

Abstract
Given most tissues are consist of abundant and diverse (sub-)cell types, an important yet unaddressed problem in bulk RNA-seq analysis is to identify at which (sub-)cell type(s) the differential expression occurs. Single-cell RNA-sequencing (scRNA-seq) technologies can answer the question, but they are often labor-intensive and cost-prohibitive. Here, we present LRcell, a computational method aiming to identify specific (sub-)cell type(s) that drives the changes observed in a bulk RNA-seq experiment. In addition, LRcell provides pre-embedded marker genes computed from putative scRNA-seq experiments as options to execute the analyses. We conduct a simulation study to demonstrate the effectiveness and reliability of LRcell. Using three different real datasets, we show that LRcell successfully identifies known cell types involved in psychiatric disorders. Applying LRcell to bulk RNA-seq results can produce a hypothesis on which (sub-)cell type(s) contributes to the differential expression. LRcell is complementary to cell type deconvolution methods.
Keywords: cell-type enrichment, cell marker genes, differential gene expression
Background
Finding differentially expressed genes (DEGs) between experimental conditions is a powerful approach to understand the molecular basis of phenotypic variation. However, most tissues consist of tens or even hundreds of diverse (sub-)cell types and DEGs may only occur in a small subset of these (sub-)cell types, which are relevant to the experimental condition. Bulk RNA-seq data alone are unable to reveal the (sub-)cell types that drive the DEGs.
The rapid development and proliferation of single-cell technologies resulted in massive accumulation of single-cell transcriptomics data (scRNA-seq) from diverse tissue types. These data reveal substantial variations in transcriptional regulation among different cell types and offer an unprecedented close-up view of the modifications underlying important biological processes, especially for disease pathology, including which cell types drive DEGs [1]. As an example, in a recent single-cell resolution analysis of Alzheimer’s disease (AD), Mathys et al. [2] identified glial–neuronal interactions in response to AD pathology. In another single-cell study, Ruzicka et al. [3] found that neurons are the most affected cell type for schizophrenia. However, steep cost and complicated protocols prevent the widespread adoption of scRNA-seq.
Over the past 10 years, many computational cell-type deconvolution methods have been developed to infer the proportions of different (sub-)cell types from bulk transcriptomic data [1, 4–10]. Benchmark studies have also been conducted to compare their performance [11, 12].
In this study, we propose a novel computational tool named LRcell. Given the result from a bulk RNA-seq differential expression (DE) study, the goal of LRcell is to delineate which (sub-)cell type(s) of the tissue underwent substantial changes between the two experimental conditions. LRcell is developed under the assumption that expression change occurred at one or few subcell type(s) between the two experimental conditions is the major contributor to the DEGs observed at the bulk tissue level. Cell-type deconvolution methods are not designed to infer such changes. Exploiting cell-type-specific marker genes identified from generic scRNA-seq available from publicly available data repositories, LRcell achieves the goal by surveying the enrichment of marker genes across all (sub-)cell types in the tissue (Figure 1). Thus, no scRNA-seq experiment matching the bulk RNA-seq experimental condition is needed. When applying LRcell to a diverse panel of bulk RNA-seq DE experiments, we successfully identify known (sub-)cell types involved in the pathogenesis of psychiatric disorders as well as produce testable new hypotheses that have the potential to produce fresh new biological insights.
Figure 1.

Overview of LRcell workflow. As input, LRcell takes in the result from a case-control bulk RNA-seq experiment conducted on specific tissue. For illustration purpose, assuming there are three (sub-)cell types within the tissue, and the marker genes derived from (unrelated) scRNA-seq experiment on the three (sub-)cell types are available and taken into account by LRcell. Here, we use the blue color to indicate cell type A, the yellow color to indicate cell type B and the green color to indicate cell type C. We map the marker genes to the entire gene list sorted by DE P-values from the most significant DE to non-DE. Next, for each tissue type, we apply a regression analysis. When using the binary indicator of marker gene as the response variable, we run a logistic regression (LR); when using the enrichment score of the marker gene produced by the Marques et al.’s method as the response variable, we run a linear regression (LiR). In both cases, the explanatory variable is the −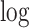 transformed DE P-value. Next, the significance of the regression analysis is calculated and converted to −
transformed DE P-value. Next, the significance of the regression analysis is calculated and converted to −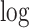 transformed FDR and plotted. In this illustrating example, LRcell result indicates cell type A is the most significant, which suggests that cell type A is likely to play a significant role in the case-control experiment.
transformed FDR and plotted. In this illustrating example, LRcell result indicates cell type A is the most significant, which suggests that cell type A is likely to play a significant role in the case-control experiment.
Results and discussion
In this work, we collect and curate a compendium of marker genes from multiple published scRNA-seq datasets. We then conduct LRcell analysis on multiple bulk RNA-seq DE experiments to demonstrate its utility.
Marker gene collection and sources
Genes that show substantial expression difference between one (sub-)cell type and others in their native state are regarded as marker genes [13]. Similar to a collection of gene set for Gene Set Enrichment Analysis (GSEA) [14], LRcell requires a compendium of high-quality cell-type marker genes. Currently, LRcell package provides users with multiple preloaded marker gene sets from human blood, human brain and mouse brain (Figure 2A), computed from scRNA-seq datasets using method introduced in Marques et al.’s [15] study. Additionally, LRcell package offers external cell markers collected by Molecular Signatures Database (MSigDB) [16] with certain criteria (for more details, see the Methods section). The external makers all originate from human species including midbrain, cord blood, ovary and skeletal muscle. We store all cell-type-specific marker gene sets into another R Bioconductor ExperimentHub package named LRcellTypeMarkers. Additional marker gene sets are being tested and will be added to the collection.
