

Pulmonary fibrosis is a pathological manifestation of dysregulated cellular and molecular responses during injury, inflammation, and oxidative stress, progressing beyond recovery and culminating in the loss of alveoli (1). As a result, the gas exchange areas become occupied by activated myofibroblasts and the extracellular matrix they generate (i.e., collagen), most often in the distal areas of the lung. Pulmonary fibrosis is observed in multiple chronic interstitial lung diseases and as a sequela of acute respiratory distress syndrome. Idiopathic pulmonary fibrosis (IPF), the most common and perhaps devastating of the fibrosing interstitial lung diseases, occurs predominantly in the elderly (2). Of unknown etiology, IPF pathogenesis is considered to involve persistent microinjuries to the alveolar epithelium and a subsequent dysregulated injury-repair response that drives fibrosis (3). Decades of fibrosis research have identified multiple signaling pathways and protein complexes that play pivotal roles in the progressive expansion of fibrotic lung lesions in IPF. Although numerous promising therapeutic targets have been identified to attenuate lung fibrosis in preclinical studies, there remains no curative treatment for IPF (4). Thus, there is a need to identify druggable therapeutic targets that are safe and efficient to attenuate ongoing pulmonary fibrosis.
ASK1 (apoptosis signal-regulating kinase 1) is a member of the MAPK (mitogen-activated protein kinase) family that regulates JNK (c-Jun N-terminal kinas) and p38 MAPK in response to tissue injury by oxidative stress, endoplasmic stress, and inflammatory cytokines (Figure 1). ASK1 is activated in a multistep process that involves reactive oxygen species (ROS), thioredoxin, and members of the TRAF (tumor necrosis factor receptor-associated factor) family, which together form the “ASK1 signalosome” (5). It has been shown that ASK1 is activated in response to pathological oxidative stress and subsequently causes fibrotic remodeling during kidney injury by inducing diverse stress-response pathways in multiple cell types (6). However, ASK1 until recently has remained unexplored as a therapeutic option against pulmonary fibrosis. In this issue of the Journal, Valenca and colleagues (pp. 484–496) provide compelling evidence that ASK1 contributes to pulmonary fibrosis by using a model in which mice are administered bleomycin by intratracheal instillation (7). ASK1 gene knockout was shown to attenuate fibrosis, as measured by histological and biochemical assays, albeit at only one time point representing the peak of fibrosis (22 days after bleomycin instillation). The pharmacological ASK1 inhibitor, selonsertib (GS-4997), when administered once daily to mice during the fibroproliferative phase, exhibited similar antifibrotic actions. Bleomycin-induced increases in lung inflammation and reduction in compliance were also attenuated by one or both interventions targeting ASK1, as were the phosphorylation of ASK1 and p38. Immunoblot analysis of markers for oxidative stress, epithelial mesenchymal transition, and type II alveolar epithelial (ATII) cell hyperplasia also suggest these processes may have some part in mediating the effects of ASK1 in bleomycin-induced lung injury and fibrosis.
Figure 1.
