

Abstract
Epithelial polyploidization after injury is a conserved phenomenon recently shown to improve barrier restoration during wound healing. Whether lung injury can induce alveolar epithelial polyploidy is not known. We show that bleomycin injury induces alveolar type 2 cell (AT2) hypertrophy and polyploidy. AT2 polyploidization is also seen in short term ex vivo cultures, where AT2-to-AT1 transdifferentiation is associated with substantial binucleation due to failed cytokinesis. Both hypertrophic and polyploid features of AT2 cells can be attenuated by inhibiting the integrated stress response using the small molecule ISRIB. These data suggest that AT2 hypertrophic growth and polyploidization may be a feature of alveolar epithelial injury. Because AT2 cells serve as facultative progenitors for the distal lung epithelium, a propensity for injury-induced binucleation has implications for AT2 self-renewal and regenerative potential upon reinjury, which may benefit from targeting the integrated stress response.
Keywords: polyploidy, integrated stress response, alveolar epithelial cell, cytokinesis, hypertrophy
Clinical Relevance
Cell polyploidy is emerging as an inducible and variable feature of tissues, where it is linked to disease physiology across a range of tissue types and organisms. Because of the complex three-dimensional organization of the lung, alveolar epithelial polyploidy has been unrecognized, and its contribution to alveolar homeostasis and repair is unknown. This study is the first, to our knowledge, to describe alveolar type 2 cell polyploidization after injury and its attenuation by an inhibitor of the integrated stress response. We discuss implications of this phenomenon for alveolar epithelial adaptation, heterogeneity, and disease.
Genome-wide association and classical genetic studies of families with pulmonary fibrosis implicate disordered epithelial repair as a driver of lung fibrosis (1–3). Although this hypothesis has been confirmed in mouse models of fibrosis that localize fibrosis-promoting effects of global mutations to the lung epithelium (4–6), the sequence of cellular events that drive fibrosis persistence remains unclear. Understanding how the distal lung epithelium repairs after injury is also relevant to patients with severe lung injury induced by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pneumonia, in which some will develop persistent and progressive lung fibrosis (7–12). Thus, although it is clear surfactant-producing alveolar type 2 pneumocytes (AT2 cells) are progenitors that give rise to alveolar type 1 pneumocytes (AT1 cells) (13, 14), the variability between individuals in the AT2-to-AT1 cell transdifferentiation program to restore normal alveolar function is unexplained.
Pathologists have long recognized that a range of injurious stimuli lead to the accumulation of abnormally large AT2 cells, described as “hypertrophic or hyperplastic” AT2 cells (15–18). Morphologically similar cells are seen in autopsy specimens or lung explants from patients with lung injury secondary to SARS-CoV-2 pneumonia (7, 11, 19), but mechanisms underlying this cell state and its consequences for lung repair after injury remain puzzling. A recent convergence of single-cell transcriptomic studies from a range of mouse injury models suggests that instead of fully progressing to an AT1 cell fate, AT2 cells become stalled at an intermediate transition state characterized by activation of cell cycle arrest pathways (p16, p21, p53), transforming growth factor-β signaling, and matrix remodeling (SERPINE1) (20–23). Serially collected single-cell RNA-sequencing data over the course of bleomycin-induced injury shows that the formation of this intermediate transition state is preceded by increased expression of genes in the integrated stress response (ISR) (22). In tissue sections, these transitional cells are characterized by elevated levels of specific intermediate filament keratins (KRT8/18 in mice; KRT17 in humans) and other structural proteins, together with an intermediate AT2-to-AT1 morphology (22, 23). These data suggest that small cuboidal AT2 cells differentiate into large and flattened AT1 cells through a morphologically stressful transition that appears prone to stalling after injury. Importantly, recent evidence suggests the AT2-to-AT1 transition state is not fixed, because therapeutic targeting of p53 or ISR signaling appears to guide AT2 cells through this vulnerable transdifferentiation step (23, 24). However, the cellular processes most vulnerable to this morphogenetic transition, which drive persistence of the AT2-to-AT1 transition state in injured lungs and can be reversed by these therapies, are unknown.
Epithelial polyploidization and consequent hypertrophy after injury is a conserved phenomenon observed from flies to vertebrates (25). It is a process by which cells increase their DNA content and bypass mitosis. Polyploid cells are larger than their diploid counterparts because cell size scales with DNA content (26). Cell hypertrophy is advantageous during epithelial repair because larger cells manifest less junctional surface area per unit of epithelium, leading to decreased permeability in an otherwise leaky barrier environment (27, 28). Increased DNA copies can also favor adaptation to cell and environmental stress in ways that are context and injury dependent (29–31). However, these benefits of polyploidization come at a cost because polyploid cells will less faithfully segregate DNA during mitosis (32–34). Thus, polyploidization of differentiated cell types—particularly those that serve also as progenitors (e.g., AT2 cells)—is likely to adversely impact future regenerative potential of the tissue. Whether AT2 polyploidy underlies injury-induced AT2 hypertrophy or is a consequence of hypertrophic growth and drives persistence of the AT2-to-AT1 cell stalled state is unknown.
In this study, we show that lung injury leads to AT2 hypertrophy and polyploidization—notably binucleated AT2s—during the acute injury response. Ex vivo analysis suggests the route to polyploidy is via failed cytokinesis during the AT2-to-AT1 flattening process. A small molecule inhibitor of the ISR, ISRIB, which we have previously shown ameliorates fibrosis development in murine models (24), inhibits the abundance of hypertrophic and polyploid AT2 cells. These data suggest that modulating stress signals during the morphogenetically challenging AT2-to-AT1 transition may improve key steps through which injured AT2 cells divide and differentiate to restore the alveolar epithelium. Implications of failing to remove and replace polyploid AT2 cells after lung injury are discussed. This article was previously published in preprint form (https://doi.org/10.1101/2021.08.10.455833).
Methods
For a list of resources used in this study, see Table E1 in the data supplement.
Mice
C57BL/6 mice were obtained from The Jackson Laboratory (stock no. 000664). SftpcCreER (35), Ai6-R26zsGreen, SftpcCreERT2;R26ReYFP (kindly provided by Ed Morrisey, University of Pennsylvania) (36, 37), and SftpcCreER;R26R-Confetti (14) (stock nos. 028054, 007906, 006148, 013731; The Jackson Laboratory). Only male mice were used in bleomycin injury experiments, based on established injury severity in males relative to females, which improves the dynamic range of assays for comparisons (38). Eight- to 12-week-old mice were used as young adult mice. All experimental protocols were approved by the institutional animal care and use committee at Northwestern University. All strains, including wild-type mice, are bred and housed in a barrier- and pathogen-free facility at the Center for Comparative Medicine at Northwestern University.
Cre Recombination, Bleomycin Administration, and ISRIB Administration
For nuclear activation of Cre-ER, tamoxifen was dissolved in sterile corn oil at 10mg/100 μl and administered via oral gavage once per day for two days to induce Cre recombination of floxed alleles for lineage tracing. For bleomycin injury, mice were anesthetized and intubated before the intratracheal administration of bleomycin (0.025 units in 50 μl of 0.9% saline). Lungs were harvested at the indicated time points for downstream analyses (e.g., fixation and frozen sectioning for immunofluorescence analysis, AT2 cell isolation by negative selection [39], or epithelial cellular adhesion molecule–positive selection [40]) and flow cytometry methods (24, 41) with modifications (see Tissue Preparation, AT2 Isolation, Flow Cytometry and Sorting). ISRIB (A14302, AdooQ BioScience) was first reconstituted in DMSO to a concentration of 4 mg/ml and then further diluted using PBS to a final concentration of 0.3125 mg/ml (42). ISRIB was prepared at 2.5 mg/kg and administered daily through intraperitoneal injection in a volume of 200 μl. Control animals were treated with vehicle (DMSO/PBS) alone.