Figure 2.

LRcell datasets and marker genes overlap between different brain regions. (A) summary of the all tissue-types in which marker genes have been pre-embedded in LRcell. In (B–D), top 100 marker genes are selected for each cell type, and thus, the maximum overlap in these figures is 100. (B) Heatmap illustrating the overlap of marker genes among cell types within the FC region derived from mouse whole brain scRNA-seq dataset. The highlighted area describes the overlap between FC_11-3.unknown, FC_11-4.unknwon and FC_11-1.Microglia as an illustration for the similarity between these three (sub-)cell types. (C) Heatmap illustrating the overlap of marker genes among cell types within the FC and cell types within the cerebellum CB. (D) Heatmap illustrating the overlap of marker genes among cell types within the FC and cell types within the hippocampus.
Properties of selected marker genes
Since the method proposed by Marques et al. [15] does not considers DEG’s fold changes, it is therefore of interest to explore the fold changes exhibit by the marker genes selected. We calculate the fold changes of each marker gene in the (sub-)cell type that they are representing versus others and plot the log10-transformed fold change for each (sub-)cell type (Supplementary Figure S1A, Supplementary data are available online at https://academic.oup.com/bib). We observe that vast majority of these marker genes show substantial (greater than five) fold changes expect for certain neuronal (sub-)cell types. We also provide a table (Supplementary Table S1, Supplementary data are available online at https://academic.oup.com/bib) listing how many marker genes having fold changes greater than five. To put the results above in context, we also calculate how many genes in the whole genome in each (sub-)cell type with fold changes larger than five (Supplementary Figure S1B, Supplementary data are available online at https://academic.oup.com/bib). As shown in the figure, the number of such marker genes again varies substantially.
Simulation settings
Because the ground truth of changes in DEGs and cell-type proportion is difficult to monitor and track, we conduct simulation studies to demonstrate the effectiveness of LRcell.
In this simulation study, we consider experiments between cases and controls involving DEGs and proportion changes. We simulate both single-cell and bulk RNA-seq data. Both types of data are generated by scDesign2 [17] using the adult mouse frontal cortex (FC) scRNA-seq dataset [18] as reference and we use the marker genes previously derived from the dataset to conduct our LRcell analysis. More details can be found in the Methods section.
For simplicity, we consider two scenarios in our simulation study: (1) the proportions for all (sub-)cell types remain the same during the condition change and DEGs are found in one specific cell type; (2) (sub-)cell-type proportions are different between case and control and no DEG is found in any (sub-)cell type. Under each scenario, we try to simulate different combinations.
Under the first scenario, we consider the following settings: (a) cell-type proportion distribution (evenly or unevenly distributed); (b) the total number of cells (1000; 5000 or 10 000 cells); (c) the number of DEGs occurred in that specific (sub-)cell type (1000; 2000 or 3000 DEGs out of 29 653 in the whole genome); and (d) fold change direction of DEGs (2 or 0.5 times of the original gene expression).
Under the second scenario, we consider the following combinations: (a) cell-type proportion distribution (evenly or unevenly distributed); (b) the total number of cells (1000; 5000 or 10 000 cells); and (c) proportion change in that specific (sub-)cell type (50; 80; 120 or 150% of the original proportion).
Additionally, to push the boundary of LRcell performance when there are many more (sub-)cell types, we simulate cases where there are 5, 10 and 15 (sub-)cell types and altering the baseline proportions which are evenly distributed in various ways.
Simulation results
For the simulation study, we take turns to alter each individual (sub-)cell type, then run LRcell or MuSiC [6] and track the rank of the altered (sub-)cell type as an indicator of the performance.
Because under the first scenario, there is no proportion change hence we do not test the performance of MuSiC. The ranking results are summarized in Supplementary Figure S2A and B, Supplementary data are available online at https://academic.oup.com/bib, and LRcell is able to correctly identify most of the (sub-)cell-type changes. The cases in which incorrect identification made are those with the smallest number of DEGs (in other word, where 1000 DEGs are simulated).
For the second scenario, we compare LRcell, MuSiC and GSEA (using marker genes as gene set). The results are summarized in Supplementary Figure S2C–E, Supplementary data are available online at https://academic.oup.com/bib. We observe that MuSiC performs steadily well under all settings while LRcell produces a few errors. This is fully expected since the scenario matches the assumption of MuSiC but not LRcell because it is not a cell-type proportion deconvolution method.
We also compare LRcell, MuSiC and GSEA under the scenario when there are more (sub-)cell types. The results are summarized in Supplementary Figure S2F–K, Supplementary data are available online at https://academic.oup.com/bib. We notice that when there are 10 (sub-)cell types, LRcell and MuSiC work equally well and when there are 15 (sub-)cell types, LRcell performs slightly better than MuSiC when adding up the ranks. In particular, for the setting of 1000 cells with 20% increase of proportion, both LRcell and MuSiC detect an incorrect but similar (sub-)cell type. A specific showcase has been presented in Supplementary Figure S3, Supplementary data are available online at https://academic.oup.com/bib, to show an overall performance regarding all (sub-)cell types. Under all settings, LRcell and MuSiC outperform GSEA.