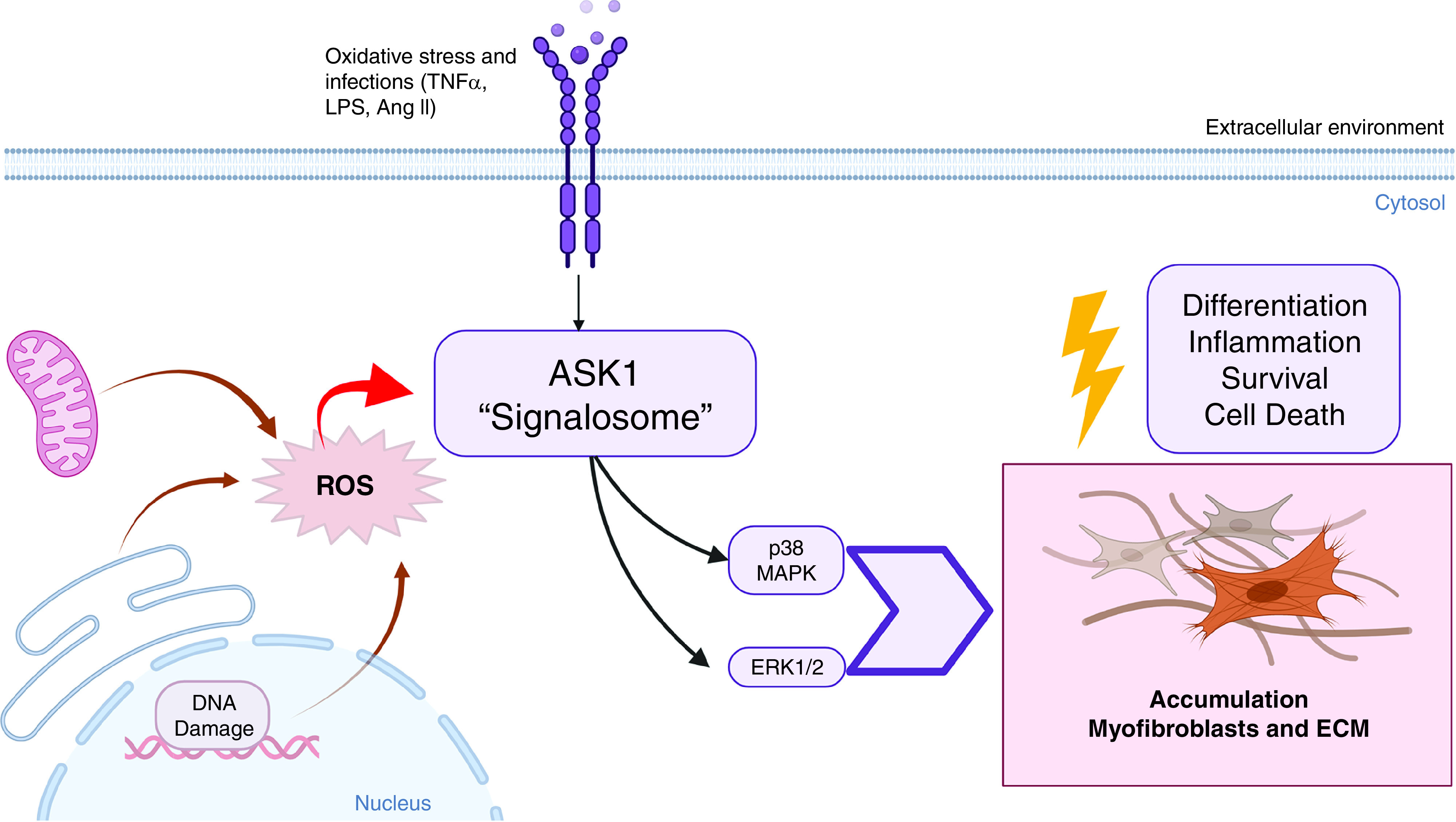
The role of ASK1 (apoptosis signal-regulating kinase 1) in the pathogenesis of pulmonary fibrosis. Lung injury, DNA damage, and oxidative stress can increase ASK1 activation in the distal areas of the lung. Furthermore, ASK1 activation contributes to increased p38 and ERK1/2 MAPK (extracellular signal-regulated protein kinase 1/2 mitogen-activated protein kinase) singling. Thus, this increase of ASK1, p38, and ERK1/2 contributes to heightened inflammation, myofibroblast differentiation, and excessive collagen deposition in the pathogenesis of pulmonary fibrosis. Ang II = angiotensin II; ECM = extracellular matrix proteins; LPS = lipopolysaccharide; ROS = reactive oxygen species; TNFα = tumor necrosis factor α.
