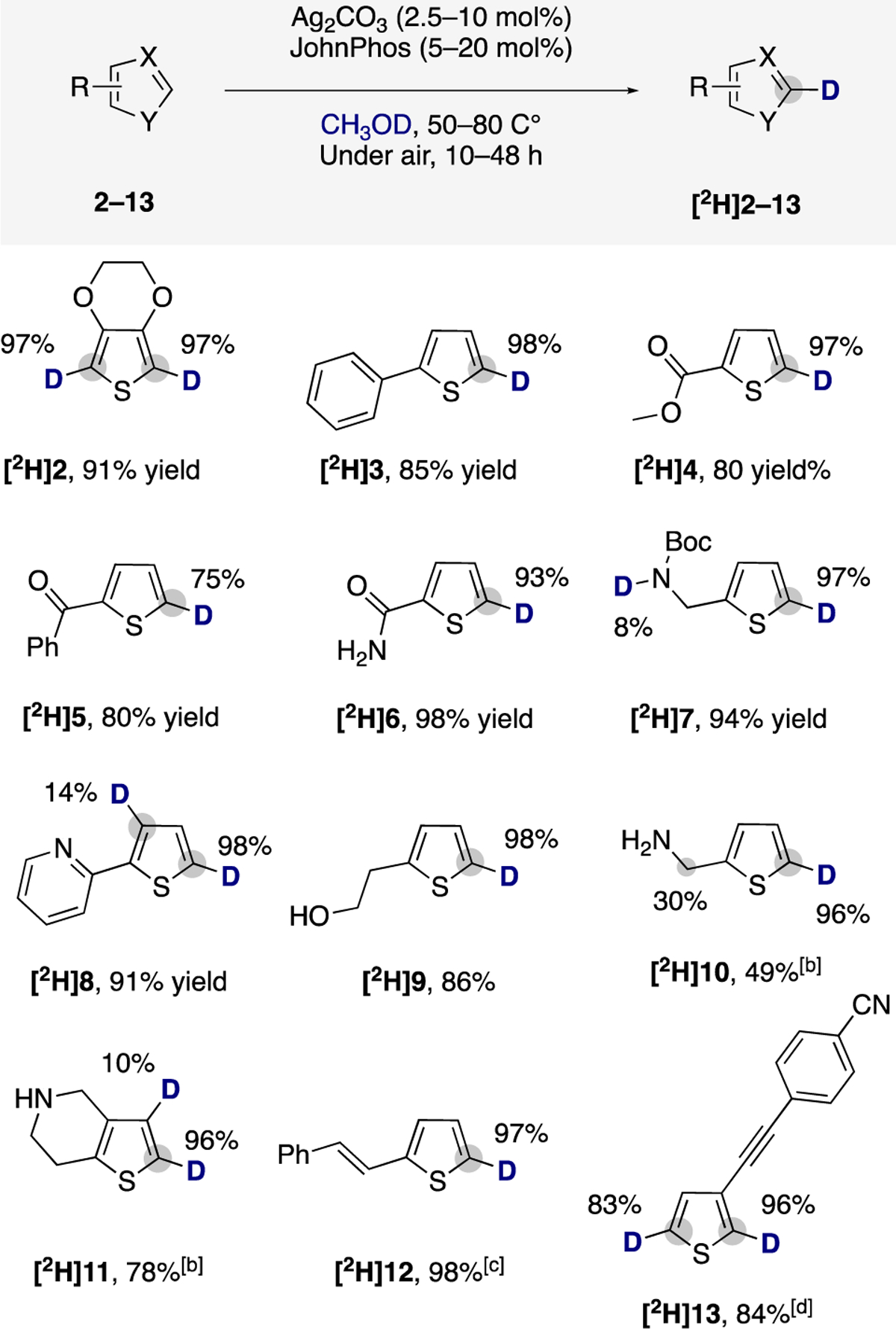
aPerformed over two reaction cycles (see SI for details). bUnder nitrogen and isolated as the hydrochloride salt. cWith THF as cosolvent. dWith CH2Cl2 as cosolvent.
Thiophenes bearing an unprotected alcohol or containing amines (9–11) underwent this HIE process, but the latter reactions required higher concentrations of silver catalysts and reaction temperatures to achieve high levels of isotopic incorporation. Accordingly, the deuterated [2H]10 product was formed from a reaction with 10 mol % of Ag2CO3 and 20 mol % of JohnPhos at 80 °C for 48 h under nitrogen to suppress partial decomposition of the starting material. Under the same reaction conditions, the thienopyridine 11, a secondary amine scaffold present in antiplatelet drugs,18 underwent deuteration at the two C–H bonds of the thiophene ring.
Finally, the broad functional group tolerance of the silver-catalyzed deuteration was further highlighted by the deuteration of 12 and 13. Existing homogeneous Ir catalysts for HIE do not tolerate unsaturated C–C bonds and nitriles, due to the sensitivity of the former toward reduction in the presence of deuterium or tritium gas and the irreversible binding of nitriles to Ir catalysts.6a,f Our developed method tolerated the aforementioned functionalities, as shown for [2H]12 and [2H]13. The synthesis of [2H]12 and [2H]13 also inspired further experiments on the use of cosolvents for catalytic HIE due to the insolubilities of 12 and 13 in CH3OD. We found that the developed silver catalyst is active for exchange of the C–H bonds with CH3OD in a range of polar and nonpolar solvents, including dimethylformamide, dimethyl sulfoxide, tetrahydrofuran, and dichloromethane (see SI for further details). Thus, the syntheses of [2H]12–[2H]13 were performed with THF or CH2Cl2 as cosolvent, respectively.
The scope of the silver-catalyzed method with a series of different classes of five-membered aromatic heterocycles is shown in Scheme 3. Thiazoles, imidazoles, and 1,2,4-triazoles are electron-poor five-membered heterocycles found in modern antibiotics, antifungal drugs, and agrochemicals.9a Despite this prevalence, homogeneous catalysts for isotopic exchange with these heterocycles without directing groups are lacking.7a,c Our data show that methyl thiazole-4-carboxylate (14) undergoes high deuterium incorporation at the 2- and 5-positions, and N-benzyl-1,2,4-triazole (15) undergoes selective deuteration at its most acidic C–H bond under the conditions we developed with the silver catalyst.
Scheme 3. Scope of Five-Membered Heterocycles That Undergo Selective C–H Deuterationa.