Microglia highly enriched in neurodegenerative dementia
After the simulation study, we conduct LRcell in real data analysis. In a recent neurodegenerative dementia study, Swarup and colleagues contrasted TPR50 mice expressing tau mutant with wild type mice using bulk RNA-seq in order to identify gene networks mediating dementia [19] (‘the mouse AD study’ afterward). To identify the cell type(s) most involved in the condition, we apply LRcell to the DEG list using pre-embedded marker genes from adult mouse FC region [18]. From LRcell result, we observe that Microglia show up as highly significant (Figure 3A) which is concordant with previous studies [20]. Additionally, the FC_11-3.unknown and FC_11–4.unknown (sub-)cell types also show high level of significance. No annotation is available for these two cell clusters in the original publication. However, pairwise comparison of marker genes among all cell clusters reveal that these two unknown cell clusters have considerable overlaps with the FC_11-1, which is also a Microglia cell type (Figure 2B), which explains the pattern we observe.
Figure 3.

Applying LRcell to real cases. (A) LRcell result of mapping the bulk neurodegenerative dementia DEGs to the mouse brain FC region. (B) GSEA result of mapping the bulk neurodegenerative dementia DEGs using the same marker genes used in LRcell (mouse brain FC) as input. (C) Cell-type proportions for control and disease samples calculated by MuSiC. Each box contains 17 individuals. The x-axis is ordered by the t-test significance between the two conditions. (D) LRcell result of mapping bulk PTSD DEGs to human PBMC. CD16+ monocytes is shown as the most significant cell type. (E) GSEA result of mapping bulk PTSD DEGs to human PBMC using the same marker genes used in LRcell (human PBMC) as input. (F) Cell-type proportions for control and disease samples calculated by MuSiC. The x-axis is ordered by the t-test significance between two conditions.
CD16+ monocytes highly enriched in posttraumatic stress disorder
In a recent study, Breen and colleagues conducted a bulk whole-transcriptome study using peripheral blood leukocytes collected from U.S. Marines, among which some developed posttraumatic stress disorder (PTSD) postdeployment [21] (‘the human PTSD study’ afterward). Using this dataset, we generate a list of DEGs that show significant difference between the PTSD group and the control group at the predeployment time point.
Using human marker genes derived from a single-cell transcriptomic study on peripheral blood mononuclear cell (PBMC) [22], LRcell analysis finds that cells annotated as CD16+ nonclassical monocytes shows up as the most significant among all cell types in PBMC (Figure 3D). Our finding makes biological sense because as stated in previous studies [23], heterogeneity exists in monocytes distinguished by CD16 surface proteins and nonclassical monocytes have been validated to regulate immune responses in trauma [24, 25].
Marker genes from different region or time points
To apply LRcell, an important question is that which marker gene sets to use, i.e. how to select single-cell RNA-seq data where the source of the tissue match the tissue type profiled in the bulk transcriptomic study. This is particularly important for complex tissues such as brain. To address this issue, we use the mouse AD study [19] as an example, which contains information from four brain regions: cortex, hippocampus (HC), cerebellum (CB) and brain stem. Brain stem is excluded from our analysis due to the lack of marker gene information from that region of the brain.
To understand how marker genes vary across brain regions, we first define marker genes in all regions of the brain to explore their spatial pattern (Figure 2C and D). We observe that glia cells, such as Astrocytes, from different regions have higher number of overlapping marker genes which indicates the homogeneity of glia cells across the brain. In contrast, neurons and interneurons share very few marker genes across different brain regions. We then apply pre-embedded adult mouse brain marker genes from FC, HC and CB to bulk DEGs obtained from cortex, HC and CB, respectively (Supplementary Figure S4, Supplementary data are available online at https://academic.oup.com/bib). We observe that Microglia cells are highly enriched in all three brain regions whereas Astrocytes are particularly highly enriched in CB (Supplementary Figure S4B, D and E, Supplementary data are available online at https://academic.oup.com/bib). Especially when applying CB marker genes to CB bulk DE experiment (Supplementary Figure S4B, Supplementary data are available online at https://academic.oup.com/bib), we notice that one (sub-)cell type of Astrocytes is highly enriched compared to others. Our observations demonstrate that selected cell types are heterogeneous spatially; meaning marker genes are highly specific not only for the cell type, but also which region the cell belongs to. Because of this finding, it is highly desirable to run LRcell using marker genes of cell types located in closely matched brain regions.
We are also curious whether marker genes curated from scRNA-seq experiments conducted on nonnormal samples is acceptable as the reference. To address this question, we use data from the HIV vaccine study [22]. We observe that the expression of cell-type-specific marker genes is mostly consistent across different time points within the same cell type (such as CD8 cells), and distinct across different cell types (Supplementary Figure S5A and B, Supplementary data are available online at https://academic.oup.com/bib). For example, B (sub-)cell types share a considerable number of marker genes across time points, while sharing fewer with other cell types. We also try using marker genes identified from samples collected from different time points to conduct LRcell analysis and observe that the enrichment signals are almost the same (Supplementary Figure S6A–C, Supplementary data are available online at https://academic.oup.com/bib). Thus, although default marker genes used in LRcell are collected from control samples, we believe that marker genes identified from nonnormal samples are acceptable when scRNA-seq data from normal samples are not available.