Tissue Preparation, AT2 Isolation, Flow Cytometry, and Sorting
Tissue preparation for AT2 isolation and analyses was performed as described (24, 41), with modifications. Briefly, mice were killed, and their lungs were perfused through the right ventricle with 10 ml of Hanks’ balanced salt solution. Lungs were removed and infiltrated with dispase and 0.2 mg/ml DNase I dissolved in Hanks’ balanced salt solution with Ca2 + and Mg2+, using a syringe with a 30-gauge needle. Lungs were chopped with scissors and incubated in Dulbecco’s modified Eagle medium containing 5% FBS for 45 minutes with agitation. The tissue suspension was filtered through a 70-μm strainer and washed in 10 ml of medium (Dulbecco’s modified Eagle medium, 5% FBS). The pellet was then suspended in red blood cell (RBC) lysis buffer for 1 minute, washed with medium, and filtered through a 40-μm strainer. The cells were incubated with epithelial cellular adhesion molecule magnetic beads, and AT2 were collected using the MultiMACS Cell24 Separator (Miltenyi Biotec). Automated cell counting was performed using the Nexcelom K2 Cellometer with acridine orange/propidium iodide reagent. Cells were incubated with Fc block (BD Biosciences) and stained with a mixture of fluorochrome-conjugated antibodies, as listed above. DNA content was quantified using Vybrant DyeCycle Violet. Single-color controls were prepared using BD CompBeads (BD Biosciences) and Arc beads (Invitrogen). Flow cytometry and cell sorting were performed at the Northwestern University Robert H. Lurie Comprehensive Cancer Center Flow Cytometry Core facility. Data were acquired on a custom BD FACSymphony instrument using BD FACSDiva software (BD Biosciences). Compensation and analysis were performed using FlowJo software (BD Biosciences). Each cell population was identified using a sequential gating strategy. The percentage of cells in the live/singlets gate was multiplied by the number of live cells using the Cellometer K2 Image cytometer to obtain cell counts. Cell sorting was performed using the BD FACSAria III instrument with a 100-μm nozzle and 40-psi pressure. After AT2 sorting, collected cells were spun at 300 × g for 10 minutes. After resuspension in PBS, cells were plated onto slides. Slides were spun in cytospin filters at 100 × g for 5 minutes, followed by staining with phalloidin (1:50) and Hoechst (1:10,000).
AT2 Isolation for Ex Vivo Two-Dimensional Culture
Lungs were inflated intratracheally with dispase and incubated for 45 minutes with agitation. Lungs were chopped and incubated in 7 ml of media containing 10% FBS and DNase I (1 mg/ml) for 10 minutes. Tissue suspensions were filtered through a 70-μm strainer. The cell pellets were resuspended in RBC lysis buffer for 2 minutes. Alveolar epithelial cells were enriched through negative selection against biotinylated CD16/32, CD45, CD90, and TER119 antibodies. The cell suspensions were then incubated at 37°C for 2 hours followed by gentle panning. Cells were counted and cultured in complete media (10% FBS, 2% penicillin-streptomycin) onto 12-mm glass coverslips. Starting 48 hours after isolation, cells received a daily media change containing either 0.025 μM ISRIB or DMSO. Cell samples were pooled from three mice. Rat AT2 cells were collected as previously described (43). AT2 cell purity was validated by cytospin analysis (as in Figure E2), with staining for SpC, keratin-8, and vimentin. Vimentin+ cells are <3%, and SpC is expressed in ∼95% of the keratin-8 population.
AT2 Organoid Culture
For organoid culture, cells were prepared as previously described (14) with modifications. Mice received tamoxifen 14 days before isolation. AT2 isolation was conducted as described above. For fibroblast isolation, lungs were inflated with 1 ml of collagenase (200 U/ml), elastase (4 U/ml), and DNase solution (0.25 mg/ml). Lungs were cut into pieces and incubated for 30 minutes at 37°C in digestion buffer. Lungs were processed in C-tubes (130-093-237, Miltenyi Biotec) using a GentleMACS/OctoMACS dissociator (Miltenyi Biotec). Suspensions were filtered through a 40-μm strainer and rinsed with magnetic-activated cell sorting buffer. The pellet was resuspended in RBC lysis buffer, washed with magnetic-activated cell sorting buffer, and incubated with CD45 magnetic beads to deplete CD45+ cells. The CD45− cell fraction was collected using the MultiMACS Cell24 Separator (Miltenyi Biotec). Cells were sorted as described above. After sorting, 5 × 103 zsGreen+ AT2 were mixed with 5 × 104 fibroblasts in 50% Matrigel (356231, Corning) and 50% organoid media (α-minimum essential medium, penicillin/streptomycin, insulin/transferrin/selenium, FBS, heparin, amphotericin B, Rock1 inhibitor, l-glutamine) in 0.4-μm Transwell inserts. Cell/Matrigel mixtures were solidified at 37°C for 5 minutes before 300 μl of media was added under the Transwell. Twenty-four hours after isolation, organoids received a media change every other day containing either 1 μM ISRIB or DMSO for 10 days.
Tissue Sectioning, Immunofluorescence, and Image Analysis
After euthanasia and perfusion, the trachea was cannulated with a Luer syringe stub blunt needle, and mouse lungs were inflated with 4% paraformaldehyde (PFA) at 15 cm H2O column pressure. Lungs were fixed in 4% PFA overnight at 4°C followed by cryoembedding in optimal cutting temperature compound. Sections were cut at 14 μm on a cryostat. For immunostaining, sections were blocked in 10% normal goat serum (NGS), 0.3%Triton X-100 in PBS for 30 minutes, followed by an additional 1:10 antimouse Fab (H+L) for 2 hours. Primary antibodies were prepared in 2% NGS, 0.3%Triton X-100 PBS, and applied to sections overnight at 4°C. Sections were washed in PBS followed by secondary antibodies for 30 minutes. Last, sections were incubated in Hoechst (1:10,000) for 5 minutes followed by a brief incubation of 0.3% Eriochrome Black before mounting. Primary antibodies were used at concentrations described below. For two-dimensional culture, cells were fixed 4 days after isolation in 4% PFA. For the alveolar organoids, organoids were fixed 10 days after isolation. Fixed cells and organoids were neutralized in 10 mM glycine followed by blocking in 10% NGS in 0.3% Triton X-100 PBS. Primary antibodies were prepared in 3% NGS in 0.3% Triton X-100 PBS and applied for 1 hour at room temperature. After PBS washes, fluorophore-conjugated secondary antibodies were prepared at 1:300 for 30 minutes in the dark at room temperature. Hoechst was diluted 1:10,000 and applied for 5 minutes at room temperature after secondary antibody incubation. Coverslips and filters were mounted using ProLong Gold antifade solution. Images were obtained using Zeiss Axioplan epifluorescence and Nikon W1 spinning disk confocal microscopes. For nuclear area quantification, five fields of view were selected for each condition per biological replicate. Images were obtained on a Zeiss microscope and analyzed in Fiji using the StarDist plugin. Primary antibodies used were as follows: SFTPC (1:100), β-catenin (1:100), podoplanin (1:30), Krt8 (1:30), and F-actin (1:50). For thick section imaging with tissue clearing, lungs were sliced in PBS using a vibratome (Leica VT1200S) in 200-μm-thick sections. Tissue slices were cleared using the LifeCanvas Technologies SHIELD method. For three-dimensional volumetric analysis, 200-μm-thick sections were imaged sequentially in 0.5-μm steps on a Nikon W1 spinning disk confocal microscope. Cell volumes were measured using AT2 lineage label (yellow fluorescent protein) via Imaris.
Live-Cell Imaging
Freshly isolated alveolar epithelial cells were plated at 500,000 per well in a 12-well plate. Forty-eight hours after isolation, medium was changed, and adherent cells were imaged every 15 minutes on a Biostation-CT system (Nikon Instruments) for a total of 5 hours. Image sequences were analyzed in Fiji (ImageJ version 2.1.0/1.53c).
Statistical Analysis
Data analysis and statistical tests were performed using GraphPad Prism software (version 9.2.0).
Results
Lung Injury Induces AT2 Hypertrophy
To understand how lung injury alters the morphogenetic sequence by which AT2 cells grow, divide, and differentiate to an AT1 fate, we used a genetic lineage-tracing system to permanently label AT2 cells in SftpcCre-ER;Lox-Stop-Lox zsGreen mice subjected to the bleomycin model of injury and fibrogenic repair (24) (Figure 1A). As with previous studies (15–18), we confirm that bleomycin injury leads to zsGreen+ cells that are larger than AT2 lineage-labeled mice exposed to saline (Figures 1B and 1C, quantification from two mice, five fields of view; frozen section image analysis pipeline in Figure E1A in the online supplement). Cell size difference may be more readily apparent in 14-μm-thick frozen sections than thinner 4-μm paraffin sections, likely because the former method favors cell visualization along a greater portion of the curved alveolus (Figure E1B). The injury-induced size increase is not an aberrant feature of cells expressing the fluorescence lineage label, because AT2 cells lacking zsGreen but expressing surfactant protein C (SpC) also increase in size (Figure E1C). Because standard sectioning (i.e., planimetry) is known to underestimate AT2 cell expansion after injury (44, 45), we also confirmed AT2 hypertrophic growth using cleared, thick-section (200-μm) confocal imaging with three-dimensional reconstruction (Figure 1D, quantification from three mice; see Video 1). Last, we can detect this size increase by flow cytometric analysis of AT2 cells (Figures E2A–E2C; median forward scatter area quantification from three mice per condition), demonstrating the injury-induced phenotype is not likely an artifact of lung fixation or processing.