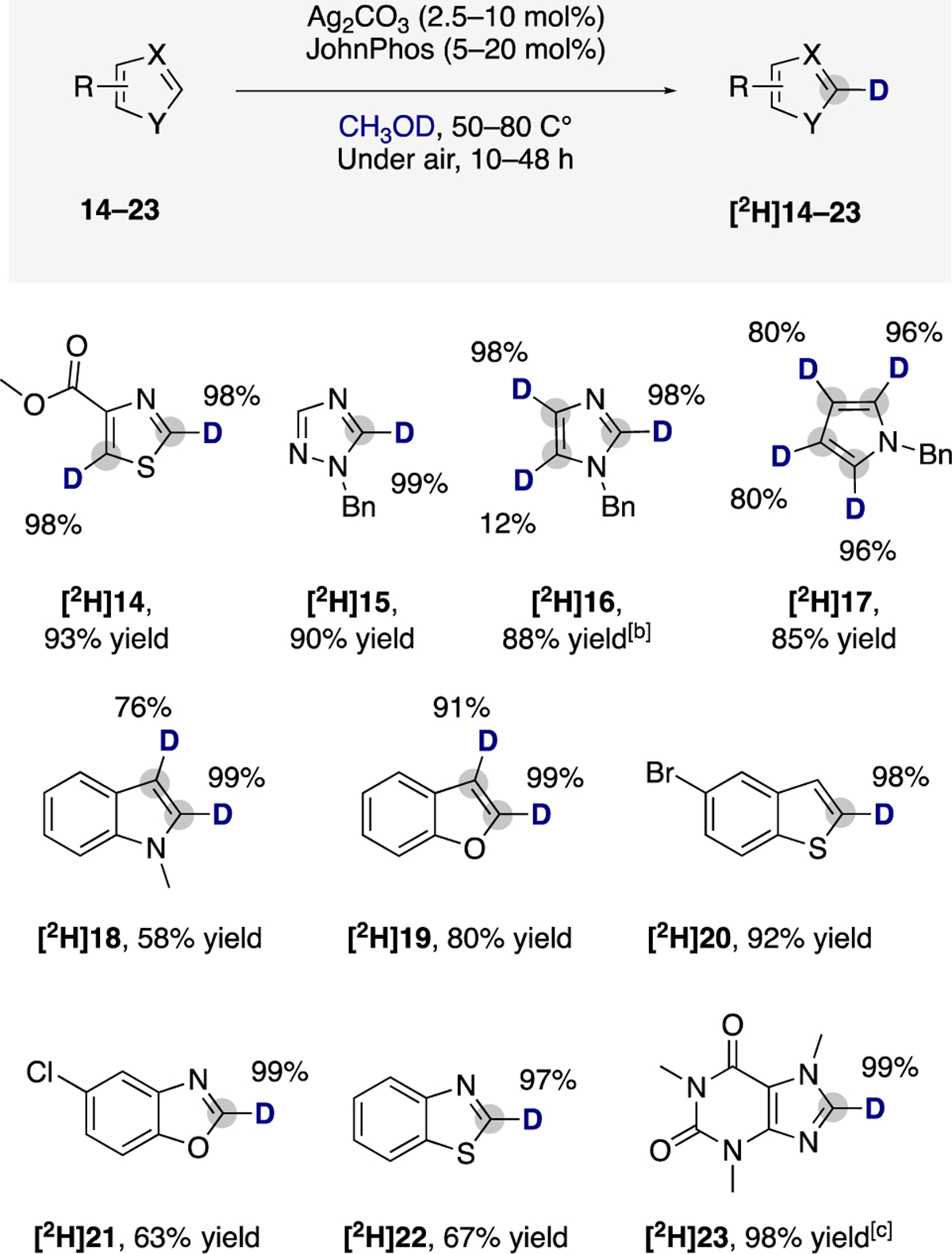
aPerformed over two reaction cycles (see SI for details). bWith di-tert-butylphenylphosphine under nitrogen. cWith CH2Cl2 as cosolvent.
N-Benzyl-imidazole (16) proved less reactive than other heteroarenes under the standard conditions. However, 16 underwent C–H deuteration at faster reaction rates when di-tert-butylphenylphosphine was used as ligand in place of JohnPhos (see SI for details). Therefore, the deuteration of 16 was performed after two reaction cycles over a period of 48 h at 65 °C under nitrogen in the presence of 10 and 20 mol % of Ag2CO3 and di-tert-butylphenylphosphine, respectively. This protocol led to deuteration of 16, with relative rates of reaction at the 2-, 4-, and 5-positions that follow the trend C2 > C4 > C5.
Indoles and pyrroles are important electron-rich heterocycles. However, isotopic labeling of these nitrogen heterocycles to high levels has required directing groups that enable C–H activation.19 In contrast, our silver-catalyzed method enabled high levels of incorporation of deuterium into 17 and 18 at the positions α and β to the nitrogen atom with the silver complex of JohnPhos as catalyst in air at 80 °C. Other fused heterocycles including benzofuran (19), benzothiophene (20), benzoxazole (21), benzothiazole (22), and caffeine (23) also underwent deuteration with a high level of deuterium incorporation at the most acidic C–H bond with the silver complex of JohnPhos as catalyst in air.
Deuteration of Pharmaceuticals.
The broad functional group tolerance and the site selectivity of the silver-catalyzed method complementary to that of existing methodologies20 prompted us to examine its applicability to the deuteration of C–H bonds in pharmaceuticals by conducting reactions with a series of active pharmaceutical ingredients. As shown in Scheme 4, pharmaceuticals containing xanthine heterocycles, namely, doxofylline (24) and pentoxyfilline (25), underwent high levels of incorporation of deuterium at the most acidic sp2 and sp3 C–H bonds at 50 °C after 24 h. The labeling of sp3 C–H bonds in 25 resulted from the facile enolization of the hydrogen atoms α to the carbonyl group. Etomidate (26), an anesthetic agent, and flumazenil (27), a GABA-antagonist, developed by Roche underwent deuteration at the imidazole core, while trace amounts of isotopic incorporation also were observed at the sp3 C–H bonds of the benzodiazepine ring of 27. It is important to note that 26 and 27 were deuterated with commercially available CH3CH2OD to avoid transesterification with CH3OD.
Scheme 4. Scope of Pharmaceuticals That Undergo Selective C–H Deuterationa.
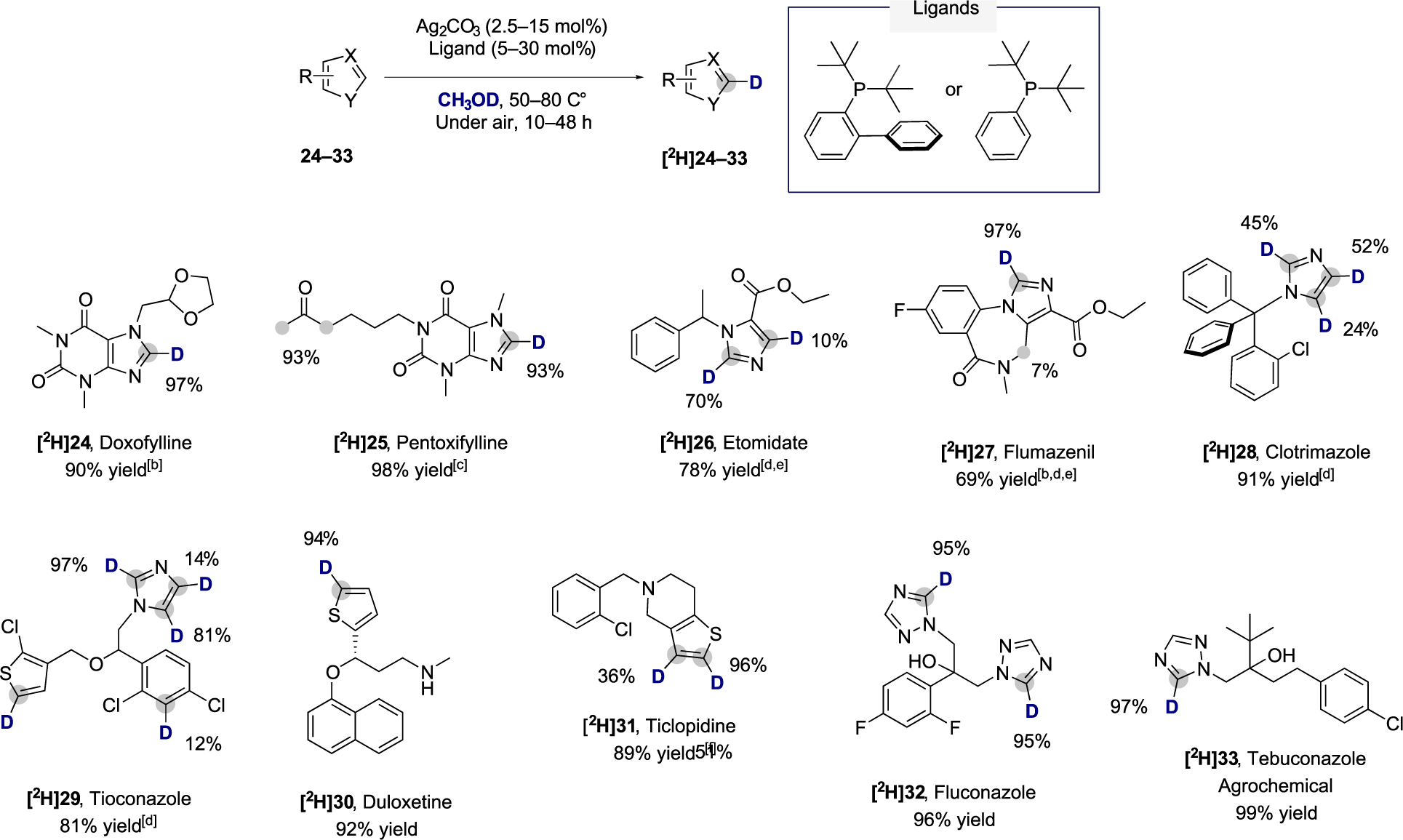
aPerformed over two reaction cycles (see SI for details), 50 equiv of deuterium was used for each of the 2 reaction cycles. For results with selected heterocycles and pharmaceuticals after one single cycle of deuteration, see Section 3 of the Supporting Information. bWith CH2Cl2 as cosolvent. cWith THF as cosolvent. dWith di-tert-butylphenylphosphine under nitrogen. eWith CH3CH2OD as deuterium source. fIsolated as the hydrochloride salt.