Comparison to GSEA
GSEA [14] is a powerful tool to determine whether a predefined gene set show concordant shift in expression when comparing two biological conditions. One could potentially replace LRcell with GSEA to identify DEG-driving cell types by treating cell-type-specific marker genes as predefined gene sets. To compare performance of the two methods, we repeat the analyses done in the mouse AD study and the human PTSD study using GSEA. The GSEA result from the mouse AD study (Figure 3B) yields several equally significant (sub-)cell types including astrocyte, endothelial, microglia, mural, oligodendrocyte and polydendrocyte. The tied significances lead to difficulties in determining which (sub-)cell type(s) potentially participated in dementia pathogenesis. Similar pattern is observed in the GSEA result on the human PTSD study (Figure 3E) which shows that monocytes, dendritic cells and some T (sub-)cell types are equally enriched. Based on the above observations, we conclude that LRcell is more effective than GSEA to identify (sub-)cell types that are most impacted by the condition change in bulk DE experiments.
Specificity, robustness and running time of LRcell
It is of interest to evaluate whether LRcell shows good specificity, i.e. low false positive rate. To do this, we simulated null scenario where there is no significant DEG in any of the (sub-)cell type. When apply LRcell to such null bulk RNA-seq data, we found that LRcell produce either no or much fewer and weaker significant result, illustrating good sensitivity of LRcell. More details can be found in the Supplementary Data.
To analyze the robustness of LRcell analysis, we run experiments from two perspectives: (i) whether the number of marker genes strongly affects LRcell results and (ii) whether a different DEGs detection method affects LRcell results.
We first conduct LRcell using different marker gene number derived from PBMC scRNA-seq dataset on the human PTSD study and we get similar enrichment performances (Supplementary Figure S7, Supplementary data are available online at https://academic.oup.com/bib). This indicates the robustness of the LRcell analysis.
In addition to DESeq2, we use Voom [26] with Limma [27] to perform DEGs analysis on the mouse AD study and the human PTSD study. Details of the usage can be found in the Supplementary Data. With the same marker genes set, we notice that the enrichment patterns are similar as FC_11-1. Microglia is highly enriched along with other (sub-)cell types (Supplementary Figure S8, Supplementary data are available online at https://academic.oup.com/bib).
In addition, we analyze the execution time among LRcell, GSEA and MuSiC under different simulation scenarios (Supplementary Figure S9, Supplementary data are available online at https://academic.oup.com/bib). We observe that LRcell and GSEA are steadily fast, while the execution time of MuSiC increases when the number of reference cells increases. LRcell takes about 3–4 s on average for each run on a typical laptop computer.
Discussions
Detecting transcriptional activity changes at the individual cell type level, especially their modifications in disease samples, is crucial for understanding the mechanisms of diseases development. In this study, we propose a novel strategy named LRcell which conducts enrichment analysis of cell-type-specific marker genes among the top (or bottom) DEGs identified by bulk transcriptome studies. Cell types that show the most enrichment are likely to play an important role in the condition alteration. When applying to real datasets, we found that LRcell can successfully identify the involvement of the Microglia and Astrocytes in the mouse AD study and rare monocytes in the human PTSD study.
Many computational methods have been developed to infer the proportions of different (sub-)cell types from bulk transcriptomic data [1, 4–12]. LRcell is not designed for estimating cell-type proportions. We assume that different proportion of (sub-)cell types in cases and control samples is not the major source of the DEGs observed at the bulk tissue level. Rather, expression changes occur at one or few (sub-)cell type(s) between case and control samples is the major contributor to the DEGs observed at the bulk tissue level. Recent studies showed supports for our assumption. For example, Segerstolpe et al. [28] showed no significant shift of cell-type proportions in pancreatic islet between type 2 diabetes patient samples and control samples, but the amount of DEGs vary substantially across (sub-)cell types. Based on this assumption, we designed LRcell to identify which (sub-)cell types may be involved in the experimental condition change and thus follow up experiments can be designed to explore the mechanisms of the involvement of the specific (sub-)cell type(s) in the experimental condition.
Although based on different assumptions, out of curiosity and also in order to put LRcell results in context, we apply MuSiC [6], a well-established deconvolution method to the mouse AD study [19] data. Because some layers of neurons are predicted to have almost zero proportion (Figure 3C) when using all 81 (sub-)cell types, we merge the original (sub-)clusters into 15 major cell types in order to achieve a better representation. Despite this, MuSiC does not detect significant difference in Microglia or Astrocyte in terms of their proportions between the two conditions. When applied to the human PTSD study [21] data, using the original cell cluster annotations, MuSiC shows that most of the T (sub-)cell types have zero proportion and the proportion of CD14+ monocytes is up to 60% (Figure 3F). In contrast, LRcell produces more sensible results because it is not limited by the number of cell types as it can detect the subtle differences among (sub-)cell types.
Interestingly, from our simulation studies, LRcell is also capable of detecting (sub-)cell types that undergo proportion changes, albeit with slightly lower accuracy comparing to state-of-the-art deconvolution methods.
A key advantage of LRcell lies in its ability to handle a large number of (sub-)cell types. This is because LRcell analyzes (sub-)cell types one-by-one, whereas deconvolution methods have to do the analysis jointly which leads to higher computation burden and poorer performance [11].
In spirit, LRcell operates similarly as GSEA, but LRcell is much more sensitive to minor differences in marker genes of (sub-)cell types, similar to the advantage of LRpath showed when comparing to GSEA [29]. This indicates LRcell’s potential to detect changes in (sub-)cell types caused by disease conditions. Simulation studies comparing LRcell with GSEA suggest very similar patterns as observed from real data analysis. Additionally, when compared to existing bulk deconvolution methods, LRcell is more stable in its ability to handle the similarities among (sub-)cell types. Thus, LRcell enables researchers to glean new biological insights from the bulk transcriptomics experiments with no need of redoing the experiment using single-cell technology. We are currently applying LRcell to a diverse set of clinical studies (Sharma, personal communication) to generate more biological insights.
