

Abstract
The 2011 EPA trichloroethylene (TCE) IRIS assessment, used developmental cardiac defects from a controversial drinking water study in rats (Johnson et al. [51]), along with several other studies/endpoints to derive reference values. An updated literature search of TCE-related developmental cardiac defects was conducted. Study quality, strengths, and limitations were assessed. A putative adverse outcome pathway (AOP) construct was developed to explore key events for the most commonly observed cardiac dysmorphologies, particularly those involved with epithelial-mesenchymal transition (EMT) of endothelial origin (EndMT); several candidate pathways were identified. A hypothesis-driven weight-of-evidence analysis of epidemiological, toxicological, in vitro, in ovo, and mechanistic/AOP data concluded that TCE has the potential to cause cardiac defects in humans when exposure occurs at sufficient doses during a sensitive window of fetal development. The study by Johnson et al. [51] was reaffirmed as suitable for hazard characterization and reference value derivation, though acknowledging study limitations and uncertainties.
Keywords: Trichloroethylene, TCE, Cardiac, Malformations, AOP
1. Introduction
Trichloroethylene (TCE), CAS No. 79–01-6, is a volatile chemical and widely used chlorinated solvent that is frequently found in ground water and in soil at contaminated sites across the U.S. TCE ranks 16th among hazardous substances most commonly found at facilities on the federal National Priorities List [4]. At sites where groundwater is contaminated and depending upon site-specific circumstances, TCE exposures and accompanying human health risks may arise from: (1) movement of TCE vapors from subsurface locations into the indoor air of overlying and nearby buildings (i.e., vapor intrusion) [5]; and/or (2) use of groundwater as a source of drinking water, process water, or irrigation water. A number of health effects have been observed after exposure to TCE during development, e.g., decreased fetal survival, impaired growth, alterations in immune and nervous system function, and structural defects, including ocular and cardiac malformations [16]. Here we report on a focused review of the published literature, conducted to update the information and critically evaluate the available data relevant to the potential for cardiac defects resulting from developmental exposures to TCE. This effort was initiated because of concerns raised about study quality and application of the reference value to short term and pregnancy exposure scenarios.
EPA completed an IRIS Toxicological Review of TCE in September 2011 [87]. The most sensitive types of noncancer health effects identified in this assessment were developmental, renal, and immunological. A reference concentration (RfC)2 of 0.0004 ppm (0.4 ppb or 2 µg/m3) is derived in U.S. EPA [87], based on route-to-route extrapolated results from oral studies for the critical effects of heart malformations in rats and immunotoxicity in mice, further supported by route-to-route extrapolated results from an oral study of nephropathy in rats. The reference dose (RfD) for non-cancer effects of 0.0005 mg/kg-day is based on the critical effects in oral studies of heart malformations in rats, adult immunological effects in mice, and developmental immunotoxicity in mice. The RfD is further supported by results from an oral study for the effect of toxic nephropathy in rats and route-to-route extrapolated results from an inhalation study for the effect of increased kidney weight in rats ([87]; pages 6–43).
After the final IRIS document was released, EPA and others realized that because fetal adverse outcomes could potentially result from short-term exposures or peaks in exposure during pregnancy, one of the two endpoints used to derive the RfC (the fetal cardiac defects) is particularly important when evaluating whether TCE exposure poses an immediate potential hazard and whether peak exposures are a potential health concern. A study by Johnson et al. [51], which reports the results of research on TCE in drinking water, including the findings of Dawson et al. [20], is included in the group of studies on which the reference values are based in the 2011 IRIS assessment, and is one of several lines of evidence regarding the hazard potential for developmental toxicity of TCE. Concerns have been raised about the Johnson et al. [51] study and EPA’s use of this study for risk evaluation [1,90,38]. Specific needs to resolve these concerns include: (1) a systematic evaluation of study quality; (2) more details in the description of the study design (e.g., the source of concurrent controls); (3) a reexamination of the dose-response relationship for cardiac defects; and (4) an evaluation of the study results in light of other studies that did not observe cardiac defects after in utero exposures. In addition, concerns have been raised regarding the interpretation of the epidemiological database for cardiac defects associated with TCE exposures [13,1,90,38].
An updated literature search and analysis of the developmental cardiac toxicity data for TCE was conducted to address the identified issues and to provide a focused, rigorous, systematic scientific review of the available data on associations between exposure to TCE and fetal cardiac defects. The scope of this update and analysis was limited to the fetal cardiac defects observed following gestational exposures to TCE and/or its oxidative metabolites, dichloroacetic acid (DCA) and trichloroacetic acid (TCA), which have been specifically associated with cardiac malformations in rats [51,49,20,27,79,78], and does not include an update on other developmental effects after TCE exposure, i.e., fetal growth retardation, embryolethality, ocular malformations, developmental neurotoxicity, and developmental immunotoxicity. This update of the fetal cardiac effects includes (1) a systematic search to identify any recently published literature; (2) a detailed evaluation of the available data; (3) a hypothesis-driven assessment of the weight of evidence (evidence integration) for the association of TCE exposures with cardiac malformations; (4) a reexamination of the dose-response relationship for cardiac malformations; and (5) a transparent description of the evaluation. This process is aligned with the [64] recommendations for systematic review, evidence integration (weight-of-evidence) evaluation, and presentation of information to increase transparency.
2. Materials and methods
2.1. Literature search update
A systematic literature search was conducted to identify all epidemiological, toxicological, and mechanistic studies relevant to cardiac defects associated with developmental exposure to TCE or its metabolites (TCA and DCA) that were published subsequent to the final systematic literature search conducted by EPA during completion of the 2011 IRIS assessment [87]. A date-delineated search of PubMed, Toxline, and Web of Science (WoS) was conducted (January 2010–January 2015), using search terms designed to identify any publications that addressed TCE or its specified metabolites. The search identified a total of 1769 unique citations, which were then screened using information contained in the title, abstract, and/or full text. Citations excluded from further consideration included studies that did not include an assessment of TCE or its metabolites, studies that did not directly assess or were not pertinent to the evaluation of cardiac development, and publications that did not include primary research data (e.g., reviews, press articles, meeting abstracts). The literature search did not identify any new experimental animal toxicology studies of fetal cardiac defects, but did identify two new epidemiological studies that assessed the association of TCE or chlorinated solvent exposures with cardiac defects [71,29] and two new studies that provided mechanistic information relevant to alterations of cardiac development following TCE (or metabolite) exposures [58,66].
2.2. Study quality review
For each epidemiological and toxicological study in the developmental toxicity database for TCE, whether previously included in the EPA TCE assessment [87] or newly identified in the updated literature search, a formal detailed review of study quality was conducted.
• Epidemiological data:
Study quality evaluation criteria and a general format for capturing epidemiological study data and characterization have previously been developed by the IRIS program and are summarized in the Guidelines for Developmental Toxicity Risk Assessment [85]. These factors include study power, potential bias in data collection, selection bias, measurement biases associated with exposure and outcome, and consideration of potential confounding and effect modification. This format was used to summarize study information and observed strengths, biases, and confounding factors for each study. An independent review of the study quality conclusions presented here was conducted by a working group that included eight EPA experts in the field of epidemiology.
• Animal toxicology data:
Study quality evaluation criteria for in vivo, in vitro, and avian in ovo developmental toxicology studies were developed specifically for this effort. These criteria included considerations described in U.S. EPA [85] and focused on the adequacy of study design and documentation of information on the test subjects (e.g., species, strain, source, sex, age/lifestage/embryonic stage), environment (e.g., husbandry, culture medium), test substance (e.g., identification, purity, analytical confirmation of stability and concentration), treatment (e.g., dose levels, controls, vehicle, group sizes, duration, route of administration), endpoints evaluated (e.g., schedule of evaluation, randomization and blinding procedures, assessment methods), and reporting (quality and completeness). Two separate reviewers conducted independent assessments of each in vivo mammalian study, and seven toxicologists independently evaluated study quality for four mammalian in vivo studies that had performed a detailed evaluation of developmental cardiac defects [15,51,28,20].
2.3. Characterization of hazard and dose-response information
• Hazard:
Critical elements of the identified epidemiological and toxicological studies were extracted and summarized in tabular format. For epidemiological studies, the exposure measure and range, outcome classification, participant selection and comparability, consideration of likely confounding, data presentation and analysis, and sample size were summarized. For animal toxicology studies, the summary included information on the test subjects (species, strain, sex, number of animals assigned per group), exposure levels, timing, and duration, no-observed-adverse-effect levels (NOAELs), lowest-observed-adverse-effect levels (LOAELs), and treatment-related effects.
• Dose-response analysis:
The cardiac malformation data [51] were reanalyzed using the Benchmark Dose Software (BMDS) nested logistic model that was used in the EPA TCE assessment [87] as well as other BMDS models to evaluate uncertainty related to model selection and modeling assumptions [88]. A benchmark response (BMR) of 0.01 (1%) extra risk was used, justified by the severity of the effect.
2.4. Mechanistic data on developmental pathways and processes
The 2011 IRIS assessment noted that many of the cardiac defects observed in humans and laboratory species (primarily rats and chickens) involved septal and valvular structures. To further characterize the potential for alterations in cardiac development, studies that evaluated aspects of valvulo-septal defects identified in the literature search, as well as mechanistic studies that had been included in the 2011 IRIS TCE assessment, were examined for relevant information. The search and data evaluation pointed to alterations in endocardial cushion formation and development. This prompted a search of the Mouse Genome Informatics (MGI) database (http://www.informatics.jax.org/) for genes associated with “abnormal cardiac epithelial to mesenchymal transition” [MP:0008825]. As a consequence, newer mechanistic concepts were explored.
2.5. Weight-of-evidence (WOE) evaluation
The WOE (evidence integration) for fetal cardiac defects was characterized according to the criteria described in the Framework for Assessing Health Risk of Environmental Exposures to Children [86], a scheme that was adapted from principles of causality assessment developed by [43]. Fig. 1 illustrates the components (key factors) included in the WOE analysis. Each participant in the review independently assessed the WOE, and through discussions arrived at a group consensus of the evidence supporting stronger and weaker weights of association for each key factor.
Fig. 1.
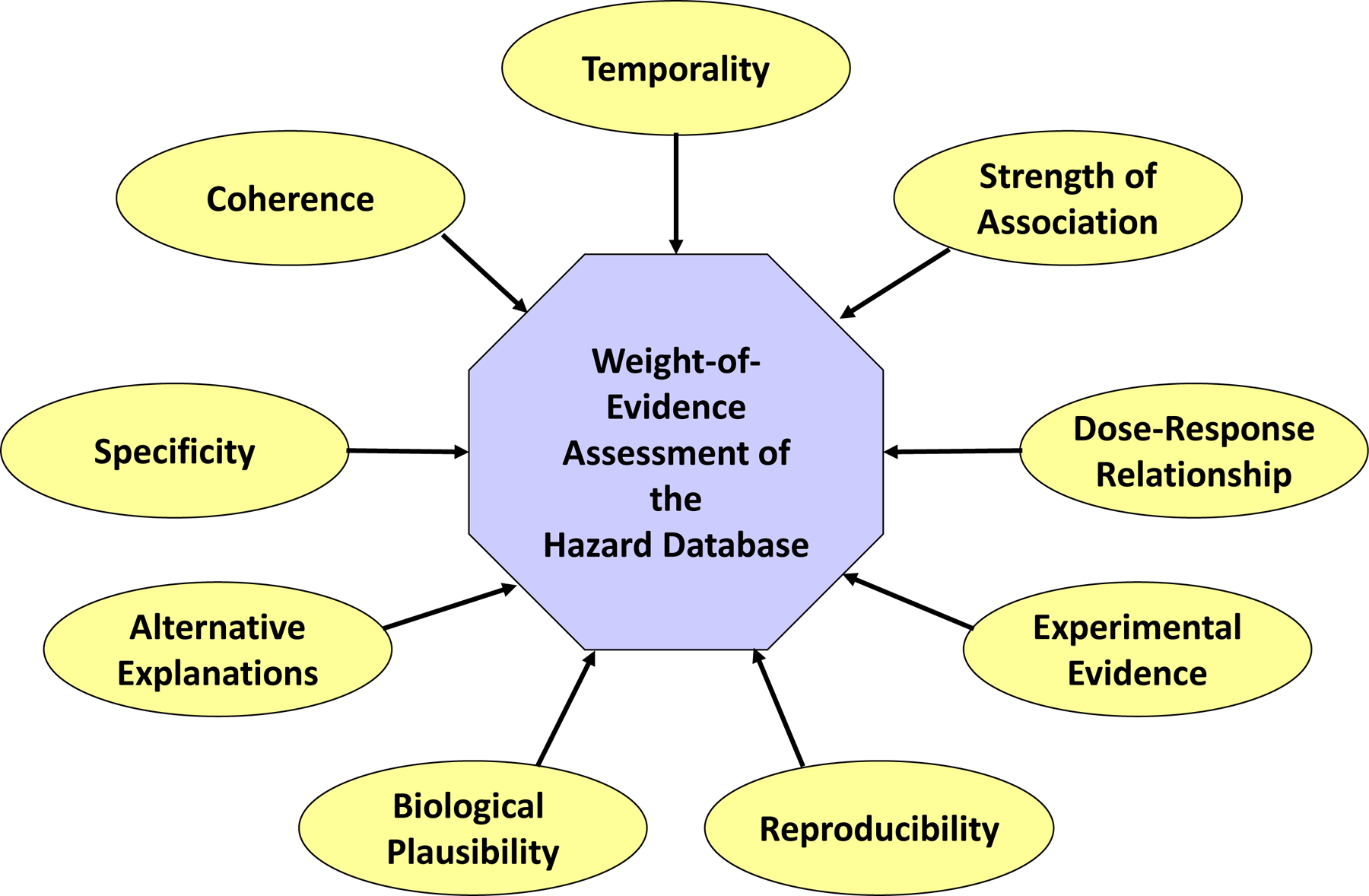
Conceptual view of a Weight-of-Evidence evaluation. Considerations within a WOE evaluation of toxicity data are shown. The relative weight of each consideration can vary, based upon the data [86], Fig. 4–4). Temporality is the premise that the exposure must occur prior to the outcome. Strength of association is the consideration of study rigor and statistical power. Variability analysis considers the source of variability within individual studies. Uncertainty analysis considers information or data gaps in individual studies and in the comprehensive database of information. Qualitative dose-response relationship is the change in an effect, and the degree of the change, as a function of exposure or dose. Experimental evidence is the alterations in response or rate of response resulting from manipulation of exposure. Reproducibility is the observation of specific effects under varied conditions. Bio-logical plausibility is the determination of whether an observed outcome could be attributed to the toxicological insult, given the currently known science. Alternative or multiple explanations are other explanations for the observed outcome(s) following the exposure of interest. Specificity refers to determination of the relationship between one exposure, the effect(s), and whether each effect is mediated through a single or alternative MOAs. Coherence is the extent to which the data are similar in outcome and exposure/dose and whether they support each biologically plausible hypothesis or MOA.
3. Results
3.1. Hazard for developmental cardiac defects
3.1.1. Epidemiological data
The epidemiological studies were reviewed for associations between maternal exposure to TCE and cardiac defects. Seven reports from six epidemiological studies that investigated developmental cardiac birth defects in relation to estimated TCE exposure during pregnancy were identified in the literature [71,29,94,8,9,35,56]; five of the seven reports were reviewed in the EPA’s 2011 Trichloroethylene Toxicological Review [87]. The publication by Forand et al. [29] analyzed the same study population described in the ATSDR [3,2] reports referenced in U.S. EPA [87]. Bove [8] and Bove et al. [9] report twice on the same study subjects. All of the studies examined outcomes in relation to oral exposures with the exception of the inhalation exposure studies from Forand et al. [29] and Yauck et al. [94]. The epidemiological study summaries and quality assessments are presented in Table 1. Consideration of bias, confounding, and chance are summarized in Table 2.
Table 1:
Study Summary and Quality Assessment for Epidemiologic Studies on TCE Exposure and Congenital Malformations
| Reference | Exposure Measure and Range | Outcome Classification | Participant Selection and Comparability | Consideration of Likely Confounding | Data Presentation and Statistical Analysis | Adequate Sample Size | Additional Comments |
|---|---|---|---|---|---|---|---|
| Ruckart et al. (2013) | Individual level. Fate and transport, and water distribution modeling, TCE, up to 1,400 ppb; other contaminates included vinyl chloride, 1,2-dichloroethylene, PCE, benzene. Average monthly concentration two months before and after conception. | Self-reported, verified by medical record; NTD, oral clefts prevalence, conotruncal heart defectsa. | United States. n=12,598 live births among mothers residing at Camp Lejeune during pregnancy, identified from birth certificates and media campaign/ referral, 1968 – 1985. Referents selected from children without a birth defect (~1:10 ratio), unmatched to cases. Excluded 54 cases due to lack of medical verification, refusal to provide medical records, and verified not to have the reported condition; 22 controls ineligible. | Bivariate analyses adjusted for mother’s age, previous pregnancy, child’s sex, child’s sibling with a birth defect, father’s occupational exposure to solvents, previous pregnancy, alcohol use, mother’s employment status, use of prenatal vitamins, or maternal fevers. | Odds ratio and 95% confidence interval; unconditional logistic regression. | 106 cases of NTDs, oral clefts and leukemia/non-Hodgkin lymphoma; medically-verified: 35 NTDs, 42 oral clefts; TCE exposed, 8 NTDs, 9 oral clefts. | Odds ratio not reported for conotruncal heart defects. Less than 3 conotruncal heart malformations observed. |
|
| |||||||
| Forand et al. (2011) | Area level. Maternal residence in one of two contaminated areas at time of birth. Sample of 25% residences affected by soil vapor intrusion: Area 1, indoor air TCE, range −0.18 – 140 ug/m3, median 16 ug/m3; Area 2, indoor PCE, range 0.1 – 24 ug/m3. | Congenital malformationsb, including cardiac (ICD-9 745.0–747.9c), in <2 year old children, NYSDOH Congenital Malformations Registry. | United States. n=1,440 live singleton births (1,090 in TCE area, 350 in PCE area); referents, 1983–2000; 3.6 million births in New York State, excluding New York City. | Adjusted for mother’s age, education, race, infant’s sex, number of previous live births, and adequate prenatal care. | Rate ratios and 95% confidence intervals, Poisson regression. | 61 children (44 in TCE area, 17 in PCE area) with at least one reportable birth defect. TCE area, 25 surveillance defects, 15 cardiac malformations (6 major, 3 conotruncal). | No births with NTDs or oral clefts. |
|
| |||||||
| Yauck et al. (2004) | Area level. Maternal residence within 1.32 miles from at least one TCE emissions source at time of birth. | Cardiac malformations, excluding patent ductus arteriosus, persistent foramen ovale, or peripheral pulmonary stenosis, hospital medical record, Milwaukee Children’s Hospital. | United States. n=4,025 infants, born 1997–1999; cases from hospital or birth records, population referents from birth certificates frequency matched by birth year; excluded infants ≤23weeks, if 24–26 weeks, died within 48 hours of birth, or Down’s syndrome diagnosis; one birth selected from multiple births. | Dichotomized by age (< 38 years, ≥ 38 years); no differences found for race, ethnicity, maternal education, parity, number of prenatal visits, or cigarette use. | Odds ratio, logistic regression. | 245 cases and 3,780 controls; TCE exposed, 46 cases, 715 controls. | Pre-existing diabetes, chronic hypertension, and alcohol associated with outcome and not included in TCE statistical model. The poorly-defined exposure surrogate and lack of TCE exposure monitoring makes interpretation of results difficult. |
|
| |||||||
| Bove et al. (1995); Bove (1996) | Area level. Maternal 1st trimester exposure to TCE, municipal water supply, 75 towns (55 ppb, maximum monthly estimate; 5% of study population above MCL of 5 ppb), other TTHMs. | Congenital malformationsd, including NTD, oral clefts and cardiac defects (ICD-9 745.0, 745.1, 745.2, 746.1, 746.3, 746.4, 746.7, 747.1, 747.3), NJDOH Birth Defects Registry and New Jersey fetal death certificates. | United States. n=80,938 singleton live-born infants and 594 singleton fetal deaths, New Jersey birth and death records, 1985–1988. | Odds ratio adjusted if differed from unadjusted by ± 15% for maternal age, race, education, parity prenatal care, previous stillbirth or miscarriage, and child’s sex. | Odds ratio, logistic regression. | 58 NTDs, 83 oral cleft, 108 major cardiac defects; TCE >10 ppb, 4 NTDs, 9 oral cleft defects, major cardiac defects, including ventricular septal defects, NR. | Effect measure estimates from univariate analysis did not differ by ±15% from multivariate analyses. |
|
| |||||||
| Goldberg et al. (1990) | Family member exposed to municipal well water contaminated with TCE (range: 6–239 ppb), DCA, chromium. | Cardiac defects, medically diagnosed closest to birth date, excluding syndromes associated with cardiac abnormalities, supraventricular tachycardia or isolated ectopic cardiac beats without gross anatomic cardiac lesions, patent ductus arteriosus in premature infants, peripheral pulmonary stenosis and bicuspid aortic value without stenosis or regurgitation. | United States. n=1,363 live births, conceived between 1969–1987 whose parents live in Tucson Valley for 1 month before and during 1st trimester of pregnancy, identified from cardiologist’s records, 218 lacking 1st trimester addresses, 406 disqualified, 31 not residing in Tucson during 1st trimester. Additional control groups: Groups 1 and 2 were current residents, selected using RDD in a) proportion to all telephone numbers or b) proportion to population with cardiac defects, | Compared to non-contaminated water area controls, more cases were Hispanic, case parents were less educated and were more likely blue-collar, and fathers were younger. No adjustment for potential confounders; possible bias introduced if differential selection between residents in contaminated area and rest of Tucson to cardiologist. | Prevalence rates, odds ratio. | 707 families (246 exposed, 461 unexposed). | Population at risk not fully elucidated because did not include cases living in study area who were treated at hospitals outside Tucson area or subjects who moved during the study period. NR if interviewers were blinded. Use of family as a control group provides estimate of the proportion of households that had at one member who worked or resided in the contaminated are, not estimate of exposure prevalence in the birth population. |
|
| |||||||
| Lagakos et al. (1986) | Maternal exposure during full period of pregnancy to 32 V0Cs detected in 1979 in two drinking water wells, including TCE: 267 µg/L, tetrachloro-ethylene: 21 μg/L, and chloroform: 12 μg/L. |
Self-reported congenital malformationse, including heart defects (ICD–9 425.3, 745.2, 745.4, 745.9, 746.6, 476.9, 747.1, 747.2, 785.2), 1960–1982. | United States. n=6,219 residences with telephones in Woburn, Massachusetts, 1,149 refused interview and 60 non-English speaking; 4,396 self-reported pregnancies. | Depending on outcome, adjusted for infant sex, maternal smoking during pregnancy, year pregnancy ended, maternal age, prior peri–natal death, prior low birth weight, and/or prior musculoskeletal anomaly. | Odds ratio, Cox proportional hazard. | 3.467 pregnancies with infant living >7 days, 177 congenital anomalies, 5 pregnancies with mother receiving water from contaminated wells. | Self-reporting of outcomes, potential recall bias, and lack of exposure data for susceptible periods during pregnancy makes interpretation of results difficult. |
Gms = grams; HCl = hydrochloric acid; IQ = interquartile; JEM = job-exposure-matrix; NR = not reported; NYSDOH = New York State Department of Health; RDD = random digit dialing; PCE = tetrachloroethylene; SES = socioeconomic status; TTHM = total trihalomethanes; VOC = voltile organic compounds;
Ruckert et al. (2013) also studied childhood leukemia and non-Hodgkin lymphoma.
Forand et al. (2012) also studied term low birthweight, pre–term birth, and fetal growth restriction.
Infants with patent ductus arteriosus (ICD-9 747.0) included if birthweight ≥2500 gms.
Bove et al. (1995) and Bove (1996) also studied low (<2,500 gms) and very low birthweight (<1,500 gms), small for gestational age, premature births examined but results not reported.
Lagakos et al. (1986) also studied perinatal death, low birth weight, and childhood disorders but did not report results.