Antifungal drugs, such as clotrimazole (28) and tioconazole (29), which contain imidazole and thiophene, underwent C–H bond deuteration in these heterocycles at 65 °C after 48 h. In addition, partial isotopic incorporation at one position of the benzene ring of 29 was observed, which presumably resulted from the moderate acidity of the labeled C–H bond. The antidepressant (S)-duloxetine (30) and the antiplatelet drug ticlopidine (31) both underwent selective deuteration at the thiophene heterocycle. Finally, fluconazole (32) and tebuconazole (33), an antifungal and agrochemical agent, respectively, underwent selective deuteration at the most acidic position of their triazole units in excellent yield and isotopic incorporation.
Mechanistic Investigation.
Having demonstrated the synthetic applications of the silver-catalyzed HIE, we performed a series of experiments to gain insight into the reaction mechanism. We first examined whether C–H bond cleavage was involved in the rate-determining step of the reaction. To do so, we measured the initial rates of deuterium and protium incorporation in parallel reactions of 34 and [2H]34 in separate vessels. As shown in Scheme 5a, a large primary kinetic isotope effect (KIE) of 4.8 ± 0.3 was measured from experiments with 2.5 mol % of Ag2CO3 and 5 mol % of JohnPhos at 40 °C under air. The large value of the measured KIE strongly suggested that C–H cleavage occurs during the rate-determining step.
Scheme 5.

Determination of KIE and Reaction Orders of the Silver-Catalyzed C–H Deuteration: (A) Determination of KIE and (B) Kinetic Orders
To gain additional kinetic data on the silver-catalyzed C–H deuteration, we measured the dependence of the reaction rate on the concentration of benzothiophene (34), silver catalyst, and CH3OD under air at 40 °C by the method of initial rates (Scheme 5b). The obtained data showed a first-order dependence on the concentration of 34 and on the concentration of the silver catalyst, and a zero-order dependence on the concentration of CH3OD (determined in THF).21 These kinetic data are consistent with the primary KIE and suggest a mechanism in which the phosphine-ligated silver catalyst cleaves the C–H bond during the rate-determining step of the reaction.
To help interpret the kinetic data, we conducted experiments to determine the resting state of the silver catalyst. We monitored the course of the deuteration of the C–H bond of 2-methylthiophene (1) by 31P NMR at 40 °C in CD3OD under air in the presence of Ag2CO3 (2.5 mol %) and JohnPhos (5 mol %). An examination of the 31P NMR spectra after different amounts of isotopic incorporation into the thiophene revealed the presence of a single species containing phosphorus in the reaction medium. This signal was observed at 44.0 ppm and comprises two superimposed doublets with large and coupling constants (657 and 758 Hz, respectively). The large 1JAg–P coupling constants indicate that the resting state of the silver catalyst consists of a metal species bearing a single coordinated phosphine per silver atom with a tricoordinated (JohnPhos)AgX2 (X = oxygen) coordination environment (Figure 1).22
Figure 1.
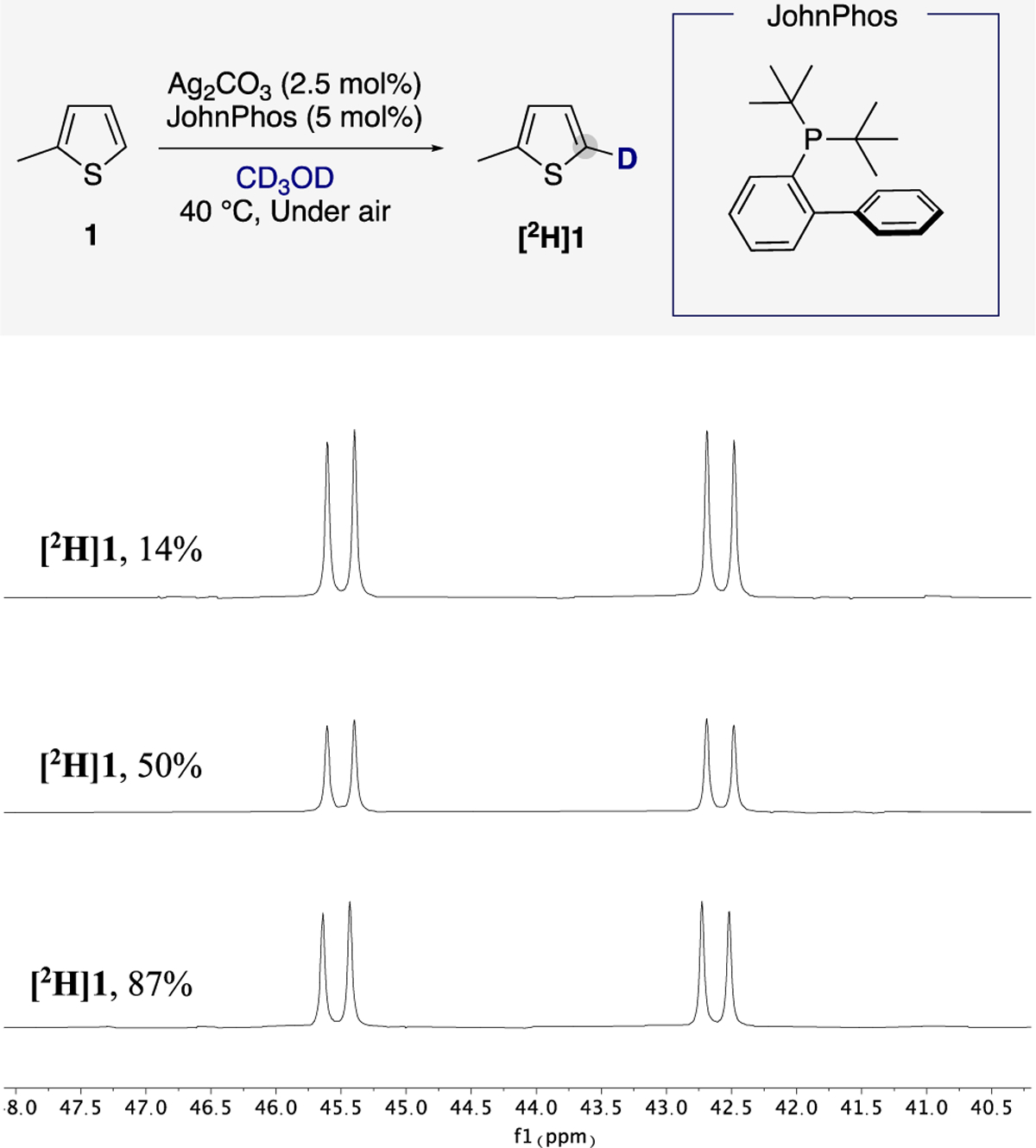
31P NMR spectra of the reaction mixture of 2-methylthiophene, Ag2CO3, and JohnPhos in CD3OD at different amounts of deuterium incorporation.