How to select marker genes representing (sub-)cell types is an important research question. Plenty of methods have been developed to optimize the selection process [15, 30, 31]. However, due to the dramatic diversity among (sub-)cell types and tissues, there is no consensus universal criteria on the selection criteria that can make the marker gene set representative and complete, which is also dependent on the goal of the study including cell clustering, cell-type calling and cell-type deconvolution, among others. For LRcell, our experience leads us to adopt the method introduced in Marques et al. for its simplicity and computation efficiency. We have performed empirical studies to illustrate the effectiveness of the marker genes selected by the adopted method. More details can be found in the Supplementary Data. Alternatively, precompiled marker gene sets from emerging databases [32] cover more and more tissue types which are great resources.
To enable straightforward comparison, currently, we select a fix number of 100 marker genes from each (sub-)cell type. Understandably, the number of marker genes for different cell types varies; it is desirable to allow flexibility in choosing the number of marker genes based on the transcriptomic patterns across cell types. However, different numbers of marker genes post challenge for conducting enrichment analyses fairly across all cell types. This will be investigated in our future studies.
LRcell currently provides embedded marker genes from human blood, human brain and mouse brain calculated from scRNA-seq experiments along with markers from 66 cell types in four tissues (midbrain, cord blood, ovary and skeletal muscle) adopted from MSigDB. We are working to include more tissue types in the future releases of LRcell which will make it more widely applicable.
Conclusions
In summary, we develop LRcell, an R Bioconductor package for identifying (sub-)cell type(s) that drive the changes observed in bulk comparative transcriptomic studies, taking advantages of newly emerged scRNA-seq data. The rationale of LRcell is that we believe marker genes of the modifying cell types tend to be enriched toward the top (or bottom) of the DEG lists. We conduct comprehensive surveys applying LRcell across various experimental conditions and successfully identify cell types that play important roles in the mouse AD study and the human PTSD study. Hence, we believe that LRcell can provide researchers important and new biological insights in terms of the source of the biological changes at the (sub-)cell-type level, without the need of conducting costly and laborious scRNA-seq experiments.
Our findings from both simulated data as well as real data suggest that LRcell is complementary to cell-type deconvolution methods. Therefore, we recommend including LRcell to bulk RNA-seq analysis to gain a holistic understanding of changes occur at the (sub-)cell-type level inside complex tissues.
Methods
Basic assumptions
The goal of LRcell is to identify the most affected (sub-)cell type(s) during the transition of experimental conditions using only bulk transcriptomic data. Based on the assumptions that cell-type-specific marker genes of key cell types tend to be overrepresented among the significant DEGs in bulk transcriptomic studies, LRcell can discover which cell type(s) is involved in certain disease or condition change. In recent years, computational methods have been developed to deconvolve bulk RNA-seq data to delineate cell-type proportion changes, which could be borrowed to answer the same question. However, whenever there are more (sub-)cell types, the results from deconvolution methods become unreliable. In contrast, LRcell enables comparison across many more cell types which is important for complex tissues such as brain.
The development of LRcell is inspired by LRpath [29], which is designed for linking experimental changes to biological pathways or a predefined gene set. In LRcell, we treat cell-type-specific marker genes as gene sets and calculate the enrichment of each cell type when comparing two biological conditions. We believe that the most enriched cell type(s) is highly likely to play an important role in the experimental condition change.
scRNA-seq data preprocessing
In this study, we include marker genes from mouse whole brain, human prefrontal cortex (pFC) and human PBMC, along with 66 cell-types’ markers from four tissues (midbrain, cord blood, ovary and skeletal muscle) adopted from MSigDB. For each scRNA-seq dataset, we first retrieve raw read count matrix. Next, we filter out low-quality cells and genes and apply column-wise normalization and log transformation on the data.
The mouse whole brain scRNA-seq dataset [18] produced using the Drop-seq technology [33] contains nine brain regions from adult mice. The data provided has already been prefiltered by the authors. For cell types other than neurons, we directly utilize the information provided on the study website (http://dropviz.org/). For neurons and interneurons, we curate the (sub-)cell types following the original study.
The human pFC scRNA-seq dataset [34], produced by 10X Genomics Chromium, is derived from the pFC region (specifically BA9). The dataset contains two conditions: healthy controls and major depressive disorder. We split the data matrix into two parts and filter out cells expressing less than 10 genes and genes expressed in less than 10 cells, respectively. We also filter out mitochondrial, ribosomal genes and genes from annotation clusters (Astros_1, Mix_1, Mix_2, Mix_3, Mix_4, Mix_5 and Inhib_4_SST).
The PBMC dataset [22], generated by CITE-seq technology [35], is derived from an HIV vaccine trial study which involves eight volunteers at three time points: immediately before, three days and seven days after the vaccine. The study contains 161 764 cells in total. To accelerate the marker gene selection, we separate the count matrix according to the time label and filter out low-quality cells and genes (mitochondrial, ribosomal genes and those expressed in less than 1000 cells). The cluster annotated as ‘Doublet’ is filtered out.
Marker gene selection
After obtaining the log-normalized gene expression matrix along with high-quality (sub-)cell-type clusters, we calculate the enrichment scores for each (sub-)cell type using the marker gene selection method described in Marques et al. [15]. The cluster-specific gene enrichment is defined as the average gene expression levels of cells in that cluster divided by the average gene expression levels in all cells. The enrichment score is adjusted by introducing a penalty representing the fraction of cells in that cluster expressing the marker gene. Combined, this score allows the identification of genes with cluster-specific high expression values to be selected as marker genes. The description below is adapted from the original publication.