Table 2:
Consideration of Biases, Confounding, and Chance in TCE – Cardiac Defect Epidemiology Studies
| Reference | Selection bias | Information bias, Exposure | Information bias, Outcome | Recall bias | Chance | Confounding |
|---|---|---|---|---|---|---|
|
Forand et al. (2012) Cohort |
Unlikely | Likely, area–level exposure assignment but soil vapor intrusion found throughout the TCE study area | Unlikely, birth defects registry study with medically-verified outcomes | Unlikely | No | Unlikely, adjusted for important maternal risk factors, including prenatal care but not folic acid intake |
|
Yauck et al. (2004) Case-control |
Unlikely | Likely, area-level exposure assignment; poorly defined exposure surrogate | Unlikely, birth certificate and birth defects registry study | Unlikely | No | Likely, univariate statistical analyses not adjusted for maternal risk factors |
|
Bove et al. (1995); Bove (1996) Cohort |
Unlikely | Likely, area–level exposure assignment | Unlikely, registry (birth, congenital malformation) study | Unlikely | Yes | Unlikely, univariate statistical analysis; effect estimate from multivariate analysis adjusted for important maternal risk factors, but not folic acid intake, not different by ±15% from univariate |
|
Goldberg et al. (1990) Prevalence |
Likely. Two of three control groups are inappropriate and sparse details on selection of 3rd control group | Likely, area-level exposure assignment | Unlikely, cases identified from cardiologists files | Likely | No | Unable to assess; study lacks details of statistical analysis |
|
Lakagos et al. (1986) Prevalence |
Unlikely | Likely, area-level exposure assessment | Likely, self-reported outcomes | Likely | Yes | Unlikely, age, education, race, prenatal care, and parity evaluated as potential confounders |
The studies were of different populations, living in different states, and of different epidemiological designs. Forand et al. [29] is a retrospective cohort study of 1440 live births among New York residents in an area contaminated with TCE via vapor intrusion. Bove [8]/Bove et al. [9] is a cross-sectional study of 80,938 singleton live-born infants and 594 singleton fetal deaths among residents in northern New Jersey receiving TCE in municipal water supplies. A strength of both studies is the use of state records, including State Birth Defects Registries with clinically verified outcomes that reduce information and subject recall bias, and the ability to control for potential confounding factors. Both of the studies observed an elevated relative risk estimate for major cardiac defects: a relative risk of 1.24 (a 50% confidence interval (CI) was reported: 0.75, 1.94) for >10 ppb TCE in municipal drinking water supplies compared to TCE exposure ≤1 ppb in Bove [8]/Bove et al. [9]; and an estimated relative risk of 2.40 (95% CI: 1.00, 5.77) compared to the rest of New York State, excluding New York City in Forand et al. [29]. Both studies report relative risk estimates for specific defects: 1.30 (50% CI: 0.88, 1.87) for ventricular septal defects and exposure to >5 ppb TCE in drinking water compared to <1 ppb (Bove [8]/Bove et al. [9]) and 4.91 (95% CI: 1.58, 15.24) for conotruncal defect in the TCE-contaminated area compared to the rest of New York State, excluding New York City [29]. Yauck et al. [94], a case-control study of 245 cases and 3780 controls, reported that living within 1.32 miles from at least one TCE emissions source in Wisconsin had a strong relative risk estimate of 6.2 (95% CI: 2.6, 14.5) for cardiac defects in infants born to mothers aged 38 years or older after controlling for potential confounding, but no association for cardiac defects was observed among infants of mothers aged less than 38 years (RR = 0.9, 95% CI: 0.6, 1.2). The original case-control study by Goldberg et al. [35] reported that the likelihood of family exposure to the contaminated water area among families with cardiac defects was three times that of exposure among randomly selected families in the same general locality. In a review article that included the Goldberg et al. [35] study, Bove et al. [7] calculated an unadjusted prevalence ratio of cardiac defects among residents of the contaminated area with first-trimester exposure compared with residents in uncontaminated areas of 2.58 (95% CI: 2.0, 3.4). Ruckart et al. [71] reported little detail on cardiac defects in a population exposed to TCE-contaminated water but noted a lower than expected number of conotruncal heart defects–although neither precise counts nor confidence intervals were reported, and the authors did not draw any conclusions concerning TCE exposure and the occurrence of all cardiac defects or conotruncal heart defects. Lagakos et al. [56] reported no association (p = 0.91) between exposure to TCE-contaminated water in Woburn, Massachusetts and a much larger categorical grouping of ‘cardiovascular anomalies’ which included heart murmurs (15 of 43 anomalies) and only 2 conotruncal heart defects.
Forand et al. [29] and Bove [8]/Bove et al. [9] provide evidence for an association between maternal TCE exposure and cardiac defects. A more mixed pattern of results is seen in three other studies with greater potential for bias and confounding [94,35,56]; however, the results of these studies are not necessarily inconsistent with the association observed by Forand et al. [29] or Bove [8]/Bove et al. [9] because, for the database as a whole, the epidemiological studies are imprecise in estimating effects due to the small number of cardiac defects. Additionally, information bias related to the exposure assessment in these studies may provide alternative explanations for the apparent heterogeneity. As the exposure assessment methods in these studies are at an aggregate level based on locality (rather than based on individual-level measurements), one can assume that the incumbent exposure measurement error (also known as information bias) is non-differential with respect to cardiac defects. That is, any errors in exposure assessment are expected to be independent of case status. Such non-differential misclassification of exposure would typically result in bias towards the null [70] and limit the ability of the studies to detect some associations and possibly exposure-response relationships. None of the studies considered maternal folic acid intake, which may reduce the risk of cardiac defects [45] and is thus a potential confounder. Because TCE has been shown to induce folate deficiency in rats [22] and in workers [36], folate concentrations may be on the direct causal pathway from TCE exposure to cardiac defects. Thus it was methodologically appropriate for these studies not to control for folic acid/folate as that would have induced bias towards the null. Rather, women with low dietary intake of folic acid may represent a susceptible sub-group. Both Forand et al. [29] and Bove [8]/Bove et al. [9] adjust for other maternal risk factors, including adequate prenatal care, as potential confounding factors. Observations in the other studies are more uncertain compared to Forand et al. [29] and Bove [8]/Bove et al. [9], and the observed heterogeneity of results may be due to alternative explanations, such as bias, chance, or potential confounding. Use of hospital cases by Yauck et al. [94] and cases identified from cardiologists’ records by Goldberg et al. [35] may introduce possible selection bias. It is difficult to evaluate control for potential confounding in Goldberg et al. [35] due to limited reporting in the publication. The self-reporting of outcome in Lagakos et al. [56] introduces uncertainty because of potential selective reporting.
In summary, epidemiologic data provide some support for the possible relationship between maternal TCE exposure and cardiac birth defects. Forand et al. [29] provide clear evidence of an association between living in an area contaminated by TCE via vapor intrusion and increased risk of conotruncal heart defects, and Bove [8]/Bove et al. [9] provide limited evidence for an association between maternal exposure to TCE, or the combination of TCE and other chlorinated solvents in drinking water, and cardiac defects. However, there are uncertainties in the interpretation of the epidemiological data on Bove [8]/Bove et al. [9] because of the small number of observed TCE-exposed cardiac defect cases, sparse reporting on TCE exposure and congenital heart defects (CHDs) in both publications, and the study’s cross-sectional design that could not establish temporality. Two other studies with potential biases also observed elevated risk estimates between TCE exposure and cardiac defects [94,35] and these provide some corroboration of the observations in Forand et al. [29]. The lack of supporting evidence from Ruckart et al. [71] may be a consequence of the small number of reported cases. Additionally, because Lagakos et al. [56] examined a much more broadly defined set of outcomes, their findings are likely much less specific than conotruncal heart defects or even cardiac defects as reported by the other investigators.
The limited finding of an association between TCE exposure and conotruncal heart defects, in particular, and cardiac defects more generally has coherence with the broader epidemiological literature that reports association between maternal occupational exposure to degreasing solvents or to organic solvents and CHDs [11,34,93,83,84]. Although the reported associations between TCE exposure and increased risks of cardiac defects were observed in several studies [29,94,8,9,35], overall, these epidemiologic studies are not sufficient to establish a causal link between TCE exposure and cardiac defects in humans. This conclusion is consistent with other reviews of the epidemiological literature for TCE exposures and CHD [13,90,38]. Additional research could better characterize human exposures and health outcomes.
3.1.2. Toxicological data
The experimental toxicology database for the assessment of developmental cardiac defects resulting from TCE exposure includes in ovo chicken studies, in vitro assays, and rodent studies that assessed fetal morphology following in utero exposures to TCE or its oxidative metabolites. Summaries of studies that assessed cardiac development in mammalian laboratory animal models are presented in Table 3a (inhalation exposure to TCE), Table 3b (oral exposures to TCE), and Table 3c (oral exposures to DCA and TCA). Studies using non-mammalian or in vitro test systems to assess cardiac development following exposures to TCE, DCA, or TCA are summarized in Table 3d. Study strengths and limitations for the mammalian inhalation and oral studies of TCE or its metabolites (DCA or TCA) are summarized in Table 4. Exposure-response arrays for general categories of adverse developmental outcomes (decreased survival, decreased growth, and altered morphological development, including cardiac defects) are presented in Figs. 2–4 for studies with gestational inhalation exposures to TCE, oral exposures to TCE, and oral exposures to DCA and TCA (respectively). Incidence data for specific developmental findings are not presented herein since that information is summarized in the IRIS assessment [87].
Table 3a.
Summary of mammalian in vivo toxicity studies assessing cardiac development — inhalation exposures
| Reference | Species/strain/ sex/number |
Exposure level/ Durationa |
NOAEL; LOAELb | Effects |
|---|---|---|---|---|
| Carney et al. (2006) | Rat, Sprague-Dawley, females, 27 dams/group | 0, 50, 150, or 600 ppm (600 ppm = 3.2 mg/L)c (268.5, 805.5, 3222 mg/m3) 6 hrs/d; GDs 6–20 |
Maternal NOAEL: 150 ppm (805.5 mg/m3) Maternal LOAEL: 600 ppm (3222 mg/m3) |
↓ Body weight gain (22% less than control) on GDs 6–9 at 600 ppm. |
| Developmental NOAEL: 600 ppm (3222 mg/m3) | No evidence of developmental toxicity, including heart defects. | |||
| Dorfmueller et al. (1979) | Rat, Long-Evans, females, 30 dams/group | 0 or 1,800 ± 200 ppm (9,674 ± 1,075 mg/m3)c 2 wks, 6 hrs/d, 5 d/wk; prior to mating and/or on GDs 0–20 |
Maternal NOAEL: 1,800 ± 200 ppm (9,674 ± 1,075 mg/m3) | No maternal abnormalities. |
| Developmental LOAEL: 1,800 ± 200 ppm (9,674 ± 1,075 mg/m3) | Statistically significant ↑ skeletal and soft tissue anomalies in fetuses from dams exposed during pregnancy only. No statistically significant treatment effects on behavior of offspring 10, 20, or 100 d postpartum. Body weight gains statistically significant ↓ in pups from dams with pre-gestational exposure. | |||
| Hardin et al. (1981) | Rat, Sprague-Dawley, female, nominal 30/group | 0 or 500 ppm (0 or 2685 mg/m3) 6–7 hrs/d; GDs 1–19 |
Maternal NOAEL: 500 ppm (2685 mg/m3) | No maternal toxicity. |
| Developmental NOAEL: 500 ppm (2685 mg/m3) | No embryonic or fetal toxicity. | |||
| Rabbit, New Zealand white, female, nominal 20/group | 0 or 500 ppm (0 or 2685 mg/m3) 6–7 hrs/d; GDs 1–24 |
Maternal NOAEL: 500 ppm (2685 mg/m3) | No maternal toxicity. | |
| Developmental LOAEL: 500 ppm (2685 mg/m3) | Hydrocephaly observed in two fetuses of two litters, considered equivocal evidence of teratogenic potential. | |||
| Healy et al. (1982) | Rat, Wistar, females, 31–32 dams/group | 0 or 100 ppm (0 or 535 mg/m3) 4 hrs/d; GDs 8–21 |
Maternal NOAEL: 100 ppm (535 mg/m3) | No maternal abnormalities. |
| Developmental LOAEL: 100 ppm (535 mg/m3) | Litters with total resorptions statistically significant ↑. Statistically significant ↓ fetal weight, and ↑ bipartite or absent skeletal ossification centers. | |||
| Schwetz et al. (1975) | Rat, Sprague-Dawley, female, 20–35/group Mouse, Swiss-Webster, females, 30–40 dams/group |
0 or 300 ppm (0 or 1611 mg/m3) 7 hrs/d; GDs 6–15 |
Maternal LOAEL: 300 ppm (1611 mg/m3) | 4–5% ↓ maternal body weight |
| Developmental NOAEL: 300 ppm (1611 mg/m3) | No embryonic or fetal toxicity; not teratogenic. | |||
| Developmental LOAEL: 150 ppm (805.5 mg/m3) | Specific gravity of brains statistically significant ↓ at PNDs 0, 10, and 20–22. Similar effects at PNDs 20–22 in occipital cortex and cerebellum. No effects at 1 mo of age. |
To convert concentrations in air (at 25ºC) from ppm to mg/m3: mg/m3 = (ppm) x (molecular weight of the compound)/(24.45). For TCE: 1 ppm = 5.37 mg/m3. 1000 mg/m3 = 1 mg/L (air). Source: U.S. EPA, Technology Transfer Network - Air Toxics Web Site, http://www.epa.gov/ttnatw01/hlthef/tri-ethy.html, last accessed 08–06-15.
NOAEL and LOAEL are based upon reported study findings.
Table 3b.
Summary of mammalian in vivo toxicity studies assessing cardiac development — oral exposures
| Reference | Species/strain/ sex/number |
Dose level/exposure durationa | Route/ vehicle | NOAEL; LOAELb | Effects |
|---|---|---|---|---|---|
| Cosby and Dukelow (1982 ) | Mouse, B6D2F1, female, 28–62 dams/group | 0, 24, or 240 mg/kg-d GDs 1–5, 6–10, or 11–15 |
Gavage in corn oil | Maternal NOAEL: 240 mg/kg-d | No maternal toxicity. |
| Developmental NOAEL: 240 mg/kg-d | No effects on embryonic or fetal development. | ||||
| Dawson et al. (1993) | Rat, Sprague-Dawley, 116 females allocated to 11 groups | 0, 1.5, or 1,100 ppm (mg/L) (0, 0.18 or 133 mg/kg-d) 2 mo before mating and/or during gestation |
Drinking water | Maternal NOAEL: 1,100 ppm (132 mg/kg-d) | No maternal toxicity. |
| Developmental LOAEL: 1.5 ppm (0.18 mg/kg-d) | Statistically significant ↑ in heart defects, primarily atrial septal defects, found at both dose levels in groups exposed prior to pregnancy and during pregnancy, as well as in group exposed to 1,100 ppm dose during pregnancy only. No statistically significant ↑ in congenital heart defects in groups exposed prior to pregnancy only. | ||||
| Fisher et al. (2001); Warren et al. (2006) | Rat, Sprague-Dawley, female, 20–25 dams/group | 0 or 500 mg/kg-d GDs 6–15 |
Gavage in soybean oil | Maternal NOAEL: 500 mg/kg-d | No maternal toxicity. |
| Developmental NOAEL: 500 mg/kg-d | No developmental toxicity. The incidence of heart malformations for fetuses from TCE-treated dams (3–5%) did not differ from negative controls. No eye defects observed. | ||||
| Johnson et al. (2003) | Rat, Sprague-Dawley, female, 9–13/group, 55 in control group | 0, 2.5, 250, 1.5, or 1,100 ppm (0, 0.00045, 0.048, 0.218, or 129 mg/kg-d)d GDs 0–22 |
Drinking water |
Developmental NOAEL: 2.5 ppb (0.00045 mg/kg-d) Developmental LOAEL: 250 ppbb (0.048 mg/kg-d) |
Statistically significant ↑ in percentage of abnormal hearts and the percentage of litters with abnormal hearts at ≥250 ppb. |
| Narotsky et al. (1995) | Rat, F344, females, 8–12 dams/group | 0, 10.1, 32, 101, 320, 475, 633, 844, or 1,125 mg/kg-d GDs 6–15 |
Gavage in corn oil | Maternal LOAEL: 475 mg/kg-d | Statistically significant dose-related ↓ dam body weight gain at all dose levels on GDs 6–8 and 6–20. Delayed parturition at ≥475 mg/kg-d; ataxia at ≥633 mg/kg-d; mortality at 1,125 mg/kg-d. |
| Developmental NOAEL: 32 mg/kg-d Developmental LOAEL: 101 mg/kg-d |
↑ full litter resorption and postnatal mortality at ≥425 mg/kg-d. Statistically significant prenatal loss at 1,125 mg/kg-d. Pup body weight ↓ (not statistically significant) on PNDs 1 and 6. Statistically significant ↑ in pups with eye defects at 1,125 mg/kg-d. Dose-related (not statistically significant) ↑ in pups with eye defects at ≥101 mg/kg-d. | ||||
| Narotsky and Kavlock (1995) | Rat, F344, females, 16–21 dams/group | 0, 1,125, or 1,500 mg/kg-d GDs 6–19 |
Gavage in corn oil | Maternal LOAEL: 1,125 mg/kg-d | Ataxia, ↓ activity, piloerection; dose-related ↓ body weight gain. |
| Developmental LOAEL: 1,125 mg/kg-d | Statistically significant ↑ full litter resorptions, ↓ live pups/litter; statistically significant ↓ pup body weight on PND 1; statistically significant ↑ incidences of microophthalmia and anophthalmia. |
For conversion of drinking water or dietary doses to mg/kg-d when no body weight or compound consumption data were available: mg/L in water x subacute conversion factor (0.121 for female rats, 0.191 for female mice); mg/L in water x subchronic conversion factor (0.093 for female rats, 0.164 for female mice); mg/kg (ppm) in feed x subacute conversion factor (0.117 for female rats, 0.224 for female mice); mg/kg (ppm) in feed x subchronic conversion factor (0.091 for female rats, 0.215 for female mice). For developmental studies, offspring duration of exposure was used; subacute conversion factor was applied unless otherwise noted. Reference: EFSA (2012).
NOAEL and LOAEL are based upon reported study findings.
Table 3c.
DCA and TCA: Summary of mammalian in vivo toxicity studies assessing cardiac development — oral exposures
| Reference | Species/strain/ sex/number |
Dose level/ exposure duration |
Route/ vehicle | NOAEL; LOAELa | Effects |
|---|---|---|---|---|---|
| DCA | |||||
| Smith et al. (1992) | Rat, Long Evans, female, 19–21 dams/group | A: 0, 14, 140, or 400 mg/kg-d B: 0, 900, 1400, 1900, or 2400 mg/kg-d GDs 6–15 |
Gavage in water | Maternal LOAEL: 14 mg/kg-d | Increased adjusted liver weight at ≥14 mg/kg-d; decreased body weight gain at ≥140 mg/kg-d; increased spleen and kidney weights at ≥400 mg/kg-d; mortality at ≥1400 mg/kg-day |
| Developmental LOAEL: 140 mg/kg-d | Increased soft tissue malformations (primarily cardiovascular, e.g., defects between ascending aorta and right ventricle) at ≥140 mg/kg-d; decreased fetal weight and length at ≥400 mg/kg-d.; increased resorptions and increased orbital anomalies at ≥900 mg/kg-d | ||||
| Fisher et al. (2001) | Rat, Sprague-Dawley, female, 20 dams/group | 0 or 300 mg/kg-d GDs 6–15 |
Gavage in water | Maternal LOAEL: 300 mg/kg-d | Decreased body weight gain |
| Developmental LOAEL: 300 mg/kg-d | Decreased fetal weight; no significant difference from control in percent fetuses with cardiovascular malformations; no increased resorptions | ||||
| Epstein et al. (1992) | Rat, Long Evans, female, 7–10 dams/group | 0 or 1900 mg/kg-d GDs 6–8, 9–11, or 12–15 0 or 2400 mg/kg-d GDs 10, 11, 12, or 13 0 or 3500 mg/kg-d GDs 9, 10, 11, 12, or 13 |
Gavage in water | Maternal LOAEL: Not characterized | Not reported |
| Developmental LOAEL: 1900 mg/kg-d | Increased interventricular septal defects, membranous type, and high interventricular septal defects; fetal weight and survival data not reported | ||||
| TCA | |||||
| Smith et al. (1989) | Rat, Long Evans, female, 20–26 dams/group | 0, 330, 800, 1200, or 1800 mg/kg-d GDs 6–15 |
Gavage in water | Maternal LOAEL: 330 mg/kg-d | Increased spleen and kidney weights; decreased body weight gain at ≥800 mg/kg-day. |
| Developmental LOAEL: 330 mg/kg-d | Decreased fetal weight and length; soft tissue malformations (primarily cardiovascular, e.g., interventricular septal defect and levocardia, at incidences ranging from 5.4–95% in treated groups); increased postimplantation loss at ≥800 mg/kg-d; skeletal malformations, mainly orbital anomalies, at 1200 and 1800 mg/kg-day. | ||||
| Fisher et al. (2001) | Rat, Sprague-Dawley, female, 19 dams/group | 0 or 300 mg/kg-d GDs 6–15 |
Gavage in water | Maternal LOAEL: 300 mg/kg-d | Decreased body weight gain |
| Developmental LOAEL: 300 mg/kg-d | Decreased fetal weight; no significant difference from control in percent fetuses with cardiovascular malformations; no increased resorptions | ||||
| Johnson et al. (1998) | Rat, Long Evans, female, 55 control, 11 TCA | 0 or 2730 ppm (291 mg/kg-d) GDs 1–22 |
Drinking water | Maternal LOAEL: 291 mg/kg-d | Decreased body weight gain |
| Developmental LOAEL: 291 mg/kg-d | Decreased fetal weight; increased resorptions per litter; increased percent fetuses with abnormal hearts (10.5% vs 2.15% in controls) | ||||
NOAEL and LOAEL are based upon reported study findings.
Table 3d.
