

Abstract
Inflammatory signaling through prostaglandin E2 receptor subtype 2 (EP2) is associated with malignant tumor growth in both experimental models and cancer patients. Thus, the absence of EP2 receptors in host tissues appears to reduce tumor growth and systemic inflammation by inducing major alterations in gene expression levels across tumor tissue compartments. However, it is not yet well-established how signaling pathways in tumor tissue relate to simultaneous signaling alterations in the surrounding tumor-stroma, at conditions of reduced disease progression due to decreased host inflammation. In the present study, wild-type tumor cells, producing high levels of prostaglandin E2 (MCG 101 cells, EP2+/+), were inoculated into EP2 knockout (EP2−/−) and EP2 wild-type (EP2+/+) mice. Solid tumors were dissected into tumor- and tumor-stroma tissue compartments for RNA expression microarray screening, followed by metabolic pathway analyses. Immunohistochemistry was used to confirm adequate dissections of tissue compartments, and to assess cell proliferation (Ki-67), prostaglandin enzymes (cyclooxygenase 2) and immunity biomarkers (CD4 and CD8) at the protein level. Microarray analyses revealed statistically significant alterations in gene expression in the tumor-stroma compartment, while significantly less pathway alterations occurred in the tumor tissue compartment. The host knockout of EP2 receptors led to a significant downregulation of cell cycle regulatory factors in the tumor-stroma compartment, while interferon γ-related pathways, chemokine signaling pathways and anti-tumor chemokines [chemokine (C-X-C motif) ligand 9 and 10] were upregulated in the tumor compartment. Thus, such gene alterations were likely related to reduced tumor growth in EP2-deficient hosts. On the whole, pathway analyses of both tumor- and tumor-stroma compartments suggested that absence of host EP2 receptor signaling reduces ‘remodeling’ of tumor microenvironments and increase local immunity, probably by decreased productions of stimulating growth factors, perhaps similar to well-recognized physiological observations in wound healing.
Keywords: prostaglandin E2 receptor subtype 2, prostaglandin E2, knockout mice, tumor stroma, tumor growth, tumor microenvironment, cell signaling, microarray
Introduction
Prostaglandins, including prostaglandin E2 (PGE2), are functionally active lipids generated from arachidonic acid by the enzymatic activity of the cyclooxygenase (COX) isoforms, COX1 and COX2 (1). It has been reported that blocking COX activity with non-steroidal anti-inflammatory drugs (NSAIDs) may reduce the risk of colorectal cancer development (1) and may even prolong the survival of patients with systemic progressive cancer (2). Thus, it is well-recognized that prostaglandins, and particularly PGE2, play a crucial role in tumor growth and progression (1,3). PGE2 exerts its effects through the interaction with E-prostanoid receptors, EP1-4. Therefore, the effects of PGE2 on tumor growth and progression may depend on the arrangement of EP receptor expression on the cell surface, since each receptor exerts distinct downstream signaling effects (1). In our previous studies on colorectal tumors, it was demonstrated that EP1 and EP2 subtype receptor proteins were highly expressed in tumor epithelial cells, with more limited expression in the tumor stroma. EP3 occurred occasionally in tumor cells, while EP4 was not detected at all (4). Additionally, tumor tissue EP2 and COX2 expression have been previously reported to predict a poor survival of patients with colorectal cancer (4,5).
The importance of EP2 receptor for tumor growth has also been previously confirmed in several mouse tumor models (6–10). Thus, our previous experiments revealed reduced tumor growth, as well as reduced systemic inflammation and altered immune responses in EP2 knockout mice (9,10). Such findings suggested that host derived factors related to the tumor microenvironment, may support reduced tumor growth, due to the fact that implanted tumor cells were wild-type in both EP2 knockout and control mice (10). Furthermore, gene expression in tumors has been mapped in order to understand signaling activities behind reduced tumor growth, linked to the expression alterations of hundreds of genes in tumors grown on EP2 knockout (10). However, such experiments were performed on tumor tissue containing mixed cell types; tumor-, stroma- and inflammatory cells (10). Therefore, the present study aimed to further extend our previous studies, by evaluating gene pathway expressions in separate tissue compartments, such as tumor tissue- and tumor stroma compartment, respectively.
Materials and methods
Animals
Adult, age-matched, male and female EP 2 knockout (EP2−/−) and EP 2 wild-type (EP2+/+) mice of strain B6.129-Ptger2tm1brey/Jackson Laboratories (11), bred on a C57BL/6 genetic background, were used in the present study (EP2−/− n=16, EP2+/+ n=17). Heterozygous, in-house breeding produced EP2−/− and EP2+/+ mice. The study was performed at a certified animal testing laboratory (Experimental Biomedicine at University of Gothenburg), and the study protocol was approved by the Gothenburg Regional Animal Ethics Committee (54–2013). All animal care and experiments were performed in accordance with national and institutional guidelines. The animals were kept under controlled ambient conditions as follows: Lights on from 7:00 a.m. to 7:00 p.m.; temperature, 21±1°C; relative humidity, 45–55%, and housed in plastic cages containing wood chip bedding material and nesting pads. The animals were provided with free access to tap water and standard laboratory rodent chow for breeding and maintenance, respectively. The animals were examined daily for signs of illness. Animal weights were recorded two to three times per week as part of the ethical protocol. No animals exhibited weight losses greater than specified in the ethical approval (−10%).
MCG 101 tumor growth and tissue dissection
The MCG 101 tumor is known to produce high concentrations of PGE2 in the systemic circulation (12). This tumor model has been used for a number of years at the Department of Surgery, University of Gothenburg (Gothenburg, Sweden) (13). The tumor originates from a chemically induced tumor on C57/BL6 mice (14), with a current appearance of an epithelial-like poorly-differentiated tumor. MCG 101 tumor growth elicits several physiological alterations similar to those observed in clinical tumor disease, such as increased inflammation (15), anorexia with reduced food intake (9), increased whole body metabolism with subsequent wasting (16) and hormone alterations (17). In the present study, MCG 101 tumor cells were cultured as previously described (14), collected by trypsin treatment (Biochrom L2143, VWR International, LLC.), centrifuged (300 × g, 10 min, room temperature) and diluted in physiological saline solution to a concentration of 100,000 cells/0.1 ml. The MCG 101 cells were then injected in the thighs of the right leg [EP2+/+ n=17 (nine females and eight males), EP2−/− n=16 (eight females and eight males)] of mice under general anesthesia (isoflurane; inhaled concentration, 2.7%). The injected tumor cells were not genetically modified to lack EP2 receptors. We have previously published that tumor growth was reduced in EP2−/− mice (10). In order to avoid additional procedures and secure RNA quality at tissue harvest, tumor growth was monitored by performing tumor size estimation using Vernier caliper readings across two dimensions (mm × mm) on day 13, the day before the end of experiment. Tumor growth was assumed to be spherical and tumor volume was calculated using the following formula: V=4/3 × π × r3. Calculated tumor volumes in EP2−/− mice were 65% compared to calculated tumor volumes in EP2+/+ mice [EP2+/+, 1103±164 mm3 (n=17); EP2−/−, 713±85 mm3 (n=16), P<0.05, Table SI]. Thus, previous observations of reduced tumor growth in EP2−/− mice (10) were consistently also observed in the present study. All mice were sacrificed on day 14 after the injection of tumor cells. The mice were anesthetized with a mixture of xylazine (5 mg/kg) and ketamine (100 mg/kg) i.p. in a volume of 0.1 ml/mouse. EDTA-blood samples were collected by cardiac puncture, tumor and tumor-stroma tissue were thereafter dissected under magnification (×4) into separate fractions for their subsequent use in microarray analysis. Tumor-stroma tissue was dissected from the tumors edge while tumor cell fractions were dissected from middle tumor area, in order to create defined tissue compartments (Fig. 1), as confirmed by immunohistochemistry on random tissue sections. Tissue samples for microarray and immunohistochemical analyses were immediately placed in RNAlater™ solution (Thermo Fisher Scientific, Inc.) and 4% neutral buffered paraformaldehyde, respectively. Tissues for microarray analysis were dissected prior to immunohistochemistry tissue sample collection. Animal death was verified by a lack of heartbeat following blood and tissue collection.
Figure 1.
