

Abstract
Voltage-gated sodium (NaV) channels play crucial roles in a range of (patho)physiological processes. Much interest has arisen within the pharmaceutical industry to pursue these channels as analgesic targets following overwhelming evidence that NaV channel subtypes NaV1.7–NaV1.9 are involved in nociception. More recently, NaV1.1, NaV1.3 and NaV1.6 have also been identified to be involved in pain pathways. Venom-derived disulfide-rich peptide toxins, isolated from spiders and cone snails, have been used extensively as probes to investigate these channels and have attracted much interest as drug leads. However, few peptide-based leads have made it as drugs due to unfavourable physiochemical attributes including poor in vivo pharmacokinetics and limited oral bioavailability. The present work aims to bridge the gap in the development pipeline between drug leads and drug candidates by downsizing these larger venom-derived NaV inhibitors into smaller, more “drug-like” molecules. Here, we use molecular engineering of small cyclic peptides to aid in the determination of what drives subtype selectivity and molecular interactions of these downsized inhibitors across NaV subtypes. We designed a series of small, stable and novel NaV probes displaying NaV subtype selectivity and potency in vitro coupled with potent in vivo analgesic activity, involving yet to be elucidated analgesic pathways in addition to NaV subtype modulation.
Keywords: Cone snail toxin, Spider toxin, Voltage gated sodium channel, Pain, Nociception, Cyclic peptide
1. Introduction
Animal venoms are a natural source of potential drug leads that have received increased attention in the last few decades [1]. Marine gastropods, including cone snails (Conus), are one of the largest single genera of living marine invertebrates. All cone snails are venomous predators and possess a very complex venom apparatus. With more that 500 species of cone snails identified [2], cone snail venoms are viewed as a largely untapped cocktail of biologically active disulfide-rich peptides (conotoxins), increasingly recognized as an emerging source of peptide-based therapeutics [3–5]. An abundance of research has been done on one class of conotoxins, the μ-conotoxins, which target a range of voltage-gated sodium channel (NaV) subtypes. It has been demonstrated that these peptides are well suited for peptide engineering involving structure modifications and amino acid replacements allowing fine-tuning of the selectivity profile and optimisation of the pharmacological properties [3,6]. In general, the μ-conotoxins are rich in basic amino acids, which are responsible for the interaction with the acidic residues of the outer vestibule within the ion-conducting pore region of the NaV channels [3,7]. μ-Conotoxin KIIIA (μ-KIIIA), from Conus kinoshitai, has only 16 amino acid residues, which makes it the smallest μ-conotoxin described to date. KIIIA, together with CnIIIC and SxIIIC, is one of the few μ-conotoxin identified that target the therapeutically relevant NaV1.7 [8,55]. However, μ-KIIIA is quite a promiscuous peptide [9], with its NaV channel subtype preference being NaV1.2 > NaV1.4 > NaV1.6 > NaV1.1 ≈ NaV1.7 > NaV1.3 > NaV1.5 [10]. Previous structure-activity and Ala replacement studies have shown that residues on the α-helix in the C-terminal part of the peptide (Lys7, Trp8, Arg10, Asp11, His12 and Arg14) are functionally important [11–13], with Lys7 of μ-KIIIA being considered a key epitope for both efficacy and potency of μ-KIIIA inhibition [14]. The cryo-electron microscopy (cryo-EM) structure of μ-KIIIA bound to human NaV1.2 confirmed the interaction of μ-KIIIA with the neurotoxin binding site 1 of NaV channels [15–17]. Analysis of this structure revealed the molecular basis for the inhibitory activity of μ-KIIIA and confirmed the key residues for interaction with NaV channels. The overall surface structure of μ-KIIIA is highly complementary to the funnel-shaped cavity formed by the extracellular segments of helix S5 and S6 in domain I–III of the pore of the channel. Specifically, Lys7 with its long side chain is crucial for channel inhibition and the structure shows the peptide binding closely to the selectivity filter of the channel with the positively charged side chain amino group of Lys7 repulsing Na+ ions, thereby preventing ion permeation through the NaV channel [16]. Overall, the results obtained from the structure-function studies, in vivo experiments and the cryo-EM experiments on μ-KIIIA render this peptide as an in-depth characterised template for further peptide engineering.
Phoneutria nigriventer are very aggressive, solitary spiders. Human envenomation involving Phoneutria spiders occurs mainly in Brazil, but sporadic cases in Central America and in neighbouring countries have been reported [18]. The venom of P. nigriventer is a complex mixture of proteins and peptides, including several neurotoxins [19]. The peptide PnTx1 represents 0.45% of the whole venom protein content and it was the first purified and sequenced neurotoxin from P. nigriventer venom [20]. PnTx1 comprises 78 amino acid residues, 14 of which are cysteines for which the disulfide connectivity is unknown (Fig. 1). The recombinant toxin, rPnTx1, inhibits mammalian NaV channel isoforms with the following order of potency: NaV1.2 > NaV1.7 ≈ NaV1.4 ≈ NaV1.3 > NaV1.6 ≈ NaV1.8 with no effect on NaV1.5 [21].
Fig. 1.
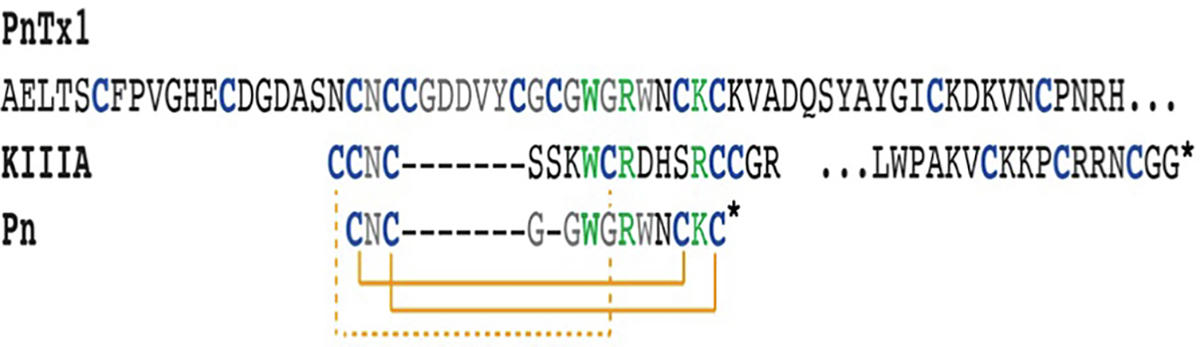
Sequence alignment of PnTx1, μ-KIIIA and the previously designed peptide Pn [24]. Key residues for NaV channel inhibition are shown in green [5,10,14,24] and cysteines are shown in blue. The asterisk indicates amidation. Orange lines (dashed and solid) signify disulfide bonds for KIIIA and Pn. Disulfide connectivity is not known for PnTx1. The sequence for PnTx1 continues after the … (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
μ-KIIIA competes with tetrodotoxin (TTX) for binding site 1, causing a blockage of the NaV channel pore [22]. PnTx1 has been reported to also be a pore blocker, and compete with μ-conotoxin GIIIB, but not with TTX for binding sites [23]. This finding suggests that PnTx1 and μ-conotoxins have different but overlapping binding sites [23]. In addition, as has been reported for μ-KIIIA and μ-conotoxin GIIIA, rPnTx1 does not achieve a complete block of the channel, even at saturating concentrations [21]. μ-KIIIA has also been shown to be analgesic in inflammatory pain models without motor impairment at a dose of 3 nmol [14].
Following identification of common sequence motifs between PnTx1 and μ-KIIIA (Fig. 1), we recently created a hybrid peptide comprising elements from both PnTx1 and μ-KIIIA resulting in the smallest cyclic peptide-based NaV channel inhibitor known to date with demonstrated activity across a range of NaV channel subtypes including NaV1.7 and the NaV1.9 chimera NaV1.9_C4 [24]. Downsizing approaches, such as recently described for the chimeric peptide Pn, could potentially allow for improved NaV subtype selective targeting by reducing cross-subtype reactivity [24], resulting in attractive cyclic peptide drug leads. The pain research community has made a considered judgment that ion channels are key pharmaceutical targets and that venom-derived toxins are a largely untapped source of molecules with potent actions on a range of ion channels. However, due to the sequence and structural similarities between different NaV channel subtypes [25]), it is imperative to tease out the molecular basis for selective inhibition in order to minimise side effects arising from off-target binding.
In the present work, we aimed to further understand the molecular mechanism driving NaV channel binding of these downsized cyclic peptides in order to improve the in vitro activity and selectivity for NaV channels of therapeutic interest. Using Multiple Attribute Positional Scanning (MAPS) to systematically evaluate the chemical space of each amino acid (excluding Cys residues) by replacing them with Lys, Glu or Tyr, we generated a fourth generation of small cyclic peptides which were subsequently assessed for activity and selectivity in vitro using electrophysiology and in vivo using validated rodent pain models. The results obtained provide exciting new insights on NaV subtype selectivity and potency in vitro and analgesic activity in vivo for a series of novel small, cyclic and stable hybrid NaV probes, taking inspiration from NaV active spider and cone snail peptides.
2. Materials and methods
2.1. Peptide synthesis
Peptides were synthesised using Fmoc-solid phase peptide synthesis protocols on a Symphony automated peptide synthesiser (Gyros Protein Technologies, AZ, USA). PnCS1 and analogues were assembled on rinkamide resin to produce an amidated C-terminal or on 2-chorotrityl (2-CTC) to produce an acid C-terminal, at 0.25 mmol scale as previously described [24] using amino-acid side-chain protecting groups Cys(Trt), Glu(tBu), Lys(Boc), Asn(Trt), Arg(Pbf), Trp(Boc). PnCS1Ac and PnCS1AcAm were acetylated in the N-terminal using 10 eq of acetic anhydride with 10 eq of N,N-diisopropylethylamine in dimethyl formamide at room temperature for 2 × 10 min. All peptides were released from the resin and amino acid side chain simultaneously deprotected by incubation with triisopropylsilane (TIPS):H2O:trifluoroacetic acid (TFA) (2:2:96, v/v/v) by stirring for 2.5 h at room temperature. TFA was evaporated under vacuum, and the peptide precipitated with ice-cold diethyl ether. The peptides were dissolved in 50% acetonitrile (ACN)/0.05% TFA and lyophilized. The crude linear peptide was purified using reversed phase high-performance liquid chromatography (RP-HPLC) (0–80% B over 80 min, flow rate 8 mL/min, solvent A; 0.05% TFA, solvent B 90% ACN/0.045% TFA on a Shimadzu Prominence RP-HPLC) and its molecular mass determined using electrospray mass spectrometry (ESI-MS). Purified peptides were oxidized at room temperature in 0.1 M ammonium bicarbonate buffer at pH 8.3 over 24 h. Peptides were >95% pure, as determined using analytical-HPLC, and 1D and 2D NMR 1H spectroscopy was used to confirm the presence of one isomer.
2.2. Nuclear magnetic resonance spectroscopy
Peptides were dissolved in 500 μL of H2O and 50 μL of D2O at concentrations of >1.5 mg/mL, and one- and two-dimensional nuclear magnetic resonance (NMR) spectroscopy experiments including TOCSY and NOESY were acquired at 298 K on a 600 MHz BrukerAvance III spectrometer equipped with a cryoprobe. Spectra were referenced to water at 4.77 ppm.