Summary of non-mammalian and in vitro studies on TCE and metabolites (DCA and TCA) assessing cardiac development
| Reference | Species/strain/ sex/number |
Dose level/ exposure duration |
Route/ vehicle | NOAEL; LOAELa | Effects |
|---|---|---|---|---|---|
| Avian In Ovo | |||||
| Bross et al. (1983) | Chicken, white leghorn, 20–24 embryos/group | TCE: 0, 1, 5, 10, or 25 µmol/egg Single injection on day 1 or 2 |
In ovo injection in mineral oil | Developmental LOAEL: 1 µmol | Decreased survival at ≥1 µmol; increased edema, light pigment, abnormal beak, club foot, and patchy feathers at ≥1 µmol; evisceration at ≥5 µmol; growth not affected; visceral (including cardiac) development was not assessed |
| Drake et al., (2006a) | Chicken, white leghorn, Babcock and Bovan strains, 32–46 embryos/group | TCE: 0, 0.2, 4, or 200 nmol/egg (0, 3, 60, or 3000 nM/egg) Single injections on HH13, HH15, HH17, and HH20; assessed on HH24 and HH30 |
In ovo injection in saline | Developmental LOAEL: 4 nmol | Decreased survival on HH30, increased proliferative index in outflow tract (OFT) and atrioventricular canal (AVC) cardiac cushion mesenchyme on HH24, increased mean cushion cellularity, and decreased blood flow on HH24 at ≥4 nmol; |
| TCA: 0 or 4 nmol/egg (0 or 60 nM/egg) Injections on HH13, HH15, HH17, and HH20; assessed on HH24 and HH30 |
In ovo injection in saline | Developmental LOAEL: 4 nmol | Decreased survival on HH30, increased proliferative index in OFT and AVC cardiac cushion mesenchyme on HH24, increased mean cushion cellularity on HH24 at ≥4 nmol | ||
| Drake et al. (2006b) | Chicken, white leghorn, Bovan strain, 35–117 embryos/group | TCE and TCA: 0, 0.2, 2, 4, 20, or 200 nmol/egg (0, 3, 30, 60, 300, or 3000 nM/egg) Single injections on HH13, HH15, HH17, and HH20; assessed on HH18, HH21, and HH23 |
In ovo injection in saline | Developmental NOAEL: 0.2 nmol | No alterations in cardiac development on HH18, HH21, or HH23 were observed, since exposure was during period of cardiac specification rather than during period of valvuloseptal morphogenesis as in Drake et al., 2006a |
| Elovaara et al. (1979) | Chicken, white leghorn, SK 12 strain, 9–14 embryos/group | TCE: 0, 5, 25, 50, or 100 µmol/egg Single injection on day 2 or 6 |
In ovo injection in olive oil | Developmental LOAEL: 5 µmol | Increased malformations (exteriorization of viscera, edema, eye abnormalities, and skeletal abnormalities) in surviving 14- or 15-day embryos at ≥5 µmol; decreased survival, weight, and length at 100 µmol; visceral (including cardiac) development was not assessed |
| Loeber et al. (1988) | Chicken, white leghorn, strain not reported, 91–128 treated embryos/ group; 266–7 control embryos/group | TCE: 0, 5, 10, 15, 20, or 25 µM/egg Single injection on day 6, 12, 18, or 23; assessed at HH29, HH 34, or HH44 |
In ovo injection in saline or mineral oil | Developmental LOAEL: 10 µM | Overall increased cardiac malformations and embryo death in all treated embryos vs. control (categorized as ectomesenchymal tissue migration abnormalities, ECM abnormalities, and cell death abnormalities); increased percent embryos with cardiac malformations at ≥10 µM; |
| Rufer et al. (2010) | Chicken, white leghorn, Hyline strain W36, 35–117 embryos/group | TCE: 0, 0.2, 4, 40, 200, or 2000 nmol/egg (0, 0.4, 8, 80, 400, or 4000 ppb/egg) Single injections on HH13, HH15, HH17, HH20, or HH24; assessed on HH24 and HH30 |
In ovo injection in saline | Developmental LOAEL: 4 nmol | Decreased survival on HH30 following exposure on HH15 or HH17 at ≥4 nmol; increased incidence of muscular ventricular septal defects (VSD) in embryos treated on HH 17 (related blood flow abnormalities confirmed by Doppler imaging); increased abnormalities of cardiac structure and function noted by echocardiography in HH28 treated embryos (incidence data not provided). |
| Zebra Fish | |||||
| Hassoun et al. (2005) | Zebrafish (Danio rerio), 30 embryos/group | DCA: 0, 4, 8, 16, or 32 mM Exposed from 4 to 144 hr post-fertilization (hpf) |
Petri dish: 20 mL buffered water, with 60 mg sea salt/L | Developmental LOAEL: 4 mM | Dose-related increased mortality at ≥8 nM on 8–55 hpf; hatching delayed at 55 hpf in ≥8 mM; increased yolk sac edema at ≥8 nM; increased craniofacial (jaw and mouth) abnormalities at 80 hpf: 5% at 4 and 8 mM, 75% at ≥16 mM; skeletal muscle deformation and notochord/muscular lordosis at ≥16 mM by 144 hpf; abnormal feeding behavior at ≥4 mM by 144 hpf; increased heart rate at ≥16 mM at 32, 55, and 80 hpf and decreased heart rate with near cessation of peripheral blood flow at ≥16 mM at 144 hpf; increased superoxide anion and nitric oxide production at ≥4 nM by 80 hpf |
| Williams et al. (2006) | Zebrafish (Danio rerio), 30 embryos/group | DCA: 0 or 32 mM (Ellagic acid groups were also conducted but are not described here) Exposed from 4 to 144 hr post-fertilization (hpf) |
Petri dish: 20 mL buffered water | Developmental LOAEL: 32 mM | At 32 mM: 100% mortality after 144 hpf; hatching rate delayed at 55 hpf; yolk sac and/or cardiac edema at 55–144 hpf; increased craniofacial (jaw and mouth) abnormalities at 80 hpf; skeletal muscle deformation and notochord/muscular lordosis by 144 hpf; abnormal feeding behavior at 144 hpf; increased heart rate at 32 and 55 hpf and decreased heart rate at 80 hpf with near cessation of peripheral blood flow at 144 hpf; increased superoxide anion at 144 hpf and nitric oxide by 55 hpf |
| In Vitro | |||||
| Hunter et al. (1996) | Mouse, CD-1, 3–6 somites, 24 control and 10–18 treated embryos/group | DCA: 0,734, 1468, 4403, 4403, 5871, 7339, 11010, or 14680 µM 24 hr exposure |
Whole embryo culture | Developmental LOAEL: 5871 uM | Increased % malformations and % neural tube defects, decreased mean number of somites at ≥5871 µM; increased pharyngeal arch defects and heart defects at ≥7339 µM; increased rotational defects, eye defects, and somite dysmorphology at ≥11010 µM |
| Mouse, CD-1, 3–6 somites, 106 control and 10–56 treated embryos/group | TCA: 0, 500, 1000, 2000, 3000, 4000, or 5000 µM 24 hr exposure |
Whole embryo culture | Developmental LOAEL: 2000 uM | Increased % malformations and % neural tube defects, decreased mean number of somites at ≥2000 µM; increased eye defects and heart defects at ≥3000 µM; increased somite dysmorphology at 4000 µM | |
| Mishima et al. (2006) | Chicken, white leghorn, strain not reported, HH13–14, 40–104 embryos/ group | TCE: 0, 10, 40, or 80 ppm 24 hr exposure |
Whole embryo culture | Developmental LOAEL: 80 ppm | Decreased mesenchymal cell number in superior and inferior AV cushions at 80 ppm |
| Saillenfait et al. (1995) | Rat, Sprague-Dawley, GD 10 explants, 4–7 somites, 20 embryos/group | TCE: 0, 2.5, 5, 10, 15, or 30 mM 46 hr exposure |
Whole embryo culture | Developmental LOAEL: 5 mM | Decreased yolk sac diameter, crown-rump length, head length, and % malformed (brain defects and reduction in embryonic axis) at ≥5 mM; increased malformations: bend in embryonic axis, reduction in first brachial arch, otic system defect, defective flexion, absence of hindlimb bud, delayed yolk sac circulation at ≥10 mM; increased eye defects and overall poor and abnormal development at ≥15 mM; no cardiac defects noted. |
NOAEL and LOAEL are based upon reported study findings.
Table 4:
Study Quality Summary for Toxicology Studies that Assessed TCE Exposure and Developmental Effects
| Reference | Exposure Quality | Test Subjects | Study Design | Endpoints | Data & Statistics | Reporting | |
|---|---|---|---|---|---|---|---|
| In Vivo Mammalian Inhalation Studies | |||||||
| Carney et al. (2006) | Strength | Ambient air control and 3 exposure groups; relevant route of administration and duration of exposure. Information on chemical source provided. Inhalation chamber characterized; dynamic airflow; mean chamber concentrations reported. |
Species, strain, source, sex, age/lifestage/BW, reported. Randomly assigned to test groups prior to mating. Adequate sample size (27 litters/group). |
GLP, guideline prenatal devtox study. All litters/fetuses evaluated. Fetal examination conducted without knowledge of treatment group. | Relevant maternal and fetal endpoints assessed. Fetal visceral exam used Staples method (examination of internal cardiac morphology) plus free-hand sectioning of head. Skeletal exam evaluated both bone and cartilage development. | Appropriate statistical methods; litter used as unit of statistical analysis. | Summary data for maternal and fetal endpoints reported. |
| Limitation | Whole body exposure. | Individual maternal and fetal data NR. | |||||
| Dorfmueller et al. (1979) | Strength | Filtered ambient air control and 3 groups with a single high dose exposure level over various durations; relevant route of administration and duration of exposure. Information on chemical source provided. Inhalation chamber characterized; dynamic airflow; chamber concentrations monitored at 13 minute intervals. |
Species, strain, source, sex, age/lifestage/BW, reported. Randomly assigned to test groups. Adequate sample size (subset of 15 litters/group assigned to c-section). | Study included groups with exposures and assessments similar to EPA guideline. | Relevant maternal and fetal endpoints assessed. Fetal visceral exam used Wilson technique (free-hand sectioning of fetuses). Skeletal exam evaluated bone development. |
Appropriate methods; litter used as unit of statistical analysis. | Summary data for maternal and fetal endpoints reported. |
| Limitation | Technical grade solvent containing 99% TCE and 0.2% epichlorhydrin. Whole body exposure. | Information on facility certification NR. Non-random assignment (based upon uterine position) of 8 fetuses/litter to either skeletal or visceral exam; disposition of additional fetuses NR | Fetal visceral exam did not include in situ dissection and examination of cardiac morphology. | Individual maternal and fetal data NR. | |||
| Hardin et al. (1981) | Strength | Control and 1 exposure group; relevant route of administration and duration of exposure. | Species, strain, sex, reported. Adequate sample size: target was 30 rats or 20 rabbits/group; report indicated difficulties in some studies resulting in 15 rabbits/group. | Study design similar to EPA guideline. | Relevant maternal and fetal endpoints assessed. Fetal visceral exam used Wilson technique (free-hand sectioning of ½ to ⅔ fetuses/litter). Skeletal exam evaluated bone development. | ||
| Limitation | Whole body exposure. Chemical characterization and source NR. Control exposure not characterized. Exposure chamber, conditions, and measurement of concentration NR. Duration of exposure not characterized for TCE. | Maternal source, age/lifestage/BW, and random assignment to test groups NR. Exact sample size NR. | Information on facility certification NR. The report summarized developmental toxicity testing for 9 chemicals including TCE; specific study design details for each test substance were NR. | The exact distribution of fetuses for visceral and skeletal evaluation NR. Random assignment to evaluation procedure NR. Whether Staples dissection method (examination of internal cardiac morphology) was used in the TCE studies was NR. | Statistical methods NR. | Study design details NR. Summary data for maternal and fetal endpoints NR. With the exception of a brain malformation in 2 rabbit fetuses, study findings for TCE were not discussed. | |
| Healy et al. (1982) | Strength | Ambient air control and 1 exposure group; relevant route of administration and duration of exposure. Information on chemical source provided. Inhalation chamber characterized; dynamic airflow; chamber concentrations monitored continuously. |
Species, strain, source, sex, age/lifestage/BW, reported. Randomly assigned to test groups after mating. Adequate sample size (31 control, 32 treated litters). | Study design similar to EPA guideline. All fetuses assessed for external, visceral, and skeletal effects. | Relevant maternal and fetal endpoints assessed. Fetal visceral exam used fresh dissection and examination of internal organs. Skeletal exam evaluated bone development. | Appropriate statistical methods for some outcomes. | Summary data for maternal and fetal endpoints reported. |
| Limitation | Whole body exposure. Chamber concentration data NR. | Information on facility certification NR. Exposure duration (4 hr/day, GD 8–21) was insufficient in daily duration and did not cover the entire period of organogenesis. No indication that fetuses were examined without knowledge of treatment group. | Fetal visceral examination did not include brain or reproductive organs. Fetal cardiac exam did not include internal morphology. | No indication that litter was used as unit of statistical analysis for fetal anomalies. | Individual maternal and fetal data NR. | ||
| Schwetz et al. (1975) | Strength | Filtered ambient air control and 1 exposure group; relevant route of administration and duration of exposure. Information on chemical source provided. Inhalation chamber characterized; dynamic airflow; chamber concentrations monitored continuously and mean concentrations reported. |
Species, strain, sex, age/lifestage/BW, reported. Adequate sample size (30 control, 18 treated rats; 26 control, 12 treated mice). | Study design similar to EPA guideline. All fetuses assessed for external, visceral, and skeletal effects. | Relevant maternal and fetal endpoints assessed. Fetal visceral exam used Wilson technique (free-hand sectioning of ½ fetuses/litter) and skeletal exam evaluated bone development in ½ fetuses/litter. One fetus/litter randomly selected for whole-body sagittal sectioning and microscopic examination. | Appropriate statistical methods; litter used as unit of statistical analysis. | Summary data for some maternal and fetal endpoints reported. |
| Limitation | Whole body exposure. 99.2% TCE; 0.76% inhibitors and impurities. | Animal source NR. Random assignment of maternal animals to test groups NR. | Information on facility certification NR. No indication that fetuses were examined without knowledge of treatment group. | Random assignment of fetuses to visceral or skeletal evaluation procedure NR. The use of in situ dissection and examination of cardiac morphology NR. | Maternal BW data NR. Individual maternal and fetal data NR. | ||
| In Vivo Mammalian Oral Studies | |||||||
| Cosby and Dukelow (1992) | Strength | Vehicle control and 2 treatment groups; relevant route of administration and duration of exposure. Information on chemical source provided. Formulations mixed immediately prior to dosing; formulation methods enhanced stability; concentration tested. Dose volume = 0.2 ml. |
Species, strain, source, sex, age/lifestage/BW, reported. Adequate sample size (7–12 litters/control cohort, 10–12litters/treated cohort). | Study designed to examine effects on reproductive success and offspring birth and postnatal outcome. | Relevant maternal and offspring endpoints assessed. All pups examined. Random selection of litters for PND 43 postmortem evaluation. | Statistical analysis of data conducted. | Summary gestation index and litter size data reported. |
| Limitation | Concentration data NR | Females non-randomly assigned to test groups (based on BW) after mating. | Corn oil vehicle. Dose duration (5 daily prenatal doses, initiating at GD 1, 6, or 11) do not cover the entire period of organogenesis. No indication that fetuses were examined without knowledge of treatment group. | Litter size standardization was implemented on PND 1 and 22; no indication of random selection of pups for culling. | Statistical methods not fully characterized. Replicate treatment data sets were pooled for tests of statistical significance. No indication that the litter was used as unit of statistical analysis. | Maternal BW and postmortem data NR. Offspring weight, length, external abnormalities, and postmortem data NR. |
|
| Dawson et al. (1990) | Strength | Vehicle control and 2 treatment groups per metabolite; relevant duration of exposure. Information on chemical source provided. |
Species, strain, source, sex, age/lifestage/BW, reported. Adequate sample size (10–17 dams/group). | Study designed specifically to assess fetal cardiac defects. Detailed fetal cardiac dissection, preservation, and examination methods provided. |
Relevant fetal endpoints assessed. Fetal cardiac exam conducted without knowledge of treatment group. Positive cardiac findings were confirmed by unanimous agreement of study authors. | Individual and summary incidences of fetal cardiac defects reported. | |
| Limitation | Route of administration = intrauterine injection (not relevant to environmental exposures). Surgery was performed on GD 7 pregnant rats to insert osmotic pump. DCA and TCA purity, stability, concentration data NR | No indication of random assignment to test groups. | No information on laboratory certification status. Duration of study conduct NR. | Maternal observations consisted of monitoring for adverse consequences of surgery. | Statistical methods NR although significance was reported. | Fetal BW and length NR; variance for mean implant and resorption data not shown in bar graph; litter incidence of cardiac defects NR. | |
| Dawson et al. (1993) a | Strength | Vehicle control and 2 treatment groups; relevant route of administration and duration of exposure. Information on chemical source provided; stability and concentration tested; formulation methods enhanced stability. Drinking water formulations mixed daily. WC measured daily. Dose calculations based on consumption, concentration, and TCE breakdown rates. |
Species, strain, source, sex, age/lifestage/BW, reported. Randomly assigned to test groups after mating. | AAALAC-certified facility. Study designed specifically to assess fetal cardiac defects. Detailed fetal cardiac dissection, preservation, and examination methods provided. |
Relevant maternal and fetal endpoints assessed. Fetal cardiac exam conducted without knowledge of treatment group. Positive cardiac findings were confirmed by unanimous agreement of study authors. | Individual fetal cardiac defect data were provided to EPA and analyzed using the litter as unit of statistical analysis. | Maternal endpoints reported for treated groups; individual fetal cardiac defects reported; litter associations for cardiac defects were provided to EPA. |
| Limitation | Vehicle = tap water (unknown contaminants); possible imprecision in WC values since dams were group housed; TCE purity, stability, concentration data NR | No. of fetuses/group reported, but not number of litters;b however, Johnson et al. reported the number of litters in the control (n = 13–15) and TCE groups (n = 9–13).a | Study conducted over period of 3 years in 2 cohorts; study dates for control animals overlapped treated groups but were not exactly concurrent. | Fetal evaluation of non-cardiac findings (visceral and skeletal) was not described. | Statistical analysis conducted under contract by study authors did not use litter as unit of statistical analysis.b | Maternal FC, WC, clin obs, placental wt, necropsy data NR; fetal BW and length, external and skeletal data, and non-cardiac visceral data NR; variance for mean implant and resorption data NR; cardiac defects were not associated with litter of origin.b | |
| Epstein et al. (1992) | Strength | 3 vehicle control groups and 3 treatment groups per exposure paradigm; relevant route of administration and durations of exposure (designed to identify critical developmental windows for cardiac defects). Information on chemical source provided; purity and stability confirmed; storage procedures enhanced stability. | Species, strain, source, sex, age/lifestage/BW, reported. Randomly assigned to test groups after mating. Adequate sample size (11–17 controls, 7–10 treated/group). | Study designed to identify critical windows of effects on cardiac development. | Relevant maternal and fetal endpoints assessed. | Statistical analysis of data conducted. | Incidence and mean (and %/litter) fetal cardiac data reported. |
| Limitation | Information on facility certification NR. Study was conducted in 3 cohorts; dates of study conduct NR. | No indication that offspring were examined without knowledge of treatment group. | No indication if the litter was used as unit of statistical analysis. | Variance NR for mean data. Maternal data NR. Individual and summary implantation, resorption, fetal BW, length, sex, and external evaluation data NR. | |||
| Fisher et al. (2001) | Strength | 2 vehicle controls and 1 treatment group per test substance; relevant route of administration and duration of exposure. Information on chemical source provided; weekly stability and concentration tested; storage procedures enhanced stability. Dose volumes were based on maternal BW. | Species, strain, source, sex, age/lifestage/BW, reported. Randomly assigned to test groups after mating. Adequate sample size (19–25 litters/vehicle control or treated group, 12 litters/positive control group). |
AAALAC-certified facility. Study designed specifically to assess fetal cardiac defects. All litters/fetuses evaluated. Fetuses were examined without knowledge of treatment group. Detailed fetal cardiac dissection, preservation, and examination methods provided. Positive control group was included. Cardiac dissection and evaluation team included Dr. Paula Johnson. |
Relevant maternal and fetal endpoints assessed. Fetal cardiac exam conducted without knowledge of treatment group. Cardiac evaluation procedures were the same as those used in Dawson et al. (1993) and Johnson et al. (1998a, 2003); hearts were also stained with hematoxylin. | Appropriate statistical methods; litter used as unit of statistical analysis. | Summary data for maternal and fetal endpoints reported. |
| Limitation | Stability schedule and data NR. Concentration not tested. | Individual maternal and fetal data NR. | |||||
| Johnson et al. (1998a) | Strength | Vehicle control and 1 treatment level for each of 7 metabolites; relevant route of administration and duration of exposure. Administration methods enhanced stability. Drinking water formulations mixed daily. WC measured daily. |
Species, strain, source, sex, age/lifestage/BW, reported. Number of dams (litters) reported: (55 controls, 10–20/ high dose groups (4); 3–4/ low dose groups (3). | AAALAC-certified facility. Study designed specifically to assess fetal cardiac defects. Detailed fetal cardiac dissection, preservation, and examination methods provided. |
Relevant maternal and fetal endpoints assessed. Fetal cardiac exam conducted without knowledge of treatment group. Positive cardiac findings were confirmed by unanimous agreement of study authors. | Statistical methods provided. The litter was used as unit of statistical analysis. | Maternal mean WC, BW and uterine data reported; fetal mean resorptions and incidence of cardiac defects reported. |
| Limitation | Information on source of test substances NR. WC values were for group housed dams(4/cage); purity, stability, concentration data NR | No indication that dams were randomly assigned to test groups. Control group was a combined cohort.b | Study dates for control animals overlapped treated groups but were not all concurrent.b | Fetal evaluation of non-cardiac visceral findings was not described. | Fetal BW and length, external data, and non-cardiac visceral data NR; litter incidence of cardiac malformations NR; variance for mean implantation and resorption data NR. | ||
| Johnson et al. (2003, 2005) | Strength | Vehicle control and 4 treatment groups; relevant route of administration and duration of exposure. Information on chemical source provided; stability and concentration tested; formulation methods enhanced stability. Drinking water formulations mixed daily. WC measured daily. Dose calculations based on consumption, concentration, and TCE breakdown rates. |
Species, strain, source, sex, age/lifestage/BW, reported. Randomly assigned to test groups after mating. Number of fetuses and litters, as well as dates of study conduct, are provided for each control and treated cohort. Control cohorts: n = 6–15); total control n = 55; TCE group cohorts: n = 9–13. Analysis of control cohort data was used to justify combining control cohorts. | AAALAC-certified facility. Study designed specifically to assess fetal cardiac defects. Detailed fetal dissection and cardiac preservation methods. All fetuses examined without knowledge of treatment group. Fetal evaluation methods were consistent across cohorts. |
Relevant maternal and fetal endpoints assessed. Fetal cardiac exam conducted without knowledge of treatment group. Positive cardiac findings were confirmed by unanimous agreement of study authors. | Individual fetal cardiac defect data were provided to EPA and analyzed using the litter as unit of statistical analysis. | Individual fetal cardiac defects reported; litter associations for cardiac defects and maternal endpoints for treated groups; were provided to EPA. |
| Limitation | TCE purity, stability, concentration NR Data derived from Dawson et al. (1993) study were treated with tap water vehicle (unknown contaminants). |
Some gaps in concurrency of treated groups and their controls resulted in part from random assignment procedures. | Animals were placed on study in small cohorts. Study conducted over period of 6 years in 5 cohorts; study data from 1994–95 were combined with Dawson et al. (1993) gestation-only data from 1989–93 plus control data from metabolite studies conducted from 1992–94. Study dates for control animals overlapped treated groups but were not exactly concurrent. | Fetal evaluation of non-cardiac findings (visceral and skeletal) was not described. | Statistical analysis conducted under contract by study authors did not consider litter effects.b | Maternal BW, FC, WC, clin obs, placental wt, necropsy data, resorptions and implantations NR; fetal BW and length, external and skeletal data, and non-cardiac visceral data NR; cardiac defects reported per 100-fetus basis, but not associated with litter of origin.b | |
| Narotsky et al. (1995) | Strength | Vehicle control and 4 treatment groups; relevant route of administration and duration of exposure. Information on chemical source provided; purity reported. Storage methods enhanced stability. Dose volumes based on GD6 BW. |
Species, strain, source, sex, age/lifestage/BW, reported. Adequate sample size (8–12 dams/group). | AAALAC-certified facility. Modified Chernoff and Kavlock devtox screening study. Visceral examination of dead pups consisted of Wilson free-hand section of head and dissection of thoracic and abdominal organs. | Relevant maternal and fetal endpoints assessed. | Appropriate statistical methods; litter used as unit of statistical analysis. | Summary data for maternal and fetal endpoints reported. |
| Limitation | Stability and concentration analysis NR | Random assignment to test group NR | Protocol did not require visceral or skeletal evaluation of live pups. | Individual maternal and fetal data NR. | |||
| Narotsky and Kavlock (1995) | Strength | Vehicle control and 2 treatment groups; relevant route of administration and duration of exposure. Information on chemical source provided; purity reported. Storage methods enhanced stability. Dose volumes based on GD6 BW. |
Species, strain, source, sex, age/lifestage/BW, reported. Assignment to test group after mating using unbiased procedure to ensure homogenous distribution of BWs. Adequate sample size (21 control, 16–17 treated/group). | AAALAC-certified facility. Modified Chernoff and Kavlock devtox screening study. Visceral examination of dead pups consisted of Wilson free-hand section of head and dissection of thoracic and abdominal organs. | Relevant maternal and fetal endpoints assessed. | Appropriate statistical methods; litter used as unit of statistical analysis. | Summary data for maternal and fetal endpoints reported. |
| Limitation | Stability and concentration analysis NR | Protocol did not require visceral or skeletal evaluation of live pups. | Individual maternal and fetal data NR. | ||||
| Smith et al. (1989) | Strength | Vehicle control and 4 treatment groups; relevant route of administration and duration of exposure. Information on chemical source provided; purity, stability and concentration confirmed. | Species, strain, source, sex, age/lifestage/BW, reported. Randomly assigned to test groups. Adequate sample size (26 controls, 20–21 treated/group). | Study design similar to EPA guideline. All fetuses assessed for external findings; 2/3 fetuses assigned to visceral exam, and 1/3 fetuses assigned to skeletal exam (bone and cartilage). | Relevant maternal and fetal endpoints assessed. | Statistical analysis of data conducted. | Maternal BW, uterine, and organ weight data reported. Mean (± SD) fetal weight, length, and malformations reported; fetal malformation incidence data reported. |
| Limitation | Stability and concentration data NR | Information on facility certification NR. No indication whether fetuses were randomly assigned to visceral or skeletal evaluation. | No indication that offspring were examined without knowledge of treatment group. | No indication if the litter was used as unit of statistical analysis. | Individual maternal and fetal data NR. | ||
| Smith et al. (1992) | Strength | Vehicle control and 7 treatment groups; relevant route of administration and duration of exposure. Information on chemical source provided; purity, stability and concentration confirmed. | Species, strain, source, sex, age/lifestage/BW, reported. Randomly assigned to test groups. Adequate sample size (20 controls, 19–21 treated/group). | Study design similar to EPA guideline. All fetuses assessed for external findings; 2/3 fetuses assigned to visceral exam, and 1/3 fetuses assigned to skeletal exam (bone and cartilage). | Relevant maternal and fetal endpoints assessed. | Statistical analysis of data conducted. | Maternal BW and uterine data reported. Mean (± SD) fetal weight, length, and malformations reported; fetal malformation incidence data reported. |
| Limitation | Stability and concentration data NR | Information on facility certification NR. No information provided regarding whether fetuses were randomly assigned to visceral or skeletal evaluation. | No indication that offspring were examined without knowledge of treatment group. | No indication if the litter was used as unit of statistical analysis. | Individual maternal and fetal data NR. | ||
NR = Not Reported; BW = body weight; FC = food consumption; WC = water consumption
For Dawson et al. (1993) and Johnson et al. (1998), a number of study details were also provided in Johnson et al. (2003, 2005, 2009, 2014).