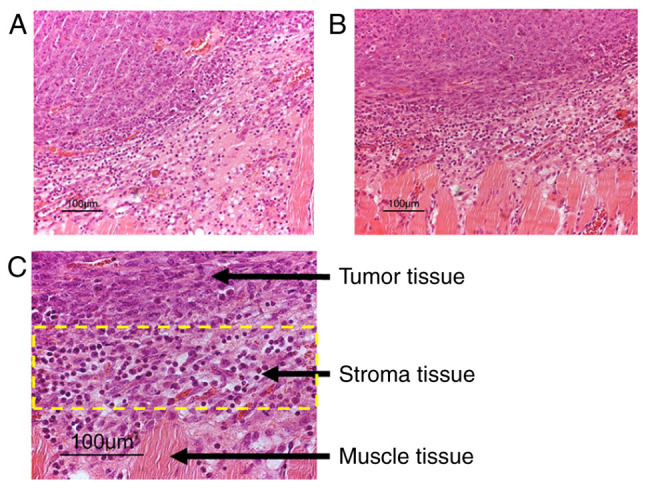
(A and B) Cell micrographs of representative hematoxylin- and eosin-stained sections of MCG101 tumor tissue with adjacent stroma and muscle tissue. Tumor and stroma tissues were dissected under magnification in two separate cell fractions, and subsequently used for microarray analyses. (C) The area within the yellow rectangle represents the tumor stroma.
RNA extraction and microarray analyses
Tumor tissue and tumor-stroma tissue from eight wild-type (EP2+/+) and eight EP2 knockout mice (EP2−/−), with four females and four males included in each group, were randomly selected from available tissues. Total RNA from each tissue was extracted using the RNeasy Fibrous Tissue kit according to the manufacturer's protocols (Qiagen GmbH). The quality of RNA was examined on an Agilent 2100 Bioanalyzer using an RNA 6000 Nano assay kit (Agilent Technologies, Inc.). RNA Integrity number was >7 in all samples. The concentrations of RNA were measured on a NanoDrop ND-1000A spectrophotometer (NanoDrop Technologies; Thermo Fisher Scientific, Inc.).
Cyanine-3 (Cy3)-labeled cRNA was prepared from 200 ng total RNA using the LowInput QuickAmp Labeling kit One-Color (Agilent Technologies, Inc.) according to the manufacturer's instructions, followed by RNeasy column purification (Qiagen, Inc.). Dye incorporation and cRNA yields were also examined using the NanoDrop ND-1000 Spectrophotometer. A total of 600 ng Cy3-labeled cRNA was fragmented at 60°C for 30 min in a reaction volume of 25 µl containing 1X Agilent fragmentation buffer and 2X Agilent blocking agent following the manufacturer's instructions. Upon completion of the fragmentation reaction, 25 µl 2X Agilent hybridization buffer were added to the fragmentation mixture and hybridized to Agilent SurePrint G3 Mouse Gene Exp v2 Array (design ID 074809) for 17 h in a rotating Agilent G2545A hybridization oven at 65°C. Following hybridization, microarrays were washed 1 min at room temperature with GE Wash Buffer 1 (Agilent Technologies, Inc.) and 1 min with 37°C GE Wash buffer 2 (Agilent Technologies, Inc.). The slides were scanned immediately after washing on an Agilent DNA Microarray Scanner (G2505C) using one color scan setting for 8×60 K array slides (scan area, 61×21.6 mm; scan resolution, 3 µm; dye channel set to green; PMT set to 100%). The scanned images were analyzed using Feature Extraction Software 10.7.3.1 (Agilent Technologies, Inc.), using default parameters to obtain background subtracted and spatially detrended processed signal intensities. The BEA Core Facility at the Karolinska Institute (Stockholm, Sweden) performed microarray hybridization and delivered Feature Extraction pre-processed data files for further evaluation. The processed signal intensity values were further analyzed using GeneSpring GX 14.9.1 software (Agilent Technologies, Inc.). The quality control of scanned data-files was performed according to standard procedures using Feature Extraction software and Genespring GX QC metrics workflow.
Data workflow in Genespring GX software
Statistical analyses and filtering options were used to generate final datasets for downstream pathway analyses. The options were: ‘filter on flags’ (present), moderated t-test (P<0.1) and fold change (FC) analyses (FC ≥2.0 and ≥5.0). The total number of entities (transcripts) present on the Agilent SurePrint G3 Mouse Gene Exp v2 Array (Agilent Technologies, Inc.; design ID: 074809) was 56,745. The number of entities remaining at each level of testing was, in the stroma tissue: Filter on flags present (31,280), moderated t-test P<0.1 (13,762), FC2 (5,669) and FC5 (388). The number of remaining entities in the tumor tissue were: Filter on flags present (29,365), moderated t-test P<0.1 (1,670), FC2 (303) and at FC5 (1). Pathway analysis was performed on the gene lists obtained after ‘fold change analysis with 2.0’. The option, in GeneSpring GX 14.9.1 software (Agilent Technologies, Inc.), of comparing gene lists with existing pathway maps at WikiPathways (https://www.wikipathways.org/) were used in pathway analyses, which were regarded statistically significant at the P<0.05 significance level. Altered entities of prostaglandin synthesis genes were searched for in gene lists created after filtering options, filter on flags-present, moderated t-test P<0.1 and FC>1.2 (Table I).
Table I.
Genes involved in prostaglandin metabolism with significantly altered gene expression in the tumor- and tumor stroma compartment in EP2−/− mice, as compared with wild-type EP2+/+ mice (P<0.1, fold change ≥1.2).
| Regulation fold change | |||
|---|---|---|---|
|
|
|||
| Gene name | Enzyme/receptor | Tumor | Stroma |
| PG metabolism | |||
| Ptgs1 | COX1 | - | ↑ 1.8 |
| Ptgs2 | COX2 | - | ↓ 4.1 |
| PG synthases | |||
| Ptges | mPGES | - | ↓ 2.4 |
| Ptges3 | cPGES | ↓ 1.3 | ↓ 1.3 |
| PGE2 | |||
| Ptger2 | EP2 | ↑ 1.8 | ↑ 2.0 |
| PGF2α | |||
| Ptgfr | FP | - | ↓ 2.8 |
| PGI2 | |||
| Ptgir | IP | ↓ 1.7 | ↑ 1.7 |
| Nuclear receptors | |||
| Ppara | PPARα | - | ↑ 3.5 |
| Pparg | PPARγ | - | ↑ 2.4 |
↑, upregulated; ↓ downregulated in EP2−/− mice compared to EP2+/+ mice. COX, cyclooxygenase; EP, E-prostanoid; PG, prostaglandin; cPGES, prostaglandin E Synthase 3; mPGES, prostaglandin E synthase; PGE2, prostaglandin E2; PGF2α, prostaglandin F2 receptor; PGI2, prostaglandin I2; PPARα and γ, peroxisome proliferator-activated receptor α and γ.
Immunohistochemistry
Tissues cut to include tumor, tumor-stroma and muscle cells were fixed in 4% neutral buffered formaldehyde solution and embedded in paraffin. Sections at a thickness of 4 µm from eight wild-type (EP2+/+) and eight knockout (EP2−/−) animals, four females and four males in each group, were prepared. The sections were deparaffinized in Histolab-Clear (Histolab Products AB), rehydrated in graded ethanol washes, and microwave heated for antigen retrieval (350 W, 5 min and 500 W, 5 min) in 10 mM sodium citrate buffer, pH 6 (COX2 and Ki-67) or 10 mM Tris, 1 mM EDTA solution, pH9 (CD4 and CD8). Immunohistochemistry was performed using the MACH 1 Universal HRP-Polymer detection kit including peroxidase and protein block, according to manufacturer's instructions (Biocare Medical, LLC, cat. no. M1U539 G, L10). Primary antibodies were diluted in Da Vinci Green antibody diluent (Biocare Medical, LLC) and incubated at 4°C overnight. The primary antibodies used were as follows: Rabbit monoclonal, anti-Ki-67 (1:200, cat. no. GTX16667, GeneTex, Inc.), rabbit polyclonal anti-COX2 (1:250; cat. no. ab15191, Abcam), rabbit monoclonal anti-CD8 alpha (1:500; cat. no. ab217344, Abcam) or rabbit polyclonal anti-CD4 antibody (1:3,000; cat. no. PA5-87425, (Invitrogen; Thermo Fisher Scientific, Inc.). Secondary antibody [MACH 1 Universal HRP-Polymer (Biocare Medical, LLC)] incubation was performed for 30 min at room temperature. Staining was visualized by ~5 min of incubation at room temperature in 3,3′-diaminobenzidine (DAB) solution, followed by washing and hematoxylin counterstaining for 1–5 min at room temperature in undiluted Mayers HTX solution (cat. no. 01820, Histolab Products AB). Negative controls, with the omission of primary antibody incubation were included for each group, EP2+/+ and EP2−/−.