Suppose there are a total of M genes, L different clusters each with 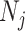 cells and the total number of cells are
cells and the total number of cells are 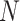 . Let
. Let 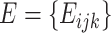 represent the gene by cell read count matrix. Here
represent the gene by cell read count matrix. Here 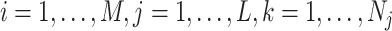 and
and 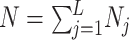 . The overall average expression of the ith gene across all cells is defined as
. The overall average expression of the ith gene across all cells is defined as
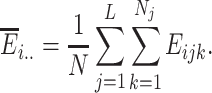 |
The average expression of gene i in the jth cluster is defined as
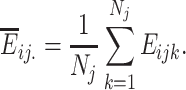 |
The enrichment for gene i in the jth cluster as
 |
Next, we consider the proportion expressing the gene i in the jth cluster as
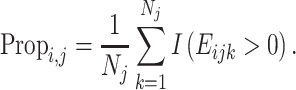 |
The 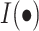 is an indicator function.
is an indicator function.
The enrichment score for gene i in the jth cluster is computed as
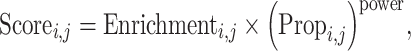 |
where ‘power’ is a hyperparameter to be tuned manually to control the penalization for the cell cluster proportion term. The power parameter is set to 1 throughout this study. After calculating the weighed gene enrichment scores in each cluster, we ranked genes based on the scores and selected the top 100 genes as the marker genes for each cluster.
MSigDB marker genes
We download cell marker gene sets from MSigDB category C8—cell type signature gene sets. Since not all tissue types are suitable for LRcell, we apply the following criteria to select tissues: (i) nonfetal tissues; (ii) have more than eight (sub-)cell types; (iii) minimum number of marker gene greater than 50 and (iv) median number of marker genes greater than 80. In the end, four tissue types—the midbrain, cord blood, ovary and skeletal muscle—remain.
Simulation strategy
Simulated scRNA-seq data are generated using scDesign2 [17], which is capable of generating synthetic scRNA-seq data using intrinsic statistical parameters learned from real scRNA-seq datasets. We generate three synthetic scRNA-seq dataset as control samples using parameters learned from the adult mouse FC scRNA-seq data. The (sub-)cell types used in the simulation study are summarized in Supplementary Table S2, Supplementary data are available online at https://academic.oup.com/bib. For the three case samples, we use the same statistical model but either alter the expression level of selected genes or the proportion of one (sub-)cell type. We then sum up corresponding read counts to obtain the bulk RNA-seq data and use them to detect DEGs between case and control samples. For LRcell analysis, we use the marker genes computed from the original scRNA-seq dataset as input. For MuSiC analysis, we use the three control replicates as the reference scRNA-seq dataset.
For implementing the scenario where only DEGs occur, we first generate three control replicates and randomly select 1000, 2000 and 3000 out of 29 653 genes. We then either double or halve the gene expressions of those genes in the specific (sub-)cell type being tested. To add certain noises, we use a normal distribution with SD as 0.1 to generate random fold change which fluctuates around 2 or 0.5.
As for the scenario where only proportion changes, we directly use the parameter named cell_type_prop from the function simulate_count_scDesign2() to change the simulated proportions. We decide two different proportion distributions when there are five (sub-)cell types: one is evenly distributed with all (sub-)cell types having 20% proportion and the other one is unevenly distributed with 40, 30, 10, 10 and 10%. When testing for the robustness of LRcell under more (sub-)cell types, we only use even distribution on cell-type proportions for illustration purpose.
Bulk RNA-seq data preprocessing
The raw count of mouse bulk RNA-seq study on neurodegenerative dementia is downloaded from Gene Expression Omnibus (GEO) (Accession number: GSE90693). DE analysis is performed using DESeq2 [36] to obtain DEGs in each brain region.
The raw count of bulk RNA-seq study on PTSD is downloaded from Recount2 [37]. We extract out the experiment contrasting PTSD cases and healthy controls with time point of preemployment and perform DESeq2 to obtain DEGs.
LRcell analysis
LRcell is inspired by LRpath, which was designed for identifying sets of predefined gene sets that show enrichment with differentially expressed transcripts in microarray experiments. LRcell uses logistic regression (LR) or linear regression to assess whether marker genes (as defined in the Marker Gene Selection subsection above) of a specific cell type are more likely to be DEGs in a particular bulk RNA-seq study. The linear regression option is added to handle the continuous enrichment scores of marker genes. Users can choose accordingly. To facilitate our analysis, we assume that the major (sub-)cell types of the tissue their marker genes are known a priori.
We apply LRcell to each cell type independently. The required input includes a list of DEGs ranked by the level of significance and a set of marker genes for each cell type. Then, LRcell runs a LR as
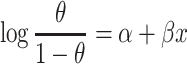 |
and
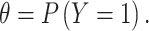 |
In which 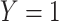 denotes that gene is a marker gene and
denotes that gene is a marker gene and 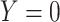 otherwise. Hence,
otherwise. Hence, 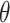 represents the chance that the gene is a marker gene. We use
represents the chance that the gene is a marker gene. We use 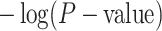 as the explanatory variable
as the explanatory variable 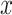 . Whether a specific cell type is involved in the experimental condition change is evaluated by testing the null hypothesis that
. Whether a specific cell type is involved in the experimental condition change is evaluated by testing the null hypothesis that 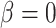 against the alternative that
against the alternative that 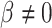 using the Wald test. Typically, we run LRcell on all (sub-)cell types found in the tissue to see which (sub-)cell type(s) drives the changes.
using the Wald test. Typically, we run LRcell on all (sub-)cell types found in the tissue to see which (sub-)cell type(s) drives the changes.