2.3. Expression of voltage-gated ion channels in Xenopus laevis oocytes
For the expression of NaV channels, including hNaV1.1, rNaV1.2, rNaV1.3, rNaV1.4, hNaV1.5, mNaV1.6, rNaV1.7, rNaV1.8, together with the auxiliary subunits rβ1 and hβ1, in Xenopus oocytes, the linearized plasmids were transcribed using the T7 or SP6 mMESSAGE-mMACHINE transcription kit (Ambion®, Carlsbad, California, USA). Stage V–VI Xenopus laevis oocytes were isolated by partial ovariectomy. The animals were anesthetized by a 15 min submersion in 0.1% tricaine methane sulfonate (Sigma®) solution (pH 7.0). Isolated oocytes were defolliculated with 1.5 mg/mL collagenase. Defolliculated oocytes were injected with 50 nL of cRNA at a concentration of 1 ng/nL using a micro-injector (Drummond Scientific®, Broomall, Pennsylvania, USA). The oocytes were incubated in a solution containing (in mM): NaCl, 96; KCl, 2; CaCl2, 1.8; MgCl2, 2 and HEPES, 5, at pH 7.4, supplemented with 50 mg/L gentamycin sulfate. The use of the frogs was in accordance with license number LA1210239 of the Laboratory of Toxicology & Pharmacology, University of Leuven. All animal care and experimental procedures agreed with the guidelines of ‘European convention for the protection of vertebrate animals used for experimental and other scientific purposes’ (Strasbourg, 18.III.1986).
2.4. Electrophysiological recordings
Two-electrode voltage-clamp recordings were performed at room temperature (18–22 °C) using a Geneclamp 500 amplifier (Molecular Devices®, Downingtown, Pennsylvania, USA) controlled by a pClamp data acquisition system (Axon Instruments®, Union City, California, USA). Whole-cell currents from oocytes were recorded 1–4 days after mRNA injection. Bath solution composition was (in mM): NaCl, 96; KCl, 2; CaCl2, 1.8; MgCl2, 2 and HEPES, 5, at pH 7.4. Voltage and current electrodes were filled with 3 M KCl. Resistances of both electrodes were kept between 0.8 and 1.5 MΩ. The elicited currents were sampled at 20 kHz and filtered at 2 kHz using a four-pole low-pass Bessel filter. Leak subtraction was performed using a – P/4 protocol. For the electrophysiological analysis of toxins, a number of protocols were applied from a holding potential of – 90 mV with a start-to-start interval of 5 s. Na+ current traces were evoked by 100 ms depolarizations to Vmax (the voltage corresponding to maximal Na+ current in control conditions). To assess the concentration response relationships, data were fitted with the Hill equation: y = 100/[1 + (EC50/[toxin])h], where y is the amplitude of the toxin-induced effect, EC50 is the toxin concentration at half maximal efficacy [toxin], is the toxin concentration and h is the Hill coefficient. All data are presented as mean ± standard deviation (SD) of at least 5 independent experiments (n ≥ 5). All data were tested for normality using a D’Agustino Pearson omnibus normality test. All data were tested for statistically significance using Bonferroni test or Dunn’s test. Data following a Gaussian distribution were analyzed for significance using one-way ANOVA. Non-parametric data were analyzed for significance using the Kruskal–Wallis test. Differences were considered significant if the probability that their difference stemmed from chance was 5% (p < 0.05). Data were analyzed using pClamp Clampfit 10.0 (Molecular Devices®, Downingtown, Pennsylvania, USA) and Origin 7.5 software (Originlab®, Northampton, Massachusetts, USA).
2.5. In vivo NaV1.7 target engagement using OD1
To assess the in vivo effect of peptide analogues, an OD1 induced model of NaV1.7 target engagement was used as previously described [26]. Male C57BL/6J mice aged 8 weeks (20–25 g) were housed in 12 h light-dark cycle with access to food and water ad libitum. Briefly, the NaV1.7 selective α-scorpion toxin OD1 (300 nM) was diluted in phosphate-buffered saline (PBS) containing 0.1% (w/v) BSA. Under brief and light (1.5% (v/v) isoflurane) anesthesia, mice were administered vehicle (0.1% (w/v) BSA in PBS) or OD1 (40 μL of 300 nM) via shallow intraplantar injection into the dorsal hind paw. Animals received OD1 alone (control, n = 5) or were co-administered OD1 with PnCS1, Pn [W4K], Pn[R6E] or Pn[W7Y] (10 μM or 100 μM, n = 5). Following injection, mice were allowed to recover in polyvinyl boxes and were video recorded for 30 min post-injection. Spontaneous nocifensive behaviours (paw lifts, licks, shakes and flinches) were counted by a blinded observer.
Animal ethics approval was obtained from The University of Queensland Animal ethics committee. All experiments were conducted in accordance with local and national regulations and the International Associations for the Study of Pain Guidelines for the Use of Animals in Research.
2.6. Algesimetric method
Male Swiss mice, weighing between 30 g and 40 g, from the Bioterism Center of Federal University of Minas Gerais (CEBIO-ICB/UFMG, Brazil), were used in all experiments. The animals were placed in standard cages, with free access to water and food. They were housed in a temperature-controlled room (24 ± 2 °C) with a 12 h light/dark cycle. After the experimental procedures, the animals were euthanized with 300 mg/kg of ketamine and 15 mg/kg of xylazine, both Sigma-Aldrich, USA. Hyperalgesia was induced using subcutaneous injection of prostaglandin E2 (PGE2; 2 μg) into the plantar surface of the hind paw (intraplantar injection). PGE2 (Sigma, EUA) was diluted in ethanol 10% whereas the peptides PnCS1, PnCS1[W4K], PnCS1[R6E] and PnCS1 [W7Y] were dissolved in sterile physiological solution (saline). All these drugs were injected into the right plantar surface of the paw in a volume of 20 μL per paw. The nociceptive threshold was measured according to Randall and Selitto [27] and adapted to mice by Kawabata et al. [28], using the mechanical paw pressure test. An analgesy-meter was used (UgoBasile, Italy) with a cone-shaped paw-presser with a rounded tip, which applies a linearly increasing force to the hind paw. The weight in grams (g) required to elicit the nociceptive response of paw withdrawal was determined as the nociceptive threshold. The nociceptive threshold was expressed in grams and it was determined in the right hind paw according to the average of three consecutive measures recorded before (0 time) and after PGE2 injection (3 h). A cut-off value of 160 g was used to reduce the possibility of damage to the paws.
To evaluate the temporal development of the dose response curve, PGE2 (2 μg) was injected into the right hind paw of the animals and the peptides were given 150 min after the local injection of PGE2 (peak of PGE2 hyperalgesia). The nociceptive threshold measurements were recorded every 5 min, from 180 to 240 min. To exclude systemic effect, PGE2 was injected into both hind paws, whereas each peptide was injected only into the right paw 150 min after PGE2 injection. The contralateral paw received vehicle (saline). Nociceptive threshold was measured in both hind paws in two moments, before any injection (time 0 min) and at 180 min after PGE2 injection, in such a way that the peak of the antinociceptive action of peptides and the peak of hyperalgesic action of PGE2 occur simultaneously at the time of measurements. The difference between these values was expressed as Δ of the nociceptive threshold. The results were shown as the mean ± SD and the data were statistically analysed using analysis of variance followed by Bonferroni test. Statistically significance was set at p < 0.05.
All animal care and experimental protocols were approved by the local Ethics Committee on the Use of Animals (CEUA) of UFMG and were in accordance with ARRIVE guidelines [29,30]. Efforts were made to minimize suffering and reduce the number of animals used in the experiments.
3. Results
Following the identification of PnCS1 as a promising scaffold for further NaV inhibitor design [24], we carried out a fourth round of structure-activity relationship studies using a Multiple Attribute Positional Scanning (MAPS) approach [31]. This approach systematically replaces every non-cysteine residue with an amino acid possessing a variety of chemical attributes. To cover a broad range of chemical space, we replaced each amino acid with alanine, lysine or glutamic acid, and replaced tryptophan residues with tyrosine. PnCS1 is cyclized via a disulfide bond and like μ-KIIIA, is amidated in the C-terminal. To investigate the importance of N- and C-terminal modifications upon NaV binding, PnCS1 with or without N-terminal acetylation and/or C-terminal amidation were also synthesised. This design cycle resulted in a series of 28 cyclic peptides (Table 1). All peptides were successfully assembled in high yield using solid phase peptide synthesis and cyclised in solution. Using NMR spectroscopy, the presence of one conformation was established prior to the peptides being subjected to in vitro and in vivo pharmacological evaluation.
Table 1.
Fourth generation MAPS mutants based on PnCS1.
| Peptide | Sequence | Peptide | Sequence |
|---|---|---|---|
|
| |||
| PnCS1 | CRRWARWNRC* | Glu mutants | |
| Ala mutants | PnCS1[R2E] | CERWARWNRC* | |
| PnCS1[R2A] | CARWARWNRC* | PnCS1[R3E] | CREWARWNRC* |
| PnCS1[R3A] | CRAWARWNRC* | PnCS1[W4E] | CRREARWNRC* |
| PnCS1[W4A] | CRRAARWNRC* | PnCS1[A5E] | CRRWERWNRC* |
| PnCS1[R6A] | CRRWAAWNRC* | PnCS1[R6E] | CRRWAEWNRC* |
| PnCS1[W7A] | CRRWARANRC* | PnCS1[W7E] | CRRWARENRC* |
| PnCS1[N8A] | CRRWARWARC* | PnCS1[N8E] | CRRWARWERC* |
| PnCS1[R9A] | CRRWARWNAC* | PnCS1[R9K] | CRRWARWNEC* |
| Lys mutants | Tyr mutants | ||
| PnCS1[R2K] | CKRWARWNRC* | PnCS1[W4Y] | CRRYARWNRC* |
| PnCS1[R3K] | CRKWARWNRC* | PnCS1[W7Y] | CRRWARYNRC* |
| PnCS1[W4K] | CRRKARWNRC* | Acid C-terminal | |
| PnCS1[A5K] | CRRWKRWNRC* | PnCS1DeAm | CRRWARWNRC |
| PnCS1[R6K] | CRRWAKWNRC* | Acetylation of N-terminal | |
| PnCS1[W7K] | CRRWARKNRC* | PnCS1AcAm | Ac-CRRWARWNRC* |
| PnCS1[N8K] | CRRWARWKRC* | Acetylation of N-terminal and acid C-terminal | |
| PnCS1[R9K] | CRRWARWNKC* | PnCS1AcDeAM | Ac-CRRWARWNRC |
All peptides were N- to C-terminal cyclised via a disulfide bond. Mutations are underlined in bold; Ala-mutants in red, Lys-mutants in blue, Glu-mutants in green, Tyr-mutations in brown and acetylation in black.
- amidated C-terminal, Ac - acetylated N-terminal.