Scoring of immunohistochemical staining
For scoring, two independent evaluators manually determined Ki-67+ and CD8+ cell numbers, and three independent evaluators determined COX2+ staining intensity from 16 immunohistochemical tissue sections. Scoring was performed in a blinded manner, without having any information on the tissue genotype. For Ki-67, a three-grade scale was used to estimate the number of KI-67+ cells in the areas of tumor and tumor stroma cells, summarized into an overall grade from each evaluator. A section presenting with the lowest number of Ki-67+ cells was ranked as grade 1, one with the highest number of Ki-67+ cells was ranked as grade 3, while when neither high nor low Ki-67+ cell levels were observed in a selected section, it was ranked as grade 2.
For COX2, a three-grade scale was also used for the overall COX2 staining intensity. In addition to the overall evaluation of staining intensity, COX2 staining was evaluated in three different areas, respectively: Areas consisting of mainly tumor cells, tumor-stroma cells, or areas that seemingly had presence of infiltrating immune cells, judged by morphological cell appearance. Such areas were graded according to high or low COX2 protein expression.
CD8+ cells were evaluated with a three-grade scoring in the tumor area. The CD4+ staining results were not graded. Approximately 5–10 selected fields in each section were evaluated for Ki-67, COX2 and CD8 staining, respectively. For statistical evaluation, the results of all evaluators were either combined into a mean score (Mann-Whitney U tests, n=8 per group) or treated as independent results from each evaluator (Fisher's exact test was applied, n=16 or n=24 per group). Images were captured using a Nikon E400 light microscope (Nikon Corporation) with magnifying power ranges between ×100 to ×400, equipped with a Nikon DS-Fi3 camera (Nikon Corporation) and NIS elements BR software version 5.30.02 (Nikon Instruments, Inc.).
Statistical analysis
The statistical evaluation of the microarray data was performed using GeneSpring GX 14.9.1 software, as described in the ‘Materials and methods’. A statistical significance level cut-off value of P<0.1 was applied in filtering options to select final datasets of altered transcripts in tumor and stroma tissue, for subsequent pathway analysis, since remaining transcript numbers in tumor tissue were below necessary items for pathway analysis at lower significance level. Pathway analyses were regarded statistically significant at P<0.05. Immunohistochemical analysis results were evaluated using the Mann-Whitney U-test and Fisher's exact test. Evaluator scores were combined into a single mean score for each tissue section in Mann-Whitney U test evaluations (n=8 per group); however, scores were treated as independent results from each evaluator in Fisher's exact test evaluation, giving n=16 or 24 per group (two or three independent evaluators). Tumor volumes were compared using a t-test. P<0.05 was considered to indicate a statistically significant difference in two-tailed tests.
Results
Immunohistochemistry
Tissue sections used for immunohistochemistry detection included regions of both the tumor and tumor-stroma, which were evaluated for Ki-67, COX2, CD4 and CD8 protein expression. Ki 67 protein was analyzed as a marker to estimate cell proliferation. Overall, a lower number of Ki-67-positive cells was observed in tissues from EP2−/− compared to EP2+/+ mice [Mann-Whitney U-test, rank-sum 48.5 in EP2−/−, and 87.5 in EP2+/+ mice, n=8/group, P<0.05 (examples of scoring intensities are shown in Fig. 2)]. The estimation of cell cycle activities by Ki-67 protein were in line with the microarray analysis results, demonstrating downregulation in cell cycle control (as shown below). Morphologically, Ki-67 staining appeared in the nucleus of most cells as was expected; however, Ki-67 expression was also observed occasionally in the cytoplasm of cells (Fig. 2).
Figure 2.
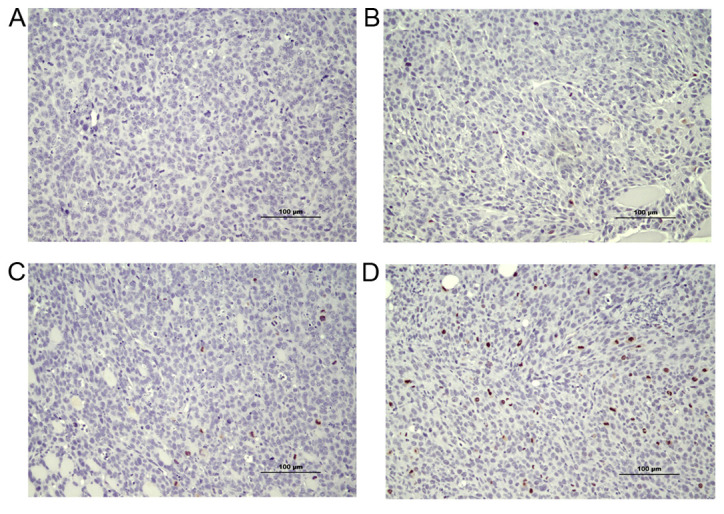
Reduced numbers of Ki-67-positive cells were observed in tumor and tumor microenvironment from EP2−/− compared to EP2+/+ mice (P<0.05). Representative micrographs of negative control and examples of Ki-67 protein staining in tissue sections which were scored as low, medium or high Ki-67 protein staining are presented. (A) Negative control. (B) Ki-67 low. (C) Ki-67 medium. (D) Ki-67 high. The majority of tumors grown in EP2−/− mice scored 1 or 2, while tumors grown in EP2+/+ mice scored 2–3. EP2, prostaglandin receptors of subtype 2.
An irregular pattern of COX2 protein staining was observed in both EP2−/− and EP2+/+ mice (Fig. 3A-F). COX2 protein expression was observed in both the tumor stroma and tumor compartments, with a trend towards an increased COX2 protein expression in stroma areas as compared with central areas of the tumor according to microscopic inspection. No statistically significant difference in COX2 protein expression was observed in tumor and stroma compartments from EP2−/− and EP2+/+ mice, according to their evaluation as a combined mean overall score of staining intensity (graded low, median, high, P>0.05 Mann-Whitney U test, n=8/group). However, significantly more tissue sections from the EP2−/− group were graded as low COX2 staining intensity (P<0.05, Fisher's exact test; EP2−/− n=10, EP2+/+ n=2, n total=24/group).
Figure 3.
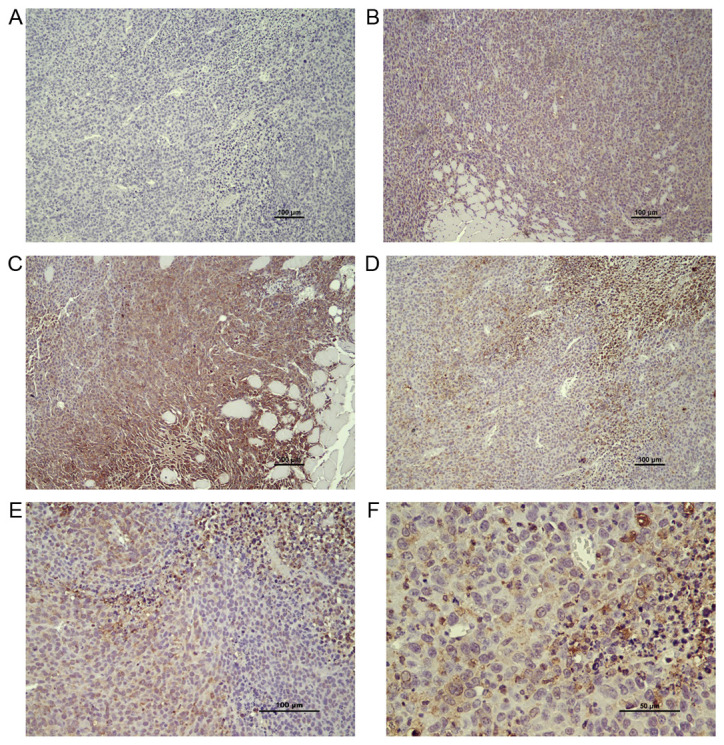
COX2 protein staining in tumor and tumor-stroma. Overall, COX2 protein expression levels were decreased in tumors grown in EP2−/− mice (P<0.05); however, COX2 protein expression was highly variable among areas in tumors from both EP2−/− and EP2+/+ mice. (A) Negative control. (B) Tumor with low COX2 expression. (C) Tumor with intense COX2 in tumor stroma areas. (D) Tumor tissue with high COX2 expression in areas with immune cell infiltration. (E) Example of highly variable COX2 expression in neighboring areas within the tumor. (F) COX2 protein expression at higher magnification. COX, cyclooxygenase; EP2, prostaglandin receptors of subtype 2.