Similar to LR, linear regression directly performs
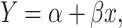 |
where Y indicates the enrichment scores of genes. Same as LR, the P-value can be obtained from testing the null hypothesis that 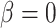 against the alternative that
against the alternative that 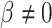 using the t-test.
using the t-test.
Once the P-values are obtained, we calculate false discover rate (FDR) using P adjust() function in R to adjust P-values with Benjamini–Hochberg method.
Input and output
LRcell requires two inputs: (i) a ranked list of genes with DE P-values in a bulk RNA-seq experiment and (ii) sets of marker genes from all (sub-)cell types of the bulk tissue acquired from scRNA-seq datasets a priori or from MSigDB C8—cell-type signature gene sets. For those cell markers derived from scRNA-seq datasets, we offer choices for users to choose between species as human or mouse and the region indicates the specific brain region or PBMC. For MSigDB cell markers, we store the marker genes into the LRcellTypeMarkers packages which can be easily downloaded. When running LRcell() function, the LR option is set as the default, while users can also set the method option as LiR if linear regression is desired. For linear regression, an enrichment score is needed as input for each gene whereas gene sets are sufficient for LR. For MSigDB cell-type signature gene set, LR option is recommended as there is no enrichment score information available. For customized input, i.e. a scRNA-seq data, we offer a LRcell_gene_enriched_scores function which takes the read counts matrix and cell annotation as input to generate enrichment scores for genes in each cell type. For further subsetting, get_markergenes can be used for generating marker genes for more specific (sub-)cell types.
The output is a list of significance P-value (or FDR), one for each (sub-)cell type. For visualization, LRcell produces Manhattan plot, which can be drawn through plot_manhattan_enrich function. We also provided a plot (plot_marker_dist function) indicating where certain cell-type-specific marker genes locate on the bulk DEGs. The bulk DEGs are sorted using log10(P-value) × sign(log2FoldChange) which could potentially give information on both up/downregulated directions. More detailed information about LRcell is available at http://bioconductor.org/packages/release/bioc/html/LRcell.html.
LRcell requires an R version beyond 4.1 and a prerequisite installation of BiocManager.
Authors’ contributions
W.M. and Z.S.Q. conceived the idea and supervised the study. W.M. realized the method and developed the R package. W.M. analyzed the real data and conducted the computational experiments. S.S. provided guidance on validating biological experiments and tested the package. W.M. and Z.S.Q. summarized the results and wrote the manuscript. All authors have read and approved the manuscript.
Key Points
We present a novel computational method named LRcell aiming to identify specific (sub-)cell type(s) that drives the changes observed in a bulk RNA-seq experiment.
LRcell provides pre-embedded marker genes of multiple tissues computed from single-cell RNA-seq experiments as options to execute the analyses.
Using real datasets, we show that LRcell successfully identifies known cell types involved in neurodegenerative dementia and posttraumatic stress disorder.
LRcell is computational efficient, capable of handling a large number of different (sub-)cell types and is complementary to cell-type deconvolution methods in the analysis of bulk RNA-seq data.
Supplementary Material
Author Biographies
Wenjing Ma is a PhD. student at the Department of Computer Science, Emory University. Her research interest is applying machine learning techniques to single-cell sequencing data analysis.
Sumeet Sharma is a resident physician in the Department of Psychiatry and Behavioral Sciences, Emory University School of Medicine.
Peng Jin is a Professor at the Department of Human Genetics, Emory University School of Medicine.
Shannon L. Gourley is an Associate Professor at the Department of Pediatrics and Department of Psychiatry and Behavioral Science, Emory University School of Medicine and a Researcher at Yerkes National Primate Research Center, Emory University.
Zhaohui S. Qin is a Professor at the Department of Biostatistics and Bioinformatics, Rollins School of Public Health, Emory University.
Funding
This work was partially supported by the National Institutes of Health R01 MH117103 and R01 DA044297 to S.L.G.; and the National Institutes of Health U01 MH116441 to P.J.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Data availability
The datasets analyzed during the current study are available in the GEO with following accession numbers: mouse whole brain [18] (GSE116470), human pFC [34] (GSE144136), human PBMC [22] (GSE164378), the mouse AD study [19] (GSE90696) and the human PTSD study [21] (GSE64814). The R package is freely available on Bioconductor (https://doi.org/doi:10.18129/B9.bioc.LRcell) and the external marker genes are stored in another R package named LRcellTypeMarkers on Bioconductor (https://doi.org/doi:10.18129/B9.bioc.LRcellTypeMarkers).