3.1. Electrophysiological characterisation of PnCS1 mutants
Two-electrode voltage clamp electrophysiology on oocytes expressing NaV subtypes was used to evaluate the activity of the 28 MAPS analogues and compared to the activity of the parent peptide PnCS1. Initially peptides were evaluated against NaV1.2, NaV1.4, NaV1.5, NaV1.6 and NaV1.8 for their % of inhibition at 100 μM and IC50 (Table 2, Hill slope coefficients in Table 3). As was observed for PnCS1, none of the MAPS mutants showed any activity at NaV1.8 and were not analysed further. None of the 28 mutants assayed were able to produce 100% inhibition (0–96%), even at 100 μM, across the subtypes investigated. The Ala-mutants and the Lys-mutants showed the highest % inhibition across NaV1.2, NaV1.4, NaV1.5 and NaV1.6 (51–96%), with analogues [N8A] and [R3K] on NaV1.2, [R2A], [R6A], [W7A] and [N8K] on NaV1.4 and [R6A] on NaV1.6 displaying inhibition of more than 95%. The Glu analogues displayed inhibition of 0–73%, however many analogues showed less than 50% inhibition. Of the Trp-mutants, analogue [W4Y] displayed only 33–43% inhibition at 100 μM, whereas [W7Y] showed 73–93% inhibition and IC50 values between 0.7 and 11.7 μM. Analogues with a modified N- or C-terminal also displayed a reduced level of inhibition, whereas the analogue with an acylated N-terminus as well as an amidated C-terminus showed no significant activity. Removing the amidated C-terminal led to a reduction in inhibition across NaV1.2, NaV1.4, NaV1.5 and NaV1.6 (10–43%) and acylation of the N-terminal while retaining the amidated C-terminal also resulted in a complete loss of inhibition (0%) across NaV1.2, NaV1.4, NaV1.5 and NaV1.6.
Table 2.
Potencya, subtype selectivity and maximum inhibitionb for the fourth generation of PnCS1 MAPS peptide analogues across NaV subtypes assessed using two-electrode voltage-clamp electrophysiology.
| Peptide | NaV1.2 | NaV1.4 | NaV1.5 | NaV1.6 | NaV1.8 |
|---|---|---|---|---|---|
|
| |||||
| PnCS1 | 1.0 ± 0.3 (97) | 0.6 ± 0.3 (94) | 2.8 ± 0.5 (94) | 0.7 ± 0.3 (96) | >100 (0) |
| PnCS1[R2A] | 2.9 ± 0.5a (84)b | 1.4 ± 0.4 (96) | 1.7 ± 0.7 (68) | 1.6 ± 0.5 (71) | >100 (0) |
| PnCS1[R3A] | 1.8 ± 0.3 (86) | 1.6 ± 0.5 (89) | >100 (51) | 1.9 ± 0.4 (87) | >100 (0) |
| PnCS1 [W4A] | 0.8 ± 0.3 (89) | 0.7 ± 0.3 (95) | 3.4 ± 0.2 (82) | 1.2 ± 0.3 (76) | >100 (0) |
| PnCS1[R6A] | 9.2 ± 1.1 (73) | 0.5 ± 0.2 (90) | 7.6 ± 2.2 (78) | 0.9 ± 0.2 (96) | >100 (0) |
| PnCS1 [W7A] | 0.8 ± 0.3 (92) | 1.1 ± 0.4 (96) | 6.8 ± 1.3 (78) | 11.4 ± 3.6 (57) | >100 (0) |
| PnCS1[N8A] | 0.9 ± 0.3 (96) | 0.9 ± 0.4 (94) | 2.2 ± 0.7 (92) | 0.8 ± 0.3 (89) | >100 (0) |
| PnCS1[R9A] | 6.7 ± 1.2 (80) | 1.1 ± 0.3 (86) | 9.2 ± 4.6 (72) | 0.9 ± 0.4 (79) | >100 (0) |
| PnCS1[R2K] | 1.1 ± 0.5 (92) | 0.8 ± 0.3 (93) | 2.5 ± 0.5 (82) | 0.6 ± 0.2 (85) | >100 (0) |
| PnCS1[R3K] | 0.8 ± 0.3 (96) | 0.6 ± 0.3 (92) | 2.7 ± 0.6 (94) | 0.8 ± 0.3 (86) | >100 (0) |
| PnCS1 [W4K] | 0.5 ± 0.4 (75) | 0.7 ± 0.4 (94) | 2.7 ± 0.5 (89) | 0.7 ± 0.4 (92) | >100 (0) |
| PnCS1[A5K] | 1.2 ± 0.4 (84) | 1.9 ± 0.8 (91) | 2.1 ± 0.2 (92) | 0.7 ± 0.3 (93) | >100 (0) |
| PnCS1[R6K] | 8.7 ± 2.4 (67) | 0.6 ± 0.4 (94) | 3.3 ± 1.5 (89) | 0.9 ± 0.3 (88) | >100 (0) |
| PnCS1 [W7K] | 2.8 ± 0.5 (71) | 1.7 ± 0.5 (87) | 3.0 ± 0.7 (90) | 8.2 ± 0.6 (82) | >100 (0) |
| PnCS1[N8K] | 4.4 ± 0.8 (81) | 0.9 ± 0.3 (96) | 2.8 ± 0.5 (86) | 0.5 ± 0.2 (92) | >100 (0) |
| PnCS1[R9K] | 10.2 ± 4.3 (62) | 6.1 ± 2.1 (66) | 9.2 ± 0.6 (67) | 6.7 ± 0.8 (72) | >100 (0) |
| PnCS1[R2E] | >100 (41) | >100 (11) | >100 (27) | >100 (5) | >100 (0) |
| PnCS1[R3E] | >100 (52) | >100 (36) | >100 (21) | >100 (24) | >100 (0) |
| PnCS1 [W4E] | 13.2 ± 5.2 (72) | 12.8 ± 4.1 (59) | >100 (34) | 14.8 ± 3.9 (67) | >100 (0) |
| PnCS1[A5E] | 1.4 ± 0.2 (81) | >100 (35) | 8.8 ± 3.7 (73) | 1.8 ± 0.5 (82) | >100 (0) |
| PnCS1[R6E] | >100 (49) | >100 (0) | >100 (19) | >100 (41) | >100 (0) |
| PnCS1 [W7E] | >100 (21) | >100 (42) | >100 (33) | >100 (27) | >100 (0) |
| PnCS1[N8E] | >100 (47) | >100 (53) | >100 (44) | >100 (27) | >100 (0) |
| PnCS1[R9E] | >100 (49) | >100 (0) | >100 (40) | >100 (11) | >100 (0) |
| PnCS1 [W4Y] | >100 (39) | >100 (33) | >100 (43) | >100 (42) | >100 (0) |
| PnCS1 [W7Y] | 7.4 ± 4.2 (76) | 7.8 ± 3.3 (73) | 11.7 ± 4.1 (75) | 0.7 ± 0.3 (93) | >100 (0) |
| PnCS1AcAm | >100 (0) | >100 (0) | >100 (0) | >100 (0) | >100 (0) |
| PnCS1DeAm | >100 (34) | >100 (43) | >100 (10) | >100 (24) | >100 (0) |
| PnCS1Ac | >100 (0) | >100 (0) | >100 (0) | >100 (0) | >100 (0) |
IC50 values in μM for n > 6, ± SD
Maximum % inhibition of peptides at 100 μM indicated in brackets with % inhibition ≥ 95 indicated in bold.
Table 3.
Hill coefficients for the concentration-response curves constructed to obtain the IC50 values in table 2.
| Peptide | NaV1.2 | NaV1.4 | NaV1.5 | NaV1.6 | NaV1.8 |
|---|---|---|---|---|---|
|
| |||||
| PnCS1 | 0.9 ± 0.2 | 1.2 ± 0.1 | 0.9 ± 0.4 | 0.9 ± 0.3 | / |
| PnCS1[R2A] | 0.9 ± 0.2 | 1.1 ± 0.2 | 0.7 ± 0.2 | 0.9 ± 0.4 | / |
| PnCS1[R3A] | 1.0 ± 0.2 | 0.8 ± 0.2 | / | 1.1 ± 0.3 | / |
| PnCS1[W4A] | 0.8 ± 0.2 | 0.9 ± 0.3 | 0.6 ± 0.2 | 0.8 ± 0.2 | / |
| PnCS1[R6A] | 0.5 ± 0.3 | 0.9 ± 0.2 | 0.9 ± 0.4 | 1.0 ± 0.2 | / |
| PnCS1[W7A] | 0.6 ± 0.2 | 0.7 ± 0.1 | 0.7 ± 0.2 | 0.9 ± 0.2 | / |
| PnCS1[N8A] | 1.0 ± 0.1 | 0.9 ± 0.4 | 1.0 ± 0.1 | 1.1 ± 0.2 | / |
| PnCS1[R9A] | 0.8 ± 0.4 | 1.0 ± 0.2 | 0.7 ± 0.2 | 1.2 ± 0.3 | / |
| PnCS1[R2K] | 1.3 ± 0.4 | 1.2 ± 0.1 | 0.6 ± 0.3 | 0.8 ± 0.3 | / |
| PnCS1[R3K] | 0.9 ± 0.2 | 0.9 ± 0.2 | 0.9 ± 0.3 | 1.2 ± 0.3 | / |
| PnCS1[W4K] | 0.6 ± 0.3 | 0.9 ± 0.2 | 1.0 ± 0.1 | 1.0 ± 0.2 | / |
| PnCS1[A5K] | 1.1 ± 0.1 | 0.8 ± 0.3 | 0.7 ± 0.3 | 0.6 ± 0.2 | / |
| PnCS1[R6K] | 0.7 ± 0.2 | 0.7 ± 0.2 | 1.1 ± 0.3 | 1.1 ± 0.1 | / |
| PnCS1[W7K] | 0.8 ± 0.1 | 0.9 ± 0.3 | 1.1 ± 0.2 | 0.7 ± 0.2 | / |
| PnCS1[N8K] | 0.7 ± 0.2 | 0.8 ± 0.2 | 0.7 ± 0.4 | 0.8 ± 0.4 | / |
| PnCS1[R9K] | 0.6 ± 0.2 | 0.7 ± 0.1 | 0.8 ± 0.3 | 0.9 ± 0.3 | / |
| PnCS1[R2E] | / | / | / | / | / |
| PnCS1[R3E] | / | / | / | / | / |
| PnCS1[W4E] | 0.9 ± 0.3 | 1.3 ± 0.4 | / | 1.4 ± 0.3 | / |
| PnCS1[A5E] | 1.2 ± 0.2 | / | 0.7 ± 0.2 | 1.8 ± 0.5 | / |
| PnCS1[R6E] | / | / | / | / | / |
| PnCS1[W7E] | / | / | / | / | / |
| PnCS1[N8E] | / | / | / | / | / |
| PnCS1[R9E] | / | / | / | / | / |
| PnCS1[W4Y] | / | / | / | / | / |
| PnCS1[W7Y] | 0.6 ± 0.2 | 0.7 ± 0.2 | 0.7 ± 0.3 | 0.6 ± 0.2 | / |
| PnCS1AcAm | / | / | / | / | / |
| PnCS1DeAm | / | / | / | / | / |
| PnCS1Ac | / | / | / | / | / |
Mutants in the Ala- and Lys-series of analogues were the most potent amongst the series of 28 peptides investigated compared to PnCS1. Across subtype NaV1.2, PnCS1 analogue [W4K] was the most potent peptide with an significant lower IC50 (0.5 ± 0.4 μM) compared to PnCS1 (IC50 of PnCS1 1.0 ± 0.3 μM), with [W4A], [W7A], [N8A], [R2K], [R3K], [W4K] and [A5K] being equipotent to PnCS1. Across NaV1.4, no peptide displayed improved activity compared to PnCS1, but several peptides including [W4A], [R6A], [R2K], [R3K], [W4K], and [R6K] were equipotent with PnCS1 (IC50 of PnCS1 0.6 ± 0.3 μM). Similarly, several analogues were equipotent with PnCS1 across NaV1.6 (IC50 of PnCS1 0.7 ± 0.3 μM), including [R6A], [N8A], [R9A], [R2K], [R3K], [W4K], [A5K], [R6K] and [N8K] with [R2K] and [N8K] being the most potent at 0.6 ± 0.2 μM and 0.5 ± 0.2 μM, respectively, but none were significantly more potent than PnCS1. All Ala- and Lys-mutants displayed reduced potency at NaV1.5 (1–3.3 fold) compared to PnCS1 with [R3A] experiencing reduced inhibition (51%) and no measurable IC50.
