

Abstract
Synthetic lethality (SL) occurs between two genes when the inactivation of either gene alone has no effect on cell survival but the inactivation of both genes results in cell death. SL-based therapy has become one of the most promising targeted cancer therapies in the last decade as PARP inhibitors achieve great success in the clinic. The key point to exploiting SL-based cancer therapy is the identification of robust SL pairs. Although many wet-lab-based methods have been developed to screen SL pairs, known SL pairs are less than 0.1% of all potential pairs due to large number of human gene combinations. Computational prediction methods complement wet-lab-based methods to effectively reduce the search space of SL pairs. In this paper, we review the recent applications of computational methods and commonly used databases for SL prediction. First, we introduce the concept of SL and its screening methods. Second, various SL-related data resources are summarized. Then, computational methods including statistical-based methods, network-based methods, classical machine learning methods and deep learning methods for SL prediction are summarized. In particular, we elaborate on the negative sampling methods applied in these models. Next, representative tools for SL prediction are introduced. Finally, the challenges and future work for SL prediction are discussed.
Keywords: synthetic lethality, computational methods, deep learning, machine learning
Introduction
Synthetic lethality (SL) is originally defined as the setting in which abnormal expression of either of two genes alone has little effect on cell viability but abnormalities in the expression of both genes concurrently lead to cell death [1]. Basically, ‘SL’ can be categorized into two classes: (i) SL, which occurs between genes with loss-of-function mutations (gene A) and their partner gene (gene B). (ii) Synthetic dosage lethality (SDL), which occurs between the overexpressed gene (gene A) and their partner gene (gene B) [2] (Figure 1). In cancer, the application of SL has the following significances: (i) SL provides an approach for targeted therapy. Abnormalities of gene A can be regarded as cancer-specific biomarkers and pharmacological inhibition of gene B leads to the selective killing of cancer cells [3]. (ii) SL expands the space of druggable targets. SL points the way to indirect targeting the genes that are not classically ‘druggable,’ owing to their molecular structure or because they are loss of function mutations [1, 4]. poly(ADP-ribose) polymerase inhibitor (PARPi) is the first successful clinical example based on SL [1, 5–7] and SL-based therapy has been regarded as one of the most effective anticancer treatments in the last decade [8]. The encouraging results of PARPi led to an increasing amount of drug candidates focusing on SL interactions. We list some recent clinical trials related to SL interactions in Table 1 [8, 9].
Figure 1.

The concept of SL and SDL.
Table 1.
Some recent clinical trials related to SL (https://clinicaltrials.gov/ct2/home). All of the listed agents are inhibitors
| Agent | Target gene | Mutate/overexpressed gene | Cancer type | Phase and ClinicalTrials.gov identifier | First posted |
|---|---|---|---|---|---|
| Olaparib | PARP | BRCA1/2 | Platinum sensitive relapsed ovarian cancer and metastatic breast cancer | IV, NCT04330040 | 1 April 2020 |
| Niraparib | Advanced pancreatic adenocarcinoma | II, NCT03601923 | 26 July 2018 | ||
| Rucaparib | Metastatic and recurrent endometrial cancer | II, NCT03617679 | 6 August 2018 | ||
| Talazoparib | Leukemia | I, NCT03974217 | 4 June 2019 | ||
| AZD6738 | ATR | TP53 | Recurrent, persistent or progressive myelodysplastic syndrome (MDS) or chronic myelomonocytic leukemia | I, NCT03770429 | 10 December 2018 |
| BAY1895344 | ATR | ATM | Advanced solid tumors and lymphomas (ATM loss and/or ATM deleterious mutations will be included) | I, NCT03188965 | 16 June 2017 |
| SRA737 | CHK1 | CCNE1, TP53, BRCA1, BRCA2, MYC, RAD50 | Advanced solid tumors or Non-Hodgkin’s Lymphoma | I and II, NCT02797964 | 14 June 2016 |
| Prexasertib (LY2606368) | BRCA | BRCA1/2 mutation associated breast or ovarian cancer, triple-negative breast cancer, and high grade serous ovarian cancer | II, NCT02203513 | 30 June 2014 | |