Additional information and/or data provided to EPA mitigated the limitations or uncertainties identified in the study report.
Fig. 2.
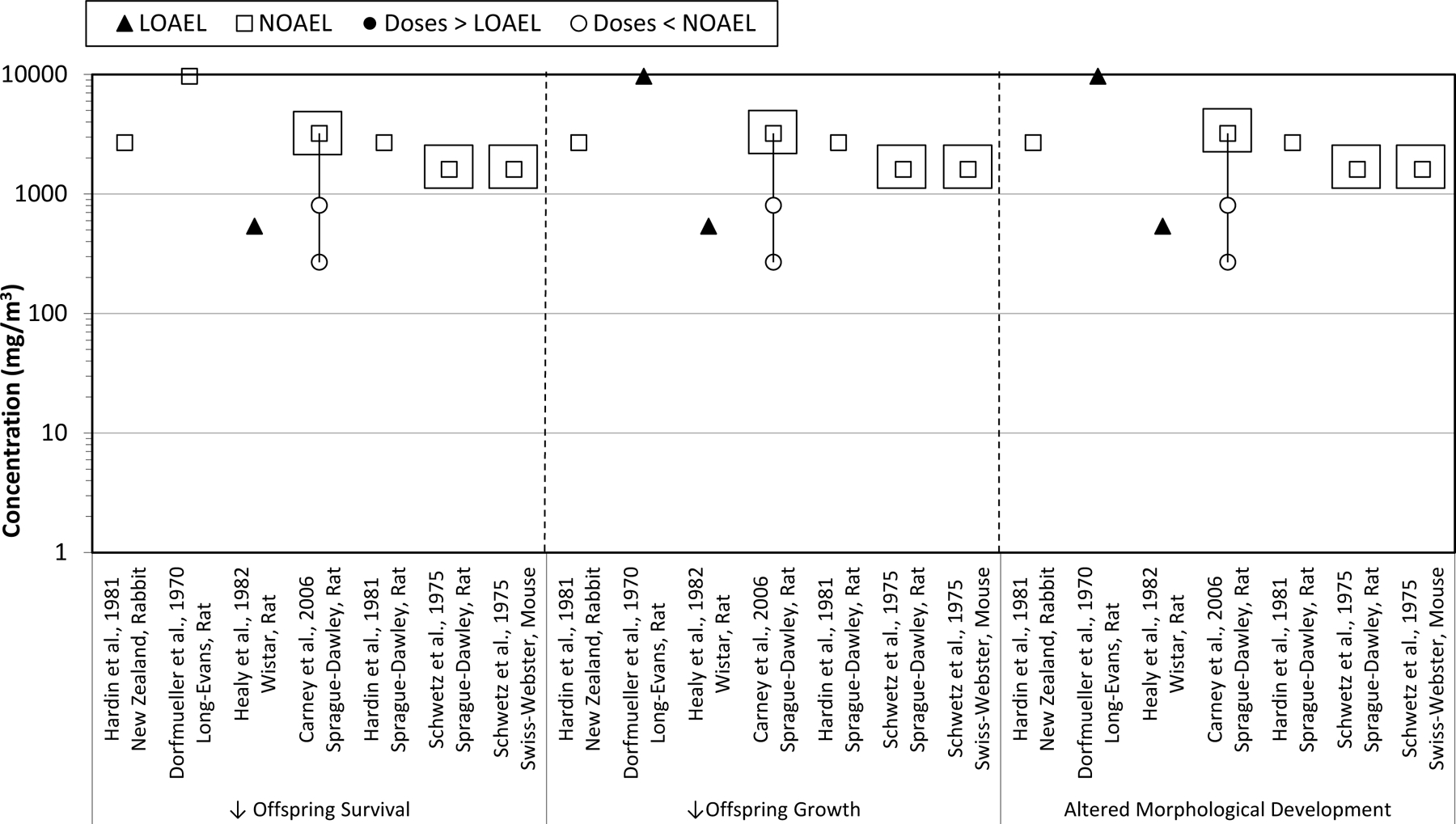
TCE inhalation developmental toxicology studies. Effects on fetal/offspring survival, growth, and morphology following maternal inhalation exposures to TCE during gestation. Boxes indicate the doses at which maternal toxicity was observed.
Fig. 4.
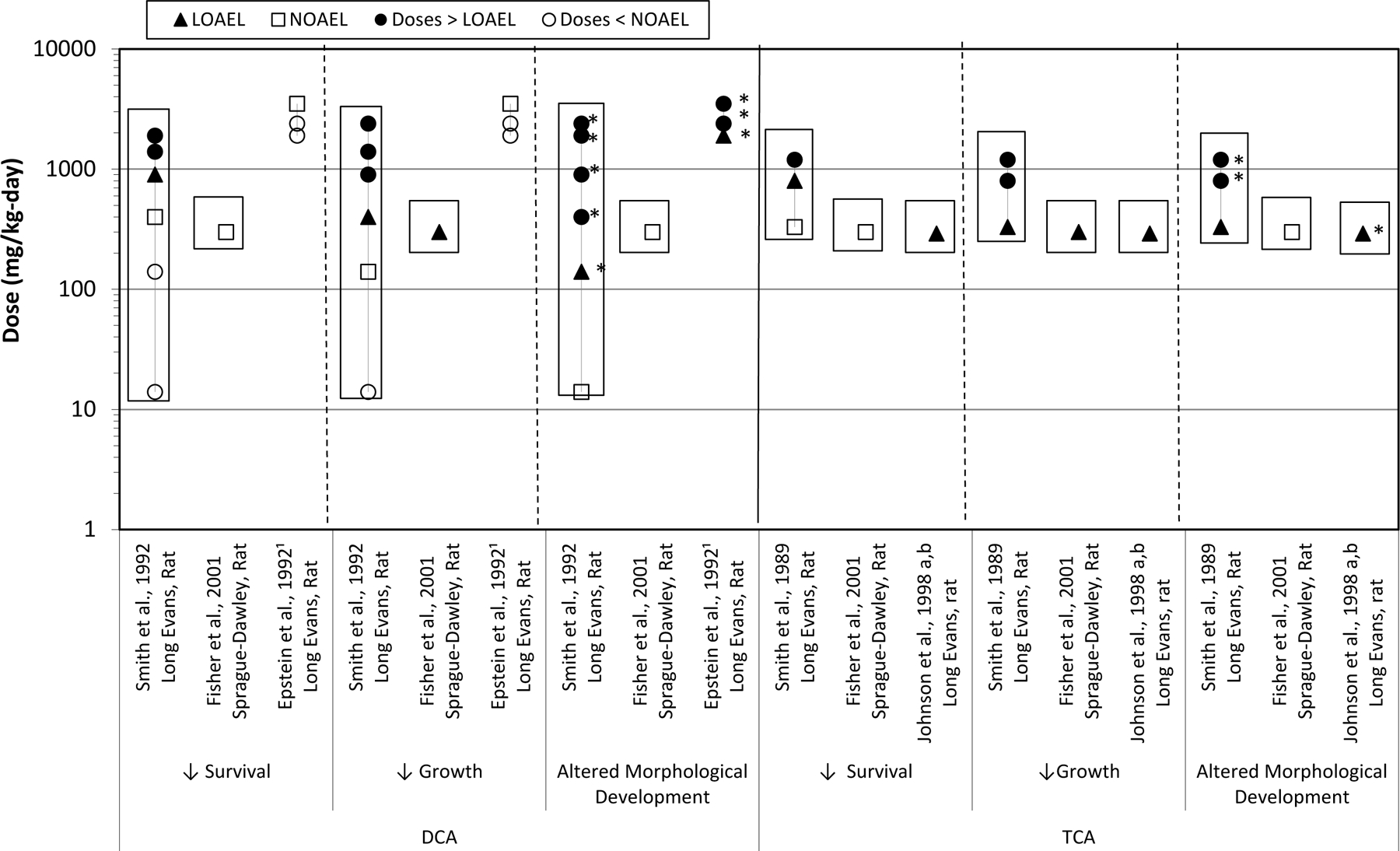
DCA and TCA oral developmental toxicology studies. Effects on fetal/offspring survival, growth, and morphology following maternal oral exposures to TCE metabolites, DCA and TCA, during gestation. Boxes indicate the doses at which maternal toxicity was observed. 1 Maternal toxicity was not reported in Epstein et al. [27]. *Doses at which cardiac defects were observed.
3.1.2.1. Inhalation rodent and rabbit TCE studies.
Five publications reported the conduct of studies in which TCE was administered by inhalation exposure to rats, using a prenatal developmental toxicity study design [15,42,39,21,74]. The studies by Hardin et al. [39] also included rabbits exposed to TCE, and the study by Schwetz et al. [74] also included mice exposed to TCE. None of these studies reported cardiac defects in fetuses following in utero exposures to TCE; however, of these, only the Carney et al. [15] and Schwetz et al. [74] provided sufficient study detail to demonstrate that they were conducted in accordance with good laboratory practices and examined the fetuses using specific methods designed to detect abnormalities of cardiac development.
3.1.2.2. Oral rodent TCE studies.
Six studies reported the results of oral administration of TCE to rodents during fetal development [51,28,61,62,20,18]. All studies were performed in rats, except Cosby and Dukelow [18] which used mice. In all of these rodent studies, TCE was administered by gavage, with the exception of the Dawson et al. [20] and Johnson et al. [51] studies, in which TCE was administered via drinking water. Only the two drinking water studies detected statistically significant treatment-related fetal cardiac defects.
The gavage studies by Fisher et al. [28], Narotsky et al. [62], and Narotsky and Kavlock [61] were conducted in accordance with good laboratory procedures. While Fisher et al. [28] conducted their cardiac evaluations with the same methods as described in Johnson et al. [51], and the first author of the Johnson et al. [51] paper participated as a member of the cardiac dissection team for the Fisher et al. [28] study, TCE-related cardiac defects were not detected. The studies by Narotsky et al. [62] and Narotsky and Kavlock [61] evaluated neonatal growth and viability, and examined cardiac and other soft tissue morphology only in pups that had died; no cardiac defects were reported. The study by Cosby and Dukelow [18] did not conduct a detailed assessment of cardiac development.
3.1.2.3. Oral rodent metabolite studies.
Detailed information on the toxicokinetics of TCE is presented in the IRIS TCE assessment [87,Chap. 3]. Data in humans and rodents indicate that TCE crosses the placenta following maternal inhalation exposure. The major route of TCE biotransformation in humans and rodents is CYP-dependent oxidative metabolism. Metabolic saturation occurs at high oral dose levels in rodents (>1000 mg/kg-day), at much higher doses than those used in the Johnson et al. [51] study (i.e., ranging from 0.048 to 129 mg/kg-day). Tissue distribution experiments using various routes of administration produced time-course data of TCE tissue concentrations that were used to develop a PBPK model for all routes of exposure. Both the applied dose and the PBPK-modeled internal dose-metrics of the oxidative metabolites relevant to cardiac defects are presented in the TCE IRIS assessment [87], Tables 5–18).
Several studies were conducted in rats to examine the effects of developmental exposures to the TCE oxidative metabolites, DCA and TCA. Studies by Smith et al. [79] and Epstein et al. [27] observed cardiac defects following gavage administration of DCA during pregnancy. Smith et al. [78] and Johnson et al. [49] reported cardiac defects with TCA exposures administered during gestation via gavage or drinking water, respectively. However, a study by Fisher et al. [28] did not detect cardiac defects following gavage administration of DCA or TCA on GD 6–15. All of these studies used dissection methods that were designed to visualize the internal structures of the fetal heart. Other TCE metabolites were evaluated by Johnson et al. [49] and found not to elicit cardiac malformations following developmental exposures (i.e., carboxymethyl cysteine, dichloroacetaldehyde, dichloroethylene, dichlorovinyl cysteine, monochloroacetic acid, trichloroacetaldehyde, and trichloroethanol). Although the proximate toxicant which causes cardiac defects has not been identified, a recent study [6] identified 5-(1,2-dichlorovinyl)-l-cysteine as a key metabolite in placental effects of TCE, suggesting that further consideration of TCE metabolites may be warranted.
3.1.2.4. In ovo avian studies.
Several studies examined cardiac development following in ovo administration of TCE to chicken embryos [72,24,23,57]. Abnormalities of cardiac structure and/or function were observed in each of these studies, at doses as low as 2–8 ppb. Defects in valvulo-septal development were similar to those that have been observed in rodents and humans, which is coherent given that early stages of cardiac development are similar across species [63].
3.1.2.5. In vitro assays.
Whole embryo culture studies that examined cardiac development were conducted by Hunter et al. [44] using 3–6 somite mouse embryos exposed to DCA or TCA and by Mishima et al. [60] using HH 13–14 chicken embryos exposed to TCE. Dose-related alterations in cardiac development were observed in both of these models, although at high (not environmentally relevant) doses.
3.1.2.6. Evaluation of cardiac defects in the animal toxicology studies.
As described, alterations in fetal cardiac development have been observed in rodent studies following in utero exposure to TCE and its oxidative metabolites. These findings are supported by the detection of cardiac anomalies in chicken embryos exposed to TCE in ovo, and in whole embryo cultures (mouse and chicken) of TCE and/or its metabolites. In spite of the concordant evidence that TCE has been associated with cardiac defects, controversy centers on the studies by Johnson et al. [51] and Dawson et al. [20], especially with respect to the study design and methods, reporting inadequacies, dose-response characteristics, and the lack of cardiac defect findings in other laboratory studies in rodents following gavage or inhalation exposures of TCE during development.
The Johnson et al. [51] publication reported the results of TCE drinking water exposures on fetal cardiac development in Sprague-Dawley rats from a 6-year-long academic research program. It included data on two TCE treatment groups studied in 1989–1991 that had previously been published by Dawson et al. [20], plus the data from two lower dose TCE treatment groups studied in 1994–1995. Cardiac malformation incidence data were compared between treated groups and combined control data from cohorts studied concurrent to treated groups over the course of the 6-year research program, including controls from studies on TCE metabolites, published in Johnson et al. [49]. Other information on the TCE studies reported in Johnson et al. [51] included published communications [40,50], errata [53,52], and individual cardiac malformation findings and evaluation methods provided to EPA by the primary study author (Dr. Paula Johnson, personal communications [47,48]). The Johnson et al. [51] paper summarized the combined results from the studies that administered TCE to pregnant rats at doses of 2.5 ppb, 250 ppb, 1.5 ppm, and 1100 ppm in drinking water throughout gestation. Fetal cardiac defects, primarily valvular and septal anomalies, were observed at ≥250 ppb.
The limitations and strengths of the toxicological studies were identified (details provided in Table 4). Limitations identified in the evaluation of the Johnson et al. [51] and Dawson et al. [20] studies presented here are consistent with the study design and reporting issues identified in the IRIS assessment [87], peer-reviewed publications such as Hardin et al. [38] and Watson et al. [90], and public comments submitted to the U.S. EPA [1]. The corresponding author for Johnson et al. [51] provided clarification on a number of topics and a detailed description of study methods beyond what had been previously published, including verification that concurrent controls were used for each of the treated groups (Fig. 5), information on fetal randomization and blinded cardiac evaluation procedures, and details of animal husbandry (Dr. Paula Johnson, personal communication, 2014). Subsequent to these discussions, the study author published an errata [53] to update the public record regarding methodological issues for Johnson et al. [51]. This information served to increase confidence in the study conduct and results. However, some study reporting and methodological details remain unknown, e.g., the precise dates that each individual control animal was on study, maternal body weight/food consumption and clinical observation data, and the detailed results of analytical chemistry testing for dose concentration. Additional possible sources of uncertainty identified for these studies include that the research was conducted over a 6-year period, that combined control data were used for comparison to treated groups, and that exposure characterization may be imprecise because tap (rather than distilled) drinking water was used in the Dawson et al. [20] study and because TCE intake values were derived from water consumption measures of group-housed animals. On the other hand, the strengths of this study include the examination of fetal hearts without knowledge of treatment (or control) group, standardized methods of fetal evaluation, examination of the gross (in situ) and internal structure of the fetal hearts by a group of three senior researchers/co-authors (P. Johnson, B. Dawson, and S. Goldberg), confirmation of cardiac anomalies by consensus agreement. In addition, individual fetal and litter cardiac abnormality data for treated groups were shared with EPA (Dr. Paula Johnson, personal communication (2008)), thereby facilitating independent statistical analysis of the data.
Fig. 5.
Control vs. TCE treatment groups and dates of exposure. During the duration of the University of Arizona (UA) research program on TCE (1989–1995), a number of developmental toxicology studies were conducted on TCE and its metabolites. Control animals (red blocks) were on study when treated animals (blue blocks) were being exposed. The blocks are general representations of time frames and are not presented to exact scale. The dates that cohorts of animals were on study (as well as dose levels and the number of dams/litters for each cohort) are shown. In three cases, information on the exact month and day of animal receipt was not available (indicated by dotted lines). Exclusively pregnancy-only TCE-treated groups are included in this figure; however, other treatment regimens were also being conducted during the time period of 6/12/89 to 10/6/95 (i.e., 3 months pre-pregnancy-only, 2 months pre-pregnancy + pregnancy). Additionally, during this time period, TCE metabolites and other toxicokinetically related chemicals were studied: dichloroacetic acid (DCA), trichloroacetic acid (TCA), monochloroacetic acid (MCAA), trichloroethanol (TCEth), trichloroacetaldehyde (TCAld), dichloroacetaldehyde (DCAld), carboxy methylcystine (CMC), dichlorovinyl cystine (DCVC), dichloroethylene (DCE). The control animal cardiac malformation incidence data were combined for statistical comparison with incidence data for pregnancy-only TCE-treated groups. Sources of information used to compile this figure: [53,52,50,51,49,20].
Inconsistencies in the results of studies that assessed cardiac development in rodents have been raised as an issue of particular concern. In the case of a number of these studies [61,62,18,42,39,21,74], a variety of animal species and strains, sources, and testing protocols were used (summarized in Table 5a), which precludes direct comparisons. For several of the older studies, information that would allow a valid comparison with the Johnson et al. [51] and Dawson et al. [20] studies is not reported. For example, detailed procedural details regarding fetal evaluation were not provided for Schwetz et al. [74], Dorfmueller et al. [21], Healy et al. [42], and Hardin et al. [39]. There is no indication whether fetuses were selected randomly for visceral evaluation, or whether they were examined without knowledge of treatment group (blinded assessment). Fetal cardiac evaluation methods were not elucidated in any detail, and the performance of fresh dissection of the heart to evaluate internal cardiac morphology was not mentioned. Hardin et al. [39] reported virtually no methodological information. In some studies, differences in overall study design limited meaningful cross-study comparison, e.g., due to limited exposure durations [18] or the evaluation of delivered PND 1 pups instead of fetuses [61,62]. The studies reported by Fisher et al. [28] and Carney et al. [15] were well-conducted developmental toxicity studies in rats and utilized procedures that facilitated evaluation of fetal cardiac morphology. Fisher et al. [28] and Carney et al. [15] did not observe treatment-related cardiac defects following TCE gavage or inhalation exposures, respectively, during gestation. Detailed examination of the study protocols (summarized in Table 5b) identified several variations in study design and conduct, including but not limited to differences in route of administration, and these differences may have contributed to the different study outcome as compared to Johnson et al. [51] and Dawson et al. [20]. In the case of the Fisher et al. [28] study, as previously noted, care was taken to follow the Johnson et al. [51] fetal evaluation procedures as closely as possible, yet a number of other differences in study design and conduct remained. For example, the source of the animals, the route of exposure, the vehicle/control substance, fetal cardiac tissue preservation methods, and some fetal cardiac evaluation procedures were different. A comparison of typical cardiac evaluation techniques used in developmental toxicology studies, illustrating some potential differences in resolution of abnormalities in the fetal heart, is presented in Table 6. This includes the procedures used by Carney et al. [15] and the procedures used by Dawson et al. [20], Johnson et al. [49], and Johnson et al. [51], as well as by Fisher et al. [28]. One possibility is that the procedural differences in fetal cardiac evaluation techniques could have contributed to differences in study outcome [90]. However, that explanation is not supported by two facts. First, the detailed description of the cardiac dissection and evaluation techniques (as reported in Dawson et al. [20] is sufficiently comparable to the procedures used by Carney et al. [15] (summarized in Table 6) to have facilitated visualization of overt cardiac malformations such as septal defects. Secondly, Fisher et al. [28] used the same cardiac evaluation techniques reported by Dawson et al. [20], Johnson et al. [49], and Johnson et al. [51], evaluated fetuses collaboratively with Dr. Johnson, and yet did not detect treatment-related incidences of cardiac defects.
Table 5a.