Although all Glu-analogue peptides produced inhibition across subtypes NaV1.2, NaV1.4, NaV1.5 and NaV1.6, only [W4E] and [A5E] were sufficiently potent to measure IC50’s across subtypes NaV1.2, NaV1.4, and NaV1.6, and NaV1.2, NaV1.5 and NaV1.6, respectively. [A5E] was 1.4- and 2.6-fold less potent at NaV1.2 and NaV1.6, respectively, with the two peptides displaying 3.1–21.1-fold loss in potency across the other subtypes. Of the Tyr-mutants, an IC50 [W4Y] could not be determined due to lack of potency, whereas [W7Y] showed 7.4-, 13-, 2.7- and no loss of potency across NaV1.2, NaV1.4, NaV1.5 and NaV1.6, respectively. Due to lack of potency for the N- and C-terminally modified peptides, IC50’s were not measurable.
3.2. Electrophysiological characterisation of PnCS1, PnCS1[W4K], PnCS1[R6E] and PnCS1[W7Y] on NaV channels involved in pain pathways
In addition to the 28 analogues being evaluated across subtypes NaV1.2, NaV1.4, NaV1.5, NaV1.6, and NaV1.8, three analogues and PnCS1 were also evaluated for inhibition and potency across the validated pain target subtypes NaV1.1, NaV1.3 and NaV1.7, prior to in vivo studies. The three peptides chosen were: [W4K] for being the most active peptide across subtypes NaV1.2, NaV1.4, NaV1.5 and NaV1.6; [R6E], for not displaying activity across any NaV subtype tested, and [W7Y] for displaying selectivity for NaV1.6 across NaV1.2, NaV1.4, NaV1.5. [W4K] was twice as potent as PnCS1 on NaV1.1, equipotent across NaV1.4 and 8.2-fold less potent on NaV1.7 compared to PnCS1 inhibiting NaV channels with the following preference: NaV1.1 ≈ NaV1.6 ≈ NaV1.7 > NaV1.3 > NaV1.8 (Table 4, Fig. 2). [W7Y] was 2.8-, 3.7- and 9.1-fold less potent across NaV1.1, NaV1.3 and NaV1.7, respectively, displaying a preference of NaV1.6 > NaV1.1 > NaV1.3 > NaV1.7 (Table 4). As observed for NaV1.2, NaV1.4, NaV1.5 and NaV1.6, [R6E] did not show any activity on subtypes NaV1.1, NaV1.3 or NaV1.7 (Table 4).
Table 4.
IC50 values of PnCS1, PnCS1[W4K], PnCS1[R6E] and PnCS1[W7Y] on NaV subtypes involved in pain.
| IC50 (μM) | PnCS1 | PnCS1[W4K] | PnCS1[R6E] | PnCS1[W7Y] |
|---|---|---|---|---|
|
| ||||
| NaV1.1 | 0.8 ± 0.3 a | 0.4 ± 0.2 | >100 | 2.2 ± 0.4 |
| NaV1.3 | 1.1 ± 0.4 | 1.3 ± 0.4 | >100 | 4.1 ± 0.8 |
| NaV1.6 | 0.7 ± 0.3 | 0.7 ± 0.4 | >100 | 0.7 ± 0.3 |
| NaV1.7 | 0.9 ± 0.2 | 7.4 ± 0.5 | > 100 | 8.2 ± 0.3 |
IC50 values for n > 5, ±SD.
Fig. 2.
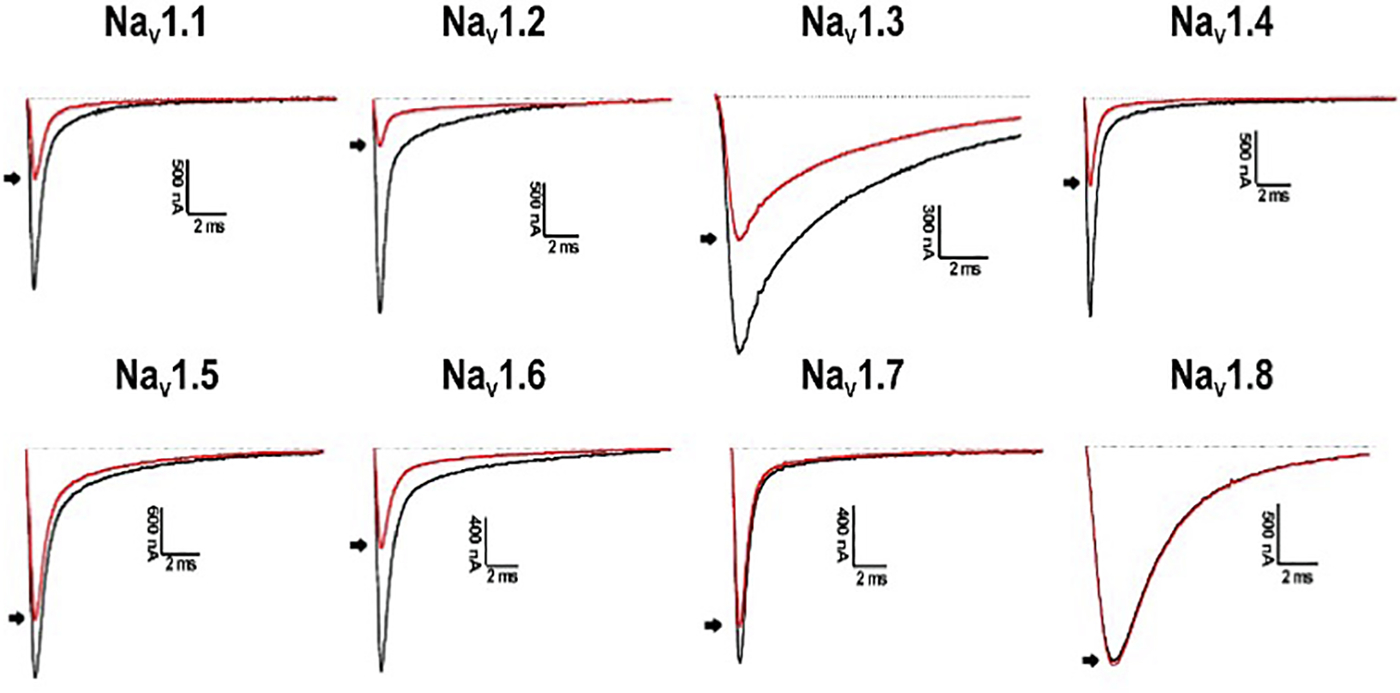
Electrophysiological characterization of PnCS1[W4K] across NaV channel subtypes NaV1.1–1.8. Representative whole-cell current traces in control (black) and toxin (red) conditions are shown. The dotted line indicates the zero-current level. The arrow marks steady-state current traces after application of 1 μM peptide. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.3. Activity of PnCS1, PnCS1[W4K], PnCS1[R6E] and PnCS1[W7Y] in a murine model of NaV1.7 mediated nociception
Despite modest activity and lack of selectivity, we were interested in examining whether a series of our analogues were efficacious in vivo. We therefore examined the effects of PnCS1, PnCS1[W4K], PnCS1[R6E] and PnCS1[W7Y] in a mouse model of NaV1.7-mediated nociception after intraplantar administration of OD1 (300 nM), an α-scorpion toxin that selectively impairs inactivation and enhances current from NaV1.7 [26,32]. OD1 was injected with or without 10 μM or 100 μM of peptide and nocifensive pain behavior was monitored by a blinded observer. At 10 μM, there was a significant reduction in nocifensive behaviour for PnCS1 and PnCS1[R6E] (nocifensive behaviour in % of OD1 control 49.8 ± 2.9%, and 47.6 ± 11.5%, respectively, p < 0.05) compared to the control (OD1; 100 ± 9.4%) whereas no significant difference in nocifensive behaviour was observed for PnCS1[W4K] and [PnCS1[W7Y] (nocifensive behaviour in % of OD1 control 72.6 ± 10.5%, and 60.1 ± 18.5%, respectively) at the same dose (Fig. 3). At a higher dose of 100 μM, all four peptides partially reduced pain behavior (PnCS1: 50.3 ± 12.7%; PnCS1[W4K]: 46.9 ± 10.6%; PnCS1[R6E]: 37.2 ± 9.7%; [PnCS1 [W7Y]: 35.7 ± 8.2%) to a similar degree as PnCs1 at 10 μM (Fig. 3).
Fig. 3.