Figure 1.

Lung injury induces alveolar type 2 cell (AT2) hypertrophy. (A) Experimental design. Young adult SftpcCreER; Lox-Stop-LoxZsGreen mice (3–5 mo) received 10 mg of tamoxifen via oral gavage 22 days before administration of bleomycin (0.025 units i.t.). Lungs from saline-treated (control) and bleomycin-treated mice were harvested on Day 10, fixed, and processed for frozen sectioning and immunofluorescence analysis. (B) Representative lung sections showing native GFP fluorescence from AT2-lineage label and immunofluorescence staining of alveolar type 1 (AT1) cells by podoplanin (Pdpn; magenta). Scale bars, 50 μm. Note lineage-labeled AT2 cells appear larger after bleomycin injury. (C) Quantification of AT2 cell area and perimeter using image analysis in Fiji (workflow in Figure E1A). Data points are from two mice, five fields of view per condition. Mean ± SD; ****P < 0.0001 by t test. (D) Quantification of lineage-labeled AT2 cell volumes using cleared, 200-μm-thick sections for optical z-stack imaging (Imaris). Data points are from three saline-treated and two bleomycin-injured mice. Mean ± SD; **P = 0.0072 by t test. Representative fields of view shown in Video 1. To ensure robustness and increase counts, injury-induced cell size increase was also validated by flow cytometry in Figure E2. Bleo = bleomycin.
Lung Injury Promotes AT2 Cell Polyploidization
Because cell size can scale with DNA context (26), we sought to examine nuclear features of hypertrophic AT2 cells. Interestingly, we found a number of lineage-labeled AT2 cells with two nuclei (Figure 2A), often observing one or two binucleated zsGreen+ cells per field of view, estimating a rate of ∼8% (Figure 2B; see also Figure 1B, inset box). To validate AT2 polyploidy via an independent method, we sought to quantify and characterize these cells by flow cytometry paired with DNA content analysis (Figures E2A–E2E). Although we readily detected AT2 cells with 2N or 4N DNA content, too few 4N cells survived the cytospin procedure to reliably quantify binucleation rate differences between saline- and bleomycin-treated mice (Figure E2E). Methodological improvements will be required to reliably isolate and quantify this apparently fragile cell state, particularly given that flow cytometry is known to underestimate expansion of AT2 numbers after injury (44).
Figure 2.

Lung injury induces polyploidization of lineage-labeled and surfactant protein C–positive (SpC+) AT2 cells. (A) Experimental design as in Figure 1. Representative confocal images of lung sections from bleomycin-treated mice; two independent fields of view from low- and high-magnification insets (boxed region) are shown (a' and a", respectively). Native GFP fluorescence from AT2-lineage label and immunofluorescence staining of AT1 cells by Pdpn (magenta). DNA labeling by Hoechst (gray). Scale bars, 50 μm and 10 μm (inset). Asterisks show suspected binucleated AT2 cells. Arrows show plan of optical z-section, revealing that nuclei appear to touch in these cells. Magenta arrow in a' indicates Pdpn signal. It is not possible in this image to know if Pdpn labels the binucleated AT2 or an adjacent AT1 cell (see Figure 6D for evidence of binucleated/Pdpn-positive cell). SpC+ binucleated AT2 cells are shown in Figure E2F. (B) Quantification of AT2 cell binucleation. Data points are from three mice (different symbol colors/mouse), five to eight fields of view per condition. Note the binucleation rate for one mouse (gray circles) was quantified 21 days after bleomycin, showing persistence of binucleation through this time point. Mean ± SD in red. ****P < 0.0001 by t test. Injury-induced binucleation was also interrogated by flow cytometry in Figures E2A–E2E.
Of interest, AT2 binucleation appears compatible with expression of the AT2 marker SpC (Figure E2F) as well as the AT1 marker podoplanin (see Attenuating the ISR Inhibits AT2 Hypertrophy section). Together with recent evidence that genetically induced AT2 binucleation is compatible with Hopx expression (46), these data suggest that alveolar epithelial polyploidy may be compatible with either the AT2 or AT1 cell state. To address whether AT2 polyploidy is a typical feature of the recently described AT2-to-AT1 cell transition state (21, 22), we interrogated Krt8 expression in our lung sections. Although we can find large binucleated cells that highly express Krt8 protein, AT2 binucleation does not exclusively correlate with a Krt8-high state (Figure 3). Remarkably, binucleation is a common feature of primary AT2 cultures grown on glass or filters (Figure 4). We are confident these ex vivo polyploid cells derive from bona fide AT2 cells, because they are uniformly Krt8 positive (Figure 4A) and can be lineage labeled using the Sftpc-CreER;Lox-stop-Lox eYFP system (Figures E4A and E4B). Even AT2-derived organoids, which can show AT2-to-AT1 cell transition state features seen in injured lungs (23), exhibit a binucleation rate similar to in vivo estimates (Figures 4B–4D vs. Figure 2B). Mechanistically, live-cell imaging reveals that binucleated cells can be generated via failed cytokinesis, further suggesting that how AT2 polyploidy is generated and maintained can be interrogated in ex vivo cultures (Figure 5).
Figure 3.

AT2 cell binucleation is not universally associated with Krt8-high expression phenotype. (A) Experimental design as in Figure 1. Confocal image of lung section from bleomycin-treated mice; same image as Figure 2A, now including Krt8 immunofluorescence channel (cyan). Asterisk denotes binucleated AT2 cell with normal levels of Krt8 protein. Native GFP fluorescence from AT2-lineage label and immunofluorescence staining of AT1 cells by Pdpn (magenta). DNA labeling by Hoechst (gray). (B) Confocal image of binucleated Krt8-high cell (asterisk) lacking AT2 lineage label. Scale bars, 50 μm and 25 μm (inset). (C) Confocal image of lineage-labeled binucleated AT2 cell adjacent to lineage-negative mononuclear Krt8-high cell (z-stack not shown). Scale bar, 25 μm. Krt8 = keratin 8.
Figure 4.

Binucleation is a feature of murine AT2 cell cultures and organoids. (A) Top: Confocal images of mouse AT2 cells grown 4 days on glass and rat AT2 cells grown 10 days on filters. Cells were fixed and stained with Hoechst (DNA in cyan or gray), phalloidin (F-actin, green), Krt8 (green), or β-catenin (red) as indicated. Top: Arrows (white) indicate two nuclei “kissing” without an intervening junction (by F-actin or β-catenin staining). Bottom: Dashed lines (white) outline binucleated cells. (B) Activation of AT2-lineage label before AT2 isolation for organoid growth. (C) AT2 cell (green), F-actin (magenta), and DNA (gray) with lineage-labeled binucleated cell (arrow). (D) Rate of AT2 binucleation in organoids. Scale bars, 50 μm. β-cat = β-catenin.
Figure 5.

AT2 cell binucleate via failed cytokinesis. Mouse AT2 cells were isolated and phase-contrast live imaged every 15 minutes (see Methods). Time stamp and evident cell-cycle phases are shown in cyan. Arrows indicate a dividing cell.