We also isolated and structurally characterized the phosphine-ligated silver complex involved in the C–H bond activation step. To isolate the observed complex, we investigated the coordination chemistry of Ag2CO3 (1 equiv) with JohnPhos (2 equiv). After allowing the two catalyst components to react under conditions of the catalytic reaction (30 min in CH3OH at 50 °C under air, Scheme 6) and appropriate workup, an air-stable, light-brown solid was isolated in 69% yield. The 31P NMR spectrum of the isolated solid in CD3OD at room temperature revealed the same set of phosphorus resonances centered at 44.0 ppm, as observed during the catalytic reaction (Figure 1). Moreover, the isolated complex catalyzed the C–H deuteration of 1 with rates that were identical to those with the catalyst formed in situ from Ag2CO3 and JohnPhos (see SI for details).
Scheme 6.
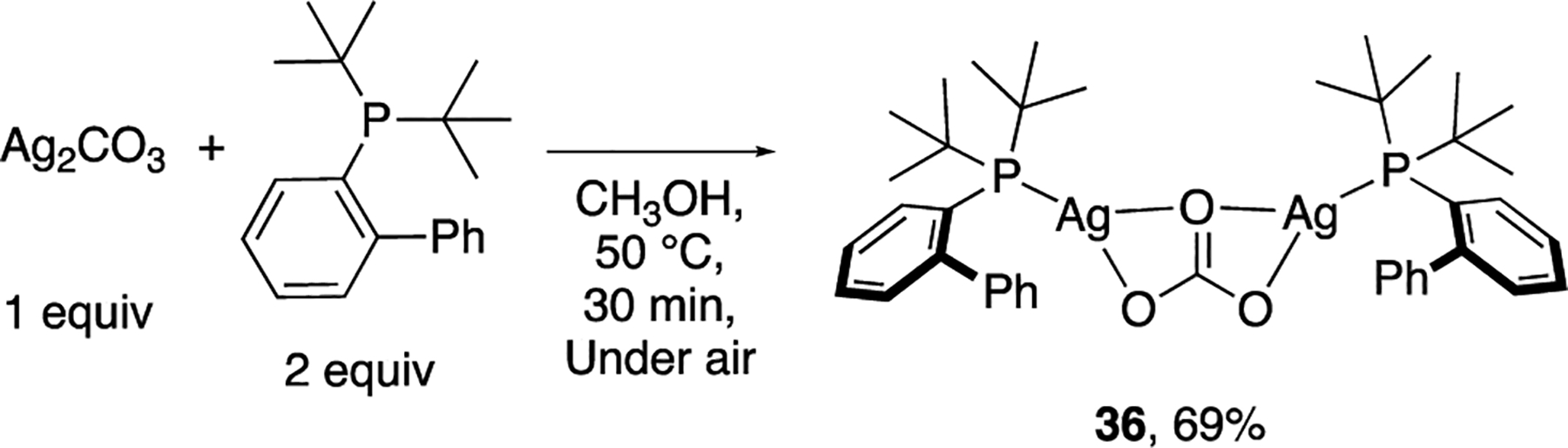
Synthesis of a Phosphine-Ligated Silver–Carbonate Complex
An analysis by solid-state IR spectroscopy of the isolated solid revealed two strong absorption bands at 1338 and 1416 cm−1, respectively, which were assigned to the E′ ν(CO) ν3 mode of the silver-coordinated CO32− ligand. Previously characterized metal-carbonato complexes have E′ ν(CO) absorption bands in the range of 1200–1600 cm−1.23 The small separation of the observed E′ ν(CO) absorption bands in 36 (Δν = 78 cm–1) is characteristic of a metal complex containing a bridging carbonate ligand and is indicative of a minimal distortion around the O–C–O angles. The Δν values of the E′ ν(CO) absorption bands in metal complexes containing bridging carbonato ligands are smaller than 250 cm−1, a difference in frequency that has been associated with small distortions of the angles around the carbonate moiety in this coordination mode.23 Solution molecular weight data indicate that the phosphine-ligated silver-carbonate complex contains two silver ions, two phosphines, and one carbonate.
The molecular weight determined by the Signer method24 was 717 g/mol in CH3OH at 24 °C. Although this measured value is lower than that for complex 36 (872 g/mol), it is clearly smaller than that expected for higher aggregates of 36 and larger than values for potential mononuclear silver compounds (e.g., 466 g/mol for LAgCO3H, L = JohnPhos). Together, the recorded characterization data suggest that a binuclear, phosphine-ligated silver-carbonate complex (36) is the resting state of the catalyst for the C–H bond deuteration. Only two phosphine-ligated silver-carbonate complexes have been isolated previously and characterized by single-crystal X-ray diffraction; their solution chemistry has not been investigated.25
Although we were unable to obtain suitable crystals of compound 36 for X-ray diffraction, we isolated and characterized an analogue 37 containing tBuXPhos (2-di-tert-butylphosphino-2′,4′,6′-triisopropylbiphenyl) by X-ray diffraction, solution NMR, and solid-state IR spectroscopy (Scheme 7a and Figure 1). The relevance of the structural studies of 37 to the C–H deuteration reaction is demonstrated by the reaction of 2-methylthiophene (1) catalyzed by 37. This reaction led to the formation of [1H]1 with 91% deuterium incorporation at the 5-position after 6 h in CH3OD (Scheme 7b).
Scheme 7.
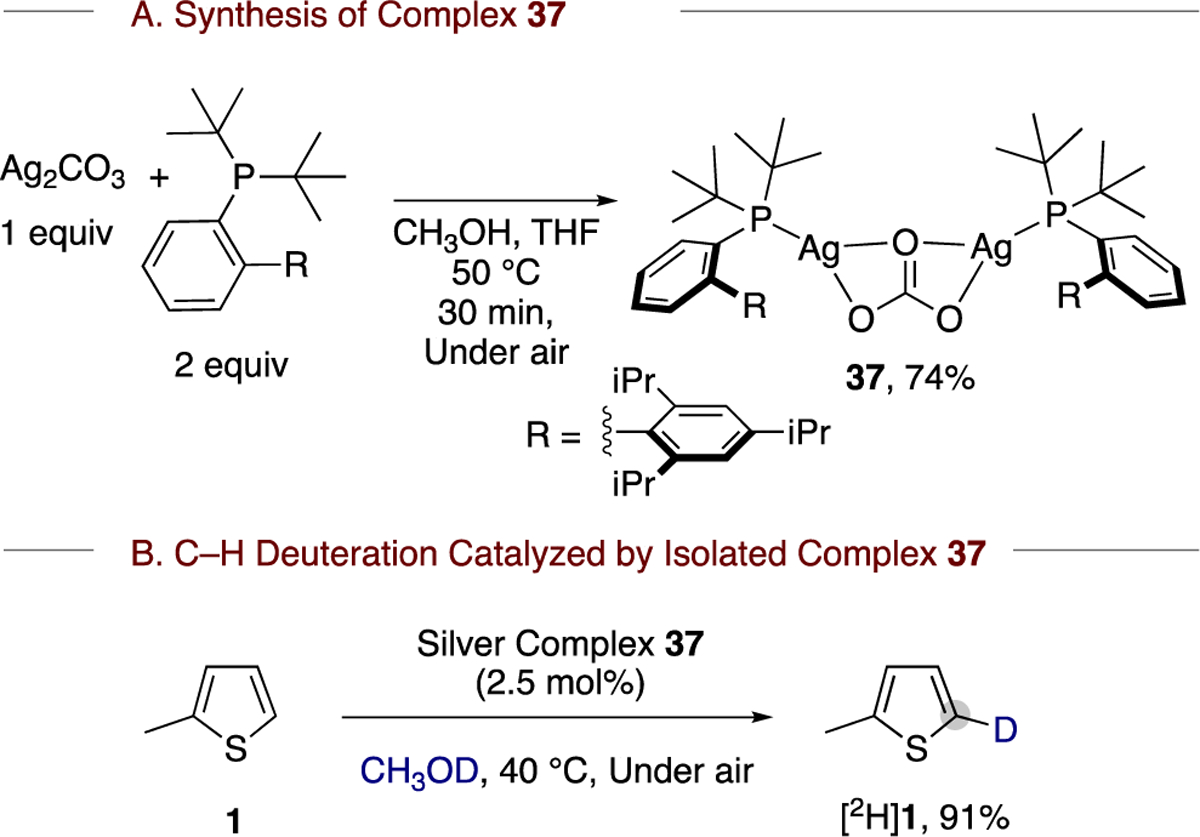
Synthesis of tBuXPhos-Ligated Silver Carbonate Complex 37 and Evaluation of Its Competence as a Catalyst for C–H Deuteration
An examination of the 31P NMR spectra of 37 in CH3OH-d4 revealed two superimposed doublets centered at 43.5 ppm, with and coupling constants of 649 and 748 Hz, respectively. The solid-state IR spectrum of 37 showed bands for the E′ ν(CO) ν3 mode of the silver-coordinated CO32− ligand at 1315 and 1440 cm−1. These NMR and IR data are comparable to the analogous data recorded for the isolated complex 36 (vide supra).