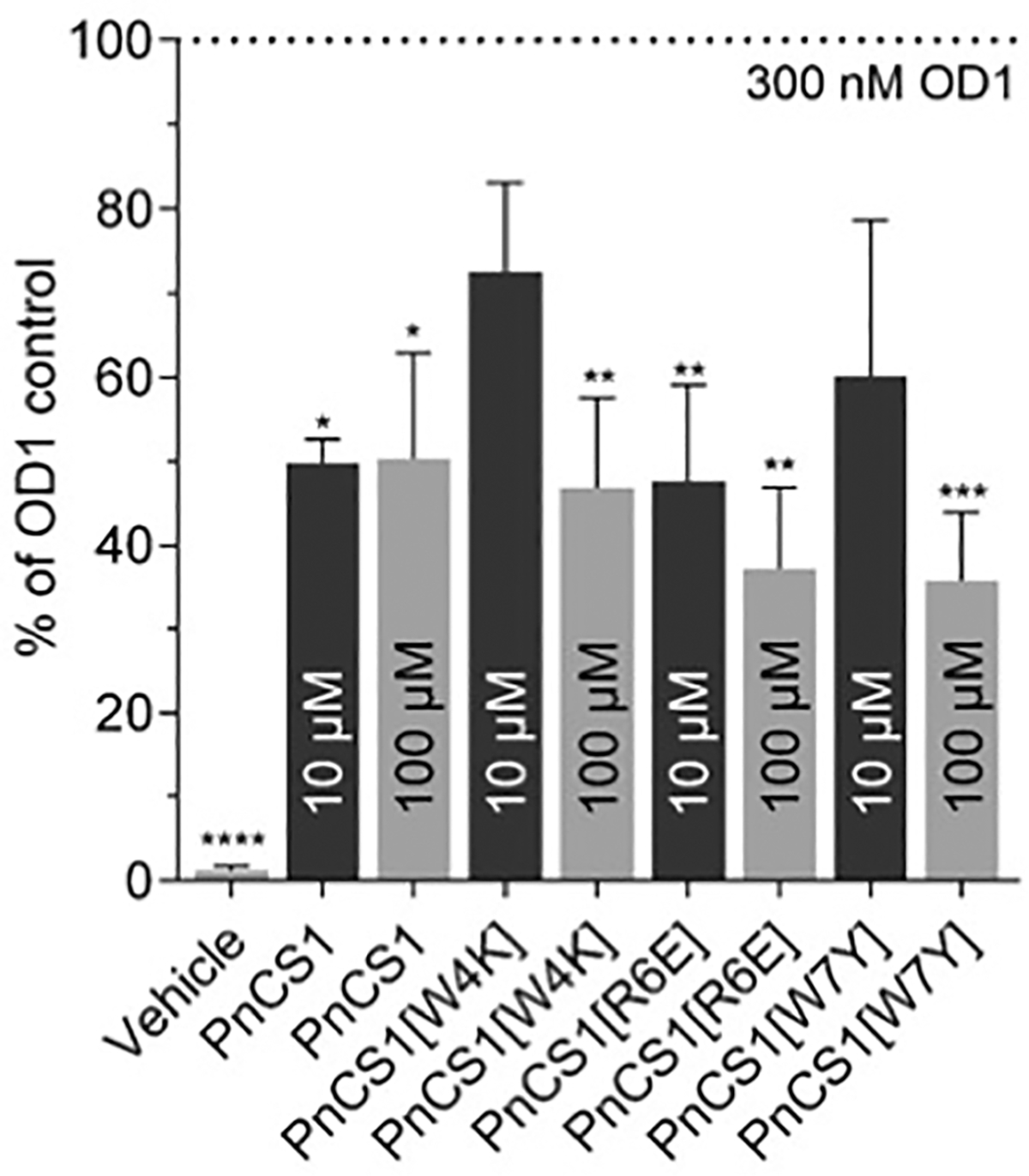
Antinocicpetive effects of PnCs1, PnCS1[W4K], PnCS1[R6E] and PnCS1 [W7Y] in a mouse model of NaV1.7 mediated nociception. Local intraplantar injection of 10 μM PnCs1 and PnCs1[R6E] partially reduced OD1 induced pain behaviours (n = 5 per group) while intraplantar injection of 100 μM of all four peptides reduced pain behaviours (n = 5 per group). Vehicle administration did not cause significant pain (1.3 ± 0.5% of OD1 control). Data are expressed as mean ± SD. Statistical significance was determined using one-way ANOVA with Dunnett’s post-test; *, p < 0.05; **, p < 0.01, ***, p < 0.001 compared to OD1 control.
3.4. In vivo activity of PnCS1 and selected mutants using a PGE2 model of nociception
The PnCS1, PnCS1[W4K], PnCS1[R6E] and PnCS1[W7Y] were evaluated separately, but similar results were observed for the four different peptides. The intraplantar injection of the four peptides, at the doses of 30 μg/paw, 15 μg/paw and 7.5 μg/paw, at the third hour after injection of PGE2 (2 μg/paw), induced a significant antinociceptive effect when compared to the control group (ethanol 10%). Increase in the nociceptive threshold was observed 5 min after peptide injection and the peak of action was noticed 30 min following injection (Fig. 4A–D). An intermediary antinociception was observed with the 15 μg/paw and 7.5 μg/paw doses and a maximum antinociception was noticed with the 30 μg/paw dose. Compared to the group treated only with prostaglandin (PGE2), no nociception was observed with the intraplantar injection of ethanol 10% (vehicle of prostaglandin) and saline (peptide vehicle). One hour following the peptide injection, the peptide treated and control groups presented similar nociceptive thresholds.
Fig. 4.
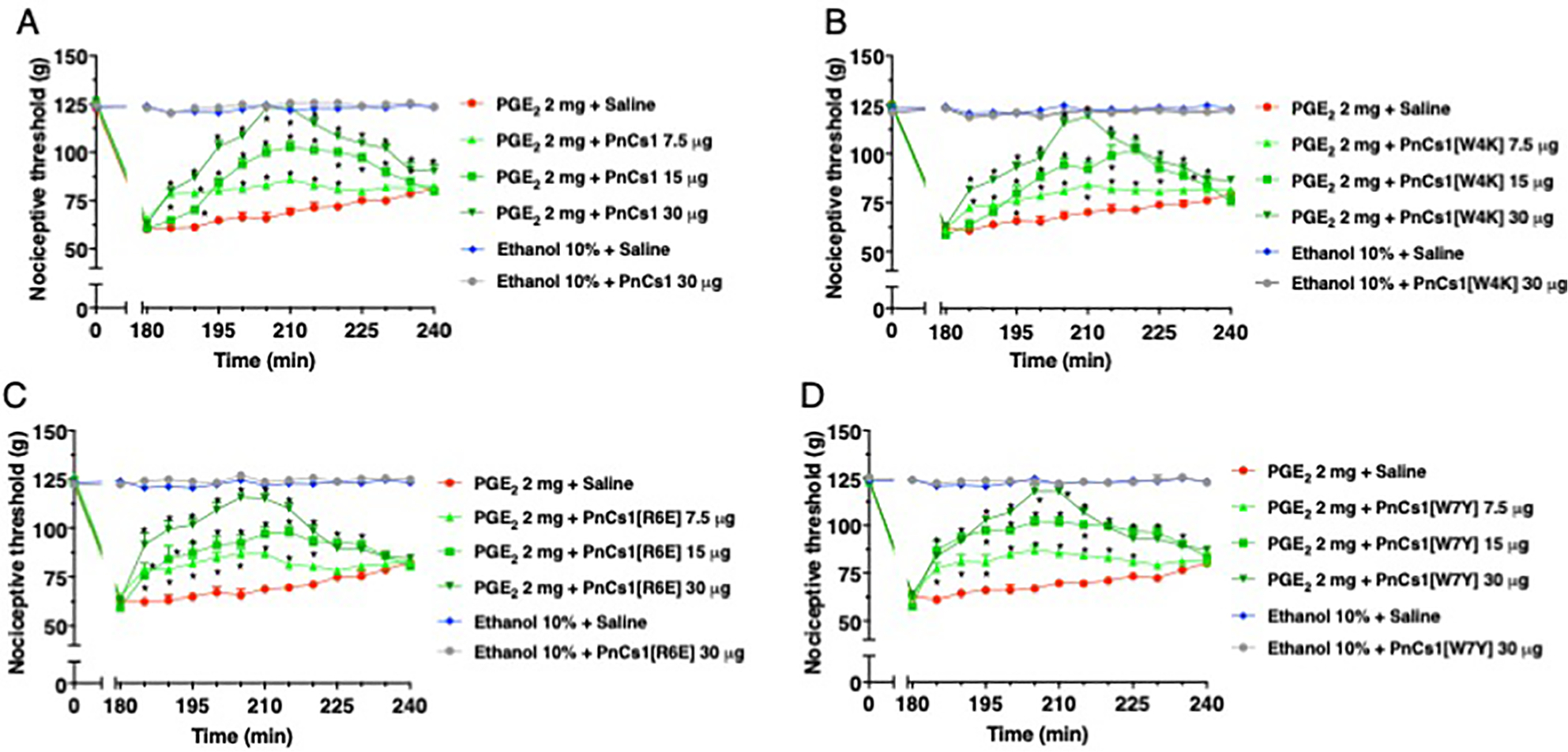
Antinociceptive effect of (A) PnCS1, (B) PnCS1[W4K], (C) PnCS1[R6E] and (D) PnCS1[W7Y] upon intraplantar injection in mice.
In order to exclude a possible systemic effect, PGE2 (2 μg/paw) was injected, at time zero, into both hind paws, whereas the peptides (30 μg/ paw) were given after 150 min, only in the right hind paw and the vehicle (saline) was injected just in the left hind paw. Nociceptive threshold measurements of both hind paws were made before and 180 min after injection of PGE2. The difference between the averages of these measurements was calculated (Δ nociceptive threshold). These assessments showed that the four peptides, at a dose of 30 μg/paw, induced antinociception restricted to the paw treated, while the contralateral paw presented nociceptive threshold without significant difference when compared to the PGE2-induced hyperalgesia (Fig. 5A–D).
Fig. 5.
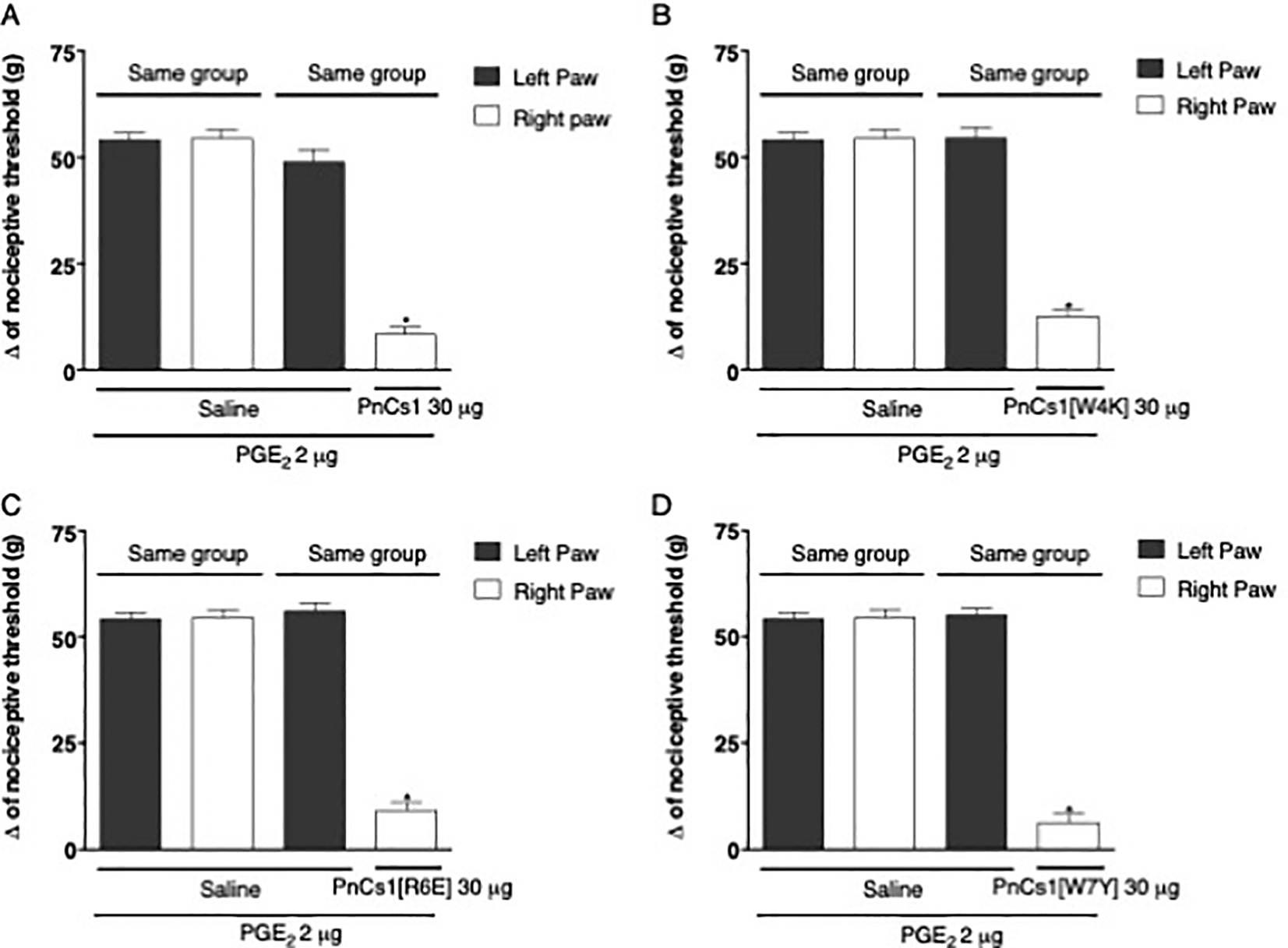
Exclusion of a possible systemic effect of (A) PnCS1, (B) PnCS1[W4K], (C) PnCS1[R6E] and (D) PnCS1[W7Y].