Given limitations of assessing polyploidy from frozen or thick sectioning (Figure E1B), together with evidence that mononuclear polyploidy also exists (47), we sought an independent method to assess the polyploidy rate in injured AT2 cells. A recent study showed that the stochastic, multicolor Confetti reporter system can be used to assess mononuclear polyploidy in mouse tissues, notably in the liver (48). Although the Confetti reporter mouse is better known for tracing clonal cell relationships in development and injury (14, 49), this tool can also validate polyploid cells in tissues with single cells expressing two or more fluorescent proteins (Figure E3A). In contrast to cell clone tracing, the lineage label must be activated by tamoxifen after genome duplication to detect single-cell polyploidy. By subjecting SftpcCre-ER × RosaConfetti mice to the bleomycin model of injury, we could identify large, partially flattened cells that express both GFP and red fluorescent protein lineage labels surrounding what appears as a single nucleus of lineage label exclusion (Figures E3B and E3C). We also identified small, round, GFP/red fluorescent protein dual-positive cells that likely reflect lineage-labeled AT2 cells that have doubled their DNA content and arrested at the G2 phase of the cell cycle (Figure E3D). Because these dual-positive cells appear rarer than the AT2 binucleation rate estimated in Figure 1 (e.g., <2% lineage-labeled AT2 cells), we may be missing polyploidization events during the AT2-to-AT1 transition, when SftpcCre expression is likely waning. Crossing the RosaConfetti reporter strain to a Cre driver whose expression increases during the AT2-to-AT1 transition may be required to assess the full extent of alveolar epithelial polyploidization during the lung injury–repair sequence. Nonetheless, these data show that lung injury can generate binucleated polyploid AT2 cells (Figure 1), and possibly polyploid mononuclear AT2 cells (Figure E3C), where polyploidization likely occurs during the postinjury reparative phase of the bleomycin model.
Inhibiting the ISR Attenuates AT2 Hypertrophy
To determine whether injury-induced AT2 hypertrophy and polyploidy can be inhibited or reversed, we considered small-molecule approaches known to target stress-activated pathways, recently suggested to guide injured AT2 cells through their vulnerable transition toward an AT1 fate (23, 24). The ISR is a highly conserved signaling network that helps cells adapt to a range of environmental stresses through activating kinases that inhibit bulk protein translation in favor of the translation of select mRNAs encoding transcription factors such as ATF4 that promote the transcription of molecular chaperones and other stress-adaptive genes (50). A central integrator of these diverse stress-responsive kinases is the eukaryotic initiation factor 2 (eIF2) complex, where phosphorylation of eIF2α at a single serine residue limits translation (51). In this way, eIF2α phosphorylation and its diverse downstream consequences compose the ISR (52, 53). ISRIB was found to render cells insensitive to eIF2α phosphorylation, thereby restoring translation capacity to normalize cell function (54–56) and limit a range of tissue pathologies in murine disease models (57–60). Our team recently discovered that ISRIB reduces fibrosis in mouse models (24). Because ISRIB therapy limited abundance of lineage-labeled AT2 cells coexpressing KRT8 but increased the abundance of lineage-labeled cells expressing podoplanin, we reason that ISRIB may attenuate fibrosis by limiting the abundance of AT2 cells stalled at the AT2-to-AT1 transition state, favoring their full transition to an AT1 fate (24). Indeed, evidence that bleomycin-injured AT2 cells show activation of ISR-associated genes during this transdifferentiation step (24) further suggests that persistent activation of the ISR in AT2 cells can thwart alveolar epithelial repair. However, the particular morphogenetic features of the AT2-to-AT1 transition that drive the persistence of this stress state remain unclear. We find that bleomycin-injured SftpcCre-ER; Lox-Stop-Lox zsGreen mice treated with ISRIB show complete inhibition of AT2 hypertrophy and no evidence of binucleation (Figures 6A and 6B). Although ISRIB did not prevent AT2 cell binucleation in ex vivo cell cultures, ISRIB treatment did limit the rate of binucleation, as well as formation of the largest cells, inferred from nuclear size within these cells (Figures 6C–6G). These data suggest that injury-induced AT2 hypertrophy and polyploidy are sensitive to inhibition of the ISR. Evidence for modest cell size attenuation in ex vivo AT2 cultures suggests that ISRIB may limit hypertrophic growth through an AT2 cell-autonomous mechanism.
Figure 6.
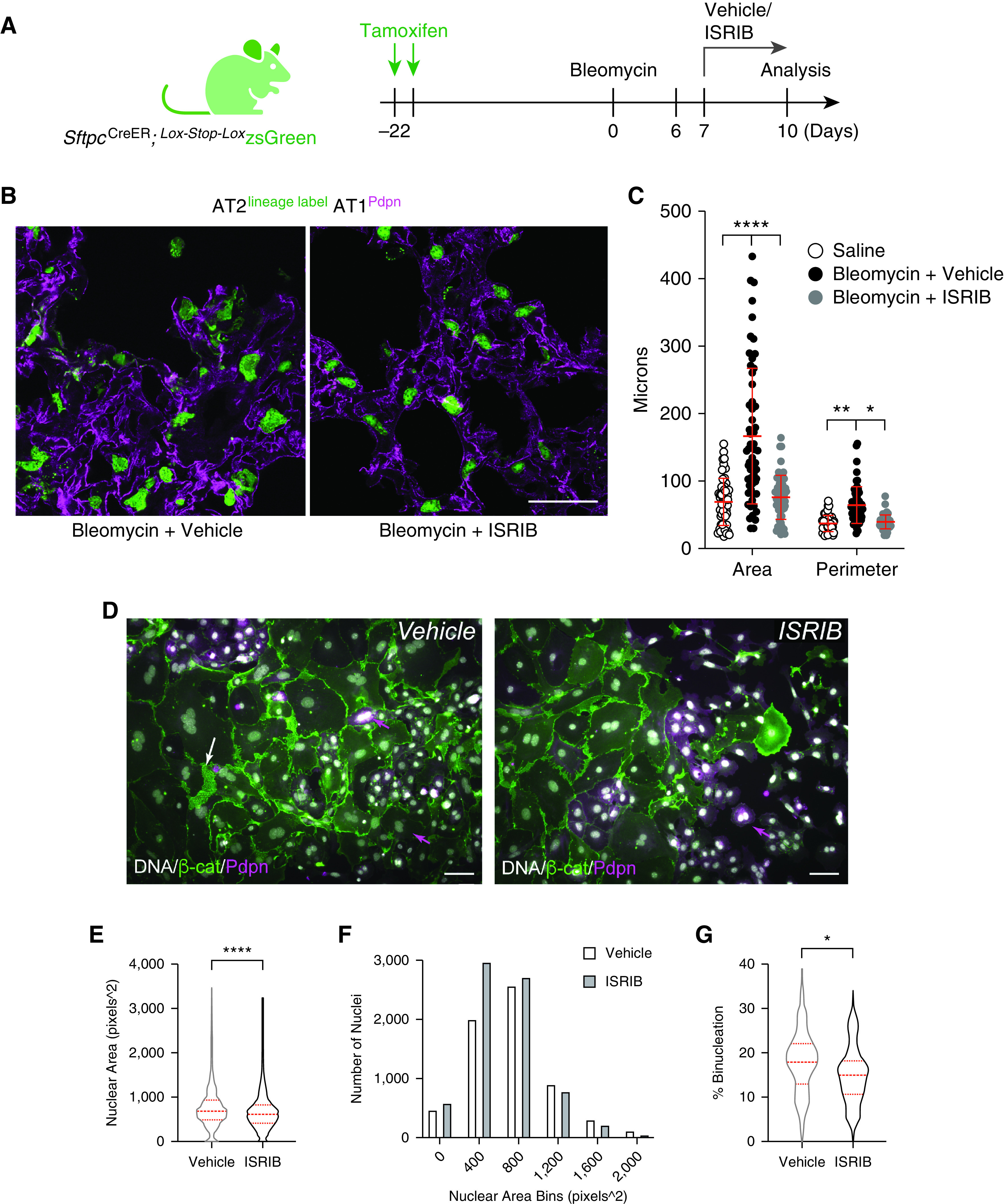
The inhibitor of the integrated stress response (ISRIB) limits AT2 hypertrophy and polyploidy cell autonomously. (A) AT2 lineage trace before bleomycin injury with and without ISRIB therapy. (B) Confocal images of fixed frozen lung sections with native zsGreen and immunofluorescence of AT1 marker Pdpn. Scale bar, 50 μm. (C) Scatterplot of zsGreen object sizes by Fiji as described in Figure E1. Graph is identical to Figure 1C with ISRIB data added. Significance was determined by ANOVA. ****P <0.0001, **P = 0.0070, and *P = 0.0122. (D) Epifluorescence overlay images of mouse AT2 cell Day 2 cultures incubated with vehicle/ISRIB (0.025 μm) through Days 4 and 5. Arrows (magenta) show Pdpn+ binucleated cells that are peripheral to large, flat, bi-/trinucleated cells with diminishing Pdpn and high levels of β-catenin (white arrow). Scale bars, 50 μm. (E) Nuclear area (DNA) measurements by ImageJ as a proxy for cell size allowed unbiased analysis. n = 6,829 (vehicle) and 7,387 (ISRIB) nuclei measured. Data were averaged from three independent AT2 isolations for cells quantified at 48 and 72 hours with or without ISRIB. ****P < 0.0001 by Mann-Whitney test. (F) Histogram of data in E. (G) Binucleation events quantified as from 44 (vehicle) and 43 (ISRIB) fields of view. *P = 0.035 by Mann-Whitney test. Dashed red lines reflect median and first and third quartiles.