An ORTEP diagram of the solid-state structure of silver complex 37 is depicted in Figure 2 and consists of a binuclear tBuXPhos-ligated silver carbonate structure, in which both silver atoms are tricoordinated with slightly distorted trigonal PAgO2 environments. The CO32− ligand in 37 bridges the two silver atoms in a highly symmetric κ2:κ2 coordination mode.25b The symmetric coordination environment around the CO32− ligand is reflected in its O–C–O angles, which are close to 120°.
Figure 2.
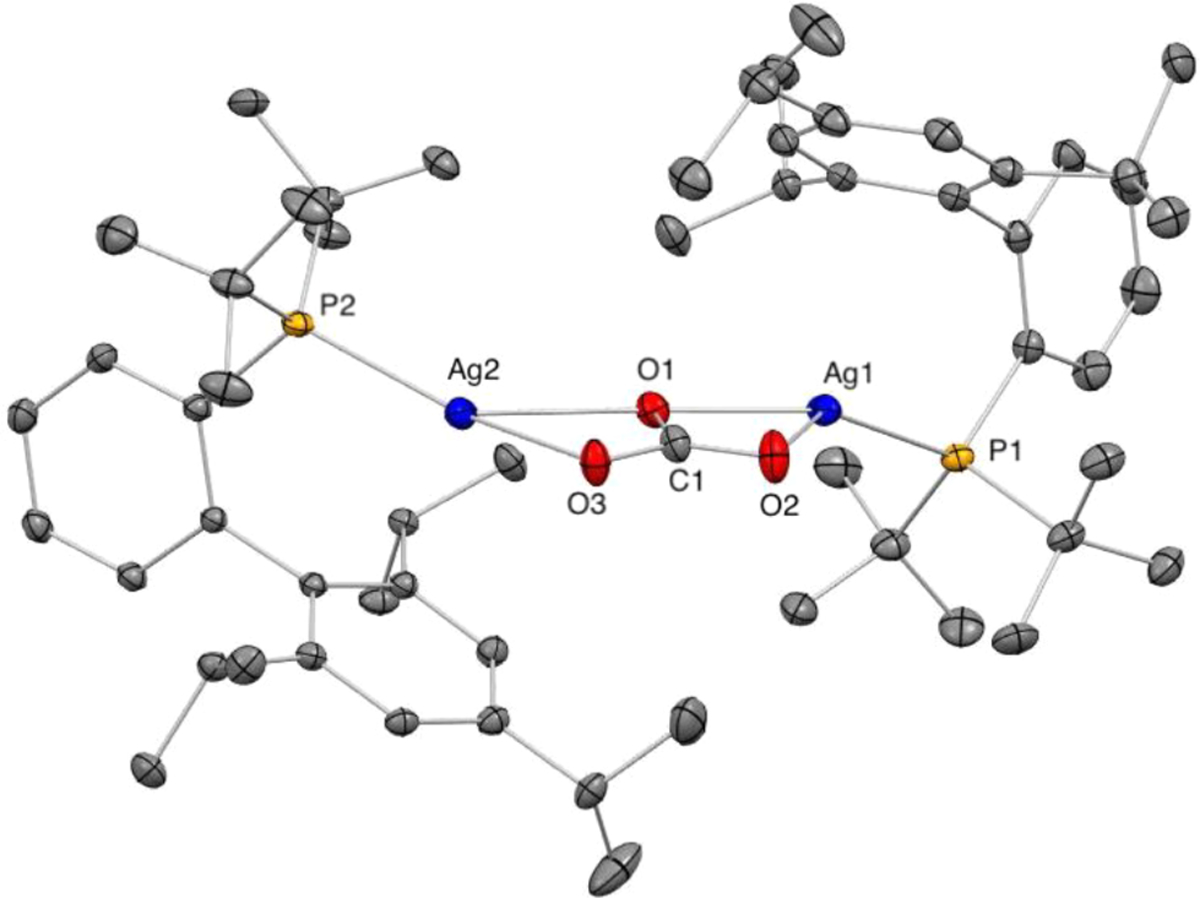
ORTEP diagram of tBuXPhos-ligated silver-carbonate complex 37. Bonds (Å): Ag1–P1, 2.3465(4); Ag1–O1, 2.2337(12), Ag1–O2, 2.4227(14); C1–O1, 1.295(2); C1–O2, 1.274(2); C1–O3, 1.2904(19); Ag2–O1, 2.4651(12); Ag2–O3, 2.1912(12); Ag2–P2, 2.3415(4). Angles (deg): O3–C1–O2, 122.49(16); O2–C1–O1, 119.51(15); O3–C1–O1, 118.00(15). Ellipsoids are shown at 50% probability. Hydrogen atoms, THF, and water within the crystal structure are omitted for clarity.
To gain information on the elementary C–H bond cleavage step, we conducted reactions with a series of thiophenes containing electron-donating and electron-withdrawing substituents (Figure 3). These studies showed that 2-bromo- and 2-methoxy-substituted thiophene underwent deuteration with comparable rates (kOMe/kBr = 0.9). However, both substrates reacted faster than 2-methylthiophene (kBr/kMe = 2.6, kOMe/kMe = 2.4). The faster rates for reaction of the more electron-rich and electron-poor thiophenes are inconsistent with an SEAr pathway for the rate-determining, C–H bond cleavage step.26 Moreover, the general selectivity of the silver-catalyzed method for H/D exchange at the most acidic C–H bonds in the five-membered heterocycles strongly suggests that the C–H bond cleavage step occurs by a concerted metalation-deprotonation mechanism.
Figure 3.

C–H Bond deuteration of 2-substituted thiophenes. C–H Deuteration of 2-substituted thiophenes. Rate constants (h−1): Me, 0.76; OMe, 1.8; Br, 2.0.
Although more research is needed to fully understand the mechanism of the silver-catalyzed deuteration of C–H bonds, an initial hypothesis that is consistent with all of our data is depicted in Scheme 8. The phosphine-ligated silver catalyst I undergoes rate-determining C–H bond activation with the heterocyclic substrate by a carbonate-assisted, concerted metalation-deprotonation step. This reaction would form one heteroarylsilver complex and one bicarbonate complex. Rapid H/D exchange between CH3OD and the bicarbonate O–H bond, followed by demetalation of the (hetero)aryl phosphine-ligated silver intermediate II by reaction with the isotopically enriched bicarbonate complex III, regenerates the starting silver complex and forms the labeled product.