4. Discussion
In this study we synthesised a fourth generation of Pn peptides consisting of 28 analogues of downsized hybrid peptides originally based on sequence homology between the potent spider-derived NaV inhibitor PnTx1 [21,23] and cone snail NaV inhibitor μ-KIIIA [13,14,16,24]. We evaluated their potency and selectivity in vitro across therapeutically interesting NaV1.1, NaV1.3, NaV1.7 and NaV1.8 subtypes, as well as off-target NaV subtypes including NaV1.2, NaV1.4, NaV1.5 and NaV1.6. Species differences in NaV subtype potency have previously been described for conotoxins, including GIIIA and GIIIB [33] as well as small molecule NaV1.7 inhibitors (https://www.biorxiv.org/content/10.1101/869206v1.full.pdf). Assessment of in vitro activity of our peptides at NaV subtypes was limited by availability of relevant clones, although the use of rodent isoforms for therapeutically relevant NaV subtypes virtually eliminates the possibility that the surprising in vivo activity in our rodent pain models arises due to species differences in NaV1.7 potency. Nevertheless, for promising candidates, selectivity across human and rodent subtypes should be assessed in more detail in future studies. We also evaluated a handful of peptides from the series in vivo using an NaV1.7 target engagement assay as well as an PGE2-mediated pain assay.
Despite an extensive MAPS analysis, exploring the chemical space, and replacing non-Cys residues with positive and negative charges as well as aromatic residues, across the full cyclic peptide, none of the peptides displayed any improvement in potency or inhibition compared to the parent peptide PnCS1 when examined using two-electrode voltage clamp electrophysiology across sodium channel subtypes expressed in oocytes (Fig. 6). Replacing Arg3 with an Ala did not significantly affect either the potency or level of inhibition across NaV1.1, NaV1.4 and NaV1.6, but did abolish potency at NaV1.5. This is surprising since Arg3 is equivalent to Lys7 in μ-KIIIA, which has been shown by us and others to be integral for binding to NaV1.2 [13,14,16,24]. This suggests that the peptide analogues are too small to make specific connections. The fact that the cyclic peptides are active on NaV1.2 and NaV1.4 is not too surprising since they are hybrids of μ-KIIIA and PnTx1, peptides known to display low IC50s at these two NaV subtypes. Subtype selectivity is one challenge that is yet to be overcome in order to design selective peptidic pore blockers for therapeutically relevant NaV subtypes.
Fig. 6.
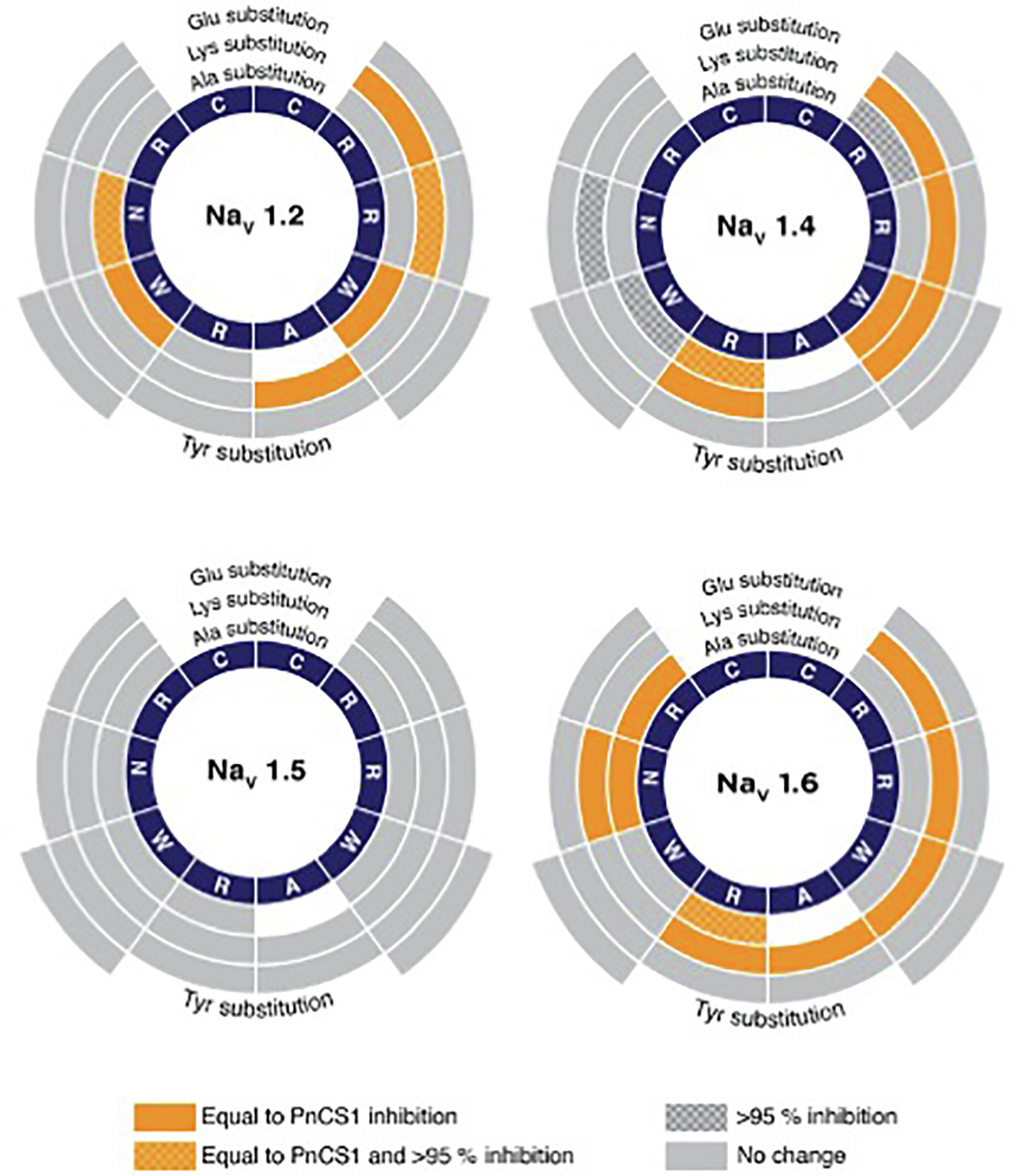
Electrophysiology analysis of PnCS1 MAPS library on NaV1.2, 1.4, 1.5 and 1.6. IC50 were calculated for each peptide and compared to the activity of parent peptide PnCS1 (NaV1.2: 1.0 ± 0.1 μM; NaV1.4: 0.6 ± 0.2 μM; NaV1.5: 2.8 ± 0.6 μM; NaV1.6: 0.7 ± 0.2 μM). Working concentrically from the centre, segments correspond to native peptide sequence (navy), effects of Ala substitution, effects of Lys substitution, effects of Glu substitution and effects of Tyr substitution. Colours and shading represent effect of substitute on IC50, equal to PnCS1 (orange plain), equal to PnCS1 and > 95% inhibition (orange chequered) and > 95% inhibition (grey chequered) and no change (grey). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Few venom-derived peptide toxins act as pore blockers, and when they do, they typically act in a promiscuous manner like TTX. This is not surprising, since there is high sequence homology across the pore of the different sodium channel subtypes [25], most likely giving rise to this observed promiscuity. However, despite the promiscuity of pore blockers, molecules like lidocaine have been proven to be very effective as local anaesthetics, nerve block agents, antiarrhythmic drugs, and to treat chronic pain and acute surgical pain [34–36]. Therefore, although drugs like lidocaine have a very narrow therapeutic window due to them targeting several subtypes, they can be extremely useful in a clinical setting.
Besides PnCS1, three peptides from the fourth generation were tested for their activity in vivo by intraplantar injection three hours after injection of PGE2 as well as in the OD1 model of NaV1.7 mediated pain. PnCS1[W4K], PnCS1[R6E] and PnCS1[W7Y] were chosen based on the initial electrophysiological data indicating interesting activity for these peptides on the tested NaV channels (Fig. 6). NaV1.7 is a well-validated and promising pain target based on genetic evidence with extensive drug discovery efforts for selective inhibitors being pursued [25]. The OD1 model provides an in vivo model to pharmacologically characterize local target engagement of NaV1.7 blockers and is therefore a great tool to investigate the translatability of in vitro to in vivo activity of NaV1.7 inhibiting compounds. PnCs1, with an IC50 of 0.9 μM at NaV1.7 expectedly reduced pain behaviours at doses of 10 μM and 100 μM in this model. The peptides with a lower activity at NaV1.7, PnCS1[W4K] and PnCS1[W7Y], only showed significant effects when a dose of 100 μM was administered; but not at 10 μM, a dose just above their IC50 values of 7.4 μM and 8.2 μM, respectively. Surprisingly, PnCS1[R6E] also showed significant antinociceptive activity (at both doses) despite inactivity at NaV1.7 channels in vitro, suggesting alternative analgesic off-targets being responsible for this result, downstream of NaV1.7 activation may be modulated, or alternatively an unexpected activity of PnCS1[R6E] at the mouse NaV1.7 orthologue. It is well documented that PGE2 has important cell signalling activities in neurons and hereby influences the pain threshold by increasing the excitability of afferent neurons innervating the area of inflammation [37]. PGE2 lowers the pain threshold in thermal, chemical and mechanical stimuli. Inflammatory mediators such as PGE2 are important contributors to the pain induced by local inflammation after tissue damage [38,39]. In fact, secondary mediators, activated by inflammatory mediators like PGE2 act directly on specific NaV channels related to nociception [37]. For example, it has been reported that inflammatory regulators mediate an up-regulation of NaV1.3, NaV1.7 and NaV1.8 channels in dorsal root ganglias (DRGs) [40]. In axotomized DRGs, the mediator glial-derived neurotrophic factor (GDNF), enhances the expression of the TTX-resistant current which largely consists of NaV1.8 and NaV1.9 current [33]. Elevated PGE2 concentrations induces protein kinase C (PKC), which in turn will also increase the TTX-resistant currents [41]. It has been reported that adenosine and bradykinin cause a NaV channel mediated alteration of the excitability of sensory neurons [37,42–44], and that PKA, induced by PGE2, alters the trafficking of NaV1.8 channels [45]. Furthermore, treatment with PGE2 resulted in an increased persistent Na+ current attributed to NaV1.9 channels for up to 1 h [46].