Discussion
For decades, clinical pathologists have described the development of “hypertrophic” AT2 cells after injury, but mechanisms that drive this state and the degree to which it benefits and/or constrains the lung injury–repair cycle is not known. We show that some of this hypertrophic growth is associated with AT2 polyploidy, most notably binucleated AT2 cells. Ex vivo analysis suggests that the route to polyploidy is via failed cytokinesis during the AT2-to-AT1 flattening process. Remarkably, attenuating the ISR with ISRIB limits the abundance of hypertrophic, polyploid AT2 cells after bleomycin injury and ex vivo culture, suggesting that the effects of ISRIB may be AT2 cell autonomous. Given recent high-resolution time-series sampling of the injured lung and analysis by single-cell RNA-sequencing revealing that the AT2-to-AT1 transition state expresses genes associated with the ISR (22), we suggest that lung injury persistently activates the ISR in AT2 cells as they divide, enlarge, and flatten to repair the alveolar epithelium, increasing their susceptibility to cytokinesis failure into polyploid AT2 cells. A key consequence of AT2 polyploidization will be loss of the AT2 stem cell daughter, which may compromise future regenerative potential of alveoli served by that AT2 progenitor. We propose that attenuating the ISR helps guide AT2 cells through this vulnerable morphogenetic sequence of cell growth, division, and transdifferentiation without polyploidy (Figure 7, model).
Figure 7.
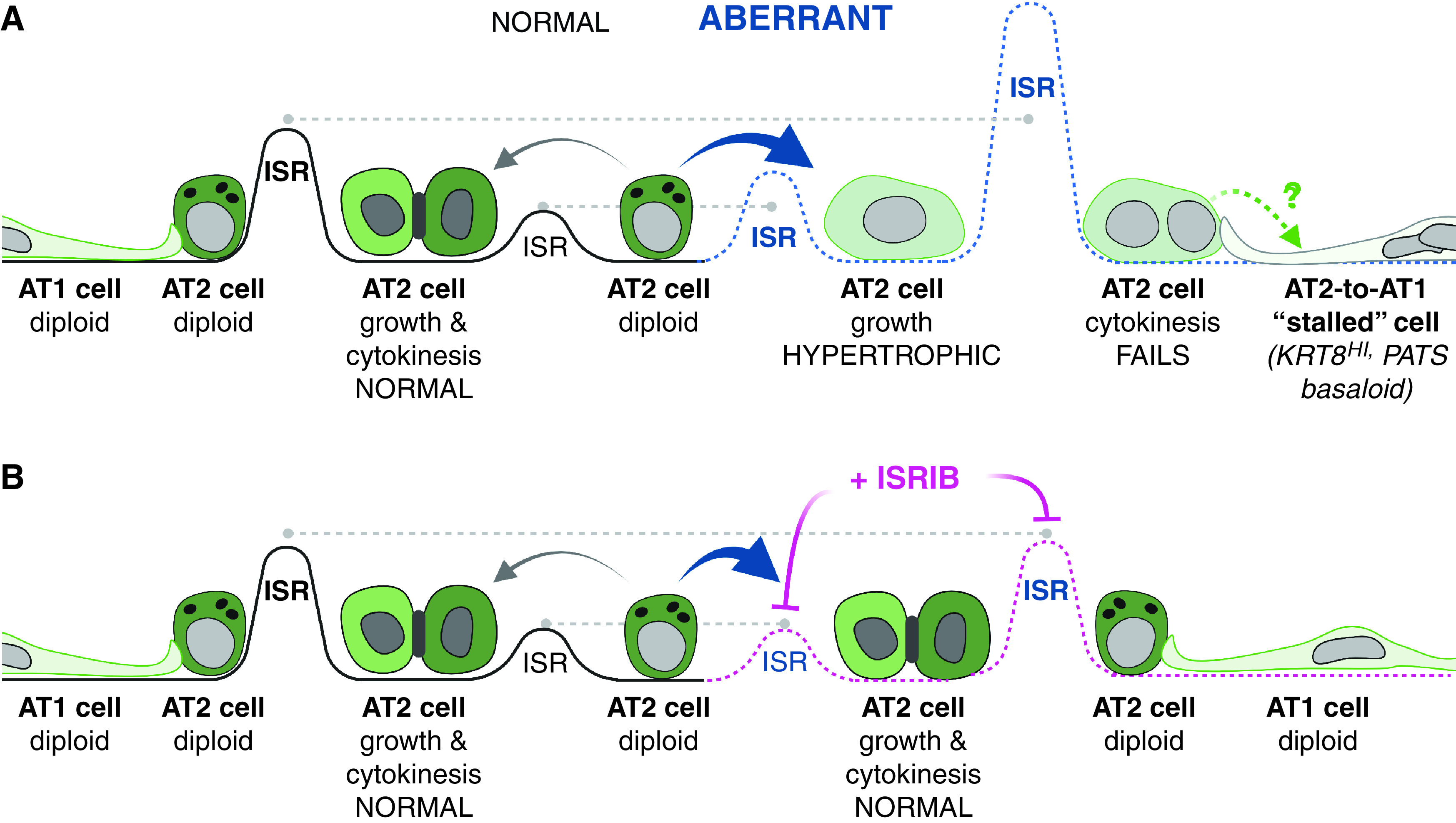
Model for alveolar epithelial stress-induced hypertrophy and polyploidization in lung injury and repair. (A) AT2 “aberrant” repair (dashed blue line) due to persistent integrated stress response (ISR), AT2 hypertrophic growth, cytokinesis failure, and stalled AT2-to-AT1 cells (right). (B) ISRIB therapy (dashed pink line) restores ISR to levels for normal AT2-to-AT1 growth, division, and differentiation (dashed gray line). Schematic created with BioRender software.
Evidence that AT2 binucleation does not exclusively correlate with cells expressing the highest levels of KRT8 protein is noteworthy. Although elevated Krt8 expression is associated with an aberrant AT2-to-AT1 transition state (21, 22), perhaps this transition is only one of a number of transcriptionally distinct routes cells take as they repair the injured alveolar epithelium. Alternatively, the Krt8-high/SftpcCreER; zsGreen-negative binucleated cells detected in Figure 3B suggests that alveolar epithelial polyploidization may also occur via Sftpc lineage-negative progenitors (35). Given the increased size and presumed fragility of polyploid alveolar cells, we suspect that they are inadequately represented in current lung single-cell RNA-sequencing pipelines. To understand how these cells specifically contribute to alveolar repair, it will be necessary to revise existing cell-sorting pipelines to quantify the abundance and degree of alveolar epithelial polyploidy, as well as isolate these cells for downstream transcriptomic analyses to establish the distinct features of the polyploid cell state (33).
Recent injury–repair studies using live-cell imaging of zebrafish epicardium as a simple epithelial model system revealed that cells along a closing wound front manifest higher tension and rates of binucleation due to failed cytokinesis (28). Although one study suggests that this may be due to excessive integrin-based extracellular matrix adhesive tension that ultimately interferes with contractility of the actin–myosin cytokinetic ring (61), there are many routes to cytokinesis failure caused by reduced or elevated Rho signaling (62). Endoplasmic reticulum stress itself can lead to cytokinesis failure (63, 64). Moreover, protein kinase R, a dsRNA-dependent protein kinase and upstream activator of the ISR, can be activated by mitosis onset, where nuclear membrane breakdown releases endogenous dsRNAs that bind and activate protein kinase R (65). This activation is required for cytokinetic fidelity, underscoring the relationship between ISR signaling and cell division. Future work will be required to determine whether cytokinesis is more vulnerable to ATF4-dependent versus -independent arms of the ISR.