Scheme 8.
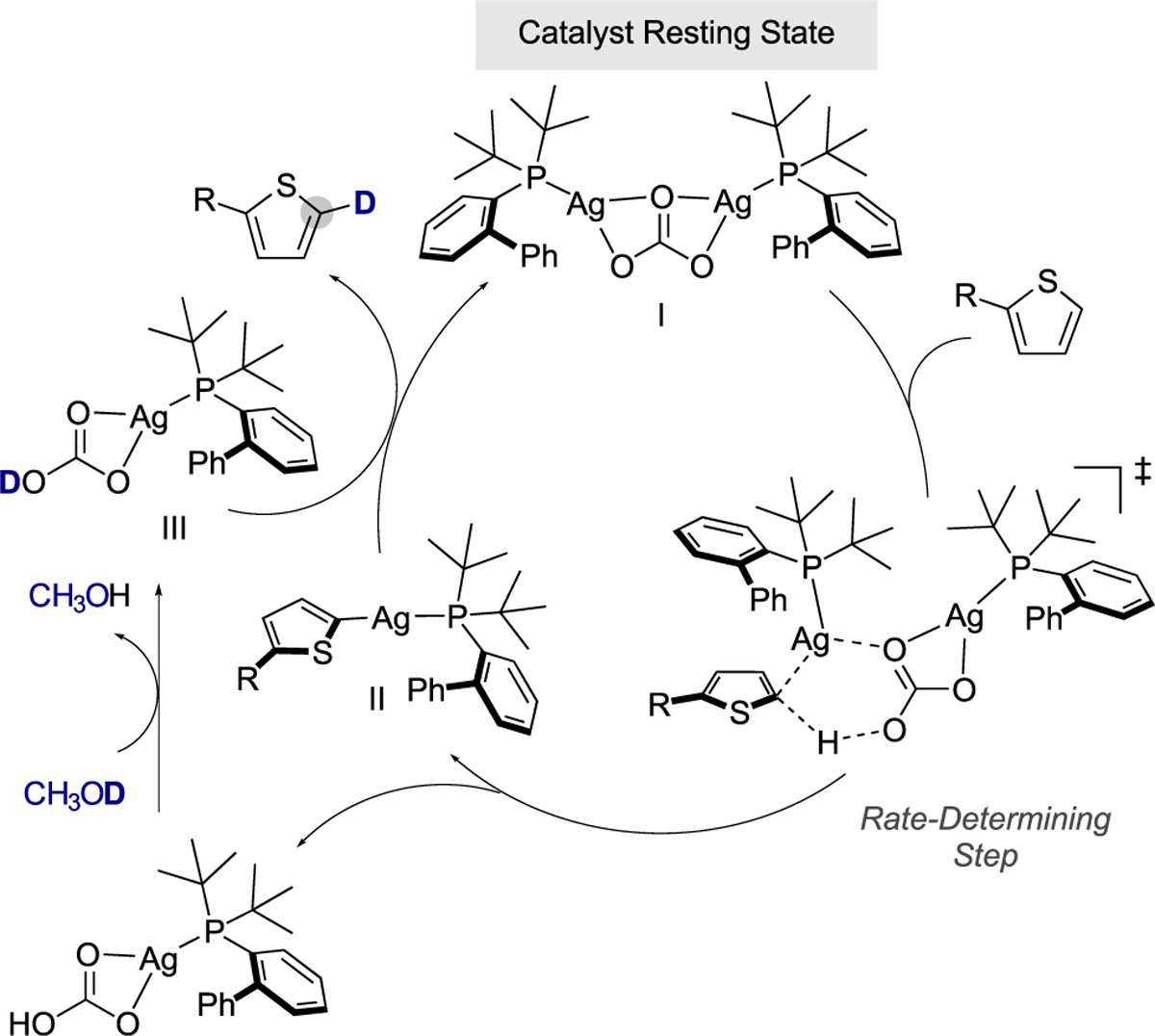
Mechanistic Hypothesis for the Site-Selective C–H Deuteration of Five-Membered Heterocycles
CONCLUSIONS
In summary, we have developed a silver-catalyzed method to conduct the previously elusive, catalytic, site-selective deuteration of C–H bonds in medicinally relevant five-membered aromatic heterocycles, including thiazole, imidazole, triazole, thiophene, and indole. These results hold promise for the synthesis of deuterated heterocyclic building blocks, an approach that can be applied for the development of pharmaceuticals with improved pharmacodynamic and pharmacokinetic properties. The developed method occurs in the absence of directing groups, under mild reaction conditions, and in the presence of a diverse array of potentially reactive functional groups, such as alcohols, amines, alkenes, alkynes, and nitriles. These attributes enable the deuterium labeling of complex pharmaceuticals ingredients,27 a result that can be extended to the incorporation of tritium because sources of this isotope in the form of T2O and CH3OT have been reported in the literature.8,28 Mechanistic experiments support turnover-limiting C–H bond activation by a new phosphine-ligated, silver-carbonate catalyst, which presumably occurs by a carbonate-assisted concerted metalation deprotonation step. The unique C–H bond activation chemistry by the herein reported phosphine-ligated, silver-carbonate complex lays the foundation for the development of a new class of catalysts that are currently being developed in our laboratory for previously elusive C–H functionalization reactions.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge financial support from the NIH (R35 GM130387) (J.F.H.). We thank Dr. Nicholas Settineri of the Berkeley X-ray Crystallography Facility for solving the crystal structure of complex 37. We thank the College of Chemistry’s NMR facility for resources provided and the staff for their assistance. Instruments in CoC-NMR are supported in part by the NIH (S10OD024998). A.T.-A. thanks UC MEXUSCONACYT for a postdoctoral research fellowship.
Footnotes
The authors declare no competing financial interest.
Complete contact information is available at: https://pubs.acs.org/10.1021/acscatal.0c04917
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.0c04917.