The role of oxidative stress, a key stimulus of ASK1 signaling, is of particular interest and warrants further investigation. Indeed, Valenca and colleagues used only surrogate measures of oxidative stress: concentrations of HO-1 (heme oxygenase-1) and Trx1 (thioredoxin 1) in their investigation. As the reduced form of Trx1 (but not its oxidized form) binds to and inhibits ASK1, evaluating the redox status of Trx1 and/or concentrations of the Trx1-ASK1 complex will provide a better understanding of the mechanisms by which ASK1 contributes to lung fibrosis (8). Interventions that selectively modulate the redox status and/or availability of Trx1 should also be considered in future investigations of ASK1 in lung fibrosis. The interactions between Trx1 and ASK1 may particularly be important in understanding the pathogenesis of IPF. Elevated concentrations of mitochondria-derived ROS and increased activities of p38 MAPK and JNK characterize a chronic state of stress in aging and age-related disease, including IPF (9–12). The dissociation of oxidized Trx1 from ASK1 is the primary mechanism that links ROS formation to the activation of p38 MAPK and JNK in aging (13). Indeed, higher concentrations of the thioredoxin-ASK1 complex in tissue of long-living Snell/Ames dwarf or Klotho-overexpressing mice suggest that this complex confers resistance to oxidative stress and prolongs survival (13). Conversely, lower concentrations of this complex are likely to increase the susceptibility of aged tissues to injury and may explain in part why age is a major predisposing factor for IPF.
The in vivo study of Valenca and colleagues is comprehensive, in that it examines a broad suite of variables. However, there is little exploration into which cell types mediate the detrimental effects of ASK1 in pulmonary fibrosis. Immunohistochemistry conducted on intact lung sections from the in vivo investigations suggests that phosphorylated ASK1 is present at low concentrations in epithelial cells under baseline conditions and at higher concentrations in regions of fibrosis in bleomycin-treated mice. However, it is difficult from the immunohistochemistry images presented to identify the cell type(s) with detectable phosphorylated ASK1. Inclusion of single-cell sequencing data mined from the IPF Cell Atlas database (https://p2med.shinyapps.io/IPFCellAtlas/) provides evidence that multiple lung cell populations—including ATII cells and fibroblasts—express ASK1 in IPF. However, single-cell transcriptomic analysis does not measure concentrations of total and phosphorylated ASK1. Future investigation using multiplex immunostaining analysis on lung tissue from patients with IPF and/or bleomycin-treated mice will help determine which cells exhibit heightened ASK1 activation in pulmonary fibrosis. Interestingly, the authors suggest the fibrotic actions of ASK1 in bleomycin-treated mice prolong lung fibroblast survival. In support of this, bleomycin-induced increases in phosphorylated ERK-1/2 and survivin, an apoptosis inhibitor, in lung homogenates were attenuated by interventions targeting ASK1. Indeed, ASK1 has previously been reported to promote cell survival, a function dependent on cell type and/or tissue context (14). Although a role of ASK1 in lung fibroblast survival is plausible, evidence showing a link between ASK1 induction, fibroblast survival (or apoptosis resistance), and fibrosis is missing in the current study.
Another possible mechanism by which ASK1 inhibition attenuates bleomycin-induced pulmonary fibrosis is by protecting ATII cells against apoptosis and the resulting denudation of the injured alveolar epithelium. Unfortunately, ATII cell apoptosis, which occurs within days after bleomycin administration in the murine model, was not assessed in the Velanca and colleagues study (15). Interestingly, increased sensitivity to ATII cell apoptosis, yet sustained fibroblast survival (referred to as the “apoptosis paradox”), is believed to underlie the aberrant wound-repair response that perpetuates fibrosis in IPF (16). Whether ASK1 contributes to this paradox through a dichotomous regulation of apoptosis/cell survival in lung epithelial and fibroblast cells needs further investigation.
Footnotes
Supported by the National Heart, Lung, and Blood Institute grant 1R01 HL134801 and 1R01 HL157176.