Although bleomycin injury leads to binucleation in only ∼5% of lineage-labeled AT2 cells, total alveolar epithelial cell polyploidy may be much greater, particularly given evidence for mononuclear polyploidy across tissues (47). Newer imaging methods that allow simultaneous assessment of DNA content and cell cycle phase will be required to distinguish true mononuclear polyploidy (i.e., greater than or equal to 4N DNA content in a G1-phase cell) from AT2 cells arrested after genome doubling at the G2 phase (i.e., also 4N DNA content) (66). Another limitation of this study is our reliance on the DNA-damaging agent bleomycin as an injury model, which could bias the alveolar repair response toward AT2 polyploidy. Future work will be required to determine the generality of this paradigm and whether AT2 polyploidy can be induced by other forms of injury (e.g., viral or pneumonectomy model of alveologenesis).
Conceptually, it will be important address whether AT2 polyploidization is restricted to the transdifferentiating daughter, sparing normal ploidy of the renewed AT2 or if lung injury promotes AT2 endocycling/endomitosis, resulting in a polyploid facultative progenitor. The former would generate polyploid AT1 cells, which might allow AT1 cells to become larger with excess DNA copies for environmental adaptation (25). Indeed, a recent proof-of-concept study, which genetically induced cytokinesis failure in AT2 cells, found that binucleation does not obviously prevent transdifferentiation toward an AT1 fate (46). These data are consistent with our own evidence that binucleation is compatible with expression of the AT1 marker podoplanin (Figure 6D). Polyploidization of AT2 cells could provide extra DNA copies for surfactant biosynthesis, similar to how mammary epithelial cells become polyploid during lactation to maximize milk production (30). However, the induction of AT2 polyploidy would likely compromise its progenitor function, which requires faithful segregation of additional chromosomes during mitosis for stem cell renewal (47). Even heart tissue, which comprises large binucleated cardiomyocytes suited to contractility, relies on a low percentage of mononuclear diploid progenitor cells (2–14%) to drive regeneration after injury. Mouse strains with a greater percentage of these mononuclear diploid cells (14%) recover from injury faster than strains with a lower percentage (2%) (67). Given this precedence, we reason that even a small percentage of AT2 cells becoming polyploid during an injury event (∼5% per injury) could ultimately contribute to substantial loss of AT2 progenitor renewal over a lifetime, absent alternative mechanisms for AT2 progenitor replacement, such as via Sftpc-negative progenitors (68).
It will be important to understand how alveolar epithelial cells sustain the polyploid state. Forced induction of polyploidy in retinal pigmented epithelial cells requires activation of p53 signaling (69). Given recent evidence that forced activation of p53 may facilitate the AT2-to-AT1 transition during lung injury (23), it is possible that induced polyploidy plays a beneficial role in the earliest stages of alveolar epithelial repair. However, evidence that ISRIB attenuates formation or duration of hypertrophic alveolar epithelial cells (this study) and promotes lung repair in the bleomycin model (24) raises the possibility that maintaining polyploid cells for too long may be detrimental.
AT2 hypertrophic growth not associated with polyploidy may also be problematic for the injury–repair sequence. Young cells manipulated to grow to a large size, even without a corresponding increase in DNA content (i.e., polyploidy), can manifest phenotypes observed in senescent cells (70, 71). Conversely, older cells are associated with volume increase, raising the possibility that cytoplasmic dilution of key factors may contribute to senescence-associated phenotypes (70). Indeed, this same research team found that cell size is a determinant of stem cell (hematopoietic) potential during aging, where cell enlargement contributes to functional decline (72). Thus, hypertrophic growth independent of polyploidy can be maladaptive. Because most of our lineage-labeled AT2 cells show size increase after injury, whereas the binucleation rate is only ∼5%, absent substantially higher levels of mononuclear polyploidy, it is likely that AT2 hypertrophy and ISR activation are upstream of and causal to the polyploidization step (Figure 7). We speculate, therefore, that an inappropriately timed hypertrophic growth spurt may interfere with successful completion of mitosis. We hypothesize that ISRIB limits hypertrophic growth (and polyploidy) through an ATF4-dependent or -independent (i.e., bulk translation) mechanism, which can be addressed using existing ATF4 gain- and loss-of-function mice. Ultimately, adaptive or maladaptive functions of hypertrophic normal (2N) versus polyploid (⩾4N) AT2 cells will be informed by transcriptomic studies that consider DNA content as a variable.
The lung needs to repair in a manner that does not massively increase cell numbers (73). Epithelial polyploidization after injury may be a way to repair such tissues where excessive cellularity may be problematic. Epithelial polyploidization consequent to injury is a process with short-term benefits (e.g., larger cells with enhanced barrier function/reduced leak, increased DNA copies for adaptation to stressful environments) but long-term consequences (e.g., reduced stem cell maintenance due to loss of self-renewed mononuclear AT2). Our data raise the possibility that modulators of the ISR may be leveraged to maintain AT2 progenitor function during injury. Future work will be required to understand the means by which ISRIB limits formation of or guides the transition of hypertrophic, polyploid AT2 cells toward an AT1 phenotype and the degree to which induced polyploidy contributes to alveolar epithelial cell heterogeneity or adaptation in lung disease.
Acknowledgments
Acknowledgment
This work relied on the following Northwestern University services and core facilities: Flow Cytometry (National Cancer Institute grants CA060553, 1S10OD011996, 1S10OD026814); Center for Advanced Microscopy (National Cancer Institute cancer center support grant P30 CA060553; National Center for Research Resources grants 1S10 RR031680, 1S10OD021704); BioCryo facility of Northwestern University’s Atomic and Nanoscale Characterization Experimental Center (National Science Foundation grants ECCS-2025633, DMR-1720139).
Footnotes
Supported by National Institute of Arthritis and Musculoskeletal and Skin Diseases grant AR073270 (M.H.); National Heart, Lung, and Blood Institute (NHLBI) grants HL135124 and HL153312, National Institute on Aging grant AG049665, and National Institute of Allergy and Infectious Diseases grant AI135964 (A.V.M.); National Institute of Environmental Health Sciences grant ES13995, NHLBI grant HL071643, and National Institute on Aging grant AG049665 (G.R.S.B.); and NHLBI grant HL134800, National Institute of Arthritis and Musculoskeletal and Skin Diseases grant AR073270, and National Institute of General Medical Sciences grant GM129312 (C.J.G.).