References
- 1. Newman AM, Steen CB, Liu CL, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol 2019;37:773–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mathys H, Davila-Velderrain J, Peng Z, et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019;570:332–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ruzicka WB, Mohammadi S, Davila-Velderrain J, et al. Single-cell dissection of schizophrenia reveals neurodevelopmental-synaptic axis and transcriptional resilience. In: medRxiv. Cold Spring Harbor Laboratory Press, 2020. [Google Scholar]
- 4. Newman AM, Liu CL, Green MR, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Method 2015;12:453–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gong T, Szustakowski JD. DeconRNASeq: a statistical framework for deconvolution of heterogeneous tissue samples based on mRNA-seq data. Bioinformatics 2013;29:1083–5. [DOI] [PubMed] [Google Scholar]
- 6. Wang X, Park J, Susztak K, et al. Bulk tissue cell type deconvolution with multi-subject single-cell expression reference. Nat Commun 2019;10:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tsoucas D, Dong R, Chen H, et al. Accurate estimation of cell-type composition from gene expression data. Nat Commun 2019;10:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gaujoux R, Seoighe C. Semi-supervised non-negative matrix factorization for gene expression deconvolution: a case study. Infect Genet Evol 2012;12:913–21. [DOI] [PubMed] [Google Scholar]
- 9. Li Z, Wu H. TOAST: improving reference-free cell composition estimation by cross-cell type differential analysis. Genome Biol 2019;20:190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhong Y, Wan Y-W, Pang K, et al. Digital sorting of complex tissues for cell type-specific gene expression profiles. BMC Bioinform 2013;14:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jin H, Liu Z. A benchmark for RNA-seq deconvolution analysis under dynamic testing environments. Genome Biol 2021;22:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cobos FA, Alquicira-Hernandez J, Powell JE, et al. Benchmarking of cell type deconvolution pipelines for transcriptomics data. Nat Comm 2020;11:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kiselev VY, Kirschner K, Schaub MT, et al. SC3: consensus clustering of single-cell RNA-seq data. Nat Method 2017;14:483–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. PNAS 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Marques S, Zeisel A, Codeluppi S, et al. Oligodendrocyte heterogeneity in the mouse juvenile and adult central nervous system. Sci Am Assoc Adv Sci 2016;352:1326–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liberzon A, Birger C, Thorvaldsdóttir H, et al. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst 2015;1:417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sun T, Song D, Li WV, et al. scDesign2: a transparent simulator that generates high-fidelity single-cell gene expression count data with gene correlations captured. Genome Biol 2021;22:1–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Saunders A, Macosko EZ, Wysoker A, et al. Molecular diversity and specializations among the cells of the adult mouse brain. Cell 2018;174:1015–1030.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Swarup V, Hinz FI, Rexach JE, et al. Identification of evolutionarily conserved gene networks mediating neurodegenerative dementia. Nat Med 2019;25:152–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Perry VH, Nicoll JA, Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol 2010;6:193. [DOI] [PubMed] [Google Scholar]
- 21. Breen MS, Maihofer AX, Glatt SJ, et al. Gene networks specific for innate immunity define post-traumatic stress disorder. Mol Psychiatry 2015;20:1538–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hao Y, Hao S, Andersen-Nissen E, et al. Integrated analysis of multimodal single-cell data. Cell 2021;184:3573–3587.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ong S-M, Teng K, Newell E, et al. A novel, five-marker alternative to CD16–CD14 gating to identify the three human monocyte subsets. Frontiers in immunology. Frontiers 2019;10:1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kratofil RM, Kubes P, Deniset JF. Monocyte conversion during inflammation and injury. Arterioscler Thromb Vasc Biol 2017;37:35–42. [DOI] [PubMed] [Google Scholar]
- 25. Kuan, Yang X, Clouston S, et al. Cell type-specific gene expression patterns associated with posttraumatic stress disorder in World Trade Center responders. Transl Psychiatry 2019;9:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Law CW, Chen Y, Shi W, et al. Voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol BioMed Central 2014;15:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ritchie ME, Phipson B, Wu DI, et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43:e47–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Segerstolpe Å, Palasantza A, Eliasson P, et al. Single-cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes. Cell Metab 2016;24:593–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sartor MA, Leikauf GD, Medvedovic M. LRpath: a logistic regression approach for identifying enriched biological groups in gene expression data. Bioinformatics 2009;25:211–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Qiu Y, Wang J, Lei J, et al. Identification of cell-type-specific marker genes from co-expression patterns in tissue samples. Bioinformatics 2021;37:3228–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. El Amrani K, Alanis-Lobato G, Mah N, et al. Detection of condition-specific marker genes from RNA-seq data with MGFR. PeerJ PeerJ Inc 2019;7:e6970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang X, Lan Y, Xu J, et al. CellMarker: a manually curated resource of cell markers in human and mouse. Nucleic Acids Res 2019;47:D721–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Macosko EZ, Basu A, Satija R, et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 2015;161:1202–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nagy C, Maitra M, Tanti A, et al. Single-nucleus transcriptomics of the prefrontal cortex in major depressive disorder implicates oligodendrocyte precursor cells and excitatory neurons. Nat Neurosci 2020;23:771–81. [DOI] [PubMed] [Google Scholar]
- 35. Stoeckius M, Hafemeister C, Stephenson W, et al. Simultaneous epitope and transcriptome measurement in single cells. Nat Method 2017;14:865–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Collado-Torres L, Nellore A, Kammers K, et al. Reproducible RNA-seq analysis using recount2. Nat Biotechnol 2017;35:319–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets analyzed during the current study are available in the GEO with following accession numbers: mouse whole brain [18] (GSE116470), human pFC [34] (GSE144136), human PBMC [22] (GSE164378), the mouse AD study [19] (GSE90696) and the human PTSD study [21] (GSE64814). The R package is freely available on Bioconductor (https://doi.org/doi:10.18129/B9.bioc.LRcell) and the external marker genes are stored in another R package named LRcellTypeMarkers on Bioconductor (https://doi.org/doi:10.18129/B9.bioc.LRcellTypeMarkers).