| MYC, CCNE1, Rb, FBXW7, BRCA1, BRCA2, PALB2, RAD51C, RAD51D, ATR, ATM, CHK2 | Advanced solid tumors | II, NCT02873975 | 22 August 2016 | ||
| Adavosertib (AZD1775) | WEE1 | TP53 | Uterine Serous Carcinoma | II, NCT04590248 | 19 October 2020 |
| SETD2 | Advanced/metastatic solid tumors | II, NCT03284385 | 15 September 2017 | ||
| BRCA | Advanced refractory cancers/lymphomas/multiple myeloma | II, NCT04439227 | 19 June 2020 | ||
| CYC140 | PLK1 | KRAS | Advanced leukemias or Myelodysplastic syndromes | I, NCT03884829 | 21 March 2019 |
| BI 6727 | Advanced, nonresectable and/or metastatic solid tumor | I, NCT01145885 | 17 June 2010 | ||
| GSK461364 | Advanced solid tumor or Non-Hodgkin’s lymphoma that has relapsed or is refractory to standard therapies | I, NCT00536835 | 28 September 2007 | ||
| Sotorasib (AMG 510) | CD274/PD-L1 | Stage IV non-small cell lung cancer | II, NCT04933695 | 22 June 2021 | |
| AZD2014 | 4EBP1 | MYC | High-risk prostate cancer | I, NCT02064608 | 17 February 2014 |
| CC-115 | Advanced solid tumors, and hematologic malignancies | I, NCT01353625 | 13 May 2011 | ||
| AZD4573 | CDK9 | Relapsed/refractory hematological malignancies | I, NCT03263637 | 28 August 2017 | |
| TP-1287 | Advanced solid tumors Sarcoma | I, NCT03604783 | 27 July 2018 | ||
| P276-00 | Stage III (unresectable) or stage IV metastatic melanoma | II, NCT00835419 | 3 February 2009 |
Despite the attractive concept of SL-based therapeutics, only PARPi has progressed to the clinic so far. A major hurdle might be the identification of clinically relevant, robust SL pairs [10]. Identifying potential SL gene pairs is mainly achieved by two methods: laboratory-based methods and computational-based methods. The most common laboratory-based methods include yeast screening, drug screening, RNA interference (RNAi) screening, clustered regularly interspaced short palindromic repeat (CRISPR) screening [8]. The limitation of yeast screening is that only a small portion of yeast genes (∼2000) have human orthologs [11], which limits the potential of this method. Drug screening tests drugs on various cell lines with specific mutations to identify SL gene pairs. SL gene pairs identified by drug screening would be easier to achieve clinical translation. However, the effect and specificity of drug inhibition tend to be lower than gene knockdown [8] and SL–gene pairs are limited in druggable gene targets. With the advent of RNAi and CRISPR/Cas9 technology, it is now possible to screen human cells for SL gene pairs. However, due to a large number of gene combinations (~200 million in a mammalian cell) [12], it is impractical to screen all potential SL pairs by these laboratory-based methods.
To overcome the abovementioned disadvantages, a variety of computational methods have been proposed, which can reduce the search space of SL gene pairs. These methods can be divided into four categories: statistics-based methods, network-based methods, classic machine learning-based methods and deep learning-based methods. Statistics-based methods are based on certain hypotheses to predict SL gene pairs. For instance, Jerby-Arnon et al. [13] developed a data-driven model called data mining synthetic lethality identification pipeline (DAISY) for SL prediction based on the assumption that SL genes tend to be co-expressed but seldom coinactivation. Network-based methods identify SL gene pairs through constructing protein–protein interaction (PPI) [14–17] network, signaling network [18, 19] or metabolism network [20–22]. With the rapid development of machine learning, various algorithms have been applied for SL prediction, including random forest (RF) [23–27], matrix factorization [28–30] and so on. Deep learning-based methods have recently emerged as useful methods to identify SL gene pairs, especially graph neural network (GNN) [31–33].
The rest of this review is organized as follows. The next section introduces SL-related databases, including label databases, feature databases and other related databases. The third section summarizes the computational methods for SL prediction. After that, negative sampling methods applied in these computational methods are explained in the fourth section. The subsequent section introduces available tools to predict SL interactions. Finally, challenges and future work are discussed in the last section.