Author Contributions: A.W., M.M.H., S.W., L.C.W., A.S.F., and R.P.A. designed and conducted experiments. A.W., M.M.H., A.S.F., A.V.M., and C.J.G. analyzed results. R.A.G. wrote a macro for image analysis. A.V.M. advised and analyzed flow cytometry. L.A.D., S.H.H., M.H., A.V.M., and G.R.S.B. advised and discussed results. C.J.G. designed and supervised study, performed analysis, and wrote manuscript. M.H., A.V.M., G.R.S.B., and C.J.G. provided funding for project.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2021-0356OC on February 24, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Yanagihara T, Sato S, Upagupta C, Kolb M. What have we learned from basic science studies on idiopathic pulmonary fibrosis? Eur Respir Rev . 2019;28:190029. doi: 10.1183/16000617.0029-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Selman M, Pardo A. The leading role of epithelial cells in the pathogenesis of idiopathic pulmonary fibrosis. Cell Signal . 2020;66:109482. doi: 10.1016/j.cellsig.2019.109482. [DOI] [PubMed] [Google Scholar]
- 3. Kropski JA, Blackwell TS. Progress in understanding and treating idiopathic pulmonary fibrosis. Annu Rev Med . 2019;70:211–224. doi: 10.1146/annurev-med-041317-102715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Povedano JM, Martinez P, Serrano R, Tejera Á, Gómez-López G, Bobadilla M, et al. Therapeutic effects of telomerase in mice with pulmonary fibrosis induced by damage to the lungs and short telomeres. eLife . 2018;7:e31299. doi: 10.7554/eLife.31299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Povedano JM, Martinez P, Flores JM, Mulero F, Blasco MA. Mice with pulmonary fibrosis driven by telomere dysfunction. Cell Rep . 2015;12:286–299. doi: 10.1016/j.celrep.2015.06.028. [DOI] [PubMed] [Google Scholar]
- 6. Vicary GW, Vergne Y, Santiago-Cornier A, Young LR, Roman J. Pulmonary fibrosis in Hermansky-Pudlak syndrome. Ann Am Thorac Soc . 2016;13:1839–1846. doi: 10.1513/AnnalsATS.201603-186FR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bharat A, Querrey M, Markov NS, Kim S, Kurihara C, Garza-Castillon R, et al. Lung transplantation for patients with severe COVID-19. Sci Transl Med . 2020;12:eabe4282. doi: 10.1126/scitranslmed.abe4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bharat A. Querrey M. Markov NS. Kim S. Kurihara C. Garza-Castillon R. et al. Lung transplantation for patients with severe COVID-19. Sci Transl Med . 2020;12(574):eabe4282. doi: 10.1126/scitranslmed.abe4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Spagnolo P, Balestro E, Aliberti S, Cocconcelli E, Biondini D, Casa GD, et al. Pulmonary fibrosis secondary to COVID-19: a call to arms? Lancet Respir Med . 2020;8:750–752. doi: 10.1016/S2213-2600(20)30222-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen J, Wu H, Yu Y, Tang N. Pulmonary alveolar regeneration in adult COVID-19 patients. Cell Res . 2020;30:708–710. doi: 10.1038/s41422-020-0369-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Delorey TM, Ziegler CGK, Heimberg G, Normand R, Yang Y, Segerstolpe Å, et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature . 2021;595:107–113. doi: 10.1038/s41586-021-03570-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McDonald LT. Healing after COVID-19: are survivors at risk for pulmonary fibrosis? Am J Physiol Lung Cell Mol Physiol . 2021;320:L257–L265. doi: 10.1152/ajplung.00238.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rock JR, Hogan BL. Epithelial progenitor cells in lung development, maintenance, repair, and disease. Annu Rev Cell Dev Biol . 2011;27:493–512. doi: 10.1146/annurev-cellbio-100109-104040. [DOI] [PubMed] [Google Scholar]
- 14. Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR, et al. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest . 2013;123:3025–3036. doi: 10.1172/JCI68782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Aso Y, Yoneda K, Kikkawa Y. Morphologic and biochemical study of pulmonary changes induced by bleomycin in mice. Lab Invest . 1976;35:558–568. [PubMed] [Google Scholar]
- 16. Miller BE, Hook GE. Regulation of phosphatidylcholine biosynthesis in activated alveolar type II cells. Am J Respir Cell Mol Biol . 1989;1:127–136. doi: 10.1165/ajrcmb/1.2.127. [DOI] [PubMed] [Google Scholar]
- 17. Miller BE, Hook GE. Hypertrophy and hyperplasia of alveolar type II cells in response to silica and other pulmonary toxicants. Environ Health Perspect . 1990;85:15–23. doi: 10.1289/ehp.85-1568321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kasper M, Haroske G. Alterations in the alveolar epithelium after injury leading to pulmonary fibrosis. Histol Histopathol . 1996;11:463–483. [PubMed] [Google Scholar]
- 19. Ting C, Aspal M, Vaishampayan N, Huang SK, Riemondy K, Wang F, et al. Fatal COVID-19 ARDS associated with incomplete AEC1 differentiation from the transitional state without senescence or fibrosis. Am J Pathol . 2022;192:454–467. doi: 10.1016/j.ajpath.2021.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wu H, Yu Y, Huang H, Hu Y, Fu S, Wang Z, et al. Progressive pulmonary fibrosis is caused by elevated mechanical tension on alveolar stem cells. Cell . 2020;180:107–121, e17. doi: 10.1016/j.cell.2019.11.027. [DOI] [PubMed] [Google Scholar]
- 21. Jiang P, Gil de Rubio R, Hrycaj SM, Gurczynski SJ, Riemondy KA, Moore BB, et al. Ineffectual type 2-to-type 1 alveolar epithelial cell differentiation in idiopathic pulmonary fibrosis: persistence of the KRT8hi transitional state. Am J Respir Crit Care Med . 2020;201:1443–1447. doi: 10.1164/rccm.201909-1726LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Strunz M, Simon LM, Ansari M, Kathiriya JJ, Angelidis I, Mayr CH, et al. Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat Commun . 2020;11:3559. doi: 10.1038/s41467-020-17358-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kobayashi Y, Tata A, Konkimalla A, Katsura H, Lee RF, Ou J, et al. Persistence of a regeneration-associated, transitional alveolar epithelial cell state in pulmonary fibrosis. Nat Cell Biol . 2020;22:934–946. doi: 10.1038/s41556-020-0542-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Watanabe S, Markov NS, Lu Z, Piseaux Aillon R, Soberanes S, Runyan CE, et al. Resetting proteostasis with ISRIB promotes epithelial differentiation to attenuate pulmonary fibrosis. Proc Natl Acad Sci USA . 2021;118:e2101100118. doi: 10.1073/pnas.2101100118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schoenfelder KP, Fox DT. The expanding implications of polyploidy. J Cell Biol . 2015;209:485–491. doi: 10.1083/jcb.201502016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Amodeo AA, Skotheim JM. Cell-size control. Cold Spring Harb Perspect Biol . 2016;8:a019083. doi: 10.1101/cshperspect.a019083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cohen E, Allen SR, Sawyer JK, Fox DT. Fizzy-related dictates a cell cycle switch during organ repair and tissue growth responses in the Drosophila hindgut. eLife . 2018;7:e38327. doi: 10.7554/eLife.38327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cao J, Wang J, Jackman CP, Cox AH, Trembley MA, Balowski JJ, et al. Tension creates an endoreplication wavefront that leads regeneration of epicardial tissue. Dev Cell . 2017;42:600–615, e4. doi: 10.1016/j.devcel.2017.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Herrtwich L, Nanda I, Evangelou K, Nikolova T, Horn V, Sagar, et al. DNA damage signaling instructs polyploid macrophage fate in granulomas. Cell . 2016;167:1264–1280, e18. doi: 10.1016/j.cell.2016.09.054. [DOI] [PubMed] [Google Scholar]
- 30. Rios AC, Fu NY, Jamieson PR, Pal B, Whitehead L, Nicholas KR, et al. Essential role for a novel population of binucleated mammary epithelial cells in lactation. Nat Commun . 2016;7:11400. doi: 10.1038/ncomms11400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang J, Batourina E, Schneider K, Souza S, Swayne T, Liu C, et al. Polyploid superficial cells that maintain the urothelial barrier are produced via incomplete cytokinesis and endoreplication. Cell Rep . 2018;25:464–477, e4. doi: 10.1016/j.celrep.2018.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Quinton RJ, DiDomizio A, Vittoria MA, Kotýnková K, Ticas CJ, Patel S, et al. Whole-genome doubling confers unique genetic vulnerabilities on tumour cells. Nature . 2021;590:492–497. doi: 10.1038/s41586-020-03133-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fox DT, Soltis DE, Soltis PS, Ashman TL, Van de Peer Y. Polyploidy: a biological force from cells to ecosystems. Trends Cell Biol . 2020;30:688–694. doi: 10.1016/j.tcb.2020.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gjelsvik KJ, Besen-McNally R, Losick VP. Solving the polyploid mystery in health and disease. Trends Genet . 2019;35:6–14. doi: 10.1016/j.tig.2018.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rock JR, Barkauskas CE, Cronce MJ, Xue Y, Harris JR, Liang J, et al. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci USA . 2011;108:E1475–E1483. doi: 10.1073/pnas.1117988108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zacharias WJ, Frank DB, Zepp JA, Morley MP, Alkhaleel FA, Kong J, et al. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature . 2018;555:251–255. doi: 10.1038/nature25786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chapman HA, Li X, Alexander JP, Brumwell A, Lorizio W, Tan K, et al. Integrin α6β4 identifies an adult distal lung epithelial population with regenerative potential in mice. J Clin Invest . 2011;121:2855–2862. doi: 10.1172/JCI57673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Redente EF, Jacobsen KM, Solomon JJ, Lara AR, Faubel S, Keith RC, et al. Age and sex dimorphisms contribute to the severity of bleomycin-induced lung injury and fibrosis. Am J Physiol Lung Cell Mol Physiol . 2011;301:L510–L518. doi: 10.1152/ajplung.00122.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen J, Chen Z, Narasaraju T, Jin N, Liu L. Isolation of highly pure alveolar epithelial type I and type II cells from rat lungs. Lab Invest . 2004;84:727–735. doi: 10.1038/labinvest.3700095. [DOI] [PubMed] [Google Scholar]