Originally Published in Press as DOI: 10.1165/rcmb.2022-0026ED on March 3, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med . 2012;18:1028–1040. doi: 10.1038/nm.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Garcia CK. Idiopathic pulmonary fibrosis: update on genetic discoveries. Proc Am Thorac Soc . 2011;8:158–162. doi: 10.1513/pats.201008-056MS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Selman M, Thannickal VJ, Pardo A, Zisman DA, Martinez FJ, Lynch JP., III Idiopathic pulmonary fibrosis: pathogenesis and therapeutic approaches. Drugs . 2004;64:405–430. doi: 10.2165/00003495-200464040-00005. [DOI] [PubMed] [Google Scholar]
- 4. Sontake V, Gajjala PR, Kasam RK, Madala SK. New therapeutics based on emerging concepts in pulmonary fibrosis. Expert Opin Ther Targets . 2019;23:69–81. doi: 10.1080/14728222.2019.1552262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Matsuzawa A, Saegusa K, Noguchi T, Sadamitsu C, Nishitoh H, Nagai S, et al. ROS-dependent activation of the TRAF6-ASK1-p38 pathway is selectively required for TLR4-mediated innate immunity. Nat Immunol . 2005;6:587–592. doi: 10.1038/ni1200. [DOI] [PubMed] [Google Scholar]
- 6. Liles JT, Corkey BK, Notte GT, Budas GR, Lansdon EB, Hinojosa-Kirschenbaum F, et al. ASK1 contributes to fibrosis and dysfunction in models of kidney disease. J Clin Invest . 2018;128:4485–4500. doi: 10.1172/JCI99768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Valenca SS, Dong BE, Gordon EM, Sun RC, Waters CM. ASK1 regulates bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol . 2022;66 doi: 10.1165/rcmb.2021-0465OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, et al. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J . 1998;17:2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hsieh CC, Rosenblatt JI, Papaconstantinou J. Age-associated changes in SAPK/JNK and p38 MAPK signaling in response to the generation of ROS by 3-nitropropionic acid. Mech Ageing Dev . 2003;124:733–746. doi: 10.1016/s0047-6374(03)00083-6. [DOI] [PubMed] [Google Scholar]
- 10. Yoshida K, Kuwano K, Hagimoto N, Watanabe K, Matsuba T, Fujita M, et al. MAP kinase activation and apoptosis in lung tissues from patients with idiopathic pulmonary fibrosis. J Pathol . 2002;198:388–396. doi: 10.1002/path.1208. [DOI] [PubMed] [Google Scholar]
- 11. Bueno M, Lai YC, Romero Y, Brands J, St Croix CM, Kamga C, et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J Clin Invest . 2015;125:521–538. doi: 10.1172/JCI74942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schuliga M, Read J, Knight DA. Ageing mechanisms that contribute to tissue remodeling in lung disease. Ageing Res Rev . 2021;70:101405. doi: 10.1016/j.arr.2021.101405. [DOI] [PubMed] [Google Scholar]
- 13. Papaconstantinou J. The role of signaling pathways of inflammation and oxidative stress in development of senescence and aging phenotypes in cardiovascular disease. Cells . 2019;8:1383. doi: 10.3390/cells8111383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Takeda K, Hatai T, Hamazaki TS, Nishitoh H, Saitoh M, Ichijo H. Apoptosis signal-regulating kinase 1 (ASK1) induces neuronal differentiation and survival of PC12 cells. J Biol Chem . 2000;275:9805–9813. doi: 10.1074/jbc.275.13.9805. [DOI] [PubMed] [Google Scholar]
- 15. Hagimoto N, Kuwano K, Nomoto Y, Kunitake R, Hara N. Apoptosis and expression of Fas/Fas ligand mRNA in bleomycin-induced pulmonary fibrosis in mice. Am J Respir Cell Mol Biol . 1997;16:91–101. doi: 10.1165/ajrcmb.16.1.8998084. [DOI] [PubMed] [Google Scholar]
- 16. Thannickal VJ, Horowitz JC. Evolving concepts of apoptosis in idiopathic pulmonary fibrosis. Proc Am Thorac Soc . 2006;3:350–356. doi: 10.1513/pats.200601-001TK. [DOI] [PMC free article] [PubMed] [Google Scholar]