In addition to the overall evaluation of COX2 staining, we performed a separate evaluation of tissue areas classified as: Areas mainly containing tumor cells, areas with tumor-stroma interactions and areas with assumed immune cell infiltration, as judged by morphological cell appearance (staining intensity graded either low or high). A high intensity of COX2 protein expression was found in several tumor areas with infiltration of immune cells (Fig. 3D). However, a statistically significant difference between genotypes was not detected for any of the specific tissue areas (P>0.05). Thus, the immunohistochemical analysis of COX2 protein expression confirmed a lower overall COX2 protein expression in tumors grown in EP2−/− mice; however, the reduced COX2 protein expression could not be related to specific cell types; tumor cells, stroma, immune cells.
CD4 and CD8 staining was performed, in order to evaluate immune cell presence in tumor tissue compartments. CD8 has been reported to appear mainly on cytotoxic T-lymphocytes, and is also found on natural killer cells (18). CD4 may occur on several types of cells, including T-helper cells, antigen presenting cells and macrophages (19). CD8+ cells appeared in clusters in the tumor-stroma compartment; in contrast, they appeared as ‘single’ infiltrating cells in the tumor compartment (Fig. 4B and C). Tumors from EP2−/− mice appeared to have more infiltrating CD8+ cells in the tumor compartment at microscopic inspection. Accordingly, significantly more tissue sections from the EP2−/− group were graded as having a high infiltration of CD8+ cells (P<0.05, EP2+/+ n=0, EP2−/− n=5; total, n=16/group, Fisher's exact test). CD4+ cells appeared in stroma areas, as well as infiltrating cells irregularly dispersed throughout the tumor compartment in both EP2+/+ and EP2−/− mice samples (Fig. 4D, IHC scoring not performed).
Figure 4.
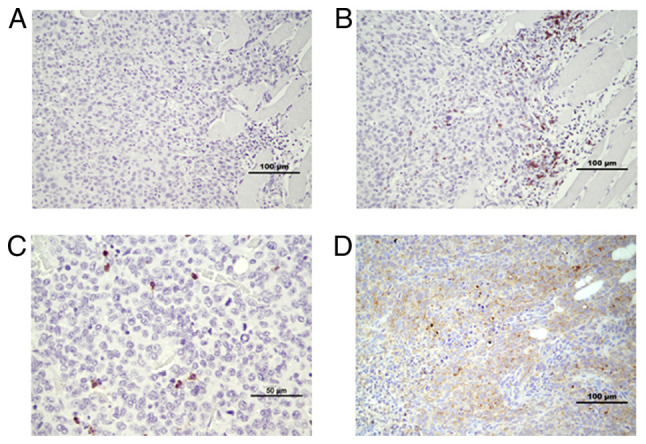
CD8+ and CD4+ cells in tumor and tumor stroma. Infiltration of CD8+ cells increased in EP2−/− mice. (A) Negative control. (B) CD8+ cells appeared in clusters in tumor stroma. (C) Example of CD8+ cell infiltration into tumor tissue from EP2−/− mice. (D) CD4+ cells appeared irregularly dispersed throughout the tumor area in both EP2-/− and EP2+/+ mice. EP2, prostaglandin receptors of subtype 2.
Microarray analyses
Overall, a significantly higher number of altered entities (transcripts; upregulated or downregulated) were observed in the stroma compartment compared to the tumor compartment in EP2−/− compared to EP2+/+ mice (Fig. 5A). In the tumor-stroma, almost 10% of all entities (transcripts) (5,669/56,745) exhibited an altered gene expression (P<0.1, FC ≥2.0), whereas only 0.5% (303/56,745) of entities from the tumor tissue displayed an altered expression with an FC >2.0. A similar pattern was also consistent at an increased FC difference (FC ≥5.0). A larger number of highly altered entities, ~0.7% (388/56,745), were observed in the stroma tissue, while only one entity from tumor tissue displayed such a large alteration in gene expression between EP2 receptor-deficient mice and wild-type mice (Fig. 5B).
Figure 5.
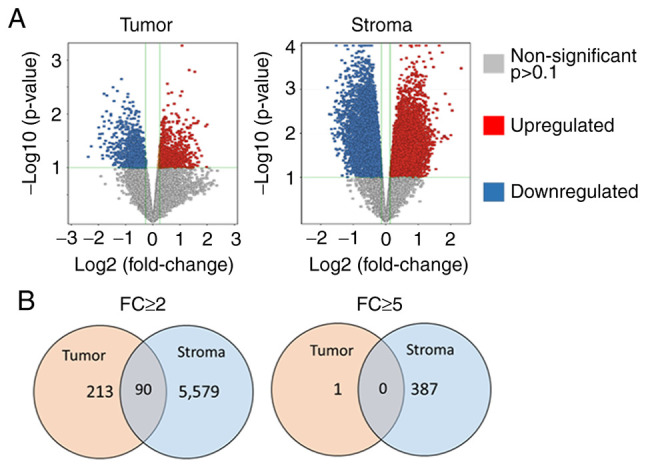
Number of transcripts (entities) with significantly altered expression in tumor and tumor stroma tissues from EP2−/− compared to EP2+/+ mice. (A) Volcano plot of all transcripts demonstrating the distribution of significantly upregulated and downregulated transcripts in tumor and tumor stroma in EP2−/− vs. EP2+/+ mice. Displayed cut-offs (green lines) are set at P<0.1 and FC 1.2. (B) Total number of significantly altered entities with Fold change ≥2.0 and ≥5.0 at P<0.1. Stroma cells displayed significantly more alterations compared to tumor cells at each fold change level. The majority of altered transcripts were specific for each tissue (tumor=orange, stroma=blue), while a lower number of altered transcripts were present in both tissue types (grey). EP2, prostaglandin receptors of subtype 2; FC, fold change.
In our previous study, gene expression was determined in ‘whole-tumor tissue’ including both tumor cells, and stroma, and also including inflammatory cells (10). In the present study, tumors were dissected into tumor- and tumor stroma compartments, and the Venn diagram illustrates that the tissue preparation technique was successful in separating the tissue types, since few altered transcripts occurred in both gene-sets. Thus, each tissue type, tumor tissue and stroma, demonstrated specific alterations in gene expression in response to host knockout of the EP2 receptor (Fig. 5B).
Alterations of prostaglandin-related genes
Transcripts of genes involved in prostaglandin metabolism were altered in both tumor and stroma compartments. More alterations, related to prostaglandin production and metabolism, occurred in the stroma in comparison with tumor compartment (Table I).
In the stroma, COX1 and COX2 transcripts displayed an inverse appearance, with upregulation of COX1 and downregulation of COX2 transcript levels in response to host knockout of EP2 receptors. However, COX1 and COX2 transcripts were not significantly altered in tumor tissue. The prostaglandin-synthase enzymes, prostaglandin E synthase 3 (cPGES; COX1-activity) and prostaglandin E synthase (mPGES; COX2-activity) were downregulated in both the stroma and tumor tissues. Among the prostaglandin specific receptors, FP receptor (PGF2α) was downregulated in the stroma, while prostaglandin I2 (PGI2) was upregulated in stroma and downregulated in tumor tissue, suggesting significance of specific transcript alterations involved in prostaglandin metabolism. Nuclear receptors, Peroxisome proliferator-activated receptor α (PPARα) and γ (PPARγ) were upregulated in the stroma tissue only (Table I).
Pathway analysis
Analysis at an FC of 2.0 resulted in 112 matched WikiPathways in stroma and 37 matching WikiPathways in tumor compartment; at an FC of 5.0, 27 pathways matched in the stroma and only one matched pathway was found in the tumor compartment (P<0.05 for all pathways (data not shown). However, several pathways represented similarities with overlapping biological functions. Significantly matched pathways, including high numbers of matched entities with known importance for tumor growth, were grouped into tables, according to tissue type and its role in cellular environment functions.
Statistically significant pathways in both tumor- and stroma compartments
Several significant pathways were observed in both stroma and tumor tissue (Table II). Selected pathways associated with intracellular signaling (PI3K, Wnt and TGFβ) and immune responses [T- and B-cell receptor signaling, chemokine signaling, double-stranded RNA-specific adenosine deaminase (Adar1)-editing] are presented in Table II. The majority of pathways displayed both upregulation and downregulation of gene transcription in response to the absence of EP2 receptor signaling. Some pathways displayed a uniform regulation pattern in the absence of EP2 receptor (Table II). Pathways that display a consistent pattern of expression may be of particular interest to find specific targets related to the reduced tumor growth observed in EP2 receptor-deficient mice. The immune response pathway ‘Adar1 editing deficiency response’, was uniformly upregulated in both stroma and tumor compartment. This pathway involved different types of interferon (IFN) activated genes and 2′-5′ oligoadenylate synthetases in the stroma, while in tumor tissue, only a few genes were altered (all upregulated); among these being chemokine (C-X-C motif) ligand 10 (Cxcl10; Table III).