Comparison of Methods Reported for Prenatal Developmental Toxicity Studies with TCE
| Schwetz et al. (1975) | Dorfmueller et al. (1979) | Hardin et al. (1981) | Healy et al. (1982) | Cosby & Dukelow (1992) | Narotsky & Kavlock (1995) | Narotsky et al. (1995) | |||
|---|---|---|---|---|---|---|---|---|---|
| Study Description/Objective | |||||||||
| Guideline-type [GT] or research [R] protocol | GT | GT | R | GT | GT | GT | R | R | R |
| Test Subjects | |||||||||
| Species | Rat | Mouse | Rat | Rat | Rabbit | Rat | Mouse | Rat | Rat |
| Strain | SD | SW | LE | WIS or SD | NZW | WIS | B6D2F1 | Fischer 344 | Fischer 344 |
| Source (company) | NR | NR | CRL | NR | NR | NR | Jackson | Harlan | Harlan |
| Source (location) | NR | NR | NR | NR | NR | NR | Bar Harbor, ME | Indianapolis, IN | Indianapolis, IN |
| Dates of study conduct | NR | NR | NR | NR | NR | NR | NR | NR | NR |
| Day of mating confirmation (GD 0 or GD 1) | GD 0 | GD 0 | GD 1 | NR | NR | NR | GD 1 | GD 0 | GD 0 |
| Day of cesarean section | GD 21 | GD 18 | GD 21 | GD 21 | GD 30 | GD 21 | Delivered | Delivered | Delivered |
| Treatment | |||||||||
| Test Substance | TCE | TCE | TCE | TCE | TCE | TCE (Trilene) | TCE | TCE | TCE |
| Source | Dow | Dow | Dow | NR | NR | ICI | Aldrich | Aldrich | Aldrich |
| Purity (%) | 99.24% | 99.24% | 99% | NR | NR | NR | NR | >99% | >99% |
| Route of administration | Inhalation a | Inhalation a | Inhalation a | Inhalation b | Inhalation b | Inhalation a | Gavage | Gavage | Gavage |
| Negative control (vehicle) | Filtered room air | Filtered room air | Filtered air | Air | Air | Ambient air | Corn oil | Corn oil | Corn oil |
| Positive control | N | N | N | N | N | N | N | N | N |
| No. of treated groups | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 2 | 4 |
| Group size (litters/group) | 30, 18 | 26, 12 | 8–12 | (30) | (20) | 31–32 | (30) | 16–21 | 8–12 |
| Random assignment of test subjects to groups | NR | NR | Y | NR | NR | NR | NR | Y | NR |
| Dose period (duration) | GD 6–15 | GD 6–15 | GD 1–20 | GD 1–19 | GD 1–24 | GD 8–21 | GD 1–5, 6–10, or 11–15 | GD 6–19 | GD 6–15 |
| Daily dosing schedule | 7 hrs/day, 7 days/wk | 7 hrs/day, 7 days/wk | 6 hrs/day, 7 days/wk | 6–7 hrs/day, 7 days/wk | 6–7 hrs/day, 7 days/wk | 4 hrs/day, days/wk NR | 1x/day | 1x/day | 1x/day |
| Maternal evaluation | |||||||||
| In-life data (BW, FC, WC, and/or clinobs) | Y | Y | Y | NR | NR | Y | Y | Y | Y |
| Postmortem data (necropsy, organ wts, pathology, and/or CL) | Y | Y | Y | Y | Y | Y | Y | N | N |
| Fetal evaluation | |||||||||
| Implantations and resorptions (early and late) | Y | Y | Y | NR | NR | Y | N | N | N |
| Fetal weight, length, sex | Y | Y | Y | NR | NR | Y | Y (PND 1) | Y (PND 1) | Y (PND 1) |
| External fetal exam | Y | Y | Y | NR | NR | Y | Y (PND 1) | Y (PND 1) | Y (PND 1) |
| Percent fetuses (litters) evaluated for external findings | 100 (100) | 100 (100) | 100 (100) | NR | NR | 100 (100) | 100 (100) | 100 (100) | 100 (100) |
| Visceral examination | Y | Y | Y | NR | Y c | Y | N | Y d | Y d |
| Percent fetuses (litters) evaluated for visceral findings | 50 (100) | 50 (100) | ~33 (100) e | NR | NR | 100 (100) | NA | NR (NR) | NR (NR) |
| Fresh dissection (in situ organ examination) | N | N | N | NR | NR | Y | N | N | Y f |
| Wilson exam (Bouins fixation, free-hand sections) | Y | Y | Y | NR | NR | N | N | Y | Y |
| Fetal cardiac examination methods | NR | NR | NR | NR | NR | NR | N | N | N |
| Fresh dissection and evaluation | NR | NR | NR | NR | NR | NR | N | N | N |
| Free-hand section of decalcified fetuses | Wilson | Wilson | Wilson | NR | NR | NR | N | Wilson d | Wilson d |
| Preservation | Bouin’s immersion | Bouin’s immersion | Bouin’s immersion | NR | NR | NR | N | Bodian’s immersion | Bodian’s immersion |
| Confirmation of findings | NR | NR | NR | NR | NR | NR | N | N | N |
| Skeletal examination | Y | Y | Y | NR | NR | Y | N | N | N |
| Percent fetuses (litters) evaluated for skeletal findings | 50 (100) | 50 (100) | ~33 (100) e | NR | NR | 100 (100) | NA | NA | NA |
| Bone development | Y | Y | Y | NR | NR | Y | N | N | N |
| Cartilage development | N | N | N | NR | NR | N | N | N | N |
| Random selection of fetuses for visceral or skeletal evaluation | NR | NR | N | NR | NR | NA | N | N | N |
| Assessment of fetuses without knowledge of treatment group | NR | NR | NR | NR | NR | NR | N | N | N |
Footnotes:
This table only includes mammalian studies with prenatal TCE exposures and an evaluation of fetal morphology.
NR = not reported; NA = not applicable; Y = yes, N = No; DW = drinking water; GD = gestation day; PND = postnatal day; RA = retanoic acid; GLA = gluteraldehyde
Test subject strain: SD = Sprague-Dawley, LE = Long Evans, WIS = Wistar, NZW = New Zealand White, SW = Swiss Webster
Test subject source: CRL = Charles River Laboratories, Jackson = Jackson Laboratories, Harlan = Harlan Laboratories
Group sizes are range of actual group size (i.e., no. of dams) on study; numbers in parentheses () indicate target group size.
= Whole-body exposure, dynamic air flow, analytical chamber concentrations.
= Whole-body exposure, inadequately characterized.
= Visceral examination was NR; however, brain malformations in TCE-treated rabbit fetuses were discussed.
= Visceral examination of dead pups only; free-hand (Wilson’s) sectioning of head only.
= Four fetuses/litter were assigned to visceral examination and 4 fetuses/litter were assigned to skeletal examination. (33% is an estimate based upon the presumption of 12 fetuses/litter.)
= Visceral evaluation of affected (i.e., abnormal) pups only.
Cardiac evaluation references: Staples exam: Stuckhardt and Poppe (1984), Staples (1974), Wilson, 1965; University of AZ exam: Johnson et al. (2003), Dawson et al. (1993).
Table 5b.
Comparison of Methods Reported for Prenatal Developmental Toxicity Studies with TCE
| Dawson et al. (1993) | Johnson et al. (2003) | Fisher et al. (2003) | Carney et al. (2006) | |
|---|---|---|---|---|
| Study Description/Objective | ||||
| GLP; guideline [G], or research [R] protocol | R | R | R | GLP, G |
| Test Subjects | ||||
| Species | Rat | Rat | Rat | Rat |
| Strain | SD | SD | SD | SD |
| Source (company) | Harlan | Harlan | CRL | CRL |
| Source (location | Indianapolis, IN | Indianapolis, IN | Raleigh, NC | Portage, MI |
| Dates of study conduct | 1989–1990 | 1989–1995 | NR | NR |
| Day of mating confirmation (GD 0 or GD 1) | NR | NR | GD 0 | GD 0 |
| Day of cesarean section | GD 22 | GD 22 | GD 21 | GD 21 |
| Treatment | ||||
| Test Substance | TCE | TCE | TCE | TCE |
| Source | Aldrich | Aldrich | Aldrich | Dow |
| Purity (%) | NR | NR | NR | 99% |
| Route of administration | DW | DW | Gavage | Inhalation a |
| Negative control (vehicle) | Tap water | Distilled water | Soybean oil | Ambient air |
| Positive control | N | N | RA | N |
| No. of treated groups | 2 | 4 | 1 | 3 |
| Group size (litters/group) | 9–15 | 9–12 | 19–25 | 27 |
| Random assignment of test subjects to groups | Y | Y | Y | Y |
| Dose period (duration, gestation-only groups) | GD 1–22 | GD 1–22 | GD 6–15 | GD 6–20 |
| Daily dosing schedule | Ad libitum, 24 hrs/day | Ad libitum, 24 hrs/day | 1x/day | 6 hrs/day, 7 days/wk |
| Maternal evaluation | ||||
| In-life data (BW, FC, WC, and/or clinobs) | Y | Y | Y | Y |
| Postmortem data (necropsy, organ wts, pathology, and/or CL) | Y | Y | Y | Y |
| Fetal evaluation | ||||
| Implantations and resorptions (early and late) | Y | Y | Y | Y |
| Fetal weight, length, sex | Y | Y | Y | Y |
| External fetal exam | Y | Y | Y | Y |
| Percent fetuses (litters) evaluated for external findings | 100 (100) | 100 (100) | 100 (100) | 100 (100) |
| Visceral examination | Y | Y | Y | Y |
| Percent fetuses (litters) evaluated for visceral findings | 100 (100) | 100 (100) | 100 (100) | 50 (100) |
| Fresh dissection (in situ organ examination) | Y (heart) | Y (heart) | Y (heart) | Y (viscera) |
| Wilson exam (Bouins fixation, free-hand sections) | N | N | N | Y (head) |
| Fetal cardiac examination methods | Y | Y | Y | Y |
| Fresh dissection and evaluation | UA method | UA method | UA method | Staples exam |
| Free-hand section of decalcified fetuses | N | N | N | N |
| Preservation | GLA flush & immersion | GLA flush & immersion | formalin immersion | NR |
| Confirmation of findings | Y b | Y b | NR | NR |
| Skeletal examination | NR | NR | NR | Y |
| Percent fetuses (litters) evaluated for skeletal findings | NR | NR | NR | 50 (100) |
| Bone development | NR | NR | NR | Y |
| Cartilage development | NR | NR | NR | Y |
| Random selection of fetuses for visceral or skeletal evaluation | NA | NA | NA | NA |
| Assessment of fetuses without knowledge of treatment group | Y | Y | Y | Y |
Footnotes:
This table only includes mammalian studies with prenatal TCE exposures and an evaluation of fetal morphology.
NR = not reported; NA = not applicable; Y = yes, N = No; DW = drinking water; GD = gestation day; RA = retanoic acid; GLA = gluteraldehyde; UA = University of Arizona
Test subject strain: SD = Sprague-Dawley
Test subject source: CRL = Charles River Laboratories; Harlan = Harlan Laboratories
Group sizes are range of actual group size (i.e., no. of dams) on study; numbers in parentheses () indicate target group size.
= Whole-body exposure, dynamic air flow, analytical chamber concentrations.
= Unanimous agreement of cardiac diagnoses by study investigators (a pathologist, a pediatric cardiologist, and a veterinarian) was required before a positive cardiac finding was diagnosed and recorded.
Cardiac evaluation references: Staples exam: Stuckhart and Poppe, 1984, Staples (1974); University of AZ exam: Johnson et al. (2003), Dawson et al. (1993).
Table 6.
Comparison of cardiac evaluation methods
| Method | Reference | Description |
|---|---|---|
| Wilson | Wilson, 1965 | • Immersion fixation of whole fetus in Bouin’s solution • Free-hand serial sectioning of fetuses (approximately 2 mm thickness), including sections through the heart and great vessels |
| Staples | Staples, 1974; Stuckhardt and Poppe, 1984 | • Dissection of unfixed decapitated or anesthetized fetus • Examination of external structure of the heart and great vessels • Examination in situ of internal structure of the heart via two cuts: ○ Incision made beginning to the right of the ventral midline surface of the heart at the apex and extending anteriorly and ventrally into the pulmonary artery (exposing the tricuspid valve between the right atrium and right ventricle and the 3 cusps of the semilunar valve of the pulmonary artery); the interventricular septum examined for defects. ○ Incision made starting to the left of the ventral midline surface at the apex and extending thorough the left ventricle into the ascending aorta (exposing the bicuspid valve between the left atrium and left ventricle and the 3 cusps of the semilunar valves of the aorta). |
| University of AZ | Dawson et al., 1993; Johnson et al., 2003 | • Dissection of unfixed fetus • Examination of the great vessels in situ, including pulmonary venous attachment to the left atrium and cranial and caudal vena caval connections to the right atrium • Removal of the heart from the thorax; the heart is flushed and then immersion fixed with 2% gluteraldehyde • Examination of external structure of the heart from dorsal and ventral aspects • Examination of internal structure of the heart and vessels: ○ Right atrial appendage excised to evaluate the atrial septum for defects (left atrial appendage removed if the atrial septum is not adequately visualized) ○ Aorta and pulmonary vessels evaluated for course, caliber, and orientation, then excised at valve rings ○ All remaining atrial tissue removed to expose pulmonary, aortic, tricuspid, and mitral valves; location of coronary ostium noted; each valve probed for patency, and formation of each valve leaflet examined. ○ Incision made ventrally through the tricuspid valve to the apex of the heart. Another incision made through the pulmonary valve toward the apex of the heart and joining the cut made through the tricuspid valve. Incision made from each edge of the mitral valve toward the apex, and the left ventricular free wall removed (allowing complete visualization of the ventricular septum for evaluation of defects). |
In summary, Johnson et al. [51] and Dawson et al. [20] observed cardiac defects in fetal rats after gestational drinking water exposures to TCE. These findings have not been confirmed in studies with exposures to TCE during gestation that were conducted by other laboratories. However, none of the other studies have repeated precisely the same study design used by Johnson et al. [51] and Dawson et al. [20]. Differences in study methods such as the route of exposure, vehicle, source or strain of animals, or other unknown factors may have contributed to differences in the detection of cardiac malformations, and at this point in time, it would be impossible to identify the specific reason. Designing and conducting an exact replica of the Johnson et al. [51] study might be very difficult, if not impossible. For example, it is possible that the study animals used by Dawson et al. [19] and Johnson et al. [51] in the University of Arizona (UA) research program on TCE in drinking water may have been particularly susceptible to perturbation of cardiac development by TCE and its metabolites. The possibility of genetic drift in the strain/source of rats over the past 10–20 years might preclude designing and conducting a study with comparable results. Yet, such a susceptibility in the animal models used by Dawson et al. [20] and Johnson et al. [51] might have rendered those studies more (or less) predictive of responses in susceptible individuals in the human population, a difficult assumption to validate. In humans, cardiovascular malformations are common birth defects with both genetic predisposition and environmental exposures contributing to the multifactorial etiology [68].
3.1.3. Mechanistic data on developmental pathways and processes
Mechanistic mode-of-action data were discussed in the 2011 IRIS assessment [87] and provided one line of evidence regarding the potential for TCE to cause cardiac defects. There was not an explicit linkage to the developmental pathways and processes driving CHD in general or valvulo-septal defects in particular. To expand upon and refine this discussion, a preliminary conceptual model based on an Adverse Outcome Pathway (AOP) framework for CHD would be useful. Although such AOP elucidation is beyond the scope of the present review, data identified in the systematic literature search and MGI database search provides motivation for that future activity. Information upon which a preliminary AOP construct is based supports the biological plausibility that TCE exposures during development could lead to disruption of key processes in the development of cardiac valves and septa.
The most commonly reported cardiac defects associated with gestational exposures to TCE and its metabolites TCA and DCA in humans, rats, and chickens were valvulo-septal defects (atrial septal defects [ASDs], muscular and membranous ventricular septal defects [VSDs]) and pulmonary and aortic stenosis [16,29,94,51,49,8,9,20,19,35,57]. In particular, the period of valvulo-septal morphogenesis defines a window of TCE vulnerability in avian systems; thus an AOP anchored to this dysmorphology could identify relevant key events and key event relationships following exposure to TCE during the vulnerable period. In normal cardiac development, valvulo-septal morphogenesis is driven by mesenchymal cells in the regions of the atrioventricular canal (AVC) and outflow tract (OFT) regions. AVC cushions are formed as mesenchymal cells are derived from squamosal endothelial cells by epithelial-mesenchymal transition [EMT], specifically of endothelial origin [EndMT], and invade and populate the cardiac jelly matrix. These mesenchymal cells subsequently proliferate and differentiate to form the AV valves and membranous septum. They also contribute to patterning the myocardium via directing vascular flow. Evidence points to a stepwise EndMT cascade involving the following key events [46,54,89]:
initiation of EndMT by signal molecules elaborated from myocardial cells into the cardiac jelly;
disassembly of cell–cell junctions between squamosal endothelial cells in the endocardium;
delamination by loss of polarity, cytoskeletal rearrangement, and breakdown of basal lamina;
invasion of cardiac jelly by newly motile mesenchymal cells;
proliferation of trans-differentiated mesenchyme to ‘cellularize’ and remodel the cardiac jelly;
patterning of the AV myocardium by flow-mediated remodeling of the looped heart;
differentiation of cardiac valves and membranous septum.
A search of the MGI database (http://www.informatics.jax.org/) for abnormalities in cardiac EMT identified mouse knockouts with developmental phenotypes similar to those reported for avian studies with TCE, implicating the possibility of disruption of the following genetic signals and responses by TCE exposure during cardiac development. Candidate genes implicated pathways such as TGF-beta signaling, ephrin signaling, Notch signaling, the VEGF pathway, and RXR signaling. Potential molecular initiating events, not yet evaluated experimentally, may involve a cellular initiation of vascular inflammatory signals, perhaps through an LXR/RXR-mediated effect on cholesterol homeostasis, vulnerability to reactive oxygen species (ROS) [91,41,28], or disruption of the downstream consequences of VEGF signaling [65].
In support of disruption of EndMT being a potential key event in TCE-induced valvulo-septal defects, embryonic TCE exposure has been associated with inhibition of cell–cell separation and mesenchymal formation [10], alterations in mesenchymal cell migration [60,75] and alterations in endocardial proliferation patterns [24]. In ovo studies have shown that TCE and TCA can alter cushion formation, cardiac function, and embryo survival [23], and cushion cellularity can be altered as a function of concentration, duration, and timing of exposure. The ephrin-EPH system might be of high relevance to an AOP for TCE-induced valvulo-septal defects. Loss of Ephrin-A1 in mice, a ligand for class A Eph receptor tyrosine kinases, results in thickened aortic and mitral valves. These embryos display hypercellularity in outflow tract endocardial cushions and elevated mesenchymal marker expression, suggesting that excessive numbers of cells undergo EMT [30]. Ephrin-A1 and its cognate receptor (EphA3) are expressed in adjacent cells in the developing endocardial cushions. In contrast to the ligand, functional inactivation of EphA3 results in hypoplasia of AVC endocardial cushions with fewer migrating mesenchymal cells [81]. As such, disruption of Ephrin-A1 ligand or EphA3 receptor function impacts endocardial cushion formation in different ways, potentially leading to hypercellularity or hypocellularity, respectively. Both effects have been described in in vitro models of TCE-induced effects on endocardial cushions. Endocardial disruption may have additional or downstream consequences on the developing heart, related to dysregulation of cellular Ca2+ fluxes and cardiac contractility [58,66,59,14,76,17] or to alterations in cardiac hemodynamics [72].
3.1.4. Weight of evidence (WOE) for hazard
The WOE (evidence integration) for fetal cardiac defects was characterized according to the criteria described in A Framework for Assessing Health Risk of Environmental Exposures to Children [86], a scheme that is derived from principles of causality assessment developed by Hill [43]. The key components (factors) of the WOE analysis were: temporality, strength of association, variability analysis, uncertainty analysis, qualitative dose-response, experimental evidence, reproducibility (consistency), biological plausibility, alternative or multiple explanations, specificity, and coherence (Fig. 1). Independent assessments of the WOE were conducted by reviewers, and a group consensus of the evidence supporting stronger and weaker weights of association for each key factor was derived. The evidence supporting stronger and weaker weight of association for each key factor is presented in Table 7.
Table 7.