SL-related databases
Due to the development of high-throughput screening technologies, a large amount of SL data have been identified. Many databases are developed to gather SL pairs, which are listed in Table 2. Among these databases, SynLethDB [34] is a unique comprehensive database for SL. Other databases are based on yeast screening, RNAi screening, CRISPR screening, computational prediction and drug screening.
Table 2.
Statistics of label databases reviewed in this paper
| Database | Methods | Description | Species and No. of SL pairs | Website | Latest update |
|---|---|---|---|---|---|
| SynLethDB V2 [34] | DAISY, text mining, large-scale screening techniques | Comprehensive database for SL | H. sapiens: 35943 S. cerevisiae: 14000 D. melanogaster: 439 M. musculus: 381 C. elegans: 105 | http://synlethdb.sist.shanghaitech.edu.cn/v2/#/ | 2020 |
| BioGRID V 4.4.201 [35–37] | Experiments and literature mining | Genetic interactions from all major model organisms and humans | Major model organisms and humans | http://www.thebiogrid.org | 1 September 2021 |
| Syn-lethality [38] | Manually curated SL pairs for human cancer from the literatures (113) SL pairs for human cancer inferred from yeast (1114) | Integrates experimentally discovered and verified human SL gene pairs into a network | H. sapiens: 1227 | http://www.ntu.edu.sg/home/zhengjie/software/Syn-Lethality/ (NTU staff’s personal web pages) | |
| GenomeRNAi [39] | RNAi | Genetic interactions detected by GenomeRNAi | H. sapiens Drosophila | http://www.genomernai.org/ | 27 November 2017 |
| DAISY [13] | Computational prediction | Statistically inferring SL pairs | H. sapiens: 2816 | http://www.cs.tau.ac.il/∼livnatje/SL_network.zip | |
| The Cellmap [40] | Yeast screening | Database of genetic interaction for S. cerevisiae | S. cerevisiae: ~10 000 (GI score < −0.35) | http://thecellmap.org | May 2016 |
| Laufer et al. study [42] | RNAi | Combinatorial RNAi and high-throughput imaging | Human cell lines: HCT116 HeLa | http://www.bioconductor.org/packages/2.12/data/experiment/html/HD2013SGI.html | |
| Vizeacoumar et al. study [43] | A negative genetic interaction map in isogenic cancer cell lines | 6 isogenic cancer cell lines (KRAS, PTTG1, PTEN, MUS81, BLM) | Support Information http://kimLab1.ccbr.utoronto.ca/projects/cancer_essential/ or http://moffatlab.ccbr.utoronto.ca/resources.php | ||
| Shen et al. study [44] | CRISPR screening | Combinatorial CRISPR screening | Human cell lines HeLa: 52 A549: 57293 T: 59 | 293 T - http://www.ndexbio.org/#/newNetwork/199f9bb1-c3eb-11e6-8e29-06603eb7f303 A549 - http://www.ndexbio.org/#/newNetwork/ec8bdae3-c3c9-11e6-8e29-06603eb7f303; HeLa - http://www.ndexbio.org/#/newNetwork/e50ee3c2-c3d4-11e6-8e29-06603eb7f303. | |
| GImap [12] | Combinatorial CRISPR screening | Human cell lines Jurkat: 454 K562:1678 | https://data.mendeley.com/datasets/rdzk59n6j4/1 | 22 July 2018 | |
| Najm et al. study [45] | Combinatorial CRISPR screening | Human cell lines A375, HT29, OVCAR8, 786O, A549, Meljuso | |||
| Zhao et al. study [46] | Metabolic gene networks through combinatorial CRISPR screening | Human cell lines A549 HeLa | Support information | ||
| GEMINI [47] | Computational prediction | A variational Bayesian approach to identify genetic interactions from combinatorial CRISPR screening | Sensitive lethal interactions and sensitive recovery interactions for four combinatorial CRISPR studies | Support information | |