Table II.
Signaling pathways with statistical significance in both the tumor- and tumor stroma compartments when performed on transcripts (entities) with a fold change >2.0.
| Tumor tissue | Stroma tissue | |||||||
|---|---|---|---|---|---|---|---|---|
|
|
|
|||||||
| Significant pathways | No. of pathways | Matched transcripts/total transcripts in pathway | Regulation | P-values | No. of pathways | Matched transcripts/total transcripts in pathway | Regulation | P-values |
| Intracellular signaling | ||||||||
| P13K Akt | 1 | 6/330 | ↓55% | 0.0074 | 1 | 61/330 | ↓35% | 2.41E-06 |
| mTOR signaling | ↑33% | ↑65% | ||||||
| Wnt signaling | 1 | 2/60 | ↑100% | 0.040 | 4 | 18/60 | ↓65% | 2.67E-05 |
| 10/37 | ↑35% | 0.003818 | ||||||
| 27/109 | 1.62E-05 | |||||||
| 24/97 | 3.98E-05 | |||||||
| TGFβ signaling | 1 | 3/52 | ↓33% | 0.00271 | 1 | 10/52 | ↓32% | 0.0411 |
| ↑66% | ↑69% | |||||||
| Immune responses | ||||||||
| T-cell receptor | 1 | 7/133 | ↑100% | 7.49E-06 | 1 | 22/133 | ↓8.3% | 0.0189 |
| signaling | ↑91.7% | |||||||
| B-cell receptor | 2 | 4/156 | ↑100% | 0.00991 | 2 | 27/156 | ↓27% | 0.0061 |
| signaling | ↑73% | |||||||
| Chemokine | 1 | 11/191 | ↓8% | 5.01E-09 | 1 | 36/191 | ↓21% | 1.35E-04 |
| signaling | ↑92% | ↑79% | ||||||
| Adar1-editing | 1 | 3/78 | ↑100% | 0.0065 | 1 | 22/78 | ↑100% | 1.95E-06 |
| deficiency response | ||||||||
↑, upregulated; ↓ downregulated in EP2−/− mice compared to EP2+/+ mice. Adar1, double-stranded RNA-specific adenosine deaminase 1; E, exponent.
Table III.
Genes in WikiPathway Adar1-editing deficiency response (WP3415_104961) with an altered expression in either the stroma- or tumor tissue from EP2−/− compared to EP2+/+ mice.
| Tissue | Significant genes | Gene ID | FC valuea | Gene name |
|---|---|---|---|---|
| Stroma tissue | ||||
| Ddx60 | 234311 | +2.5 | DEAD (Asp-Glu-Ala-Asp) box polypeptide 60 | |
| Dhx58 | 80861 | +2.2 | DEXH (Asp-Glu-X-His) box polypeptide 58 | |
| F830016B08Rik | 240328 | +3.0 | RIKEN cDNA F830016B08 gene | |
| Gbp6 | 100702 | +2.7 | Guanylate binding protein 6 | |
| Gm12185 | 620913 | +2.7 | Predicted gene 12185 | |
| Gm4951 | 240327 | +2.5 | Predicted gene 4951 | |
| Ifi213 | 623121 | +3.5 | Interferon activated gene 213 | |
| Iigp1 | 60440 | +2.8 | Interferon inducible GTPase 1 | |
| Irf7 | 54123 | +2.6 | Interferon regulatory factor 7 | |
| Ly6a | 110454 | +2.0 | Lymphocyte antigen 6 complex, locus A | |
| Ly6c1 | 17067 | +2.1 | Lymphocyte antigen 6 complex, locus C1 | |
| Mx2 | 17858 | +2.3 | MX dynamin-like GTPase 2 | |
| Oas1a | 246730 | +2.1 | 2′-5′ Oligoadenylate synthetase 1A | |
| Oas1g | 23960 | +2.3 | 2′-5′ Oligoadenylate synthetase 1G | |
| Oas2 | 246728 | +3.8 | 2′-5′ Oligoadenylate synthetase 2 | |
| Oas3 | 246727 | +2.6 | 2′-5′ Oligoadenylate synthetase 3 | |
| Oasl1 | 231655 | +2.3 | 2′-5′ Oligoadenylate synthetase-like 1 | |
| Oasl2 | 23962 | +2.4 | 2′-5′ Oligoadenylate synthetase-like 2 | |
| Sp100 | 20684 | +2.1 | Nuclear antigen Sp100 | |
| Traf6 | 22034 | +2.0 | TNF receptor-associated factor 6 | |
| Xaf1 | 327959 | +2.0 | XIAP associated factor 1 | |
| Zbp1 | 58203 | +2.5 | Z-DNA binding protein 1 | |
| Tumor tissue | ||||
| Iigp1 | 60440 | +2.6 | Interferon inducible GTPase 1 | |
| Cxcl10 | 15945 | +2.5 | Chemokine (C-X-C motif) ligand 10 | |
| Tgtp2 | 100039796 | +2.4 | T-cell specific GTPase 2 |
The pathway matched at P<1.95 E-06.
Average FC value was considered for transcripts with replicate probes on microarray. Positive and negative sign with FC value indicated the regulation of the genes; the positive sign (+) indicates upregulation. FC, fold change.
Statistically significant pathways in stroma compartment, only
Pathway maps related to Cell cycle control was among the most statistically significant matches (P<0.001; Table IV). Almost every cell cycle control-associated gene was downregulated with only few of those genes upregulated. Cell-dependent kinases (Cdks) and minichromosome maintenance complex components (Mcms) formed the majority of decreased transcripts, while increased expression was displayed for RB transcriptional corepressor 1 (RB1), growth arrest and DNA-damage-inducible 45a (Gadd45a), and cyclin-dependent kinase inhibitor 1C (P57) (Cdkn1c) in G1 to S cell cycle pathway (Table V). Thus, decreased cell division in stroma may be a major observation related to reduced tumor growth in EP2-deficient hosts.
Table IV.
Statistically significant pathways in either the tumor- or stroma tissue only. Pathway analysis were performed on transcripts (entities) with a fold change >2.
| Tissue Pathway function Pathway name | No. of pathways | P-value range | Regulation | Matched transcripts/total transcripts in pathway |
|---|---|---|---|---|
| Tumor | ||||
| Intracellular signaling | ||||
| Type II interferon signaling (IFNG) | 1 | 5.59E-10 | ↑100% | 7/34 |
| Immune response | ||||
| Cytokine and inflammatory responses | 1 | 1.25E-05 | ↓50 % ↑50% | 4/30 |
| Inflammatory response pathway | 1 | 5.44E-04 | ↓25% ↑75% | 3/30 |
| Stroma | ||||
| Intracellular signaling | ||||
| MAPK signaling | 2 | 4.37E-05 | ↓29% ↑71% | 35/167 |
| 4.37E-04 | 31/159 | |||
| miR-193a and MVP in colon cancer metastasis | 1 | 0.0179 | ↓100% | 3/7 |
| Metabolic signaling | ||||
| Prostaglandin synthesis | 1 | 3.56E-05 | ↓67% ↑33% | 12/31 |
| Cell cycle control | ||||
| Cell cycle | 2 | 4.95E-22 | ↓83% ↑17% | 45/88 |
| Cell cycle | 1 | 9.21E-22 | 45/88 | |
| G1 to S-cell cycle control | 2 | 2.84E-20 | ↓91% ↑9% | 36/62 |
| Receptor signaling | ||||
| TGFβ receptor signaling | 2 | 7.08E-05 | ↓50% ↑50% | 32/150 |
| EGFR1 signaling | 1 | 0.0013 | ↓30% ↑70% | 32/176 |
| Nuclear receptors | ||||
| PPAR signaling | 1 | 1.69E-04 | ↓18% ↑82% | 20/85 |
| Nuclear receptors signaling | 1 | 0.00133 | ↓5.5% ↑94.4% | 11/38 |
| Nuclear receptor in lipid metabolism and toxicity | 1 | 0.01015 | ↑100% | 8/30 |
↑, upregulated; ↓ downregulated in EP2−/− mice as compared to EP2+/+ mice. MVP, major vault protein; PPAR, peroxisome proliferator-activated receptor; E, exponent.
Table V.