Experimental procedures and spectroscopic data on the reaction products (PDF)
Crystallographic data (CIF)
REFERENCES
- (1).(a) Gant TG Using Deuterium in Drug Discovery: Leaving the Label in the Drug. J. Med. Chem 2014, 57, 3595–3611. [DOI] [PubMed] [Google Scholar]; (b) Elmore CS; Bragg RA Isotope Chemistry; a Useful tool in the Drug Discovery Arsenal. Bioorg. Med. Chem. Lett 2015, 25, 167–171. [DOI] [PubMed] [Google Scholar]; (c) Harbeson SL; Tung RD Deuterium in Drug Discovery and Development. Annu. Rep. Med. Chem 2011, 46, 403–417. [Google Scholar]; (d) Harbeson SL; Tung RD Deuterium Medicinal Chemistry: A New Approach to Drug Discovery and Development. MedChem. News 2014, 8, 8–22. [Google Scholar]; (e) Mullard A Deuterated Drugs Draw Heavier Backing. Nat. Rev. Drug Discovery 2016, 15, 219–221. [DOI] [PubMed] [Google Scholar]
- (2).(a) Marathe PH; Shyu WC; Humphreys WG The Use of Radiolabeled Compounds for ADME Studies in Discovery and Exploratory Development. Curr. Pharm. Des 2004, 10, 2991–3008. [DOI] [PubMed] [Google Scholar]; (b) Lockley WJS; McEwen A; Cooke R Tritium: A Coming of Age for Drug Discovery and Development ADME Studies. J. Labelled Compd. Radiopharm 2012, 55, 235–257. [Google Scholar]; (c) Isin EM; Elmore CS; Nilsson GN; Thompson RA; Weidolf L Use of Radiolabeled Compounds in Drug Metabolism and Pharmacokinetic Studies. Chem. Res. Toxicol 2012, 25, 532–542. [DOI] [PubMed] [Google Scholar]
- (3).(a) Atzrodt J; Derdau V; Kerr WJ; Reid M Deuterium- and Tritium-Labelled Compounds: Applications in the Life Sciences. Angew. Chem., Int. Ed 2018, 57, 1758–1784. [DOI] [PubMed] [Google Scholar]; (b) Isotope Effects in Chemistry and Biology; Kohen A, Limbach H-H, Eds.; CRC Press: Boca Raton, FL, 2006. [Google Scholar]
- (4).(a) Voges R; Heys JR; Moenius T Preparation of Compounds Labelled with Tritium and Carbon-14; John Wiley & Sons: Chichester, UK, 2009. [Google Scholar]
- (5).(a) Atzrodt J; Derdau V; Fey T; Zimmermann J The Renaissance of H/D Exchange. Angew. Chem., Int. Ed 2007, 46, 7744–7765. [DOI] [PubMed] [Google Scholar]; (b) Atzrodt J; Derdau V; Kerr WJ; Reid M C–H Functionalisation for Hydrogen Isotope Exchange. Angew. Chem., Int. Ed 2018, 57, 3022–3047. [DOI] [PubMed] [Google Scholar]
- (6).(a) Hesk D; Das PR; Evans B Deuteration of Acetanilides and Other Substituted Aromatics Using [Ir(COD)(Cy3P)(Py)]PF6 as Catalyst. J. Labelled Compd. Radiopharm 1995, 36, 497–502. [Google Scholar]; (b) Heys JR Organoiridium Complexes for Hydrogen Isotope Exchange Labeling. J. Labelled Compd. Radiopharm 2007, 50, 770–778. [Google Scholar]; (c) Klei SR; Golden JT; Tilley TD; Bergman RG Iridium-catalyzed H/D Exchange into Organic Compounds in Water. J. Am. Chem. Soc 2002, 124, 2092–2093. [DOI] [PubMed] [Google Scholar]; (d) Zhou J; Hartwig JF Iridium-catalyzed H/D Exchange at Vinyl Groups Without Olefin Isomerization. Angew. Chem., Int. Ed 2008, 47, 5783–5787. [DOI] [PubMed] [Google Scholar]; (e) Nilsson GN; Kerr WJ The Development and Use of Novel Iridium Complexes as Catalysts for ortho-Directed Hydrogen Isotope Exchange Reactions. J. Labelled Compd. Radiopharm 2010, 53, 662–667. [Google Scholar]; (f) Salter R The Development and Use of Iridium(I) Phosphine Systems for ortho-Directed Hydrogen-isotope Exchange. J. Labelled Compd. Radiopharm 2010, 53, 645–657. [Google Scholar]; (g) Valero M; Kruissink T; Blass J; Weck R; Güssregen S; Plowright AT; Derdau V C–H Functionalization–Prediction of Selectivity in Iridium(I)-Catalyzed Hydrogen Isotope Exchange Competition Reactions. Angew. Chem., Int. Ed 2020, 59, 5626–5631. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Kerr WJ; Knox GJ; Reid M; Tuttle T; Bergare J; Bragg RA Computationally Guided Development of a Chelated NHC-P Iridium(I) Complex for the Directed Hydrogen Isotope Exchange of Aryl Sulfones. ACS Catal. 2020, 10, 11120–11126. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Kerr WJ; Knox GJ; Paterson LC Recent Advances in Iridium(I) Catalysis Towards Directed Hydrogen Isotope Exchange. J. Labelled Compd. Radiopharm 2020, 63, 281–295. [DOI] [PubMed] [Google Scholar]
- (7).(a) Yu RP; Hesk D; Rivera N; Pelczer I; Chirik PJ Iron-catalysed Tritiation of Pharmaceuticals. Nature 2016, 529, 195–199. [DOI] [PubMed] [Google Scholar]; (b) Palmer WN; Chirik PJ Cobalt-Catalyzed Stereoretentive Hydrogen Isotope Exchange of C(sp3)–H Bonds. ACS Catal. 2017, 7, 5674–5678. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Yang H; Zarate C; Palmer WN; Rivera N; Hesk D; Chirik PJ Site-Selective Nickel-Catalyzed Hydrogen Isotope Exchange in N-Heterocycles and Its Application to the Tritiation of Pharmaceuticals. ACS Catal. 2018, 8, 10210–10218. [Google Scholar]; (d) Zarate C; Yang H; Bezdek MJ; Hesk D; Chirik PJ Ni(I)–X Complexes Bearing a Bulky α-Diimine Ligand: Synthesis, Structure, and Superior Catalytic Performance in the Hydrogen Isotope Exchange in Pharmaceuticals. J. Am. Chem. Soc 2019, 141, 5034–5044. [DOI] [PubMed] [Google Scholar]; (e) Corpas J; Viereck P; Chirik PJ C(sp2)–H Activation with Pyridine Dicarbene Iron Dialkyl Complexes: Hydrogen Isotope Exchange of Arenes Using Benzene-d6 as a Deuterium Source. ACS Catal. 2020, 10, 8640–8647. [Google Scholar]; For a recent example:; (f) Garhwal S; Kaushansky A; Fridman N; Shimon LJW; de Ruiter G Facile H/D Exchange at (Hetero)Aromatic Hydrocarbons Catalyzed by a Stable Trans-Dihydride N-Heterocyclic Carbene (NHC) Iron Complex. J. Am. Chem. Soc 2020, 142, 17131–17139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Loh YY; Nagao K; Hoover AJ; Hesk D; Rivera NR; Colletti SL; Davies IW; MacMillan DWC Photoredox-catalyzed Deuteration and Tritiation of Pharmaceutical Compounds. Science 2017, 358, 1182–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Vitaku E; Smith DT; Njardarson JT Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]; (b) Taylor RD; MacCoss M; Lawson ADG Rings in Drugs. J. Med. Chem 2014, 57, 5845–5859. [DOI] [PubMed] [Google Scholar]
- (10).(a) Gomtsyan A Heterocycles in Drugs ans Drug Discovery. Chem. Heterocycl. Compd 2012, 48, 7–10. [Google Scholar]; (b) Dalvie DK; Kalgutkar AS; Khojasteh-Bakht SC; Obach RS; O’Donnell JP Biotransformation Reactions of Five-membered Aromatic Heterocyclic Rings. Chem. Res. Toxicol 2002, 15, 269–299. [DOI] [PubMed] [Google Scholar]
- (11).(a) Lipinski CA Bioisosterim in Drug Design. Annu. Rep. Med. Chem 1986, 21, 283–291. [Google Scholar]; (b) Patani GA; LaVoie EJ Bioisosterism: A Rational Approach in Drug Design. Chem. Rev 1996, 96, 3147–3176. [DOI] [PubMed] [Google Scholar]