| Wan et al. study [41] | Application of GEMINI to identify genetic interactions | Human cell lines A549: 126 A375: 18 HT29: 18 | https://github.com/FangpingWan/EXP2SL/tree/master/GEMINI | ||
| Slorth [25] | Predict SL pairs in a RF classifier | H. sapiens: 518636 S. cerevisiae: 372560 D. melanogaster: 93002 S. pombe: 52594 C. elegans: 56908 | http://slorth.biochem.sussex.ac.uk | Jun, 2019 | |
| CGIdb [48] | Identify potential SL pairs for specific cancer types from TCGA and functional screen data | H. sapiens: 10 637 | http://www.medsysbio.org/CGIdb | 2019 | |
| Srivas et al. study [49] | Drug screening | Evaluate thousands of TSG-drug combinations | Yeast: 1420 HeLa: 127 | Support information | 2016 |
Note: BioGRID, Biological General Repository for Interaction Datasets; DAISY, Data mining SL identification pipeline; TCGA, The Cancer Genome Atlas; TSG, tumor suppressor genes; H. sapiens, Homo sapiens; S. cerevisiae, Saccharomyces cerevisiae; D. melanogaster, Drosophila melanogaster; M. musculus, Mus musculus; C. elegans, Caenorhabditis elegans; S. pombe, Schizosaccharomyces pombe.
In addition, we list 12 commonly used feature databases to be fed into computational models in Table 3. These databases comprise genes’ or proteins’ sequence property (GenBank, Unitprot), functional property [gene ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), molecular signatures database (MSigDB), Comparative Toxicogenomics Database (CTD), LINCS, PhyloGene and comprehensive resource of mammalian protein complexes (CORUM)] and topological property in PPI (Search Tool for the Retrieval of Interacting Genes/Proteins (STRING), Human Protein Reference Database (HPRD) and Human Integrated Protein-Protein Interaction Reference (HIPPIE)).
Table 3.
Statistics of feature databases reviewed in this paper
| Database | Statistics | Website | Latest update |
|---|---|---|---|
| GenBank release 246.0 [112] | Gene sequence data: 233 642 893 | www.ncbi.nlm.nih.gov/genbank/ | 15 October 2021 |
| Unitprot release 2021_03 [113] | Protein sequence data: 219 740 215 | https://www.uniprot.org/ | 2 June 2021 |
| GO release 2021-10-26 [114] | 43 832 GO terms 7 827 176 annotations | http://geneontology.org/ | 26 October 2021 |
| KEGG Release 100.0 [115] | Pathway maps: seven categories, 548 maps | http://www.kegg.jp/ | 1 October 2021 |
| MSigDB V7.4 [116] | Pathway comembership | http://www.broadinstitute.org/msigdb | April 2021 |
| CTD [117] | Gene-pathway annotations: 135 789 | http://ctdbase.org/ | 5 October 2021 |
| LINCS Data Portal 3.0 [118] | 978 landmark genes under different perturbations | https://lincsproject.org/LINCS/ | June 2021 |
| PhyloGene [119] | http://genetics.mgh.harvard.edu/phylogene/ | 2015 | |
| CORUM 3.0 [120] | Mammalian protein complexes: 4274 | http://mips.helmholtz-muenchen.de/corum/ | 9 March 2018 |
| STRING 11.5 [121] | PPIs: more than 20 billion | https://string-db.org/ | 12 August 2021 |
| HPRD release 9 [122] | PPIs: 41 327 | http://www.hprd.org/ | 13 April 2010 |
| HIPPIE v2.0 [123] | Confidence scored and annotated PPIs: over 270 000 | http://cbdm.uni-mainz.de/hippie/ | 14 February 2019 |
Note: UniProt, The Universal Protein Resource; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; MsigDB, The Molecular Signatures Database; CTD, The Comparative Toxicogenomics Database; CORUM, The comprehensive resource of mammalian protein complexes; STRING, Search Tool for the Retrieval of Interacting Genes/Proteins database; HPRD, Human Protein Reference Database; HIPPIE, Human Integrated Protein–Protein Interaction reference; PPI, Protein–protein interaction.