Genes in WikiPathway G1 to S cell cycle (WP413_84705) with an altered expression in stroma tissue from EP2−/− compared to EP2+/+ mice.
| Significant genes | Gene ID | FCa | Known function/name |
|---|---|---|---|
| Ccnd1 | 12443 | −3.1 | Cyclin D1 |
| Ccnd2 | 12444 | −2.0 | Cyclin D2 |
| Ccne1 | 12447 | −2.9 | Cyclin E1 |
| Cdc45 | 12544 | −2.8 | Cell division cycle 45 |
| Cdk1 | 12534 | −4.5 | Cyclin-dependent kinase 1 |
| Cdk2 | 12566 | −2.3 | Cyclin-dependent kinase 2 |
| Cdk4 | 12567 | −2.4 | Cyclin-dependent kinase 4 |
| Cdk6 | 12571 | −2.5 | Cyclin-dependent kinase 6 |
| Cdkn1c | 12577 | +2.6 | Cyclin-dependent kinase inhibitor 1C (P57) |
| Cdkn2a | 12578 | −5.1 | Cyclin-dependent kinase inhibitor 2A |
| E2f5 | 13559 | −2.3 | E2F transcription factor 5 |
| Gadd45a | 13197 | +3.3 | Growth arrest and DNA-damage-inducible 45 alpha |
| Mcm2 | 17216 | −2.4 | Minichromosome maintenance complex component 2 |
| Mcm3 | 17215 | −3.0 | Minichromosome maintenance complex component 3 |
| Mcm4 | 17217 | −2.5 | Minichromosome maintenance complex component 4 |
| Mcm5 | 17218 | −3.4 | Minichromosome maintenance complex component 5 |
| Mcm6 | 17219 | −3.1 | Minichromosome maintenance complex component 6 |
| Mcm7 | 17220 | −2.4 | Minichromosome maintenance complex component 7 |
| Myc | 17869 | −3.1 | Myelocytomatosis oncogene |
| Orc1 | 18392 | −3.9 | Origin recognition complex, subunit 1 |
| Orc2 | 18393 | −2.0 | Origin recognition complex, subunit 2 |
| Orc5 | 26429 | −2.0 | Origin recognition complex, subunit 5 |
| Orc6 | 56452 | −3.2 | Origin recognition complex, subunit 6 |
| Pcna | 18538 | −2.1 | Proliferating cell nuclear antigen |
| Pkmyt1 | 268930 | −2.8 | Protein kinase, membrane associated tyrosine/threonine 1 |
| Pola2 | 18969 | −2.2 | Polymerase (DNA directed), alpha 2 |
| Pole | 18973 | −3.0 | Polymerase (DNA directed), epsilon |
| Pole2 | 18974 | −3.8 | Polymerase (DNA directed), epsilon 2 (p59 subunit) |
| Prim1 | 19075 | −3.6 | DNA primase, p49 subunit |
| Prim2 | 19076 | −3.2 | DNA primase, p58 subunit |
| Rb1 | 19645 | +2.4 | RB transcriptional corepressor 1 |
| Rbl1 | 19650 | −2.5 | Retinoblastoma-like 1 (p107) |
| Rpa3 | 68240 | −2.1 | Replication protein A3 |
| Tfdp1 | 21781 | −2.2 | Transcription factor Dp 1 |
| Trp53 | 22059 | −2.2 | Transformation related protein 53 |
| Wee1 | 22390 | −2.2 | WEE 1 homolog 1 (S. pombe) |
The pathway matched at P<2.84E-20.
Average FC value was considered for transcripts with replicate probes on microarray. The positive and negative sign with FC value indicated the regulation of the genes; a positive sign (+) indicates upregulation and a negative sign (−) indicates the downregulation of gene expression. FC, fold change.
Statistically significant pathways in tumor compartment, only
Fewer numbers of significant pathways were observed in the tumor compartment; however, several pathways were associated with immune activation (Table IV). Gene transcripts in the type II IFN-γ signaling pathway exhibited a uniform pattern of upregulation in EP2-deficient mice (Table VI). IFN-γ (Ifng; +3.8), IFN regulatory factor 8 (Irf8, +2.1), Cxcl9 (+2.5) and CxclL10 (+2.5) demonstrated increased expression in tumor tissue (Table VI), suggesting activation of type II IFN immune response in tumors in the absence of host EP2 receptor signaling.
Table VI.
Genes in WikiPathway type II interferon signalling (IFNG) (WP1253_71753) with altered expression in tumor tissue from EP2−/− compared to EP2+/+ mice.
| Significant gene | Gene ID | FCa | Gene name/function |
|---|---|---|---|
| Ciita | 12265 | +2.3 | Class II transactivator |
| Cxcl10 | 15945 | +2.5 | Chemokine (C-X-C motif) ligand 10 |
| Cxcl9 | 17329 | +2.5 | Chemokine (C-X-C motif) ligand 9 |
| Cybb | 13058 | +2.3 | Cytochrome b-245, beta polypeptide |
| Ifng | 15978 | +3.8 | Interferon gamma |
| Irf8 | 15900 | +2.1 | Interferon regulatory factor 8 |
| Tap1 | 21354 | +3.1 | Transporter 1, ATP-binding cassette, sub-family B (MDR/TAP) |
The pathway matched at P<5,59E-10.
Average FC value was considered for transcripts with replicate probes on microarray. The positive sign (+) with FC values indicates the upregulation of gene expression. FC, fold change.
Discussion
Inflammation with increased production of prostaglandin E2 has been linked to tumor growth in different types of cancer, and several experimental, as well as clinical studies have demonstrated decreased carcinogenesis and cancer progression following provision of NSAIDs (1). Our previous research has also provided evidence of prolonged survival of patients on anti-inflammatory treatment, despite the occurrence of various types of metastatic tumor diseases (2). In addition, a brief 3-day pre-operative NSAID treatment was shown to increase tumor immunity in colorectal tumors, inducing increased antigen presenting cell numbers, including B-cells and macrophages, in tumor epithelial areas, while increased infiltration of cytotoxic CD8 positive T-lymphocytes was observed in both tumor epithelium and stromal tissues (20).
NSAIDs inhibit COX-enzymes, which are upstream events in the production of prostanoids (1). Thus, COX inhibition suppresses formation of PGE2 as well as other prostanoids [Prostaglandin D2 (PGD2), PGI2, PGF2α and thromboxane A2 (TXA2)]. However, the final overall effects of PGE2 are dependent on the expression and combination of receptors on cell surfaces, where PGE2 normally activates four subtype receptors, EP1-4 (21). As previously reported, the absence of host EP1 or EP2 subtype receptors decreased MCG 101 tumor growth, while the absence of host EP3 receptors promoted tumor growth, demonstrating that each receptor may contribute to the sum of PGE2 effects on tumor growth (9,10,16,22). The particular importance of EP2 receptor signaling in tumor growth was further demonstrated in human colorectal tumors, with COX2 and EP2 receptor expression predicting patient survival (4).
The present study expands our previously published research concerning EP2 knockout mice, with the absence of host EP2 subtype receptor-signaling reducing tumor growth, altering tumor gene expression and reducing systemic inflammation (9,10). Traditionally, studies of tumor host-interaction have generally focused on genetic alterations and signaling within tumor cells in the tumor compartment. However, more recently the tumor microenvironment has gained increased attention for its role in tumor progression (23). In the model used in the present study, only host tissues instead of tumor cells, were genetically modified to lack EP2 receptors. Therefore, alterations in tumor growth and intrinsic tumor gene expression should be secondary to changes in the host response. It was thus logical to extend previous findings by analyzing gene expression levels separately in tumor stroma- and tumor compartments, in an attempt to enhance the understanding of prostaglandin crosstalk between stroma and tumor cells.
The downregulation of cell cycle control genes in the tumor-stroma was among the most statistically significant alterations in the present study, implicating reduced cell division in stroma due to the lack of EP2 receptors (Tables IV and V), as confirmed by the reduced Ki-67 protein expression (Fig. 2). Mcm and origin recognition complex genes, required for the initiation of DNA synthesis, as well as cdk 4/6 and cyclin D transcripts were among the decreased transcripts, while RB1 that can act as a brake in the cell cycle, increased (Table V). Type D cyclins are responsive to growth factors, therefore the observed transcript alterations may indicate that lack of EP2 receptor-induced signaling reduced release of growth factors or other mediators needed for ‘tissue remodeling’ of the tumor microenvironment. IL-6 is probably a factor, with a 50% reduction in plasma levels, in EP2 knockouts as demonstrated in our earlier study (10). A similar downregulation of cyclin D has also been found in tumors from colorectal cancer patients receiving a 3-day pre-operative treatment with NSAID (20).