- 40. Jansing NL, McClendon J, Kage H, Sunohara M, Alvarez JR, Borok Z, et al. Isolation of rat and mouse alveolar type II epithelial cells. Methods Mol Biol . 2018;1809:69–82. doi: 10.1007/978-1-4939-8570-8_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GR, Perlman H. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am J Respir Cell Mol Biol . 2013;49:503–510. doi: 10.1165/rcmb.2013-0086MA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Halliday M, Radford H, Sekine Y, Moreno J, Verity N, le Quesne J, et al. Partial restoration of protein synthesis rates by the small molecule ISRIB prevents neurodegeneration without pancreatic toxicity. Cell Death Dis . 2015;6:e1672. doi: 10.1038/cddis.2015.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Flozak AS, Lam AP, Russell S, Jain M, Peled ON, Sheppard KA, et al. β-Catenin/T-cell factor signaling is activated during lung injury and promotes the survival and migration of alveolar epithelial cells. J Biol Chem . 2010;285:3157–3167. doi: 10.1074/jbc.M109.070326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jansing NL, Patel N, McClendon J, Redente EF, Henson PM, Tuder RM, et al. Flow cytometry underestimates and planimetry overestimates alveolar epithelial type 2 cell expansion after lung injury. Am J Respir Crit Care Med . 2018;198:390–392. doi: 10.1164/rccm.201709-1838LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hsia CC, Hyde DM, Ochs M, Weibel ER, ATS/ERS Joint Task Force on Quantitative Assessment of Lung Structure An official research policy statement of the American Thoracic Society/European Respiratory Society: standards for quantitative assessment of lung structure. Am J Respir Crit Care Med . 2010;181:394–418. doi: 10.1164/rccm.200809-1522ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liberti DC, Kremp MM, Liberti WA, III, Penkala IJ, Li S, Zhou S, et al. Alveolar epithelial cell fate is maintained in a spatially restricted manner to promote lung regeneration after acute injury. Cell Rep . 2021;35:109092. doi: 10.1016/j.celrep.2021.109092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Øvrebø JI, Edgar BA. Polyploidy in tissue homeostasis and regeneration. Development . 2018;145:dev156034. doi: 10.1242/dev.156034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Matsumoto T, Wakefield L, Tarlow BD, Grompe M. In vivo lineage tracing of polyploid hepatocytes reveals extensive proliferation during liver regeneration. Cell Stem Cell . 2020;26:34–47, e3. doi: 10.1016/j.stem.2019.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nabhan AN, Brownfield DG, Harbury PB, Krasnow MA, Desai TJ. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science . 2018;359:1118–1123. doi: 10.1126/science.aam6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Emanuelli G, Nassehzadeh-Tabriz N, Morrell NW, Marciniak SJ. The integrated stress response in pulmonary disease. Eur Respir Rev . 2020;29:200184. doi: 10.1183/16000617.0184-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sudhakar A, Ramachandran A, Ghosh S, Hasnain SE, Kaufman RJ, Ramaiah KV. Phosphorylation of serine 51 in initiation factor 2α (eIF2α) promotes complex formation between eIF2α(P) and eIF2B and causes inhibition in the guanine nucleotide exchange activity of eIF2B. Biochemistry . 2000;39:12929–12938. doi: 10.1021/bi0008682. [DOI] [PubMed] [Google Scholar]
- 52. Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. The integrated stress response. EMBO Rep . 2016;17:1374–1395. doi: 10.15252/embr.201642195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Costa-Mattioli M, Walter P. The integrated stress response: from mechanism to disease. Science . 2020;368:eaat5314. doi: 10.1126/science.aat5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sidrauski C, McGeachy AM, Ingolia NT, Walter P. The small molecule ISRIB reverses the effects of eIF2α phosphorylation on translation and stress granule assembly. eLife . 2015;4:e05033. doi: 10.7554/eLife.05033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sidrauski C, Tsai JC, Kampmann M, Hearn BR, Vedantham P, Jaishankar P, et al. Pharmacological dimerization and activation of the exchange factor eIF2B antagonizes the integrated stress response. eLife . 2015;4:e07314. doi: 10.7554/eLife.07314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zyryanova AF, Kashiwagi K, Rato C, Harding HP, Crespillo-Casado A, Perera LA, et al. ISRIB blunts the integrated stress response by allosterically antagonising the inhibitory effect of phosphorylated eIF2 on eIF2B. Mol Cell . 2021;81:88–103, e6. doi: 10.1016/j.molcel.2020.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Anand AA, Walter P. Structural insights into ISRIB, a memory-enhancing inhibitor of the integrated stress response. FEBS J . 2020;287:239–245. doi: 10.1111/febs.15073. [DOI] [PubMed] [Google Scholar]
- 58. Chou A, Krukowski K, Jopson T, Zhu PJ, Costa-Mattioli M, Walter P, et al. Inhibition of the integrated stress response reverses cognitive deficits after traumatic brain injury. Proc Natl Acad Sci USA . 2017;114:E6420–E6426. doi: 10.1073/pnas.1707661114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Krukowski K, Nolan A, Frias ES, Boone M, Ureta G, Grue K, et al. Small molecule cognitive enhancer reverses age-related memory decline in mice. p. eLife 2020;9:e62048. [DOI] [PMC free article] [PubMed]
- 60. Rabouw HH, Langereis MA, Anand AA, Visser LJ, de Groot RJ, Walter P, et al. Small molecule ISRIB suppresses the integrated stress response within a defined window of activation. Proc Natl Acad Sci USA . 2019;116:2097–2102. doi: 10.1073/pnas.1815767116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Uroz M, Garcia-Puig A, Tekeli I, Elosegui-Artola A, Abenza JF, Marín-Llauradó A, et al. Traction forces at the cytokinetic ring regulate cell division and polyploidy in the migrating zebrafish epicardium. Nat Mater . 2019;18:1015–1023. doi: 10.1038/s41563-019-0381-9. [DOI] [PubMed] [Google Scholar]
- 62. Normand G, King RW. Understanding cytokinesis failure. Adv Exp Med Biol . 2010;676:27–55. doi: 10.1007/978-1-4419-6199-0_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bicknell AA, Babour A, Federovitch CM, Niwa M. A novel role in cytokinesis reveals a housekeeping function for the unfolded protein response. J Cell Biol . 2007;177:1017–1027. doi: 10.1083/jcb.200702101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Niwa M. A cell cycle checkpoint for the endoplasmic reticulum. Biochim Biophys Acta Mol Cell Res . 2020;1867:118825. doi: 10.1016/j.bbamcr.2020.118825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kim Y, Lee JH, Park JE, Cho J, Yi H, Kim VN. PKR is activated by cellular dsRNAs during mitosis and acts as a mitotic regulator. Genes Dev . 2014;28:1310–1322. doi: 10.1101/gad.242644.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zielke N, Edgar BA. FUCCI sensors: powerful new tools for analysis of cell proliferation. Wiley Interdiscip Rev Dev Biol . 2015;4:469–487. doi: 10.1002/wdev.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Patterson M, Barske L, Van Handel B, Rau CD, Gan P, Sharma A, et al. Frequency of mononuclear diploid cardiomyocytes underlies natural variation in heart regeneration. Nat Genet . 2017;49:1346–1353. doi: 10.1038/ng.3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Vaughan AE, Brumwell AN, Xi Y, Gotts JE, Brownfield DG, Treutlein B, et al. Lineage-negative progenitors mobilize to regenerate lung epithelium after major injury. Nature . 2015;517:621–625. doi: 10.1038/nature14112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Potapova TA, Seidel CW, Box AC, Rancati G, Li R. Transcriptome analysis of tetraploid cells identifies cyclin D2 as a facilitator of adaptation to genome doubling in the presence of p53. Mol Biol Cell . 2016;27:3065–3084. doi: 10.1091/mbc.E16-05-0268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Neurohr GE, Terry RL, Lengefeld J, Bonney M, Brittingham GP, Moretto F, et al. Excessive cell growth causes cytoplasm dilution and contributes to senescence. Cell . 2019;176:1083–1097. doi: 10.1016/j.cell.2019.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Demidenko ZN, Blagosklonny MV. Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell Cycle . 2008;7:3355–3361. doi: 10.4161/cc.7.21.6919. [DOI] [PubMed] [Google Scholar]
- 72. Lengefeld J, Cheng CW, Maretich P, Blair M, Hagen H, McReynolds MR, et al. Cell size is a determinant of stem cell potential during aging. Sci Adv . 2021;7:eabk0271. doi: 10.1126/sciadv.abk0271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Leach JP, Morrisey EE. Repairing the lungs one breath at a time: how dedicated or facultative are you? Genes Dev . 2018;32:1461–1471. doi: 10.1101/gad.319418.118. [DOI] [PMC free article] [PubMed] [Google Scholar]