Other databases provide data of large-scale single-gene knockout, cancer genomics and mutations and orthology analysis. SL interactions can be identified from the first two kinds of databases and can be inferred with the help of orthology analysis databases. The statistics of these databases are listed in Table 4.
Table 4.
Statistics of other related SL databases reviewed in this paper
| Database | Description | Website | Latest update |
|---|---|---|---|
| The cancer dependency map [124] | Databases based on large-scale single gene knockout | https://depmap.org/portal/ | 19 August 2021 |
| TCGA | Cancer genomics and mutation databases | https://www.cancer.gov/tcga | 29 October 2021 |
| CCLE [125] | https://sites.broadinstitute.org/ccle | 2019 | |
| COSMIC v95 [126] | https://cancer.sanger.ac.uk/cosmic | 24 November 2021 | |
| InParanoid 8 [127] | Orthology analysis | https://inparanoid.sbc.su.se/cgi-bin/index.cgi | December 2013 |
| OrthoMCL-DB [128, 129] | https://orthomcl.org/orthomcl/app/ | 8 September 2021 |
Note: TCGA, The Cancer Genome Atlas; CCLE, Cancer Cell Line Encyclopedia; COSMIC, Catalogue of Somatic Mutations in Cancer.
A brief introduction of each feature database and other database is shown in the supplementary file.
Comprehensive label databases
SynLethDB
SynLethDB [34] is a comprehensive database for SL and it has two versions so far. SynLethDB 1.0 was released in 2015 and SynLethDB 2.0 was updated in 2020. SL pairs are collected from multiple sources, including manual curations from literatures, three SL-related databases (BioGRID [35–37], Syn-lethality [38] and GenomeRNAi [39]), bispecific shRNA screening (DECIPHER), computational predictions (DAISY [13]) and text mining data for five species (human, mouse, fruit fly, worm and yeast). A brief introduction of the integrated databases is provided in the supplementary file.
SynLethDB provides a webserver to calculate the confidence score for each SL pair by integrating individual scores derived from different evidence sources. In addition, the latest version adds SynLethKG. It is a comprehensive knowledge graph (KG) of SL including 11 types of biomedical entities and 27 types of relationships, representing features and relationships between genes, cancers and drugs.
Label databases based on yeast screening
The CellMap
The CellMap [40] is a web-based database of genetic interaction for Saccharomyces cerevisiae released in 2016. Through constructing over 23 million double mutants, ~350 000 positive and ~550 000 negative genetic interactions are identified. Three different interaction maps are constructed: nonessential × nonessential (N×N), essential × nonessential (E×N) and essential × essential (E×E) genetic network. The number of SL pairs between nonessential genes (~10 000) is estimated by applying an extreme negative interaction score threshold (<−0.35) to the N × N dataset.
Label databases based on RNAi screening
SL gene pairs can be experimentally obtained by the comparison of single and dual mutants in the same assay. Compared to double mutant yeast strains, which can be developed through high-throughput mating methodologies, it is more challenging to develop human cell lines with double mutations [10, 41]. Early human double perturbation screening used combinatorial siRNA knockdowns or siRNA knockdown under specific mutations to obtain genetic interaction data. SL pairs can be extracted from these data based on a specific indicator.
Laufer et al.’s study
Laufer et al. [42] performed 51 680 combinatorial RNAi experiments and identified genetic interactions for one or more of 11 phenotypes between 2376 gene pairs in human colon cancer cells.
Vizeacoumar et al.’s study
Vizeacoumar et al. [43] identified negative genetic interaction partners of five specific driver-mutated genes across a set of isogenic cancer cell lines through pooled shRNA screening. A total of 826 genetic interactions are tested and 200 negative genetic interactions (24.2%) are confirmed. They generate a genetic interaction network consisting of 2014 nodes and 2617 edges.
Label databases based on CRISPR screening
With the advances in CRISPR technology, it is now possible to systematically map SL networks in human cancer cells using combinatorial CRISPR screening.
Shen et al.’s study
Shen et al. [44] developed a high-throughput CRISPR screening approach for targeting single and pairwise genes. They screened all possible pairs of 73 cancer genes in three human cell lines, with totally 152 SL gene pairs were identified.