Chemokine signaling through the CXCL9-10-11 and C-X-C chemokine receptor type 3 (CXCR3) axis is involved in the recruitment of immune cells, including NK and cytotoxic T-cells in response to IFN-γ (24). It has been reported that PGE2 may inhibit IFN-γ-induced CXCL9 and CXCL10 secretion from epithelial breast and ovarian cancer cells, and conversely that unselective NSAIDs, including indomethacin, may promote this release (25,26). Chemokines have also been reported as predictors of survival in colorectal cancer (27) and advanced ovarian cancer (25) and are assumed to play a crucial role in angiogenesis (28,29). Accordingly, the knockout of EP2 receptors upregulated transcripts of IFN related genes in the tumor compartment, including CXCL9, CXCL10 and IFN-g (Table VI), in spite of continuous PGE2 production in tumor tissue and increased PGE2 levels in blood, in both EP2 knockouts and wild type controls (9). Thus, the mechanisms involved in prostaglandin regulation of chemokines, appear to not be dependent on reduced PGE2 levels only, supported by previously published findings that selective COX2 inhibitors and unselective NSAIDS had opposite effects on chemokine secretion in breast and ovarian cancer cells (25,26). Nevertheless, an improved immune response may be a significant factor behind reduced tumor growth in EP2 knockouts, further supported by the findings in the present study demonstrating increased infiltration of CD8+ cells in the tumor compartment (Fig. 4). This may have been induced by the altered prostaglandin synthesis in stroma (Table I) or by alternative EP receptor-induced signaling in the absence of host EP2 receptors. EP2 receptors and EP receptors of subtype 3 and 4 have been reported to be expressed on fibroblasts and several types of immune cells, including macrophages, dendritic cells, NK cells and T-cells (23).
CXCL10 has also been reported as a target in the ‘Adar1 editing deficiency response pathway’. Adar1 is an RNA editing enzyme, deaminating adenosine bases to inosine in cellular RNA, adding further complexity in genomic regulation by exerting direct effects on RNA transcripts (30). Therefore, the finding of a consistent upregulation of transcripts in the ‘ADAR 1 editing deficiency response pathway’ is interesting (Table III). This pathway was associated with upregulation of IFN-related gene expression in the stroma, including IFN regulatory factor 7 (Irf7) and IFN activated gene 213 (Ifi213), known to operate as a significant barrier to tumor formation and progression (30). The 2′,5′-oligoadenylate synthases (Oas1, Oas2, Oas3 and Oasl) group of enzymes are mainly known as immune regulators by IFN; however, they have also been reported to control apoptosis and tumorigenesis (31,32).
The expression of PPARα and PPARγ were also observed increased in the EP2 knockout tumor stroma. In previous studies, it has been reported that PPARγ is involved in COX-regulated cell differentiation, apoptosis, inflammation and carcinoma development (33,34), and that ligands for PPARγ may inhibit the induction of the apoptosis of several carcinoma cell types (35,36). Previous experiments evaluating the interactions of EP2 receptor and nuclear receptors have revealed that reduced EP2 expression by PPARγ ligands may be related to inhibition of cellular proliferation (37).
In conclusion, the results of the present study confirmed our previous findings that the absence of host EP2 receptor signaling reduces tumor growth, probably through major alterations in gene expressions (10). However, it was demonstrated that cells in the tumor stroma compartment exhibited quantitatively more, and larger (increased fold change) alterations in gene expression, compared to the tumor tissue compartment. The overall findings, related to processes of crosstalk between cells in stroma and tumor compartments, suggested that both immune cells and conventional stroma cells (macrophages and fibroblasts) may affect tumor growth by PGE2 signaling on EP2 receptors, including dependency of both immune- and growth-related factors (Fig. 6). The observed pathway alterations in the tumor stroma may thus reflect physiological alterations in normal wound healing. It would be important to discover cellular interactions of this type for treatment and control of tumor progression in patients without indications for surgery.
Figure 6.

Proposed mechanism based on findings in the present study. Host EP2 knockout disrupts PGE2 signaling and causes major alterations in tumor stroma with inflammatory cell response to prostaglandins, illustrated by X-factors. This leads to the downregulation of cell cycle control factors in the stroma compartment and increased expression of interferon regulatory factors in the tumor compartment. The upregulation of IFN-γ in tumor cells induces expression of CXCR3 receptors and its ligands CXCL9 and CXCL10 (chemokines), leading to the accumulation of immune cells (CD4, CD8). This may be a major mechanism behind reduced tumor growth in absence of EP2 receptor signaling. EP2, prostaglandin receptors of subtype 2; CXCR3, C-X-C chemokine receptor type 3; CXCL, Chemokine (C-X-C motif) ligand.
Supplementary Material
Acknowledgements
The authors would like to acknowledge the expert technical skills of research engineer Susann Fält (Dr) at NEO, BEA (Bioinformatics and Expression Analysis core facility) at Karolinska Institute who performed microarray hybridization.
Funding Statement
The present study was supported by grants from the Swedish Cancer Society (CAN 2015/400 and 200776 PjF).
Availability of data and materials
The datasets generated and analyzed during the current study are available in the Gene Expression Omnibus (GEO) under accession no. GSE193098.
Authors' contributions
BMI and KL contributed to the conception and design of the experiments. MK, CE, JBF, US and BMI contributed to the acquisition and analysis of the data. CE, US and BMI performed the animal experiments. MK and BMI evaluated the microarrays. MK and BMI performed the immunohistochemical analysis, and MK, JBF and BMI evaluated the results of the immunohistochemical analysis. MK drafted the study, which was revised by BMI and KL. MK and BMI confirm the authenticity of all raw data. All authors critically reviewed, and have read and approved the final version of the manuscript. KL provided funding.
Ethics approval and consent to participate
The Regional Animal Ethics Committee at University of Gothenburg approved the protocol (54–2013).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Wang D, DuBois RN. Role of prostanoids in gastrointestinal cancer. J Clin Invest. 2018;128:2732–2742. doi: 10.1172/JCI97953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lundholm K, Gelin J, Hyltander A, Lönnroth C, Sandström R, Svaninger G, Körner U, Gülich M, Kärrefors I, Norli B, et al. Anti-inflammatory treatment may prolong survival in undernourished patients with metastatic solid tumors. Cancer Res. 1994;54:5602–5606. [PubMed] [Google Scholar]
- 3.Aoki T, Narumiya S. Prostaglandin E2-EP2 signaling as a node of chronic inflammation in the colon tumor microenvironment. Inflamm Regen. 2017;37:4. doi: 10.1186/s41232-017-0036-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gustafsson A, Hansson E, Kressner U, Nordgren S, Andersson M, Wang W, Lönnroth C, Lundholm K. EP1-4 subtype, COX and PPAR gamma receptor expression in colorectal cancer in prediction of disease-specific mortality. Int J Cancer. 2007;121:232–240. doi: 10.1002/ijc.22582. [DOI] [PubMed] [Google Scholar]
- 5.Gustafsson A, Hansson E, Kressner U, Nordgren S, Andersson M, Lönnroth C, Lundholm K. Prostanoid receptor expression in colorectal cancer related to tumor stage, differentiation and progression. Acta Oncol. 2007;46:1107–1112. doi: 10.1080/02841860701403061. [DOI] [PubMed] [Google Scholar]
- 6.Sonoshita M, Takaku K, Sasaki N, Sugimoto Y, Ushikubi F, Narumiya S, Oshima M, Taketo MM. Acceleration of intestinal polyposis through prostaglandin receptor EP2 in Apc(Delta 716) knockout mice. Nat Med. 2001;7:1048–1051. doi: 10.1038/nm0901-1048. [DOI] [PubMed] [Google Scholar]