A NaV channel inhibiting activity, as observed for the PnCS peptides, will contribute to a reduced Na+ current and thus hereby induce an antinociceptive effect in a model of inflammatory pain. Therefore, it is no surprise that PnCS1, PnCS1[W4K], and PnCS1[W7Y] induce antinociception in a PGE2 induced model of pain. Indeed, the antinociceptive effect seen for these peptides can be explained by inhibition of NaV1.3 (PnCS1, PnCS1[W4K]), NaV1.7 (PnCS1, PnCS1[W4K], PnCS1 [W7Y]) and NaV1.9 (PnCS1) [24]. Furthermore, the inhibition of NaV1.1 (PnCS1, PnCS1[W4K], PnCS1[W7Y]) and NaV1.6 (PnCS1, PnCS1[W4K], PnCS1[W7Y]) might contribute as well, although the involvement of these channels in mechanical pain pathways has been reported [47], caution is still needed when interpreting these results. Nevertheless, caution is required when interpreting the in vivo data since there is no obvious correlation between the in vitro observed electrophysiological data and the in vivo observed analgesia. This indicates that these peptides might exert their analgesic effect by targeting other ion channels or receptors involved in analgesic pathways.
Rather unexpected was the observation that in both the OD1 and the PGE2 model of nociception, PnCS1[R6E] seems to be as active as the other peptides. Based on the available electrophysiological data in X. laevis oocytes, this peptide has a reduced affinity for NaV1.1-NaV1.8, but yet it appears to be active in vivo. Further experiments are needed to confirm PnCS1[R6E] activity on other targets. Moreover, these peptides need to be tested on other ion channels and receptors in order to exclude that the observed nociceptive effect is a resultant of off-target activity on other nociceptors such as, e.g., CaV, TRP channels or opioid and cannabinoid receptors.
It has been well recognized that NaV channels play a crucial role in inherited diseases, such as cardiovascular arrhythmias, central nervous system disorders and pain syndromes. This knowledge highlights NaV channel isoforms as targets of novel compounds that will hopefully fulfil the unmet therapeutic need to successfully treat these disorders [48,49]. Therefore, molecules capable of selective targeting and modulation of NaV channel isoforms represent attractive pharmacological tools, either to identify the specific isoform involved in different channelopathies or as potential therapeutics. Drugs currently used in humans can roughly be divided in either small molecules or large biologics, including antibodies. The small organic molecules tend to display the desirable physicochemical property of oral bioavailability, but on the other hand may suffer from reduced target selectivity that is manifest in unwanted side effects. TTX is an interesting example of a low molecular weight compound targeting Nav channels. Despite being characterized as a NaV channel blocker for many years, TTX is still one of the most efficient NaV channel inhibitors known to date. TTX is selective for NaV channels and has a preference for what is known as TTX-sensitive NaV channels over the cardiac NaV1.5 channel and NaV1.8 and NaV1.9, and importantly, does not cross the blood-brain barrier. Not surprising, TTX is under heavy investigation for development of analgesic therapeutics as evidenced by the existing 76 patents related to TTX applications [50]. Nevertheless, several hurdles need to be overcome before TTX can be further developed into a druggable compound. Clinical trials on TTX revealed several occurring side effects, mainly due to toxicity upon systemic distribution of TTX and analogues. Among the most severe side effects reported were ataxia, aspiration pneumonia, hypertension and nausea [13,50,51]. This demonstrates the difficulties and challenges that are involved in the development of NaV channel inhibitors into usable therapeutics.
By contrast with small molecules, large biologics on the other hand, tend to be exquisitely specific for their targets due to their larger surface area. However, this advantage usually comes at the cost of low bioavailability, poor membrane permeability, and metabolic instability [52,53]. Peptides have emerged with the promise to bridge the gap between small molecules and large biologics, and the field of drug development is now refocusing its efforts to pursue peptides as lead molecules that fit between these two molecular weight extremes and at the same time, exhibit the advantageous characteristics of both [54]. Indeed, molecules combining advantages of small molecules (cost, conformational restriction, membrane permeability, metabolic stability, oral bioavailability) with those of large biologics (natural components, target specificity, high potency) might represent the novel tools to overcome the hurdles experienced today in drug discovery [54]. It is within this philosophy of combining the better of two worlds that we decided to combine the sophisticated evolutionary peptide chemistry of cone snails and spiders in order to design small, cyclic and bioactive peptides. The resulting peptides do represent the first and the smallest (ten residues) cyclic NaV modulators to date. These peptides are unique pharmacological tools to investigate disease pathways including, but not limited to, neuropathic and nociceptive pain. Moreover, they represent promising starting scaffolds for further development of peptide-based therapeutics. Notwithstanding, a major challenge in developing these cyclic Pn peptides in therapeutics will be creating ligands that target a single NaV channel subtype. Moreover, future studies are required to elucidate which other pain targets are also recognized by these peptides in order to understand the potent analgesia observed in vivo. Pharmacological interactions of the cyclic Pn peptides with membrane receptors and ion channels other than their NaV channel target cannot be underestimated and should be investigated in order to validate the therapeutic effectiveness of these peptides.
Funding
This study was supported by grants G0E7120N, GOC2319N and GOA4919N from the F.W.O. Vlaanderen awarded to J.T, and a National Health a Medical Research Council (NHMRC) Project Grant (APP1080405) awarded to C.I.S. S.P. was supported by Coordenação de Aperfeiçoamento de Pessoal de Nivel Superior (CAPES) and KU Leuven funding (PDM/19/164), C.I.S. was supported by an Australian Research Council (ARC) Future Fellowship (FT160100055), D.J.C is an ARC Australian Laureate fellow (FL150100146), I.V. is supported by a NHMRC Career Development Fellowship (APP1162503), and K.L.M and A.M were supported by Australian Government Research Training Program Scholarships. The authors declare no conflict of interest.
Abbreviations:
- μ-KIIIA
μ-conotoxin KIIIA
- PnTx1
Phoneutria nigriventer toxin Tx1
- NaV
voltage gated sodium channels
- TTX
tetrodotoxin
- MAPS
multiple attribute positional scanning
Footnotes
CRediT authorship contribution statement
Steve Peigneur: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Software, Validation, Visualization, Writing - original draft, Writing - review & editing. Cristina da Costa Oliveira: Formal analysis, Investigation, Methodology, Visualization. Flávia Cristina de Sousa Fonseca: Data curation, Formal analysis, Investigation, Methodology, Visualization. Kirsten L. McMahon: Formal analysis, Investigation, Visualization, Writing - review & editing. Alexander Mueller: Data curation, Formal analysis, Investigation, Visualization, Writing - review & editing. Olivier Cheneval:. Ana Cristina Nogueira Freitas: Data curation, Project administration, Software. Hana Starobova: Formal analysis, Investigation, Writing - review & editing. Igor Dimitri Gama Duarte: Funding acquisition, Project administration, Resources, Supervision. David J. Craik: Conceptualization, Funding acquisition, Resources, Writing - review & editing. Irina Vetter: Formal analysis, Funding acquisition, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing - review & editing. Maria Elena de Lima: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing - review & editing. Christina I. Schroeder: Conceptualization, Formal analysis, Funding acquisition, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing - original draft, Writing - review & editing. Jan Tytgat: Conceptualization, Funding acquisition, Project administration, Resources, Software, Supervision, Validation, Writing - review & editing.
References
- [1].Robinson SD, Undheim EAB, Ueberheide B, King GF, Venom peptides as therapeutics: advances, challenges and the future of venom-peptide discovery, Expert Rev. Proteomics 14 (10) (2017) 931–939. [DOI] [PubMed] [Google Scholar]
- [2].Prashanth JR, Dutertre S, Lewis RJ, Pharmacology of predatory and defensive venom peptides in cone snails, Mol. Biosyst 13 (12) (2017) 2453–2465. [DOI] [PubMed] [Google Scholar]
- [3].Green BR, Olivera BM, Venom peptides from cone snails: pharmacological probes for voltage-gated sodium channels, Curr. Top. Membr 78 (2016) 65–86. [DOI] [PubMed] [Google Scholar]
- [4].Israel MR, Tay B, Deuis JR, Vetter I, Sodium channels and venom peptide pharmacology, Adv. Pharmacol 79 (2017) 67–116. [DOI] [PubMed] [Google Scholar]
- [5].Lewis RJ, Dutertre S, Vetter I, Christie MJ, Conus venom peptide pharmacology, Pharmacol. Rev 64 (2) (2012) 259–298. [DOI] [PubMed] [Google Scholar]
- [6].Green BR, Bulaj G, Norton RS, Structure and function of mu-conotoxins, peptide-based sodium channel blockers with analgesic activity, Future Med. Chem 6 (15) (2014) 1677–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lipkind GM, Fozzard HA, KcsA crystal structure as framework for a molecular model of the Na(+) channel pore, Biochemistry 39 (28) (2000) 8161–8170. [DOI] [PubMed] [Google Scholar]
- [8].Markgraf R, Leipold E, Schirmeyer J, Paolini-Bertrand M, Hartley O, Heinemann SH, Mechanism and molecular basis for the sodium channel subtype specificity of micro-conopeptide CnIIIC, Br. J. Pharmacol 167 (3) (2012) 576–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bulaj G, West PJ, Garrett JE, Watkins M, Zhang MM, Norton RS, Smith BJ, Yoshikami D, Olivera BM, Novel conotoxins from Conus striatus and Conus kinoshitai selectively block TTX-resistant sodium channels, Biochemistry 44 (19) (2005) 7259–7265. [DOI] [PubMed] [Google Scholar]
- [10].Wilson MJ, Yoshikami D, Azam L, Gajewiak J, Olivera BM, Bulaj G, Zhang MM, mu-Conotoxins that differentially block sodium channels NaV1.1 through 1.8 identify those responsible for action potentials in sciatic nerve, PNAS 108 (25) (2011) 10302–10307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].McArthur JR, Singh G, McMaster D, Winkfein R, Tieleman DP, French RJ, Interactions of key charged residues contributing to selective block of neuronal sodium channels by mu-conotoxin KIIIA, Mol. Pharmacol 80 (4) (2011) 573–584. [DOI] [PubMed] [Google Scholar]