GI map
Horlbeck et al. [12] systematically screened 222 784 gene pairs from two human cancer cell lines through the CRISPR interference method, and constructed a large genetic interaction (GI) map.
Najm et al.’s study
Najm et al. [45] developed a dual-Cas9 platform to screen genetic interactions across six human cell lines and examined SL interactions among them.
Zhao et al.’s study
Zhao et al. [46] probed metabolic gene networks through combinatorial CRISPR screening developed by Shen et al. [44]. They interrogated a set of 51 genes in A549 and HeLa cells, which are involved in glycolysis and pentose phosphate pathways.
Label databases based on computational prediction
GEMINI and Wan et al.’s study
Zamanighomi et al. develop GEMINI refer to section Tools and applications to identify sensitive lethal and sensitive recovery interactions from combinatorial CRISPR screening. Wan et al. [41] used GEMINI to identify SL interactions from the combinatorial CRISPR experiments in three cell lines. They provide SL gene pairs with both SL relationships and L1000 gene expression profiles.
Slorth
Benstead-Hume et al. [25] extracted various features from PPI networks for use in a RF classifier to predict SL and SDL pairs both within and across five species. All predicted pairs can be obtained in the Slorth database released in 2019.
Cancer genetic interaction database
Han et al. [48] developed an algorithm to identify potential SL interactions for specific cancer types from The Cancer Genome Atlas (TCGA) refer to the Supplementary data and functional screening data. As a result, 10 637 SL interactions are detected. They integrate SL interactions predicted by other studies and construct the Cancer Genetic Interaction database (CGIdb).
Label databases based on drug screening
Srivas et al.’s study
Srivas et al. [49] exploited ~169 000 potential interactions between tumor suppressor genes (TSG) orthologs and druggable genes in yeast. Under the guidance of the strongest signal, they screened thousands of TSG–drug pairs in HeLa cells and construct conserved SL interaction networks.
Computational methods for SL prediction
The increasing volume of biological data and the rapid development of computer technology have paved the way to develop computational methods for SL prediction. The principle behind computational methods is to utilize biological knowledge that is confirmed to be able to determine known SL interactions, thus providing valuable insights into identifying more SL interactions from genes of interest [50]. Moreover, they show an impressive ability in SL prediction. In general, computational methods can be divided into (i) statistical-based methods, (ii) network-based methods, (iii) classic machine learning (ML) methods and (iv) deep learning methods. Due to the various principles of these methods, they have their own merits and demerits, which are listed in Table 5. Summary of studies involved in this review are shown in Table 6 and their performance scores are summarized in Table 7.
Table 5.
Summary of SL prediction methods and representative models
| Methods and representative models | Description | Advantages | Disadvantages | Application scenarios |
|---|---|---|---|---|
| Statistical-based methods | Fit existing data based on certain hypothesis | From the perspective of systems biology Do not require known SL data | The selection of hypothesis or threshold is highly subjective and unstable | There are insufficient known SL data |
| DAISY [13] | Identifies SL interactions in cancer through three statistical procedures in parallel | Comprehendible to biologists Mining data from clinical cancer samples | The biological data are at times noisy and inaccurate | Identification of clinical-related SL interactions in cancer |
| Network-based methods | Study SL pairs from the perspective of biological network | Add network structure information to gain a more comprehensive understanding of genes globally | Network data are incomplete and contains a lot of noises | There are insufficient known SL data |