WOE Evaluation of the Potential for Development Exposures to TCE to Result in Cardiac Defects
| Key Factor a,b | Type of evidence considered | Data | Evidence for stronger weight of association |
Evidence for weaker weight of association |
Comments or Null Evidence |
|---|---|---|---|---|---|
| Temporality | Timing of exposures and response | Tox | Studies in various species in which TCE (or metabolites DCA or TCA) were administered during a sensitive period of in utero cardiac development resulted in morphological and/or functional alterations. • Drinking water administration of TCE to rats on GD 1–22 resulted in a statistically significant treatment-related increase in the incidence of cardiac defects (Johnson et al., 2003; Dawson et al., 1993). • Drinking water administration of TCA (the TCE oxidative metabolite) to rats on GD 1–22 resulted in a statistically significant treatment-related increase in the incidence of cardiac defects (Johnson et al., 1998). Gavage administration of TCE metabolites (DCA and TCA) on GD 6–15 (Smith et al., 1992, 1989) or of DCA during discrete windows of time within GD 6–15 (Epstein et al., 1992) resulted in treatment-related increases in the incidences of cardiac defects. • Avian in ovo studies that administered TCE or TCA during the period of valvuloseptal morphogenesis (e.g., HH 15–20) resulted in altered cardiac morphology and/or function (Rufer et al., 2010; Drake et al., 2006a; Loeber et al, 1988). • A study of DCA exposure to zebra fish (Hassoun et al., 2005) demonstrated evidence of a disruption in cardiac development (pericardial edema and altered heart rate). • Mouse whole embryo culture studies of DCA and TCA administered at the period of 3–6 somites detected cardiac defects (Hunter et al., 1996); a chicken whole embryo culture study of TCE administered at HH 13–14 detected alterations in AV cushion (Mishima et al., 2006). • Avian atrioventricular canal cell culture (HH 16) study found evidence of inhibited endothelial cell separation and early events of mesenchymal cell formation in the heart following TCE exposures (Boyer et al., 2000). |
Some in vivo or in vitro studies rodent studies in which TCE (or metabolites DCA or TCA) was administered during a sensitive period of in utero cardiac development resulted in no morphological alterations. • Gavage administration of TCE or metabolites (DCA and TCA) to rats on GD 6–15 did not result in treatment-related cardiac defects (Fisher et al., 2001). • Inhalation exposures of TCE to rats on GD 6–20 (Carney et al., 2006) or to rats and mice on GD 6–15 (Schwetz et al., 1975) did not result in treatment-related cardiac defects. |
• NE |
| Exposure occurs before outcomes onset | Epi |
• Four cohort or case-control studies consider temporality (Ruckart et al., 2013; Forand et al., 2012; Yauck et al., 2004; Goldberg et al., 1990). Three studies observed an association between the TCE exposure surrogate and major cardiac defects (Forand et al., 2012; Yauck et al., 2004; Goldberg et al., 1990). An association with conotruncal defects, specifically, was observed in Forand et al. (2012). | • Temporality was not considered in Bove (1996)/Bove et al. (1990), Goldberg et al. (1990), or Lagakos et al. (1986). | • The small numbers of conotruncal heart defects in Ruckart et al. (2013) precluded any analysis of this endpoint and TCE exposure. | |
| Strength of association | Study quality, including study strengths and limitations | Tox | • For Johnson et al. (2003), Dawson et al. (1993), and Johnson et al. (1998),, all of which detected cardiac malformations, study quality strengths include randomized assignment to test group, detailed description of fetal cardiac dissection and evaluation procedures, evaluation of fetal hearts without knowledge of treatment group, and confirmation of all cardiac defects by consensus of 3 experts. Statistical analysis of data from this study was appropriately conducted by EPA statisticians using individual fetal and litter data that were provided by the study author. • The power of detection in the Johnson et al. (2003) study was enhanced by the use of historical controls that did not demonstrate a temporal shift in cardiac defects. A significant dose related trend in cardiac defects was observed even without large group sizes. • A strong association of exposure to response was observed at high dose levels in multiple studies that identified cardiac defects. In Johnson et al. (2003) there was a highly significant positive trend for cardiac defects. • Potential confounding factors exist in studies that did not identify cardiac defects (e.g., different routes of exposure, the use of different rodent strains or suppliers across studies, and the use of soybean oil as a vehicle in Fisher et al. (2001). |
• For Johnson et al. (2003) major study quality limitations include the use of data pooled from separate study cohorts conducted over an approximately 6-year period, the use of tap water as the vehicle for some of control and treated groups (as reported by Dawson et al. (1993) with no characterization of possible contaminants and incomplete reporting of study methods and results. • While Dawson et al. (1993) indicated that levels of TCE in dose formulations were tested by gas chromatography, the analytical findings were not reported. Johnson et al. (2003) did not report whether dose formulations were analyzed. Further, levels of TCE were not assessed in the vehicle control water; therefore, it is plausible that TCE contaminated the water and that doses were actually higher than measured. • The Dawson et al. (1993) and Johnson et al. (2003) studies estimated doses based on the average water consumption. This method does not provide precise information to calculate TCE dose because variability in drinking water consumption among dams is not characterized. • The dose selection for Johnson et al. (2003) resulted in a NOAEL that is approximately 700-fold lower than the next highest dose. • Some studies that did not identify treatment-related cardiac defects following developmental exposures to TCE, e.g., Carney et al. (2006), Fisher et al. (2001), and Schwetz et al. (1975), were well-conducted and adequately-reported GLP and/or guideline studies with no substantive limitations identified. • One study (Fisher et al., 2001) attempted to replicate the methods used in the Johnson et al. (2003) study, utilizing the same fetal cardiac dissection and evaluation techniques, and including one of the Johnson et al. (2003) study authors in the assessment team, yet found no treatment-related cardiac defects. |
• Some studies that reported no cardiac defects following TCE gestational exposures (Narotsky and Kavlock, 1995; Narotsky et al., 1995; Healy et al., 1982; Hardin et al., 1981) or avian in ovo studies (Bross et al., 1983; Elovaara et al., 1979) did not indicate that detailed evaluation of fetal hearts was conducted. • A rat whole embryo culture study of TCE administered at the period of 4–7 somites detected no cardiac defects in a study by (Saillenfait et al., 1995); however, the study methods indicate that there was no evaluation of the embryonic heart. |
| Magnitude of the effect measure | Epi | • Increased risk estimates between all or major cardiac defects ranged from 1.24 (95% CI: 0.75, 1.94) to 2.40 (95% CI: 1.27, 3.62) observed in 3 studies (Forand et al., 2012; Bove et al, 1995; Goldberg et al., 1990). Stronger associations, observed with the TCE exposure surrogate for conotruncal defects and ventricular septal defects than for major cardiac defects, a broader category (Forand et al., 2012; Bove et al., 1995). A fourth study observed an increased risk estimate of 6.2 (95% CI: 2.6, 14.5) for cardiac defects in infants of mothers aged >38 years and maternal residence within 1.32 miles from at least one TCE emissions source Yauck et al., 2004). | • No association in Yauck et al. (2004) in mothers <38 years of age and maternal residence within 1.32 miles from at least one TCE emissions source nor in Lagakos et al., (1986), which does not observe an association with cardiac defects. Alternative reasons such as lower statistical power may explain these observations. | • NE | |
| Variability analysis | Sources of within- and cross-study variability that contribute to uncertainty | Tox | • Johnson et al. (2003) test subject source, husbandry, and randomization procedures were consistent across all cohorts, i.e., including Dawson et al. (1993) and metabolite studies Johnson et al. (2003). Fetal cardiac evaluation methodology, which included evaluation without knowledge of treatment group and confirmation of all cardiac anomalies by 3 expert scientists, was also consistently applied across cohorts and studies from the UA laboratory. This had the result of reducing intra- and inter-study variability in the assessment. • Johnson et al. (2003) reported that cardiac defect incidences were consistent across all control cohorts (55 litters over approximately 6 years). An EPA review of the available control data did not observe unusual heterogeneity in prevalence of malformations. • Studies that reported cardiac defects following administration of metabolites (DCA and TCA) used randomized assignment of maternal animals to test group, thus reducing intra-study variability. • Although Dawson et al. (1993( and Johnson et al. (2003) identified cardiac defects following exposures to TCE during development, Carney et al. (2006), Fisher et al. (2001), and Schwetz et al. (2006) did not find treatment-related cardiac abnormalities. This may be the result of differences in the study design and assessment methods. This includes such aspects as animal strain, age, source, exposure route and vehicle, duration of exposure, and cardiac evaluation methods. |
• The Johnson et al. (2003) study reported data from several cohorts of animals, which were on study over a period of approximately 6 years. The data included control cohorts, some of which were concurrent and some that were non-concurrent to the TCE-treated groups (Johnson et al., 2014, 2005). Data that definitively link the individual control litter response data with each particular cohort are no longer available for independent examination. • Different study outcomes were observed in studies that had many similarities in study design and conduct, i.e., Dawson et al. (1993) and Johnson et al. (2003) identified exposure related cardiac defects while Fisher et al. (2001) did not. In the Fisher et al. (2001) study, care was taken to ensure that the same cardiac evaluation methods were used as in the Dawson et al. (1993) and Johnson et al. (2003) studies, including fetal evaluation with knowledge of treatment group, and one of the study authors of Johnson et al. (2003) participated in the fetal examination. • The use of soy bean oil in the Fisher et al. (2001) study vs. water vehicle and control for Johnson et al. (2003) and Dawson et al. (1993) studies. • The Johnson et al. (2003) and Dawson et al. (1993) studies did not calculate variability in TCE dose by measuring individual dam water consumption. |
• Based upon the toxicokinetic profile of TCE (U.S. EPA, 2011), it is considered unlikely that toxicokinetic factors contributed significantly to differences in response across study protocols. |
| Sources of within- and cross-study variability that contribute to uncertainty | Epi | • NE (not considered in Hill analysis) | • NE (not considered in Hill analysis) | • Studies examined different populations, exposure levels, gradients, and media. Additionally, different sets of strengths and uncertainties in this set of studies would contribute to observed cross-study variability. | |
| Uncertainty analysis | Missing information or data gaps, within and across studies | Tox | • For the studies conducted by the UA laboratory (Johnson et al., 2003; Dawson et al., 1993) that identified cardiac defects following exposures to TCE, DCA, or TCA, detailed descriptions of evaluation methods for assessment of cardiovascular effects were provided. • Individual fetal and litter cardiac findings data, as well as detailed information on study conduct and fetal evaluation methods, were provided to the EPA for Johnson et al. (2003) and Dawson et al. (1993). |
• The publications for studies conducted by the UA laboratory that identified cardiac defects following exposures to TCE, DCA, or TCA (Johnson et al., 2003; Johnson et al., 1998; Dawson et al., 1993) did not report essential study details, and generally did not include summaries of maternal data or fetal data for endpoints other than cardiac defects. • For well-conducted studies that did not detect cardiac defects following developmental exposures to TCE or metabolites (Carney et al., 2006; Fisher et al., 2001) adequate descriptions of study methodology and summary data for maternal and fetal findings were reported. • Mechanistic data for alterations in cardiac development are limited and do not identify initiating events for the putative AOP. |
• NE |
| Missing information or data gaps, within and across studies | Epi | • NE (not considered in Hill (1965) analysis) | • NE (not considered in Hill analysis) | • NE | |
| Qualitative dose-response | Association between exposure/dose and degree of effect | Tox | • Alterations in cardiac development were observed in multiple studies at high dose levels following TCE, DCA, or TCA exposures (Johnson et al., 2003; Johnson et al., 1998; Dawson et al., 1993; Smith et al., 1992, 1989). • The incidence of cardiovascular effects increased as a function of dose in Johnson et al. (2003). • An association between exposure to TCE (or DCA or TCA) and alterations in cardiac development was reported in various animal models, i.e., LE and SD rats, CD-1 mice, chicken embryos, and zebra fish (Drake et al., 2006a,b; Williams et al., 2006; Hassoun et al., 2005; Johnson et al., 2003; Dawson et al., 1993; Smith et al., 1992, 1989). • A BMDL for Johnson et al. (2003) was derived by EPA statisticians from individual cardiac defect data provided to EPA. Litter contribution to the outcome of interest was incorporated in the analysis. A significant dose-response trend was identified, whether or not the high dose value was included in the analysis. |
• The dose response for cardiac defects identified by Johnson et al. (2003) could only be fit to a model with elimination of the high dose data from the analysis. The lowest dose tested had a zero response for cardiac defects, below the historical control incidence. The doses tested were spaced over several orders of magnitude, with wide gaps. • Carney et al. (2006) was the only other study in the database that evaluated developmental effects of TCE over multiple dose levels. In that study, no fetal toxicity and minimal maternal toxicity was reported. |
• TCE doses tested in Johnson et al. (2003) and Dawson et al. (1993) (drinking water): 2.5 ppb, 250 ppb, 1.5 ppm, or 1100 ppm (0, 0.00045, 0.048, 0.218, or 129 mg/kg-day) • TCE doses tested in Fisher et al. (2001) (gavage): 500 mg/kg-day • TCE doses tested in Carney et al. (2006) (inhalation): 50, 150, or 600 ppm (268.5, 805.5, or 3222 mg/m3) |
| Exposure-response gradient: Association between exposure/dose and degree of effect | Epi | • NE | • Goldberg et al. (1990) and Lagakos et al. (1986) examined exposure-response; none observed. | • NE | |
| Experimental evidence | Hypothesis testing: manipulation of exposure scenario with resulting alterations in response | Tox | • A study by (Epstein et al., 1992) administered the metabolite DCA to rats on varied days of gestation and identified critical windows of exposure for eliciting cardiac developmental defects. • No statistically significant increases in congenital heart defects were observed in groups of rats that were exposed to TCE prior to pregnancy only (Dawson et al., 1993). • Drake et al. (2006b) demonstrated that cardiac defects did not occur in chick embryos exposed to TCE and TCA during the period of cardiac specification (approximately GD 6 in rats) rather than the period of valvuloseptal morphogenesis. |
• Studies in rodents that administered TCE via drinking water detected an increase in fetuses with cardiac defects (Johnson et al., 2003; Dawson et al., 1993); studies that administered TCE via other routes (gavage and inhalation) were negative for this response (Carney et al., 2006; Fisher et al., 2001; Schwetz et al., 1975). • In a whole embryo culture (WEC) study of DCA and TCA (Hunter et al., 1996), that identified cardiac defects, the acid nature of DCA and TCA may have impacted dysmorphogenesis. |
• Studies that manipulated the gestational exposure period were not conducted with TCE. |
| Association not observed once exposure ceases | Epi | • NE | • No differences between observed and expected numbers of cardiac defect cases once wells were closed in contaminated area (Goldberg et al., 1990). | • NE | |
|
Reproducibility
[Consistency] |
Reproducibility: Corroboration across studies, labs, routes of exposure, species, etc. | Tox | • Studies that administered TCE in drinking water to rats on GD 1–22 were conducted over a period of approximately 6 years by researchers at the same academic facility (UA, Tucson) used the same cardiac evaluation methods and identified treatment and dose-related cardiac malformations (Johnson et al., 2003); Johnson et al., 1998; Dawson et al., 1993). A preliminary screening study that utilized intrauterine administration of TCE also detected cardiac defects (Dawson et al., 1993). The types of cardiac malformations observed were similar across study cohorts and treatment groups throughout the duration of the research program. • Studies on TCE metabolites (TCA and TCA) conducted in other laboratories (Epstein et al., 1992; Smith et al., 1992, 1989) identified cardiac defects similar to those observed in the UA studies. • Cardiac septal anomalies were observed in avian in ovo studies (Rufer et al., 2010; Drake et al., 2006a), and in WEC assays (Mishima et al., 2006; Hunter et al., 1996) with TCE and/or metabolite exposures. Zebrafish studies also demonstrated evidence of alterations in cardiac development (Williams et al., 2006; Hassoun et al., 2005). |
• Studies conducted in other laboratories than UA and that administered TCE by gavage or inhalation (Carney et al., 2006; Fisher et al., 2001; Schwetz et al., 1975) did not identify statistically significant increases in cardiac defects. Fisher et al. (2001) used the same cardiac evaluation methods as the UA lab. | • Studies that did not identify cardiac defects with TCE and/or metabolite exposures (Carney et al., 2006; Fisher et al., 2001; Schwetz et al., 1975) did not replicate all aspects of the Johnson et al. (2003) study, even though Fisher et al. (2001) used the same cardiac evaluation techniques as (Johnson et al., 2003) and Dawson et al. (1993), and therefore provide only limited evidence of lack of reproducibility. |
| Consistency: Association observed in different populations, places, time and circumstances. | Epi | • Association between cardiac defects and TCE exposure surrogate observed in four studies. These studies were of different populations living in different states (NY, NJ) and covered slightly different time periods (1983–2000, 1985–1988) (Forand et al., 2012; Bove, 1996; Bove et al., 1995). Two other studies of weaker designs were of different populations and carried out in two different locations in the United States, and provide supporting evidence (Yauck et al., 2004; Goldberg et al., 1990). | • Lagakos et al. (1986) compared a pregnancy receiving contaminated residential well water to a pregnancy not receiving residential water from contaminated wells and does not observed an association between cardiac defects and contaminated drinking water. | • NE | |
| Biological plausibility | Observed outcome can be attributed to toxic insult given the known science | Tox | • Avian in ovo studies and atrioventricular cell culture studies support the biological plausibility of effects of TCE on cardiac development, given that early chick heart development is similar to mammalian (including human), particularly regarding the role of the cardiac cushion in septation (NRC, 2006). • Preliminary exploration of a possible adverse outcome pathway (AOP) has resulted in a reasonable conceptual model for TCE-induced congenital heart defects. In this construct, the vulnerable period is defined by endocardial morphogenesis. Endothelial–mesenchyme transition (EMT) is disrupted in the area of the atrioventricular canal, leading to septal defects. Studies in knockout mice have suggested the possible disruption of genetic signals and response by TCE exposure during cardiac development. Candidate genes have implicated pathways such as TGF-beta, ephrins, Notch signaling, VEGF pathway, and RXR signaling. Potential molecular initiating events may involve a cellular initiation of vascular inflammatory signals, perhaps through an LXR/RXR-mediated effect on cholesterol homeostasis, vulnerability to reactive oxygen species or disruption of the downstream consequences of VEGF signaling. |
• A definitive AOP for TCE-induced cardiac defects, including a putative initiating event, has not yet been characterized. Additional mechanistic data are needed to support the hypothesized AOP. • There are insufficient mechanistic data to characterize additional potential MOAs other than that hypothesized in the AOP construct. |
• It is possible that multiple modes of action are involved in alterations to cardiac development. |
| Observed association plausible given the known science | Epi | • NE | • NE | • In vitro and in vivo animal studies report cardiac defects with TCE and TCE-metabolite exposure. | |
| Alternative or multiple explanations | Other possible explanations for observed outcome after the exposure of interest | Tox | • Given the presumed contribution of both environmental exposures and genetic predisposition in human congenital heart disease (Richards and Garg, 2010), it is possible that the test subjects used in the Johnson et al. (2003) study and others conducted in that laboratory may have been particularly susceptible to alterations in cardiac development. • Other contributing factors or confounding factors were not specifically identified in the evaluated in-vivo studies. • It is possible that the absence of treatment-related cardiac defects in well-conducted TCE studies (Carney et al., 2006; Fisher et al., 2001) or metabolite studies (Fisher et al., 2001) was due to confounding variables such as differences in strain/source of animal model, route of exposure, toxicokinetics, vehicle [e.g., soybean oil in Fisher et al. (2001)], or differences in cardiac evaluation methods. • It is unlikely that the cardiac defects observed by Johnson et al. (2003) were an artifact of the evaluation procedures used, since a study by Fisher et al. (2001, using the same fetal cardiac evaluation procedures, did not identify an association between TCE exposure and the incidence of cardiac defects. |
• There is a possibility that cardiac defects detected in the Dawson et al. (1993) study were associated in part with the use of tap water as a control vehicle (i.e., possible presence of contaminants). | • NE |
| Other possible explanations for observed outcome after the exposure of interest (not considered in Hill analysis) | Epi | • Potential maternal risk factors were adjusted in statistical analysis in Forand et al. (2012) and Yauck et al. (2004) or were not found in statistical analyses to influence observed association by +15% (Bove, 1996; Bove et al., 1995). | • Potential for confounding from another exposure given the poor exposure definition in Yauck et al. (2004). The positive association in Goldberg et al. (1990) may result from likely selection biases in controls. | • NE | |
| Specificity | Single cause and effect relationship resulting from exposure to test substance | Tox | • Cardiac defects in rats appear to be attributable to direct chemical exposure to TCE or metabolites (DCA or TCA) and are unlikely to be the result of secondary effect of maternal toxicity. Johnson et al. (2003) reported that TCE exposure via drinking water to pregnant rats did not result in maternal toxicity. Carney et al. (2006) reported minimal decreases in body weight gain in dams, with no adverse fetal outcomes. In fetuses, there was no indication of TCE-related fetal weight deficits, external or skeletal anomalies, or of soft tissue alterations other than cardiac defects in Johnson et al. (2003), nor in any other study. • The majority of the cardiac malformations following TCE exposures to rats (Johnson et al., 1993; Dawson et al., 1993) or chicks (Rufer et al., 2010; Drake et al., 2006a) during sensitive periods of cardiac development were ventricular septal defects, valve defects, or outflow tract abnormalities. Mechanistic data suggest a common etiology (disruption of the cardiac cushion formation) for the observed cardiac defects (Boyer et al., 2000). |
• Studies conducted in other laboratories than UA and that administered TCE by gavage or inhalation (Carney et al., 2006; Fisher et al., 2001; Schwetz et al., 1975) did not identify cardiac defects. Fisher et al. (20001) used the same cardiac evaluation methods as the UA lab. • The cardiac defects detected in the Dawson et al. (1993) study might have been related to the use of tap water as a vehicle (i.e., possible contaminants). |
• NE |
| Single cause and effect relationship resulting from exposure to test substance | Epi | • NE | • Specificity not as critical compared to other Hill aspects since outcomes may have several risk factors. Maternal risk factors, specifically chemical risk factors, associated with cardiac defects in infants have not been well studied. | • NE | |
| Coherence | Summary: Extent to which data are similar in outcome and exposure across database | Tox | • Multiple studies were conducted at UA (Johnson et al., 2003; Johnson et all, 1998; Dawson et al., 1993), in which rats were administered TCE or metabolites DCA or TCA in drinking water on GD 1–22 and for which study design and cardiac evaluation methodologies were consistent. The outcomes of these studies (detection of cardiac defects, particularly septal defects, valve abnormalities, and outflow tract anomalies) are consistent across these studies. Additionally, these outcomes are supported by the results of avian in ovo and in vitro studies, studies with TCE metabolites (DCA and TCA) in rodents, in vitro whole embryo culture studies, and mechanistic data. | • Developmental toxicity studies with TCE that were conducted in other laboratories (Carney et al., 2006; Fisher et al., 2001; Schwetz et al., 1975) administered TCE to rats of other strains or sources, using different routes of exposure (inhalation or gavage), administered on different days of gestation (i.e., not including GD 1–6) than the UA studies and did not identify cardiac defects. No other study in the TCE database reported cardiac defects at the low dose levels reported by Johnson et al. (2003). | • NE |
| Cause and effect interpretation should not conflict with the generally known facts of the natural history and biology of the disease | Epi | • Associations in epidemiologic studies of cardiac defects and maternal occupational exposure to degreasing solvents or to organic solvents (Gilboa et al., 2012); Wilson et al., 1998; Tikkanen and Heinonen, 1991, 1988). | • NE | • NE |
NE = No relevant evidence
HH = Hamburger-Hamilton stages of chick development (Hamburger and Hamilton, 1951)
UA = University of Arizona
Tox = Animal toxicology studies; Epi = Epidemiological studies
Key Factor references:
Despite the recognized uncertainties and limitations in the TCE database, the evidence supports a conclusion that TCE has the potential to cause cardiac defects in humans when exposure occurs at sufficient doses during a sensitive period of fetal development. This conclusion is warranted by the data that demonstrate or suggest a potential hazard to cardiac development, including epidemiological studies, developmental toxicology studies in rodents with TCE and its metabolites (DCA and TCA), avian in ovo studies, in vitro assays, and mechanistic data that form the basis of a preliminary conceptual model of an AOP for valvuloseptal defects resulting from TCE exposures. Limitations within the database that increase the uncertainties regarding this conclusion are acknowledged. These limitations are described in detail above. The epidemiological studies provide evidence of associations between TCE, or TCE and other chlorinated solvents, and cardiac defects, but these studies have limitations related mainly to exposure measurement error and lower statistical power due to the rarity of cardiac defects. The rodent developmental toxicology studies conducted by Dawson et al. [20], Johnson et al. [51], and Johnson et al. [49] that reported cardiac defects resulting from TCE (and metabolite) drinking water exposures have study design and reporting limitations. Additionally, two good quality (GLP) inhalation and gavage rodent studies conducted in other laboratories, Carney et al. [15] and Fisher et al. [28], respectively, have not detected cardiac defects.
In accordance with the Guidelines for Developmental Toxicity Risk Assessment [85], the database is considered to be adequate to support categorization of the health-related database for hazard and dose-response, with the determination that there is “Sufficient Experimental Animal Evidence” and “Limited Human Data” for developmental cardiac toxicity. This category “includes data from experimental animal studies and/or limited human data that provide convincing evidence for the scientific community to judge that a potential for developmental toxicity exists.” The minimum evidence that would be necessary to determine whether there is or is not sufficient evidence of developmental toxicity is the existence of appropriate, well-conducted animal studies. The overall TCE database met this criterion, although limitations and uncertainties in the primary study used in dose response [51] are acknowledged.
3.2. Dose-response assessment for developmental cardiac defects
Given the hazard conclusion that (despite uncertainties and limitation in the database) TCE has the potential to cause cardiac defects in humans when exposure occurs at sufficient doses during a sensitive period of fetal development, the next critical issue addressed by this update is the dose-response assessment.
3.2.1. Suitability of Johnson et al. [51] study for deriving a point of departure for a reference value
The Johnson et al. [51] study is the only available study potentially useable for dose-response analysis of fetal cardiac defects. On the whole, the Johnson et al. [51] study is considered suitable for use in deriving a POD for the following reasons. The study has an appropriate design. It was conducted by a relevant route of exposure (drinking water), covered the entire period of gestation which subsumes the developmental window for the initiation of cardiac defects, and tested multiple exposure levels. Further support was derived from the finding of a robust, statistically significant dose-response relationship. Additionally, this judgement took into consideration the strengths and limitations of the study and uncertainties identified in the WOE analysis.
The study was conducted over a period of 6 years, with exposed animals and their concurrent controls distributed across time. This design is not problematic per se; clinical trials and epidemiological studies are frequently conducted similarly, with staggered entry of subjects [31,69]. An important consideration to address is the potential for increased variability among litters owing to temporal drift and other possible factors.
Overdispersion, or greater variation among litters than is expected based on within-litter variation among offspring, can be dealt with by a standard method for clustered data [77,33,55,32,67]. This method deals effectively with between-litter variation from all sources, assuming that within-litter variation (conditional on the litter-mean) is approximately binomial. This method was applied for significance tests and dose-response analyses (discussed below).
Another concern about the study design is that the two highest exposure levels and their associated controls were observed in 1989–93, and the two lowest exposures and their controls were observed during 1993–1995 (Table 8; [53,52]). This raises a question whether temporal change rather than exposures can account for the observed responses. We also note that the two highest TCE doses and their controls, reported originally in Dawson et al. [20], used tap water as a vehicle and drinking water source. Hypothetically, if teratogens in tapwater did increase cardiac defects, that would likely increase the control response and perhaps impede the ability to observe a significant increase.
Table 8.
Data analysis of cardiac abnormalities reported byJohnson et al. (2003)
| Conc. in drinking water, ppm | 0 | 0 | 0 | 0.0025 | 0.250 | 1.5 | 1100 |
|---|---|---|---|---|---|---|---|
| Dose, mg/kg-d | 0 | 0 | 0 | 0.00045 | 0.048 | 0.218 | 129 |
| Internal dose metrica | 0 | 0 | 0 | 0.00031 | 0.033 | 0.15 | 88 |
| Dates | 1989–93 | 1993–95 | all | 1994–95 | 1994–95 | 1989–90 | 1989–90 |
| N (litters)b | 20 | 35 | 55 | 12 | 9 | 13 | 9 |
| N (fetuses)b | 232 | 374 | 606 | 144 | 110 | 181 | 105 |
| N (fetuses with cardiac defect)b | 7 | 6 | 13 | 0 | 5 | 9 | 11 |
| p (fetuses with cardiac defect) | 0.0302 | 0.0160 | 0.0215 | 0 | 0.0455 | 0.0497 | 0.1048 |
Total Oxidative Metabolism per unit (body weight)3/4; units are mg/wk-kg3/4
For the purpose of this analysis, the control litters (fetuses) were designated as belonging to the 1989–93 or 1993–95 cohorts based upon an analysis of the numbers of control animals assigned to study, the incidences of cardiac malformations reported, and individual animal identification numbers Dawson et al. (1993); Johnson (2003, 2005, 2014); Johnson (2009); Paula Johnson, personal communication.
Employing all of the data, there is a highly significant (P < 0.001) increasing dose-response trend (Fig. 6) based on a Cochran-Armitage trend test after adjusting for overdispersion. The trend is also significant (P < 0.04) when the highest dose is dropped. The temporal disjunction between the middle and high dose groups prompts further examination. There is no significant trend for the two low-dose groups and their controls. When the two high dose groups and related controls (Table 8) were considered separately, a significant trend (P < 0.03) was found.
Fig. 6.