- [12].Stevens M, Peigneur S, Dyubankova N, Lescrinier E, Herdewijn P, Tytgat J, Design of bioactive peptides from naturally occurring mu-conotoxin structures, J. Biol. Chem 287 (37) (2012) 31382–31392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Van Der Haegen A, Peigneur S, Tytgat J, Importance of position 8 in mu-conotoxin KIIIA for voltage-gated sodium channel selectivity, FEBS J 278 (18) (2011) 3408–3418. [DOI] [PubMed] [Google Scholar]
- [14].Zhang MM, Green BR, Catlin P, Fiedler B, Azam L, Chadwick A, Terlau H, McArthur JR, French RJ, Gulyas J, Rivier JE, Smith BJ, Norton RS, Olivera BM, Yoshikami D, Bulaj G, Structure/function characterization of micro-conotoxin KIIIA, an analgesic, nearly irreversible blocker of mammalian neuronal sodium channels, J. Biol. Chem 282 (42) (2007) 30699–30706. [DOI] [PubMed] [Google Scholar]
- [15].French RJ, Yoshikami D, Sheets MF, Olivera BM, The tetrodotoxin receptor of voltage-gated sodium channels–perspectives from interactions with micro-conotoxins, Mar. Drugs 8 (7) (2010) 2153–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Pan X, Li Z, Huang X, Huang G, Gao S, Shen H, Liu L, Lei J, Yan N, Molecular basis for pore blockade of human Na(+) channel Nav1.2 by the mu-conotoxin KIIIA, Science 363 (6433) (2019) 1309–1313. [DOI] [PubMed] [Google Scholar]
- [17].Stevens M, Peigneur S, Tytgat J, Neurotoxins and their binding areas on voltage-gated sodium channels, Front. Pharmacol 2 (2011) 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Peigneur S, de Lima ME, Tytgat J, Phoneutria nigriventer venom: a pharmacological treasure, Toxicon 151 (2018) 96–110. [DOI] [PubMed] [Google Scholar]
- [19].Richardson M, Pimenta AM, Bemquerer MP, Santoro MM, Beirao PS, Lima ME, Figueiredo SG, Bloch C Jr., Vasconcelos EA, Campos FA, Gomes PC, Cordeiro MN, Comparison of the partial proteomes of the venoms of Brazilian spiders of the genus Phoneutria, Comp. Biochem. Physiol. C Toxicol. Pharmacol 142 (3–4) (2006) 173–187. [DOI] [PubMed] [Google Scholar]
- [20].Diniz CR, Cordeiro Mdo N, Junor LR, Kelly P, Fischer S, Reimann F, Oliveira EB, Richardson M, The purification and amino acid sequence of the lethal neurotoxin Tx1 from the venom of the Brazilian ’armed’ spider Phoneutria nigriventer, FEBS Lett 263 (2) (1990) 251–253. [DOI] [PubMed] [Google Scholar]
- [21].Silva AO, Peigneur S, Diniz MR, Tytgat J, Beirao PS, Inhibitory effect of the recombinant Phoneutria nigriventer Tx1 toxin on voltage-gated sodium channels, Biochimie 94 (12) (2012) 2756–2763. [DOI] [PubMed] [Google Scholar]
- [22].Cestele S, Catterall WA, Molecular mechanisms of neurotoxin action on voltage-gated sodium channels, Biochimie 82 (9–10) (2000) 883–892. [DOI] [PubMed] [Google Scholar]
- [23].Martin-Moutot N, Mansuelle P, Alcaraz G, Dos Santos RG, Cordeiro MN, De Lima ME, Seagar M, Van Renterghem C, Phoneutria nigriventer toxin 1: a novel, state-dependent inhibitor of neuronal sodium channels that interacts with micro conotoxin binding sites, Mol. Pharmacol 69 (6) (2006) 1931–1937. [DOI] [PubMed] [Google Scholar]
- [24].Peigneur S, Cheneval O, Maiti M, Leipold E, Heinemann SH, Lescrinier E, Herdewijn P, De Lima ME, Craik DJ, Schroeder CI, Tytgat J, Where cone snails and spiders meet: design of small cyclic sodium-channel inhibitors, FASEB J. 33 (3) (2019) 3693–3703. [DOI] [PubMed] [Google Scholar]
- [25].Vetter I, Deuis JR, Mueller A, Israel MR, Starobova H, Zhang A, Rash LD, Mobli M, NaV1.7 as a pain target - From gene to pharmacology, Pharmacol. Ther 172 (2017) 73–100. [DOI] [PubMed] [Google Scholar]
- [26].Deuis JR, Wingerd JS, Winter Z, Durek T, Dekan Z, Sousa SR, Zimmermann K, Hoffmann T, Weidner C, Nassar MA, Alewood PF, Lewis RJ, Vetter I, Analgesic effects of GpTx-1, PF-04856264 and CNV1014802 in a mouse model of NaV1.7-mediated pain, Toxins 8 (3) (2016) 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Randall LO, Selitto JJ, A method for measurement of analgesic activity on inflamed tissue, Arch. Int. Pharmacodyn. Ther 111 (4) (1957) 409–419. [PubMed] [Google Scholar]
- [28].Kawabata A, Nishimura Y, Takagi H, L-leucyl-L-arginine, naltrindole and D-arginine block antinociception elicited by L-arginine in mice with carrageenin-induced hyperalgesia, Br. J. Pharmacol 107 (4) (1992) 1096–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG, Animal research: reporting in vivo experiments: the ARRIVE guidelines, Br. J. Pharmacol 160 (7) (2010) 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].McGrath JC, Lilley E, Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP, Br. J. Pharmacol 172 (13) (2015) 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Murray JK, Long J, Zou A, Ligutti J, Andrews KL, Poppe L, Biswas K, Moyer BD, McDonough SI, Miranda LP, Single residue substitutions that confer voltage-gated sodium ion channel subtype selectivity in the NaV1.7 inhibitory peptide GpTx-1, J. Med. Chem 59 (6) (2016) 2704–2717. [DOI] [PubMed] [Google Scholar]
- [32].Durek T, Vetter I, Wang CI, Motin L, Knapp O, Adams DJ, Lewis RJ, Alewood PF, Chemical engineering and structural and pharmacological characterization of the alpha-scorpion toxin OD1, ACS Chem. Biol 8 (6) (2013) 1215–1222. [DOI] [PubMed] [Google Scholar]
- [33].Cummins TR, Dib-Hajj SD, Black JA, Waxman SG, Sodium channels and the molecular pathophysiology of pain, Prog. Brain Res 129 (2000) 3–19. [DOI] [PubMed] [Google Scholar]
- [34].Dokken K, Fairley P, Sodium Channel Blocker Toxicity, StatPearls, Treasure Island (FL), 2020. [PubMed] [Google Scholar]
- [35].Beecham GB, Bansal P, Goyal A, Lidocaine, StatPearls, Treasure Island (FL), 2020. [Google Scholar]
- [36].Hermanns H, Hollmann MW, Stevens MF, Lirk P, Brandenburger T, Piegeler T, Werdehausen R, Molecular mechanisms of action of systemic lidocaine in acute and chronic pain: a narrative review, Br. J. Anaesth 123 (3) (2019) 335–349. [DOI] [PubMed] [Google Scholar]
- [37].Cardoso FC, Lewis RJ, Sodium channels and pain: from toxins to therapies, Br. J. Pharmacol 175 (12) (2018) 2138–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Aley KO, Levine JD, Role of protein kinase A in the maintenance of inflammatory pain, J. Neurosci 19 (6) (1999) 2181–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Fitzgerald EM, Okuse K, Wood JN, Dolphin AC, Moss SJ, cAMP-dependent phosphorylation of the tetrodotoxin-resistant voltage-dependent sodium channel SNS, J. Physiol 516 (Pt 2) (1999) 433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Black JA, Liu S, Tanaka M, Cummins TR, Waxman SG, Changes in the expression of tetrodotoxin-sensitive sodium channels within dorsal root ganglia neurons in inflammatory pain, Pain 108 (3) (2004) 237–247. [DOI] [PubMed] [Google Scholar]
- [41].Gold MS, Levine JD, Correa AM, Modulation of TTX-R INa by PKC and PKA and their role in PGE2-induced sensitization of rat sensory neurons in vitro, J. Neurosci 18 (24) (1998) 10345–10355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Binshtok AM, Wang H, Zimmermann K, Amaya F, Vardeh D, Shi L, Brenner GJ, Ji RR, Bean BP, Woolf CJ, Samad TA, Nociceptors are interleukin-1beta sensors, J. Neurosci 28 (52) (2008) 14062–14073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Dib-Hajj SD, Binshtok AM, Cummins TR, Jarvis MF, Samad T, Zimmermann K, Voltage-gated sodium channels in pain states: role in pathophysiology and targets for treatment, Brain Res. Rev 60 (1) (2009) 65–83. [DOI] [PubMed] [Google Scholar]
- [44].Griswold DE, Douglas SA, Martin LD, Davis TG, Davis L, Ao Z, Luttmann MA, Pullen M, Nambi P, Hay DW, Ohlstein EH, Endothelin B receptor modulates inflammatory pain and cutaneous inflammation, Mol. Pharmacol 56 (4) (1999) 807–812. [PubMed] [Google Scholar]
- [45].Liu C, Li Q, Su Y, Bao L, Prostaglandin E2 promotes Na1.8 trafficking via its intracellular RRR motif through the protein kinase A pathway, Traffic 11 (3) (2010) 405–417. [DOI] [PubMed] [Google Scholar]
- [46].Khasar SG, Gold MS, Levine JD, A tetrodotoxin-resistant sodium current mediates inflammatory pain in the rat, Neurosci. Lett 256 (1) (1998) 17–20. [DOI] [PubMed] [Google Scholar]
- [47].Osteen JD, Herzig V, Gilchrist J, Emrick JJ, Zhang C, Wang X, Castro J, Garcia-Caraballo S, Grundy L, Rychkov GY, Weyer AD, Dekan Z, Undheim EA, Alewood P, Stucky CL, Brierley SM, Basbaum AI, Bosmans F, King GF, Julius D, Selective spider toxins reveal a role for the Nav1.1 channel in mechanical pain, Nature 534 (7608) (2016) 494–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Peigneur S, Paolini-Bertrand M, Gaertner H, Biass D, Violette A, Stocklin R, Favreau P, Tytgat J, Hartley O, delta-Conotoxins synthesized using an acid-cleavable solubility tag approach reveal key structural determinants for NaV subtype selectivity, J. Biol. Chem 289 (51) (2014) 35341–35350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Peigneur S, Zula A, Zidar N, Chan-Porter F, Kirby R, Madge D, Ilas J, Kikelj D, Tytgat J, Action of clathrodin and analogues on voltage-gated sodium channels, Mar. Drugs 12 (4) (2014) 2132–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Melnikova DI, Khotimchenko YS, Magarlamov TY, Addressing the issue of tetrodotoxin targeting, Mar. Drugs 16 (10) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Nieto FR, Cobos EJ, Tejada MA, Sanchez-Fernandez C, Gonzalez-Cano R, Cendan CM, Tetrodotoxin (TTX) as a therapeutic agent for pain, Mar. Drugs 10 (2) (2012) 281–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Bhardwaj G, Mulligan VK, Bahl CD, Gilmore JM, Harvey PJ, Cheneval O, Buchko GW, Pulavarti SV, Kaas Q, Eletsky A, Huang PS, Johnsen WA, Greisen PJ, Rocklin GJ, Song Y, Linsky TW, Watkins A, Rettie SA, Xu X, Carter LP, Bonneau R, Olson JM, Coutsias E, Correnti CE, Szyperski T, Craik DJ, Baker D, Accurate de novo design of hyperstable constrained peptides, Nature 538 (7625) (2016) 329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Craik DJ, Fairlie DP, Liras S, Price D, The future of peptide-based drugs, Chem. Biol. Drug Des 81 (1) (2013) 136–147. [DOI] [PubMed] [Google Scholar]
- [54].Yu R, Kompella SN, Adams DJ, Craik DJ, Kaas Q, Determination of the alpha-conotoxin Vc1.1 binding site on the alpha9alpha10 nicotinic acetylcholine receptor, J. Med. Chem 56 (9) (2013) 3557–3567. [DOI] [PubMed] [Google Scholar]
- [55].McMahon KL, Tran HNT, Deuis JR, Lewis RJ, Vetter I, Scrhoeder CI, Discovery, Pharmacological Characterisation and NMR Structure of the Novel μ-Conotoxin SxIIIC, a Potent and Irreversible NaV Channel Inhibitor, Biomedicines 8 (Oct 2) (2020) E391. [DOI] [PMC free article] [PubMed] [Google Scholar]