| IDLE [21] | Predicts enzymatic SDLs from a GSMM | The first computational method that captures enzymatic SDL effects in metabolic networks Uncovers the mechanisms behind SDLs | Does not integrate more data source such as patient-specific omics data | Identifies SDLs that have a significant impact on tumor in clinical settings |
| Fast-SL [22] | Rapidly identifies SL pairs in metabolic networks | Overcomes the issue of computational complexity | Does not identify human SL gene pairs | Identifies higher order SL pairs in metabolic network |
| Classic ML methods | Learn general patterns from a limited set of known SL data and use those patterns to make predictions about unknown or unobserved SL gene pairs | Good performance on small data sets Effectively integrate multidimensional feature data | Manually generated features and need to understand the features that represent the data Lacks of negative samples | Require known SL data and feature data of high quality |
| De Kegel et al. study [26] | RF-based model to predict paralog SL pairs | Makes interpretable predictions for paralog SL pairs | Restricted in the identification of paralog SL pairs | Identifies context-specific paralog SL pairs |
| GRSMF [28] | A GRSMF model | Has the ability of data-adaptiveness and avoids determining the dimension of the latent space | Focuses on mapping genes to latent representations and cannot aggregate information from neighbor genes | There are not enough negative samples |
| Deep learning methods | Use a multistep feature transformation to obtain a feature representation of the original data, and further input into the prediction function to obtain the final result | Discover deep features for representation learning and pattern recognition from large dataset Does not require manual feature extraction. | Demand a large amount of data and computational resources. Limited by the quality and quantity of the data, which contain many false positives and false negatives. It is hard to train the model. Poor interpretability Lack of negative samples | Require sufficient known SL data and feature data of high quality |
| EXP2SL [41] | A semisupervised neural network method | Utilizes unlabeled SL data to predict cell-line-specific SL pairs Demonstrates that L1000 expression profiles are effective features data for SL prediction | Limited sample space and cell lines | Predicts cell-line specific SL pairs There are insufficient labeled SL samples |
| DDGCN [31] | A dual-dropout GCN method | Uses SL dataset with better quality Aggregates information from neighbor genes | Focuses solely on known SL pairs and ignores other data sources of genes | There are sufficient SL samples of high quality and insufficient feature data |
Table 6.
Summary of studies involved in this review
| Category | Study | Published year | Algorithms | SL data | Feature data | Program code |
|---|---|---|---|---|---|---|
| Statistical-based methods | Li et al. [51] | 2011 | MLE | SGD [130] | Domain relationships | |
| Zhang et al. [52] | 2012 | MLE | SGD [130] | Protein sequences | ||
| Conde-Pueyo et al. [53] | 2009 | Homologous mapping | BioGRID [35–37] | Somatic mutations, GO annotation, drugs and their gene targets | ||
| Lee et al. [54] | 2013 | Homologous mapping | BioGRID [35–37] | Homology information, gene expression information | ||
| Deshpande et al. [55] | 2013 | Homologous mapping | Literatures [56] | Homology information | ||
| Kirzinger et al. [16] | 2019 | Homologous mapping | Gene expression data, homology information | |||
| Jerby-Arnon et al. [13] | 2014 | DAISY | SCNA and mutation profiles, gene essentiality profiles, gene expression profiles | |||
| Srihari et al. [58] | 2015 | Statistical analysis | Genomic copy-number and gene expression | |||
| Guo et al. [34] | 2016 | Statistical analysis | BioGRID [35–37], Syn-Lethality [38] GenomeRNAi [39] DAISY [13] The DECIPHER Project, | http://histone.sce.ntu.edu.sg/SynLethDB/ | ||
| Wang et al. [59] | 2019 | Statistical analysis | SynLethDB [34] and Literatures [15, 49, 58, 61, 131] | Somatic mutation information, shRNA data, yeast genetic interactions | ||
| Lee et al. [60] | 2018 | ISLE | SCNA, gene expression, mutation and survival data | https://github.com/jooslee/ISLE/ | ||