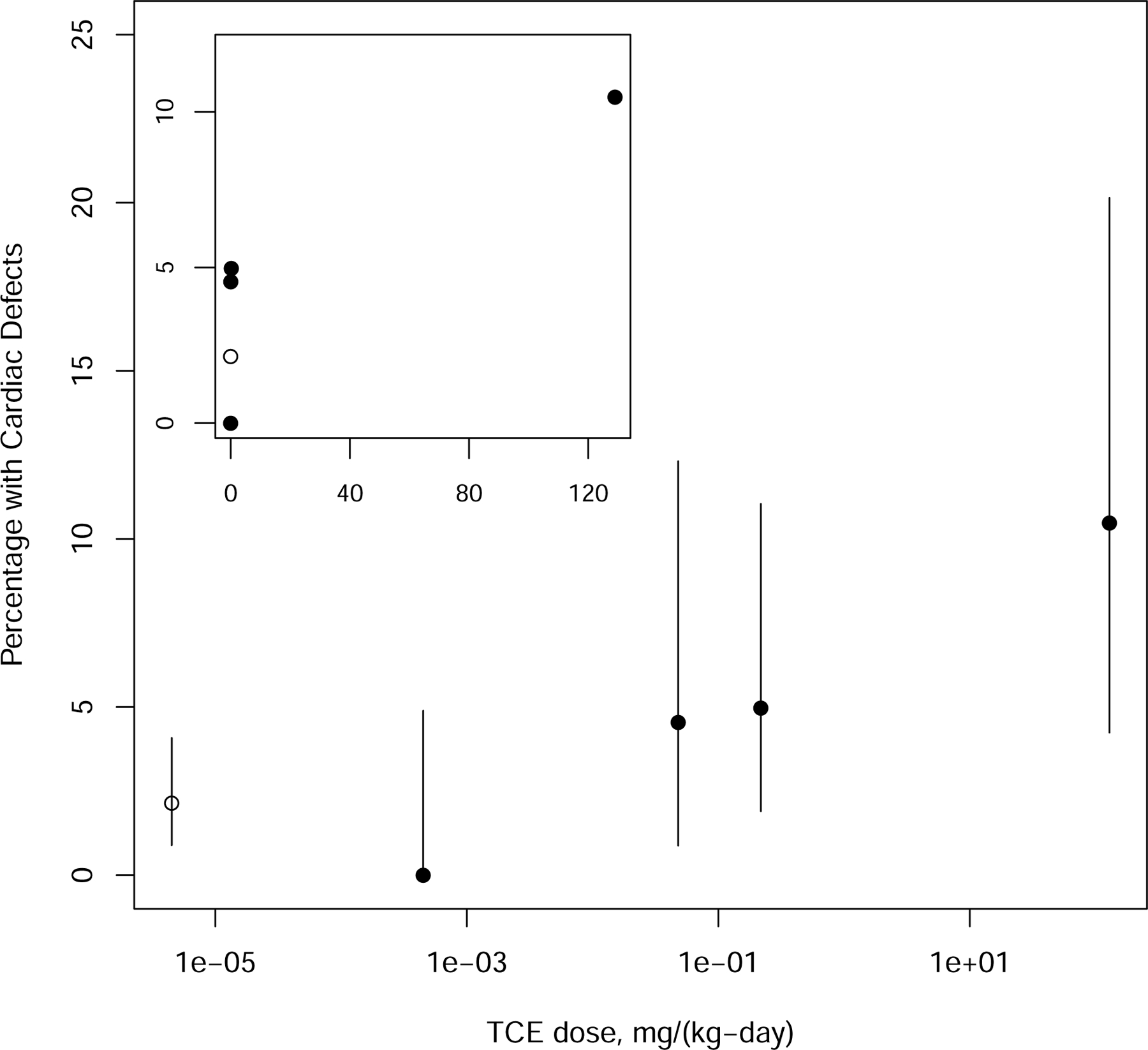
Percent of Offspring with Cardiac Defects [51]. The dose is on log scale. The inset figure shows the same data on the untransformed scale. Confidence limits (95%) for percentages are also shown. The solid points identify the treated groups and the open points identify the control.
Confidence that data from Johnson et al. [51] represent a real response is supported by the increasing trend in response (Fig. 6), and the observations of higher percentages of cardiac malformations elicited by higher doses (500 mg/kg-day and higher) in studies of rats exposed to TCE metabolites, TCA and DCA [27,79,78]. The highest dose in the Johnson et al. [51] study lies at the lower end of doses that elicited substantial responses in these other studies. Thus, a hypothesis that the Johnson data represent a false positive or an anomalous dose-response pattern seems implausible, based on trend tests and comparison with studies that used higher doses.
3.2.2. Dose-response modeling of the data from Johnson et al. [51]
Dose-response modeling of the cardiac malformation data from Johnson et al. [51] was conducted using the nested log-logistic and other BMDS models (http://www.epa.gov/ncea/bmds/) and a BMR of 0.01 (1%) extra risk, the BMR level that was used in the EPA 2011 TCE assessment [87].
The nested dose-response model accounts for overdispersion using a beta-binomial model [88]. To confirm that approach, we also applied a suite of models for dichotomous binomial data after adjusting the data for clustering, using an estimated design effect of 1.53 [77,33,55,67].
Given the uncertainties in the dose-response analysis related to the nature of the data, the confidence in the POD based on Johnson et al. [51] has limitations. Overall, however, the POD derived in the 2011 TCE assessment [87], which used an approach consistent with standard U.S. EPA dose-response practices, remains a reasonable choice.
Several sources of uncertainty related to modeling assumptions were examined:
Do the data have a plateau at less than 100% response? The evidence is equivocal and does not permit a clear answer. Considering the confidence intervals for responses in Fig. 6, it is not clear whether the response reaches a plateau or increases more gradually. A number of National Toxicology Program (NTP) studies of developmental toxicity also have a low but significant maximum response, although they differ in apparent pattern of response.3 A model with a plateau is plausible, but would not substantially change the general conclusion and results. (We used the dichotomous-Hill model in BMDS, which allows a plateau to be estimated.)
Is it better to drop or retain the high dose? For the 2011 TCE assessment [87], the high dose was dropped on the strength of an examination of residuals at the low doses for the nested model. The decision to drop the high dose is confirmed in this re-examination, using non-nested dichotomous models (adjusted for intralitter correlation using estimated design effects [33]). Dropping the high dose leads to higher model goodness of fit and better fit in the region of the BMD01 and BMD05.
Are there sufficient data in the low-dose region and near the BMD01 to permit reliable inference about the dose-response curve shape (which influences the BMD and BMDL)? BMD inference at the 1% extra-risk level is highly uncertain, because BMD and BMDL values vary by several orders of magnitude depending on the modeling assumptions. This is attributed in part to the lack of monotonicity at the lowest dose and the apparent supralinearity of the overall exposure-response relationship. Additional doses would be required to better specify the curve shape in the low-dose region. More reliable inference can be made for higher BMRs.
3.2.3. Uncertainty in the point of departure (POD)
There is substantial model and parameter uncertainty at the 1% level of extra risk, although 1% is the appropriate BMR based on severity of the effect (i.e., cardiac malformations). These uncertainties can be attributed primarily to having too few data points in the low-dose range, where more data would be required to adequately characterize the dose-response shape. Uncertainty decreases for higher BMR levels (5% and 10% extra risk), although 10% exceeds the range of the data for some models.
The BMDL01 0.0207 mg/kg-day (BMD01 0.0646) for the nested log-logistic model selected in the 2011 TCE assessment (with slope constrained and without the high dose group) [87] provides a compromise value from the range of BMDLs derived from the variety of models examined.
With a 5% BMR (i.e., 5-fold greater), the BMDL for the nested log-logistic model (BMDL05 0.108 mg/kg-day; BMD05 0.337) [87] is about 5-fold higher than the 2011 BMDL01.
Model-averaged BMDL01 or BMDL05 for dichotomous models (using a Rao-Scott transformation to adjust for intra-litter correlation; [33]) with the high dose dropped to achieve better fit in the low-dose range yielded the following values: BMD01 0.0809 mg/kg-day and BMDL01 0.0225 mg/kg-day, BMD05 0.282 mg/kg-day and BMDL05 0.178 mg/kg-day. This option yields results similar to that of the modeling approach used in the 2011 TCE assessment [87].
The LOAEL/NOAEL approach, although there is also uncertainty about defining a POD with this approach, uses either the second highest dose (0.218 mg/kg-day) or the next lower dose (0.048 mg/kg-day) as a POD. These are biologically plausible as LOAELs because the apparent extra risk values calculated from the observed responses of 2.9% and 2.5%, respectively, exceed 1%, the level identified as a suitable BMR.
In summary, additional dose-response analyses were performed to characterize the uncertainty in the POD. Alternative PODs were derived based on use of alternative models, alternative BMR levels, or alternative procedures (such as a LOAEL/NOAEL approach), each with different strengths and limitations. These alternatives were within about an order of magnitude of the POD derived in the 2011 TCE assessment [87].
Overall, taking into account the Johnson et al. [51] study design, strengths and limitations, and uncertainties in the WOE, and in spite of any reservations based upon considerations pertaining to confidence in the dose response, a majority of the expert participants in this update project agreed that the Johnson et al. [51] study was suitable for use in deriving a POD. The majority of the participants agreed that the results of the present analysis are consistent with and further support the dose-response conclusions of the 2011 IRIS TCE assessment [87].
4. Discussion/conclusions
This updated systematic review and analysis was conducted to address the potential for exposure to TCE and its metabolites during critical windows of development to result in cardiac defects. The review developed: (1) an updated characterization of the available data and uncertainties in the TCE database for cardiac defects, (2) an expanded consideration of the mechanistic database that may support future research to develop an AOP for cardiac defects resulting from TCE exposures, (3) documentation of data and WOE evaluations (evidence integration) for hazard, and (4) an extended characterization of the dose-response modeling.
4.1. Updated characterization of available data and uncertainties
One of the goals of this review was to identify any new data (i.e., postdating the last literature search performed for the EPA 2011 TCE document [87]) that address cardiac malformations associated with exposures to TCE, DCA, and TCA. A total of 1769 unique citations were identified and screened for relevance. Of these, only two additional epidemiological studies and two mechanistic studies met the established inclusion criteria. We found no animal toxicology studies (in vivo, in vitro, or in ovo) that evaluated cardiac defects with TCE (or metabolite) exposures and that had been published since January 2010.
The epidemiological and toxicological studies that had been considered in the 2011 TCE document [87] and the new studies that were identified were evaluated for study quality in a transparent and consistent manner, utilizing multiple reviewers with relevant expertise. Study strengths were identified. The epidemiological studies were examined in detail for considerations of bias, confounding, and chance. Study flaws, inadequacies, and limitations were described for the toxicological studies. These analyses formed the basis for characterizing uncertainties in the epidemiological and toxicological databases.
Several epidemiological studies observed evidence of an association between TCE exposures and CHDs. This was found to be coherent with broader epidemiological literature reporting an association between maternal occupational exposure to degreasing solvents or organic solvents and cardiac defects. The available database of epidemiologic studies provided some support for an association but is not sufficient to establish a causal link.
Evaluation of the toxicological data included targeted attention given to studies and issues that have been portrayed as controversial in the published literature. This was particularly in regard to the findings of cardiac defects identified by Dawson et al. [20] and Johnson et al. [51]. A number of potential concerns associated with these studies were dispelled, e.g., that inadequate or inappropriate cardiac evaluation methods were used, control animals were not on study concurrently with treated animals, fetuses were not randomly assigned to evaluations, cardiac examinations were conducted with knowledge of treatment group, and statistical analysis of cardiac malformation data was inappropriate. Detailed comparisons of methods used in the various developmental toxicology studies to evaluate potential cardiac defects helped to facilitate this analysis as well as to identify differences between the studies that found cardiac defects with TCE exposures [51,20] and similarly-conducted studies that did not [15,28]. The detailed methodological evaluation led to the conclusion that differences in study methods (e.g., route of exposure, vehicle, animal source or strain, or other factors) may have contributed to differences in the detection of cardiac malformations, an issue that can no longer be definitively resolved. As noted previously in the 2011 TCE document [87], some limitations of these studies were found to be unresolvable, yet resulting uncertainties were not judged to compromise the use of the studies for hazard characterization and dose-response assessment.
4.2. Expanded consideration of the mechanistic database
Mechanistic data were considered as part of the WOE analysis for the 2011 TCE assessment [87]. However, those data did not provide a linkage to the developmental pathways and processes responsible for observed cardiac defects. Further consideration of data identified in the literature search and the MGI database motivated exploration of the potential for identifying a preliminary conceptual model of an AOP framework. It was proposed that an AOP anchored to the primary dysmorphologies associated with gestational TCE, DCA, and TCA exposure (i.e., valvulo-septal defects, muscular and membranous ventral septal defects, and pulmonary and aortic stenosis) might identify key events and relationships. In this construct, the vulnerable period is defined by endocardial morphogenesis. Endothelial–mesenchyme transition (EMT) is disrupted in the area of the atrioventricular canal, leading to septal defects. Studies in knockout mice have suggested the possible disruption of genetic signals and response by TCE exposure during cardiac development. Candidate genes have implicated pathways such as TGF-beta signaling, ephrin signaling, Notch signaling, the VEGF pathway, and RXR signaling. Potential molecular initiating events may involve a cellular initiation of vascular inflammatory signals, perhaps through an LXR/RXR-mediated effect on cholesterol homeostasis, vulnerability to reactive oxygen species or disruption of the downstream consequences of VEGF signaling. Although these hypothetical initiating events have not yet been experimentally investigated, the disruption of EndMT is well-supported as a potential key event in valvulo-septal defects induced by TCE exposures. Even at this preliminary stage of AOP development, the potential construct provides support for the biological plausibility of TCE exposures resulting in cardiac defects, and it is a significant achievement in defining research needs. Further research can provide opportunities to improve understanding of the mechanism, including exploring linkages between proposed AOPs for molecular targets and cellular processes underlying early heart development, and using alternative experimental models and methods to evaluate effects of TCE and its metabolites.
4.3. Documentation of the WOE evaluation (evidence integration)
A structured approach to the WOE evaluation for both epidemiological and toxicological hazard was conducted according to the precepts of a published EPA evidence integration framework [86] that is based upon criteria established by Hill [43]. The hypothesis-based evidence for stronger and weaker weight of association was summarized and evaluated.
Overall, the WOE supported the conclusion that TCE exposure at sufficient doses during prenatal development has the potential to cause cardiac defects in humans. In Johnson et al. [51], the lowest dose to rats that resulted in these outcomes was 0.048 mg/kg-day TCE in drinking water.
This conclusion is based upon multiples lines of evidence:
Epidemiological studies that identified a clear association between cardiac defects and maternal TCE exposures via vapor intrusion [29] and limited evidence for an association of TCE, or TCE in combination with other solvents, in drinking water (Bove [8]/Bove et al. [9]).
Toxicology studies with TCE from one laboratory [51,20] that identified treatment and dose-related defects in cardiac development in rats following maternal drinking water exposures, although study design and reporting deficiencies were noted, and other laboratories were unable to replicate the findings using different routes of exposure [15,28].
Toxicology studies with metabolites of TCE from two laboratories that observed defects in cardiac development in rats after maternal high-dose gavage or drinking water exposure to DCA [79,27] or TCA [78,49].
In ovo studies from two laboratories [72,23,24,57] that found defects in cardiac structure or function in chicken embryos resulting from low-dose TCE exposures that disrupted valvulo-septal development (a process highly conserved across species, including humans)
In vitro assays (whole embryo culture studies) from two laboratories that identified alterations in cardiac development with high doses of TCE [60] or its metabolites DCA and TCA [44] exposures to chicken or mouse embryos, respectively.
Mechanistic data, including a putative AOP construct, that is consistent with the potential for TCE to cause cardiac defects and supports the biological plausibility of an effect on cardiac development with exposure to TCE.
The evidence was characterized as “Sufficient Experimental Animal Evidence” and “Limited Human Evidence” in accordance with the Guidelines for Developmental Toxicity Risk Assessment [85].
4.4. Extended characterization of the dose-response modeling
The dose-response relationship for cardiac defects in the Johnson et al. [51] study is robust and statistically significant. The study design is unusual when compared with standard guideline developmental toxicology protocols. Treated and concurrent control animals were evaluated over a 6-year period, there was a temporal gap between the 2 lower dose groups and the 2 higher dose groups. The possibility of increased variability among litters due to temporal drift and perhaps other factors across time (overdispersion), was dealt with by using a standard method for clustered data. The dose-response trend was found to be highly significant after adjusting for overdispersion. Because the maximal observed response was 10%, models with plateaus of less than 100% were investigated and were found to not substantially change the general conclusions and results. Confidence in the dose-response relationship is supported by the increasing trend in response and by metabolite studies that demonstrate findings at higher dose levels. Despite uncertainties in the dose-response analysis, the use of the Johnson et al. [51] study for dose-response assessment remains a reasonable choice.
Fig. 3.
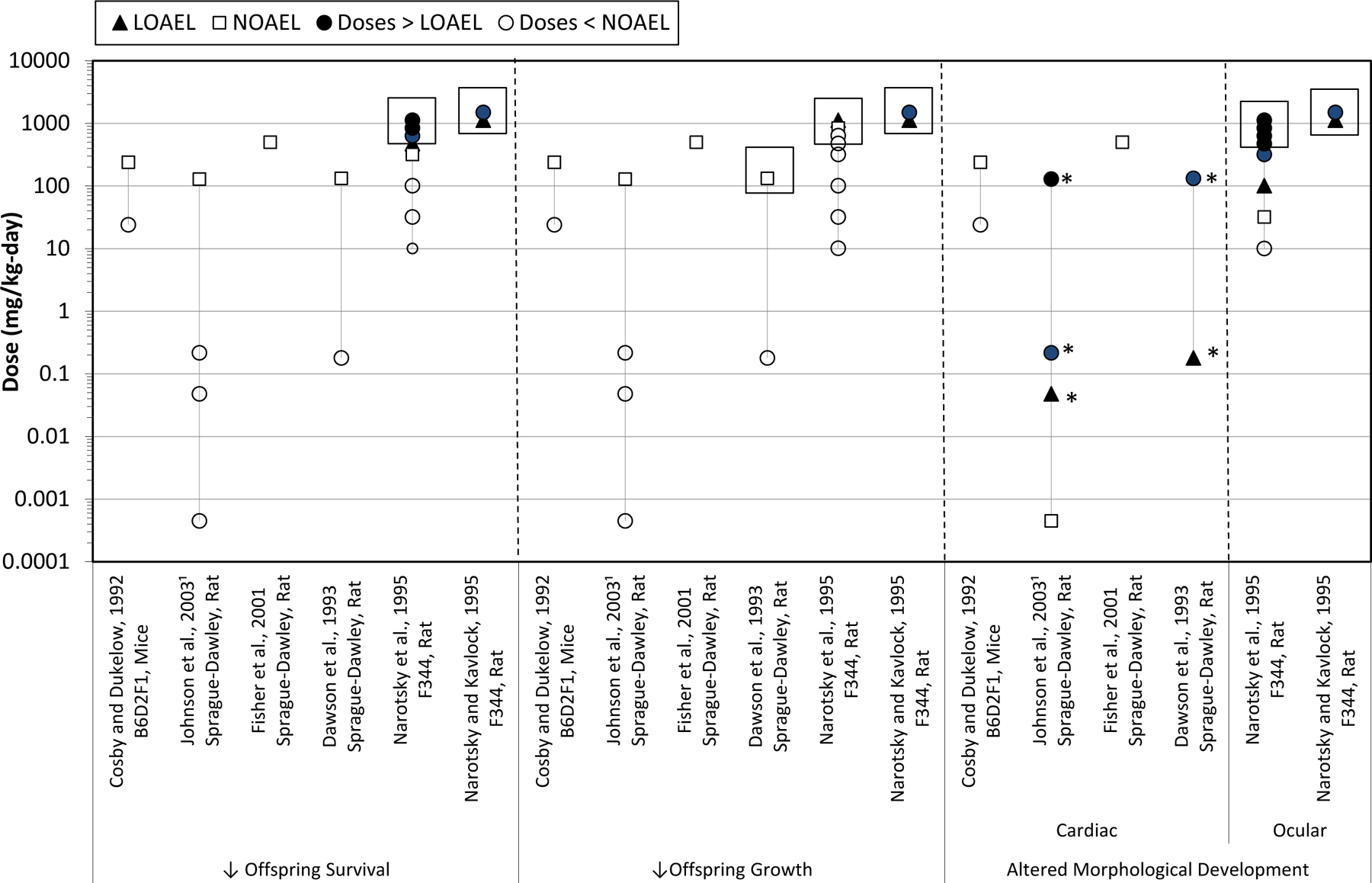
TCE oral developmental toxicology studies. Effects on fetal/offspring survival, growth, and morphology following maternal oral exposures to TCE during gestation. Boxes indicate the doses at which maternal toxicity was observed. 1Maternal toxicity was not reported in Johnson et al. [51]. * Doses at which cardiac defects were observed.
Acknowledgements
This manuscript is dedicated to the memory of Cheryl Siegel Scott (1955–2014). In her role as an epidemiologist at the Environmental Protection Agency, National Center for Environmental Assessment, Cheryl contributed significantly to the 2011 IRIS TCE assessment, and to the developmental epidemiology evaluation that forms a cornerstone of this review.
The authors thank Weihsueh Chiu (Texas A&M University, College of Veterinary Medicine and Biomedical Sciences, College Station, TX 77843) and Jamie Strong (EPA/OW, Washington, DC) for contributions to earlier drafts of the manuscript and Mafruz Haque (EPA/ORD/NCEA, Washington, DC) and Robert Kavlock (EPA/ORD, Washington, DC) for contributions to figures.
Abbreviations:
- AOP
adverse outcome pathway
- BMD
benchmark dose
- BMDS
Benchmark Dose Software
- BMDL
95% lower confidence limit on the benchmark dose
- BMR
benchmark response
- CHD
congenital heart defects
- CI
confidence interval
- DCA
dichloroacetic acid
- IRIS
Integrated Risk Information System
- EPA
U.S. Environmental Protection Agency
- MGI
Mouse Genome Informatics
- NTP
National Toxicology Program
- POD
point of departure
- TCA
trichloroacetic acid
- TCE
trichloroethylene
- WOE
weight of evidence (evidence integration)
Footnotes
A reference concentration (RFC) or dose (RfD) is an estimate of a continuous inhalation exposure (daily oral exposure) for a chronic duration (up to a lifetime) to the human population (including sensitive subgroups) that is likely to be without an appreciable risk of deleterious effects during a lifetime.
These NTP studies have a significant increase in malformations and maximum response less than 10%: TER86091, mice, MeDOPA; TER84054, rabbits, Carbon disulphide; TER82079, rats, Gentian Violet; TER84063, rats, DEHP; TER84111, mice, theophylline. http://tools.niehs.nih.gov/ntptox/index.cfm?fuseaction=ntpsearch.allchemicalsforstudy&searchterm=Developmental.
Conflict of interest
The authors declare that there are no conflicts of interest.
References
- [1].AIA (Aerospace Industries Association), Comments on the EPA toxicological review of TCE: developmental effects, Project No. 0904539.00, Available online at http://www.regulations.gov/#!documentDetail;D=EPA-HQ-ORD-2009-0791-0009.