- (12).(a) Junk T; Catallo WJ Preparative Supercritical Deuterium Exchange in Arenes and Heteroarenes. Tetrahedron Lett. 1996, 37, 3445–3448. [Google Scholar]; (b) Junk T; Catallo WJ Hydrogen Isotope Exchange Reactions Involving C–H (D, T) bonds. Chem. Soc. Rev 1997, 26, 401–406. [Google Scholar]; (c) Junk T; Catallo WJ; Elguero J Synthesis in Superheated Aqueous Media: Preparation of Fully Deuterated Pyrazolesand Quinoxazolines. Tetrahedron Lett. 1997, 38, 6309–6312. [Google Scholar]; (d) Patel M; Saunthwal RK; Verma AK Base-Mediated Deuteration of Organic Molecules: A Mechanistic Insigth. ACS Omega 2018, 3, 10612–10623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Lee SY; Hartwig JF Palladium-Catalyzed, Site-Selective Direct Allylation of Aryl C–H Bonds by Silver-Mediated C–H Activation: A Synthetic and Mechanistic Investigation. J. Am. Chem. Soc 2016, 138, 15278–15284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).For an example of a silver-perfluoroalkoxide that cleaves the C–H bond of π-arene complexes, see:; (a) Panigrahi A; Whitaker D; Vitorica-Yrezabal IJ; Larrosa I Ag/Pd Cocatalyzed Direct Arylation of Fluoroarene Derivatives with Aryl Bromides. ACS Catal. 2020, 10, 2100–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Batuecas M; Luo J; Gergelitsová I; Krämer K; Whitaker D; Vitorica-Yrezabal IJ; Larrosa I Catalytic Asymmetric C–H Arylation of (η6-Arene)chromium Complexes: Facile Access to Planar-Chiral Phosphines. ACS Catal. 2019, 9, 5268–5278. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Colletto C; Panigrahi A; Fernández-Casado J; Larrosa I Ag(I)–C–H Activation Enables Near-Room-Temperature Direct α-Arylation of Benzo[b]thiophenes. J. Am. Chem. Soc 2018, 140, 9638–9643. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Whitaker D; Burés J; Larrosa I Ag(I)-Catalyzed C–H Activation: The Role of the Ag(I) Salt in Pd/Ag-Mediated C–H Arylation of Electron-Deficient Arenes. J. Am. Chem. Soc 2016, 138, 8384–8387. [DOI] [PubMed] [Google Scholar]
- (15).Lotz MD; Camasso NM; Canty AJ; Sanford MS Role of Silver Salts in Palladium-Catalyzed Arene and Heteroarene C–H Functionalization Reactions. Organometallics 2017, 36, 165–171. [Google Scholar]
- (16).Li W; Yuan D; Wang G; Zhao Y; Xie J; Li S; Zhu C Cooperative Au/Ag Dual-Catalyzed Cross-Dehydrogenative Biaryl Coupling: Reaction Development and Mechanistic Insight. J. Am. Chem. Soc 2019, 141, 3187–3197. [DOI] [PubMed] [Google Scholar]
- (17).On the basis of our mechanistic findings, Zhang and Huang reported the deuteration of a handful of five-membered heterocycles catalyzed by the combination of Ag2CO3 and JohnPhos in DMSO with D2O as a source of isotope in the presence of stoichiometric K2CO3 as base, but these conditions did not lead to H/D exchange of most medicinally relevant heterocycles, such as imidazoles, triazoles, and even indoles.; Li E-C; Hu G-Q; Zhu Y-X; Zhang H-H; Shen K; Hang X-C; Zhang C; Huang W Ag2CO3-Catalyzed H/D Exchange of Five-Membered Heteroarenes at Ambient Temperature. Org. Lett 2019, 21, 6745–6749. [DOI] [PubMed] [Google Scholar]
- (18).Testa L; Biondi Zoccai GGL; Valgimigli M; Latini RA; Pizzocri S; Lanotte S; Laudisa ML; Brambilla N; Ward MR; Figtree GA; Bedogni F; Bhindi R Current Concepts on Antiplatelet Therapy: Focus on the Novel Thienopyridine and Non-Thienopyridine Agents. Adv. Hematol 2010, 2010, 595934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Kerr WJ; Lindsay DM; Owens PK; Reid M; Tuttle T; Campos S Site-Selective Deuteration of N-Heterocycles via Iridium-Catalyzed Hydrogen Isotope Exchange. ACS Catal. 2017, 7, 7182–7186. [Google Scholar]
- (20). For a comparison of the reactivity of Crabtree’s catalyst, which is one of the most widely used in industry for isotope exchange, with that of our silver catalyst, see the SI Section 7.
- (21). Despite the zero-order in CH3OD, the overall yield of C–H bond deuteration is higher when the concentration of CH3OD was increased. Indeed, when the C–H bond deuteration reaction of 34 approached 15% conversion, the rate began to decrease at lower concentrations of CH3OD (see S12 in the SI). We hypothesized that partial decomposition of the silver catalyst occurred at lower concentrations of CH3OD on the basis of the observation of a black precipitate in these experiments.
- (22).(a) Goel RG; Pilon P Tri-tert-butylphosphine Complexes of Silver(I). Preparation, Characterization, and Spectral Studies. Inorg. Chem 1978, 17, 2876–2879. [Google Scholar]; (b) Herberhold M; Milius W; Akkus N The Molecular Structure of the 1:1 Adduct of Silver Cyanide with Tri(1-cyclohepta-2,4,6-trienyl)phosphane, Ag(CN)[P(C7H7)3]. Z. Anorg. Allg. Chem 2006, 632, 97–100. [Google Scholar]; (c) Grirrane A;Álvarez E; García H; Corma A Catalytic Activity of Cationic and Neutral Silver(I)–XPhos Complexes with Nitrogen Ligands or Tolylsulfonate for Mannich and Aza-Diels–Alder Coupling Reactions. Chem. - Eur. J 2016, 22, 340–354. [DOI] [PubMed] [Google Scholar]; (d) Muetterties EL; Alegranti CW Solution Structure and Kinetic Study of Metal-Phosphine and-Phosphite Complexes. I. The Silver (I) System. J. Am. Chem. Soc 1972, 94, 6386–6391. [Google Scholar]; (e) Yuan Z; Dryden NH; Vittal JJ; Puddephatt RJ Chemical Vapor Deposition of Silver. Chem. Mater 1995, 7, 1696–1702. [Google Scholar]; (f) Siegert U; Hahn H; Lang H Investigation of the Solution Behavior of tri-n-butylphosphine copper(I) and silver(I) acetates using 31P{1H} NMR Spectroscopy. Inorg. Chim. Acta 2010, 363, 944–948. [Google Scholar]
- (23).Greenaway AM; Dasgupta TP; Koshy KC; Sadler GG A Correlation Between Infrared Stretching Mode Absorptions and Structural Angular Distortions for the Carbonato Ligand in a Wide Variety of Complexes. Spectrochim. Acta, Part A 1986, 42, 949–954. [Google Scholar]
- (24).Zoellner RW A Reusable Apparatus for the Convenient Determination of the Molecular Weight of Air- or Moisture-sensitive Compounds. J. Chem. Educ 1990, 67, 714–715. [Google Scholar]
- (25).(a) Bowmaker GA; Effendy; Hanna JV; Healy PC; King SP; Pettinari C; Skelton BW; White AH Solution and Mechanochemical Syntheses, and Spectroscopic and Structural Studies in the Silver(I) (bi-)carbonate: Triphenylphosphine System. Dalton Trans. 2011, 40, 7210–7218. [DOI] [PubMed] [Google Scholar]; (b) Zhang J; Yang Q; Zhu Y; Liu H; Chi Z; Su C-Y Tetraphenylethylene-based Phosphine: Tuneable Emission and Carbon Dioxide Fixation. Dalton Trans. 2014, 43, 15785–15790. [DOI] [PubMed] [Google Scholar]
- (26).For similar observations with Pd(II) catalysts in arene and heteroarene arylation, see:; (a) Gorelsky SI; Lapointe D; Fagnou K Analysis of the Concerted Metalation-Deprotonation Mechanism in Palladium-Catalyzed Direct Arylation Across a Broad Range of Aromatic Substrates. J. Am. Chem. Soc 2008, 130, 10848–10849. [DOI] [PubMed] [Google Scholar]; (b) Gorelsky SI; Lapointe D; Fagnou K Analysis of the Palladium-Catalyzed (Aromatic)C–H Bond Metalation–Deprotonation Mechanism Spanning the Entire Spectrum of Arenes. J. Org. Chem 2012, 77, 658–668. [DOI] [PubMed] [Google Scholar]
- (27).A search in SciFinder only provided results for the synthesis of [2H]24 and [2H]25, whereas [2H]26–32 have not been reported in the literature.
- (28).Koniarczyk JL; Hesk D; Overgard A; Davies IW; McNally A A General Strategy for Site-Selective Incorporation of Deuterium and Tritium into Pyridines, Diazines and Pharmaceuticals. J. Am. Chem. Soc 2018, 140, 1990–1993. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.