- 7.Qiu J, Li Q, Bell KA, Yao X, Du Y, Zhang E, Yu JJ, Yu Y, Shi Z, Jiang J. Small-molecule inhibition of prostaglandin E receptor 2 impairs cyclooxygenase-associated malignant glioma growth. Br J Pharmacol. 2019;176:1680–1699. doi: 10.1111/bph.14622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang L, Yamagata N, Yadav R, Brandon S, Courtney RL, Morrow JD, Shyr Y, Boothby M, Joyce S, Carbone DP, Breyer RM. Cancer-associated immunodeficiency and dendritic cell abnormalities mediated by the prostaglandin EP2 receptor. J Clin Invest. 2003;111:727–735. doi: 10.1172/JCI16492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iresjö BM, Wang W, Nilsberth C, Andersson M, Lönnroth C, Smedh U. Food intake, tumor growth, and weight loss in EP2 receptor subtype knockout mice bearing PGE2-producing tumors. Physiol Rep. 2015;3:e12441. doi: 10.14814/phy2.12441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Asting AG, Iresjö BM, Nilsberth C, Smedh U, Lundholm K. Host knockout of E-prostanoid 2 receptors reduces tumor growth and causes major alterations of gene expression in prostaglandin E2-producing tumors. Oncol Lett. 2017;13:476–482. doi: 10.3892/ol.2016.5448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kennedy CR, Zhang Y, Brandon S, Guan Y, Coffee K, Funk CD, Magnuson MA, Oates JA, Breyer MD, Breyer RM. Salt-sensitive hypertension and reduced fertility in mice lacking the prostaglandin EP2 receptor. Nat Med. 1999;5:217–220. doi: 10.1038/5583. [DOI] [PubMed] [Google Scholar]
- 12.Lonnroth C, Svaninger G, Gelin J, Cahlin C, Iresjo B, Cvetkovska E, Edstrom S, Andersson M, Svanberg E, Lundholm K. Effects related to indomethacin prolonged survival and decreased tumor-growth in a mouse-tumor model with cytokine dependent cancer cachexia. Int J Oncol. 1995;7:1405–1413. doi: 10.3892/ijo.7.6.1405. [DOI] [PubMed] [Google Scholar]
- 13.Lundholm K, Edström S, Karlberg I, Ekman L, Scherstén T. Relationship of food intake, body composition, and tumor growth to host metabolism in nongrowing mice with sarcoma. Cancer Res. 1980;40:2516–2522. [PubMed] [Google Scholar]
- 14.Axelsson H, Lönnroth C, Andersson M, Lundholm K. Mechanisms behind COX-1 and COX-2 inhibition of tumor growth in vivo. Int J Oncol. 2010;37:1143–1152. doi: 10.3892/ijo_00000766. [DOI] [PubMed] [Google Scholar]
- 15.Andersson C, Gelin J, Iresjö BM, Lundholm K. Acute-phase proteins in response to tumor growth. J Surg Res. 1993;55:607–614. doi: 10.1006/jsre.1993.1192. [DOI] [PubMed] [Google Scholar]
- 16.Wang W, Andersson M, Lönnroth C, Svanberg E, Lundholm K. Anorexia and cachexia in prostaglandin EP1 and EP3 subtype receptor knockout mice bearing a tumor with high intrinsic PGE2 production and prostaglandin related cachexia. J Exp Clin Cancer Res. 2005;24:99–107. [PubMed] [Google Scholar]
- 17.Svaninger G, Lundberg PA, Lundholm K. Thyroid hormones and experimental cancer cachexia. J Natl Cancer Inst. 1986;77:555–561. [PubMed] [Google Scholar]
- 18.McKinney EF, Cuthbertson I, Harris KM, Smilek DE, Connor C, Manferrari G, Carr EJ, Zamvil SS, Smith KGC. A CD8+ NK cell transcriptomic signature associated with clinical outcome in relapsing remitting multiple sclerosis. Nat Commun. 2021;12:635. doi: 10.1038/s41467-020-20594-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hansen T. Inhibition of T-cell responses by CD4 on an antigen-presenting cell. APMIS. 2006;114:32–38. doi: 10.1111/j.1600-0463.2006.apm_339.x. [DOI] [PubMed] [Google Scholar]
- 20.Lönnroth C, Andersson M, Arvidsson A, Nordgren S, Brevinge H, Lagerstedt K, Lundholm K. Preoperative treatment with a non-steroidal anti-inflammatory drug (NSAID) increases tumor tissue infiltration of seemingly activated immune cells in colorectal cancer. Cancer Immun. 2008;8:5. [PMC free article] [PubMed] [Google Scholar]
- 21.Woodward DF, Jones RL, Narumiya S. International union of basic and clinical pharmacology. LXXXIII: Classification of prostanoid receptors, updating 15 years of progress. Pharmacol Rev. 2011;63:471–538. doi: 10.1124/pr.110.003517. [DOI] [PubMed] [Google Scholar]
- 22.Axelsson H, Lönnroth C, Wang W, Svanberg E, Lundholm K. Cyclooxygenase inhibition in early onset of tumor growth and related angiogenesis evaluated in EP1 and EP3 knockout tumor-bearing mice. Angiogenesis. 2005;8:339–348. doi: 10.1007/s10456-005-9023-8. [DOI] [PubMed] [Google Scholar]
- 23.Mizuno R, Kawada K, Sakai Y. Prostaglandin E2/EP signaling in the tumor microenvironment of colorectal cancer. Int J Mol Sci. 2019;20:6254. doi: 10.3390/ijms20246254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tokunaga R, Zhang W, Naseem M, Puccini A, Berger MD, Soni S, McSkane M, Baba H, Lenz HJ. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation-a target for novel cancer therapy. Cancer Treat Rev. 2018;63:40–47. doi: 10.1016/j.ctrv.2017.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bronger H, Singer J, Windmüller C, Reuning U, Zech D, Delbridge C, Dorn J, Kiechle M, Schmalfeldt B, Schmitt M, Avril S. CXCL9 and CXCL10 predict survival and are regulated by cyclooxygenase inhibition in advanced serous ovarian cancer. Br J Cancer. 2016;115:553–563. doi: 10.1038/bjc.2016.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bronger H, Kraeft S, Schwarz-Boeger U, Cerny C, Stöckel A, Avril S, Kiechle M, Schmitt M. Modulation of CXCR3 ligand secretion by prostaglandin E2 and cyclooxygenase inhibitors in human breast cancer. Breast Cancer Res. 2012;14:R30. doi: 10.1186/bcr3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kistner L, Doll D, Holtorf A, Nitsche U, Janssen KP. Interferon-inducible CXC-chemokines are crucial immune modulators and survival predictors in colorectal cancer. Oncotarget. 2017;8:89998–90012. doi: 10.18632/oncotarget.21286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dufour JH, Dziejman M, Liu MT, Leung JH, Lane TE, Luster AD. IFN-gamma-inducible protein 10 (IP-10; CXCL10)-deficient mice reveal a role for IP-10 in effector T cell generation and trafficking. J Immunol. 2002;168:3195–3204. doi: 10.4049/jimmunol.168.7.3195. [DOI] [PubMed] [Google Scholar]
- 29.Angiolillo AL, Sgadari C, Taub DD, Liao F, Farber JM, Maheshwari S, Kleinman HK, Reaman GH, Tosato G. Human interferon-inducible protein 10 is a potent inhibitor of angiogenesis in vivo. J Exp Med. 1995;182:155–162. doi: 10.1084/jem.182.1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hartner JC, Walkley CR, Lu J, Orkin SH. ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat Immunol. 2009;10:109–115. doi: 10.1038/ni.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Domingo-Gil E, Esteban M. Role of mitochondria in apoptosis induced by the 2-5A system and mechanisms involved. Apoptosis. 2006;11:725–738. doi: 10.1007/s10495-006-5541-0. [DOI] [PubMed] [Google Scholar]
- 32.Castelli JC, Hassel BA, Maran A, Paranjape J, Hewitt JA, Li XL, Hsu YT, Silverman RH, Youle RJ. The role of 2′-5′ oligoadenylate-activated ribonuclease L in apoptosis. Cell Death Differ. 1998;5:313–320. doi: 10.1038/sj.cdd.4400352. [DOI] [PubMed] [Google Scholar]
- 33.Koeffler HP. Peroxisome proliferator-activated receptor gamma and cancers. Clin Cancer Res. 2003;9:1–9. [PubMed] [Google Scholar]
- 34.Houseknecht KL, Cole BM, Steele PJ. Peroxisome proliferator-activated receptor gamma (PPARgamma) and its ligands: A review. Domest Anim Endocrinol. 2002;22:1–23. doi: 10.1016/S0739-7240(01)00117-5. [DOI] [PubMed] [Google Scholar]
- 35.Han S, Inoue H, Flowers LC, Sidell N. Control of COX-2 gene expression through peroxisome proliferator-activated receptor gamma in human cervical cancer cells. Clin Cancer Res. 2003;9:4627–4635. [PubMed] [Google Scholar]
- 36.James SY, Lin F, Kolluri SK, Dawson MI, Zhang XK. Regulation of retinoic acid receptor beta expression by peroxisome proliferator-activated receptor gamma ligands in cancer cells. Cancer Res. 2003;63:3531–3538. [PubMed] [Google Scholar]
- 37.Han S, Roman J. Suppression of prostaglandin E2 receptor subtype EP2 by PPARgamma ligands inhibits human lung carcinoma cell growth. Biochem Biophys Res Commun. 2004;314:1093–1099. doi: 10.1016/j.bbrc.2004.01.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and analyzed during the current study are available in the Gene Expression Omnibus (GEO) under accession no. GSE193098.