- [2].ATSDR (Agency for Toxic Substances and Disease Registry), Health Consultation: Endicott Area Investigation: Health Statistics Review: Cancer and Birth Outcome Analysis, Endicott Area, Town of Union, Broome County, New York, U.S. Department of Health and Humans Services, Atlanta, GA, 2006. http://www.atsdr.cdc.gov/HAC/pha/EndicottAreaInvestigation/EndicottHealthStatsReviewHC052606.pdf. [Google Scholar]
- [3].ATSDR (Agency for Toxic Substances and Disease Registry), Health Consultation: Health Statistics Review Follow-up: Cancer and Birth Outcome Analysis: Endicott Area Investigation, Endicott Area, Town of Union, Broome County, New York, U.S. Department of Health and Human Services, Atlanta, GA, 2008. http://www.atsdr.cdc.gov/hac/pha//EndicottAreaInvestigationFollowUp/EndicottAreaHC051508.pdf. [Google Scholar]
- [4].ATSDR (Agency for Toxic Substances and Disease Registry), The Priority List of Hazardous Substances That Will Be the Subject of Toxicological Profiles [ATSDR Tox Profile], CDC Agency for Toxic Substances and Disease Registry, Atlanta, GA, 2013. http://www.atsdr.cdc.gov/SPL/index.html. [Google Scholar]
- [5].ATSDR (Agency for Toxic Substances and Disease Registry), Public Health Statement on Trichloroethylene [ATSDR Tox Profile], CDC Agency for Toxic Substances and Disease Registry, Atlanta, GA, 2014. http://www.atsdr.cdc.gov/ToxProfiles/tp19-c1-b.pdf. [Google Scholar]
- [6].Boldenow E, Hassan I, Chames MC, Xi C, Loch-Caruso R, The trichloroethylene metabolite S-(1,2-dichlorovinyl)-l-cysteine butnot trichloroacetate inhibits pathogen-stimulated TNF-in humanextraplacental membranes in vitro, Reprod. Toxicol 52 (2015) 1–6, 10.1016/j.reprotox.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bove F, Shim Y, Zeitz P, Drinking water contaminants and adverse pregnancy outcomes: a review [Review], Environ. Health Perspect 110 (2002) 61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bove FJ, Public drinking water contamination and birthweight, prematurity, fetal deaths, and birth defects, Toxicol. Ind. Health 12 (1996) 255–266. [PubMed] [Google Scholar]
- [9].Bove FJ, Fulcomer MC, Klotz JB, Esmart J, Dufficy EM, Savrin JE, Public drinking water contamination and birth outcomes, Am. J. Epidemiol 141 (1995) 850–862. [DOI] [PubMed] [Google Scholar]
- [10].Boyer A, Finch W, Runyan R, Trichloroethylene inhibits development of embryonic heart valve precursors in vitro, Toxicol. Sci 53 (2000) 109–117. [DOI] [PubMed] [Google Scholar]
- [11].Brender JD, Shinde MU, Zhan FB, Gong X, Langlois PH, Maternal residential proximity to chlorinated solvent emissions and birth defects in offspring: a case-control study, Environ. Health 13 (96) (2014), 10.1186/1476-069x-13-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bross G, Difranceisco D, Desmond ME, The effects of low dosages of trichloroethylene on chick development, Toxicology 28 (1983) 283–294, 10.1016/0300-483x(83)90002-1. [DOI] [PubMed] [Google Scholar]
- [13].Bukowski J, Critical review of the epidemiologic literature regarding the association between congenital heart defects and exposure to trichloroethylene, Crit. Rev. Toxicol 44 (2014) 581–589, 10.3109/10408444.2014.910755. [DOI] [PubMed] [Google Scholar]
- [14].Caldwell PT, Thorne PA, Johnson PD, Boitano S, Runyan RB, Selmin O, Trichloroethylene disrupts cardiac gene expression and calcium homeostasis in rat myocytes, Toxicol. Sci 104 (2008) 135–143, 10.1093/toxsci/kfn078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Carney EW, Thorsrud BA, Dugard PH, Zablotny CL, Developmental toxicity studies in Crl:CD (SD) rats following inhalation exposure to trichloroethylene and perchloroethylene, Birth Defects Res. B Dev. Reprod. Toxicol 77 (2006) 405–412, 10.1002/bdrb.20091. [DOI] [PubMed] [Google Scholar]
- [16].Chiu WA, Jinot J, Scott CS, Makris SL, Cooper GS, Dzubow RC, Bale AS, Evans MV, Guyton KZ, Keshava N, Lipscomb JC, Barone S Jr., Fox JF, Gwinn MR, Schaum J, Caldwell JC, Human health effects of trichloroethylene: key findings and scientific issues [Review], Environ. Health Perspect 121 (2013) 303–311, 10.1289/ehp.1205879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Collier JM, Selmin O, Johnson PD, Runyan RB, Trichloroethylene effects on gene expression during cardiac development, Birth Defects Res. A Clin. Mol. Teratol 67 (2003) 488–495, 10.1002/bdra.10073. [DOI] [PubMed] [Google Scholar]
- [18].Cosby NC, Dukelow WR, Toxicology of maternally ingested trichloroethylene (TCE) on embryonal and fetal development in mice and of TCE metabolites on in vitro fertilization, Fundam. Appl. Toxicol 19 (1992) 268–274. [DOI] [PubMed] [Google Scholar]
- [19].Dawson B, Johnson P, Goldberg S, Ulreich J, Cardiac teratogenesis of trichloroethylene and dichloroethylene in a mammalian model, J. Am. Coll. Cardiol 16 (1990) 1304–1309. [DOI] [PubMed] [Google Scholar]
- [20].Dawson B, Johnson P, Goldberg S, Ulreich J, Cardiac teratogenesis of halogenated hydrocarbon-contaminated drinking water, J. Am. Coll. Cardiol 21 (1993) 1466–1472, 10.1016/0735-1097(93)90325-U. [DOI] [PubMed] [Google Scholar]
- [21].Dorfmueller MA, Henne SP, York RG, Bornschein RL, Manson JM, Evaluation of teratogenicity and behavioral toxicity with inhalation exposure of maternal rats to trichloroethylene, Toxicology 14 (1979) 153–166. [DOI] [PubMed] [Google Scholar]
- [22].Dow J, Green T, Trichloroethylene induced vitamin B(12) and folate deficiency leads to increased formic acid excretion in the rat, Toxicology 146 (2000) 123–136, 10.1016/S0300-483X(00)00156-6. [DOI] [PubMed] [Google Scholar]
- [23].Drake V, Koprowski S, Lough J, Hu N, Smith S, Trichloroethylene exposure during cardiac valvuloseptal morphogenesis alters cushion formation and cardiac hemodynamics in the avian embryo, Environ. Health Perspect 114 (2006) 842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Drake VJ, Koprowski SL, Hu N, Smith SM, Lough J, Cardiogenic effects of trichloroethylene and trichloroacetic acid following exposure during heart specification of avian development, Toxicol. Sci 94 (2006) 153–162, 10.1093/toxsci/kfl083. [DOI] [PubMed] [Google Scholar]
- [25].EFSA (European Food Safety Authority), Guidance on selected default values to be used by the EFSA Scientific Committee, Scientific Panels and Units in the absence of actual measured data, EFSA J 10 (2012) 2579, 10.2903/j.efsa.2012.2579. [DOI] [Google Scholar]
- [26].Elovaara E, Hemminki K, Vainio H, Effects of methylene chloride, trichloroethane, trichloroethylene, tetrachloroethylene and toluene on the development of chick embryos, Toxicology 12 (1979) 111–119, 10.1016/0300-483x(79)90037-4. [DOI] [PubMed] [Google Scholar]
- [27].Epstein DL, Nolen GA, Randall JL, Christ SA, Read EJ, Stober JA, Smith MK, Cardiopathic effects of dichloroacetate in the fetal Long-Evans rat, Teratology 46 (1992) 225–235, 10.1002/tera.1420460306. [DOI] [PubMed] [Google Scholar]
- [28].Fisher J, Channel S, Eggers J, Johnson P, MacMahon K, Goodyear C, Sudberry G, Warren D, Latendresse J, Graeter L, Trichloroethylene, trichloroacetic acid, and dichloroacetic acid: do they affect fetal rat heart development, Int. J. Toxicol 20 (2001) 257–267. [DOI] [PubMed] [Google Scholar]
- [29].Forand SP, Lewis-Michl EL, Gomez MI, Adverse birth outcomes and maternal exposure to trichloroethylene and tetrachloroethylene through soil vapor intrusion in New York State, Environ. Health Perspect 120 (2012) 616–621, 10.1289/ehp.1103884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Frieden LA, Townsend TA, Vaught DB, Delaughter DM, Hwang Y, Barnett JV, Chen J, Regulation of heart valve morphogenesis by Eph receptor ligand, ephrin-A1, Dev. Dyn 239 (2010) 3226–3234, 10.1002/dvdy.22458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Friedman LM, Furberg CD, DeMets D, Fundamentals of Clinical Trials, 4th ed., Springer-Verlag, New York, NY, 2010, 10.1007/978-1-4419-1586-3. [DOI] [Google Scholar]
- [32].Fung KY, Krewski D, Rao JN, Scott AJ, Tests for trend in developmental toxicity experiments with correlated binary data, Risk Anal 14 (1994) 639–648, 10.1111/j.1539-6924.1994.tb00277.x. [DOI] [PubMed] [Google Scholar]
- [33].Fung KY, Marro L, Krewski D, A comparison of methods for estimating the benchmark dose based on overdispersed data from developmental toxicity studies, Risk Anal 18 (1998) 329–342, 10.1111/j.1539-6924.1998.tb01299.x. [DOI] [PubMed] [Google Scholar]
- [34].Gilboa SM, Desrosiers TA, Lawson C, Lupo PJ, Riehle-Colarusso TJ, Stewart PA, van Wijngaarden E, Waters MA, Correa A, Stud NBDP, Association between maternal occupational exposure to organic solvents and congenital heart defects, National Birth Defects Prevention Study, 1997–2002, Occup. Environ. Med 69 (2012) 628–635, 10.1136/oemed-2011-100536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Goldberg SJ, Lebowitz MD, Graver EJ, Hicks S, An association of human congenital cardiac malformations and drinking water contaminants, J. Am. Coll. Cardiol 16 (1990) 155–164. [DOI] [PubMed] [Google Scholar]
- [36].Green T, Dow J, Ong C, Ng V, Ong H, Zhuang Z, Yang X, Bloemen L, Biological monitoring of kidney function among workers occupationally exposed to trichloroethylene, Occup. Environ. Med 61 (2004) 312–317, 10.1136/oem.2003.007153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hamburger V, Hamilton HL, A series of normal stages in the development of the chick embryo, J. Morphol 88 (1951) 49–92, 10.1002/jmor.1050880104. [DOI] [PubMed] [Google Scholar]
- [38].Hardin B, Kelman B, Brent R, Trichloroethylene and dichloroethylene: a critical review of teratogenicity [Review], Birth Defects Res. A Clin. Mol. Teratol 73 (2005) 931–955, 10.1002/bdra.20192. [DOI] [PubMed] [Google Scholar]
- [39].Hardin BD, Bond GP, Sikov MR, Andrew FD, Beliles RP, Niemeier RW, Testing of selected workplace chemicals for teratogenic potential, Scand. J. Work. Environ. Health 7 (1981) 66–75. [PubMed] [Google Scholar]
- [40].Hardin BD, Kelman BJ, Brent RL, Trichloroethylene and cardiac malformations [Letter], Environ. Health Perspect 112 (2004) A607–A608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Hassoun E, Kariya C, Williams F, Dichloroacetate-induced developmental toxicity and production of reactive oxygen species in zebrafish embryos, J. Biochem. Mol. Toxicol 19 (2005) 52–58, 10.1002/jbt.20051. [DOI] [PubMed] [Google Scholar]
- [42].Healy TEJ, Poole TR, Hopper A, Rat fetal development and maternal exposure to trichloroethylene 100 ppm, Br. J. Anaesth 54 (1982) 337–341. [DOI] [PubMed] [Google Scholar]
- [43].Hill AB, The environment and disease: association or causation? Proc. R. Soc. Med 58 (1965) 295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hunter E, Rogers E, Schmid J, Richard A, Comparative effects of haloacetic acids in whole embryo culture, Teratology 54 (1996) 57–64, . [DOI] [PubMed] [Google Scholar]
- [45].Jenkins KJ, Correa A, Feinstein JA, Botto L, Britt AE, Daniels SR, Elixson M, Warnes CA, Webb CL, Noninherited risk factors and congenital cardiovascular defects: current knowledge: a scientific statement from the American Heart Association Council on cardiovascular disease in the young, Circulation 115 (2007) 2995–3014, 10.1161/CIRCULATIONAHA.106.183216. [DOI] [PubMed] [Google Scholar]
- [46].Jensen B, Wang T, Christoffels VM, Moorman AFM, Evolution and development of the building plan of the vertebrate heart [Review], BBA—Mol. Cell Res 1833 (2013) 783–794, 10.1016/j.bbamcr.2012.10.004. [DOI] [PubMed] [Google Scholar]
- [47].Johnson P, Personal communication from Paula Johnson, University of Arizona, to Susan Makris, U.S. EPA, 26 August 2008. Available online, 2008. [Google Scholar]
- [48].Johnson P, Personal communication from Paula Johnson, University of Arizona, to Susan Makris, U.S. EPA, 21 February 2014. Available online, 2014. [Google Scholar]
- [49].Johnson PD, Dawson BV, Goldberg SJ, Cardiac teratogenicity of trichloroethylene metabolites, J. Am. Coll. Cardiol 32 (1998) 540–545, 10.1016/s0735-1097(98)00232-0. [DOI] [PubMed] [Google Scholar]
- [50].Johnson PD, Dawson BV, Goldberg SJ, Mays MZ, Trichloroethylene: Johnson et al.’s response [Letter], Environ. Health Perspect 112 (2004) A608–A609, 10.1289/ehp.112-a608. [DOI] [Google Scholar]
- [51].Johnson PD, Goldberg SJ, Mays MZ, Dawson BV, Threshold of trichloroethylene contamination in maternal drinking waters affecting fetal heart development in the rat, Environ. Health Perspect 111 (2003) 289–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Johnson PD, Goldberg SJ, Mays MZ, Dawson BV, Erratum: threshold of trichloroethylene contamination in maternal drinking waters affecting fetal heart development in the rat [Erratum], Environ. Health Perspect 113 (2005) A18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Johnson PD, Goldberg SJ, Mays MZ, Dawson BV, et al. , Erratum: Erratum for Johnson [Environ Health Perspect 113:A18 (2005)], Environ. Health Perspect 122 (2014) A94, 10.1289/ehp.122-a94. [DOI] [Google Scholar]
- [54].Kovacic JC, Mercader N, Torres M, Boehm M, Fuster V, Epithelial-to-mesenchymal and endothelial-to-mesenchymal transition: from cardiovascular development to disease [Review], Circulation 125 (2012) 1795–1808, 10.1161/CIRCULATIONAHA.111.040352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Krewski D, Zhu Y, A simple data transformation for estimating benchmark doses in developmental toxicity experiments, Risk Anal 15 (1995) 29–39, 10.1111/j.1539-6924.1995.tb00090.x. [DOI] [PubMed] [Google Scholar]
- [56].Lagakos SW, Wessen BJ, Zelen M, An analysis of contaminated well water and health effects in Woburn, Massachusetts, J. Am. Stat. Assoc 81 (1986) 583–596. [Google Scholar]
- [57].Loeber C, Hendrix M, Diez De Pinos S, Goldberg S, Trichloroethylene: a cardiac teratogen in developing chick embryos, Pediatr. Res 24 (1988) 740–744, 10.1203/00006450-198812000-00018. [DOI] [PubMed] [Google Scholar]
- [58].Makwana O, Ahles L, Lencinas A, Selmin OI, Runyan RB, Low-dose trichloroethylene alters cytochrome P450–2C subfamily expression in the developing chick heart, Cardiovasc. Toxicol 13 (2013) 77–84, 10.1007/s12012-012-9180-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Makwana O, King NM, Ahles L, Selmin O, Granzier HL, Runyan RB, Exposure to low-dose trichloroethylene alters shear stress gene expression and function in the developing chick heart, Cardiovasc. Toxicol 10 (2010) 100–107, 10.1007/s12012-010-9066-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Mishima N, Hoffman S, Hill EG, Krug EL, Chick embryos exposed to trichloroethylene in an ex ovo culture model show selective defects in early endocardial cushion tissue formation, Birth Defects Res. A Clin. Mol. Teratol 76 (2006) 517–527, 10.1002/bdra.20283. [DOI] [PubMed] [Google Scholar]
- [61].Narotsky MG, Kavlock RJ, A multidisciplinary approach to toxicological screening: II. Developmental toxicity, J. Toxicol. Environ. Health 45 (1995) 145–171, 10.1080/15287399509531987. [DOI] [PubMed] [Google Scholar]
- [62].Narotsky MG, Weller EA, Chinchilli VM, Kavlock RJ, Nonadditive developmental toxicity in mixtures of trichloroethylene, di(2-ethylhexyl) phthalate, and heptachlor in a 5×5×5 design, Fundam. Appl. Toxicol 27 (1995) 203–216, 10.1093/toxsci/27.2.203. [DOI] [PubMed] [Google Scholar]
- [63].NRC (National Research Council), Assessing the Human Health Risks of Trichloroethylene: Key Scientific Issues, The National Academies Press, Washington, DC, 2006. http://www.nap.edu/catalog.php?recordid=11707. [Google Scholar]
- [64].NRC, Chapter 7: a roadmap for revision, in: Review of the Environmental Protection Agency’s Draft IRIS Assessment of Formaldehyde, The National Academies Press, Washington, D.C, 2011. http://www.nap.edu/catalog.php?recordid=13142. [Google Scholar]
- [65].Ou J, Ou Z, Mccarver D, Hines R, Oldham K, Ackerman A, Pritchard K, Trichloroethylene decreases heat shock protein 90 interactions with endothelial nitric oxide synthase: implications for endothelial cell proliferation, Toxicol. Sci 73 (2003) 90–97, 10.1093/toxsci/kfg062. [DOI] [PubMed] [Google Scholar]
- [66].Palbykin B, Borg J, Caldwell PT, Rowles J, Papoutsis AJ, Romagnolo DF, Selmin OI, Trichloroethylene induces methylation of the Serca2 promoter in H9c2Cells and embryonic heart, Cardiovasc. Toxicol 11 (2011) 204–214, 10.1007/s12012-011-9113-3. [DOI] [PubMed] [Google Scholar]
- [67].Rao JNK, Scott AJ, A simple method for the analysis of clustered binary data, Biometrics 48 (1992) 577–585, 10.2307/2532311. [DOI] [PubMed] [Google Scholar]
- [68].Richards AA, Garg V, Genetics of congenital heart disease, Curr. Cardiol. Rev 6 (2010) 91–97, 10.2174/157340310791162703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Rothman K, Greenland S, Lash T, Modern Epidemiology, 3rd ed., Lippincott Williams & Wilkins, Philadelphia, PA, 2008. [Google Scholar]
- [70].Rothman KJ, Greenland S, Modern epidemiology, in: Modern Epidemiology, 2nd ed., Lippincott, Williams, & Wilkins, Philadelphia, PA, 1998. [Google Scholar]
- [71].Ruckart PZ, Bove FJ, Maslia M, Evaluation of exposure to contaminated drinking water and specific birth defects and childhood cancers at Marine Corps Base Camp Lejeune, North Carolina: a case-control study, Environ. Health 12 (104) (2013), 10.1186/1476-069x-12-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Rufer ES, Hacker TA, Flentke GR, Drake VJ, Brody MJ, Lough J, Smith SM, Altered cardiac function and ventricular septal defect in avian embryos exposed to low-dose trichloroethylene, Toxicol. Sci 113 (2010) 444–452, 10.1093/toxsci/kfp269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Saillenfait AM, Langonne I, Sabate JP, Developmental toxicity of trichloroethylene, tetrachloroethylene and four of their metabolites in rat whole embryo culture, Arch. Toxicol 70 (1995) 71–82, 10.1007/bf02733666. [DOI] [PubMed] [Google Scholar]
- [74].Schwetz BA, Leong BKJ, Gehring PJ, The effect of maternally inhaled trichloroethylene, perchloroethylene, methyl chloroform, and methylene chloride on embryonal and fetal development in mice and rats, Toxicol. Appl. Pharmacol 32 (1975) 84–96, 10.1016/0041-008x(75)90197-0. [DOI] [PubMed] [Google Scholar]
- [75].Selmin O, Thorne P, Caldwell P, Johnson P, Runyan R, Effects of trichloroethylene and its metabolite trichloroacetic acid on the expression of vimentin in the rat H9c2 cell line, Cell Biol. Toxicol 21 (2005) 83–95, 10.1007/s10565-005-0124-3. [DOI] [PubMed] [Google Scholar]
- [76].Selmin OI, Thorne PA, Caldwell PT, Taylor MR, Trichloroethylene and trichloroacetic acid regulate calcium signaling pathways in murine embryonal carcinoma cells p19, Cardiovasc. Toxicol 8 (2008) 47–56, 10.1007/s12012-008-9014-2. [DOI] [PubMed] [Google Scholar]
- [77].Shoukri MM, Chaudhary MA, Analysis of Correlated Data with SAS and R: Third Edition, Chapman & Hall/CRC, Boca Raton, FL, 2007. https://www.crcpress.com/Analysis-of-Correlated-Data-with-SAS-and-R-Third-Edition/Shoukri-Chaudhary/9781584886198. [Google Scholar]
- [78].Smith MK, Randall JL, Read EJ, Stober JA, Teratogenic activity of trichloroacetic acid in the rat, Teratology 40 (1989) 445–451, 10.1002/tera.1420400506. [DOI] [PubMed] [Google Scholar]
- [79].Smith MK, Randall JL, Read EJ, Stober JA, Developmental toxicity of dichloroacetate in the rat, Teratology 46 (1992) 217–223, 10.1002/tera.1420460305. [DOI] [PubMed] [Google Scholar]
- [80].Staples RE, Detection of visceral alterations in mammalian fetuses [Abstract], Teratology 9 (1974) A37A38. [Google Scholar]
- [81].Stephen LJ, Fawkes AL, Verhoeve A, Lemke G, Brown A, A critical role for the EphA3 receptor tyrosine kinase in heart development, Dev. Biol 302 (2007) 66–79, 10.1016/j.ydbio.2006.08.058. [DOI] [PubMed] [Google Scholar]
- [82].Stuckhardt JL, Poppe SM, Fresh visceral examination of rat and rabbit fetuses used in teratogenicity testing, Teratog. Carcinog. Mutagen 4 (1984) 181–188, 10.1002/tcm.1770040203. [DOI] [PubMed] [Google Scholar]
- [83].Tikkanen J, Heinonen O, Maternal exposure to chemical and physical factors during pregnancy and cardiovascular malformations in the offspring, Teratology 43 (1991) 591–600, 10.1002/tera.1420430614. [DOI] [PubMed] [Google Scholar]
- [84].Tikkanen J, Heinonen OP, Cardiovascular malformations and organic solvent exposure during pregnancy in Finland, Am. J. Ind. Med 14 (1988) 1–8, 10.1002/ajim.4700140102. [DOI] [PubMed] [Google Scholar]
- [85].U.S. EPA (U.S. Environmental Protection Agency), Guidelines for Developmental Toxicity Risk Assessment. (EPA/600/FR-91/001), U.S. Environmental Protection Agency, Risk Assessment Forum, Washington, DC, 1991. http://cfpub.epa.gov/ncea/cfm/recordisplay.cfm?deid=23162. [Google Scholar]
- [86].U.S. EPA (U.S. Environmental Protection Agency), A Framework for Assessing Health Risk of Environmental Exposures to Children. (EPA/600/R-05/093F), U.S. Environmental Protection Agency, Office of Research and Development, National Center for Environmental Assessment, Washington, DC, 2006. http://cfpub.epa.gov/ncea/cfm/recordisplay.cfm?deid=158363. [Google Scholar]
- [87].U.S. EPA (U.S. Environmental Protection Agency), Toxicological review of trichloroethylene (CASRN 79–01-6) in support of summary information on the Integrated Risk Information System (IRIS) [EPA Report]. (EPA/635/R-09/011F), 2011, Washington, DC: http://www.epa.gov/iris/supdocs/0199index.html. [Google Scholar]
- [88].U.S. EPA (U.S. Environmental Protection Agency), Benchmark dose software (BMDS). Version 2.5.0.82 [build: 5/17/2014] [BMDS], U.S. Environmental Protection Agency, National Center for Environmental Assessment, Washington, DC, 2014, Retrieved from http://www.epa.gov/ncea/bmds/. [Google Scholar]
- [89].von Gise A, Pu WT, Endocardial and epicardial epithelial to mesenchymal transitions in heart development and disease [Review], Circ. Res 110 (2012) 1628–1645, 10.1161/circresaha.111.259960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Watson RE, Jacobson CF, Williams AL, Howard WB, DeSesso JM, Trichloroethylene-contaminated drinking water and congenital heart defects: a critical analysis of the literature [Review], Reprod. Toxicol 21 (2006) 117–147, 10.1016/j.reprotox.2005.07.013. [DOI] [PubMed] [Google Scholar]
- [91].Williams FE, Sickelbaugh TJ, Hassoun E, Modulation by ellagic acid of DCA-induced developmental toxicity in the zebrafish (Danio rerio), J. Biochem. Mol. Toxicol 20 (2006) 183–190, 10.1002/jbt.20135. [DOI] [PubMed] [Google Scholar]
- [92].Wilson JG, Methods for administering agents and detecting malformations in experimental animals, in: Warkany J (Ed.), Teratology: Principles and Techniques, 8 ed., University of Chicago Press, Chicago, IL, 1965, pp. 262–267. [Google Scholar]
- [93].Wilson PD, Loffredo CA, Correa-Villaseñor A, Ferencz C, Attributable fraction for cardiac malformations, Am. J. Epidemiol 148 (1998) 414–423. [DOI] [PubMed] [Google Scholar]
- [94].Yauck JS, Malloy ME, Blair K, Simpson PM, Mccarver DG, Proximity of residence to trichloroethylene-emitting sites and increased risk of offspring congenital heart defects among older women, Birth Defects Res. A Clin. Mol. Teratol 70 (2004) 808–814, 10.1002/bdra.20060. [DOI] [PubMed] [Google Scholar]