| Wang et al. [61] | 2013 | The univariate F-test or t-test | Gene expression | |||
| Chang et al. [62] | 2016 | Statistical analysis | Literatures [5, 6, 132, 133] | Gene expression | ||
| Feng et al. [63] | 2019 | Statistical analysis | Genomics and patient survival data | |||
| Sinha et al. [65] | 2017 | MiSL | Mutation, copy number and gene expression | https://purl.stanford.edu/ny450yx7231 | ||
| Yang et al. [64] | 2021 | SiLi | Large-scale sequencing data | |||
| Network-based methods | Kranthi et al. [15] | 2013 | PPI networks | PPIs | ||
| Jacunski et al. [14] | 2015 | PPI networks | BioGRID [35–37] | PPIs, functional annotations | ||
| Ku et al. [17] | 2020 | PPI networks | PPIs, pathways | |||
| Zhang et al. [19] | 2015 | Signaling networks | Signaling data | |||
| Liu et al. [18] | 2018 | Signaling networks | SynLethDB [34] | PPIs | ||
| Apaolaza et al. [20] | 2017 | Metabolic networks | Gene expression data | |||
| Megchelenbrink et al. [21] | 2015 | IDLE | The human metabolic network | |||
| Pratapa et al. [22] | 2015 | Fast-SL | Genome-scale metabolic networks | https://github.com/RamanLab/FastSL | ||
| Classic ML methods | Paladugu et al. [67] | 2008 | SVM | Literatures [134] [134–136] | PPI network | |
| Wu et al. [71] | 2021 | k-NN | SynLethDB [34] | Seven similarities of gene pairs (gene expression, protein sequence, PPI, copathway, GO biological process, GO cellular component and GO molecular function) | ||
| Yin et al. [69] | 2019 | DT | SynLethDB [34] | Mutation, CNV and clinical data of breast cancer | ||
| Pandey et al. [72] | 2010 | MNMC | SGD [130] | PPIs, functional annotations, Pathways, mutant phenotype, proteins phylogenetic profiles, sequence similarity of genes and proteins | ||
| Wu et al. [73] | 2014 | Ensemble learning | BioGRID [35–37] | Semantic similarity, PPIs, sequence orthologs, semantic similarity, co-complex membership, co-pathway membership, gene expression correlation, Common/interacting domains, the number of domains | ||
| Das et al. [23] | 2019 | DiscoverSL (RF) | SynLethDB [34] | Mutation, gene expression, copy number alteration, gene-pathway information | https://github.com/shaoli86/DiscoverSL/releases/tag/V1.0 | |
| Li et al. [24] | 2019 | RF | Shen et al. study [44] | GO term and KEGG pathway | ||
| Benstead-Hume et al. [25] | 2019 | RF | BioGRID [35–37] | PPIs | ||
| De Kegel et al. [26] | 2021 | RF | Shared PPIs, evolutionary conservation, etc. | https://github.com/cancergenetics/paralog_SL_prediction; https://doi.org/10.5281/zenodo.5139973 | ||
| Benfatto et al. [27] | PARIS (RF) | CRISPR screens with genomics and transcriptomics data | https://github.com/sbenfatto/PARIS | |||
| Huang et al. [28] | 2019 | GRSMF (Matrix factorization) | SynLethDB [34] | GO similarity matrix | https://github.com/Oyl-CityU/GRSMF | |
| Liany et al. [30] | 2020 | CMF (Matrix factorization) | SynLethDB [34] | Essentiality Profile, mRNA gene expression, SCNA level, pairwise coexpression | https://github.com/lianyh | |
| Liu et al. [29] | 2020 | SL2MF (Matrix factorization | SynLethDB [34] | PPI similarity, GO similarity | ||
| Deep learning methods | Wan et al. [41] | 2020 | Neural network | Shen et al. study [44] GI map [12] Najm et al. study [45] Zhao et al. study [46] | L1000 gene expression profiles [118] | https://github.com/FangpingWan/EXP2SL |
| Cai et al. [31] | 2020 | GCN | SynLethDB [34] | https://github.com/CXX1113/Dual-DropoutGCN | ||
| Long et al. [32] | 2021 | GAT | SynLethDB [34], SynLethDB- v2.0 (http://synlethdb.sist.shanghaitech.edu.cn/v2) | GO semantic similarity, PPIs | https://github.com/longyahui/GCATSL | |
| Hao et al. [33] | 2021 | GAE | SynLethDB [34] | GO similarity matrix, PPIs, coexpression、mutual exclusion score、copathway | https://github.com/DiNg1011/SLMGAE | |
| Zhang et al. [2] | 2021 | KG | SynlethDB [34], Jerby-Arnon et al. [13] | Three relationships (different cancer types and their mutant genes, drugs and targets, drugs and their indications) | ||
| Wang et al. [80] | 2021 | KG | SynLethDB [34], SynLethDB- v2.0 (http://synlethdb.sist.shanghaitech.edu.cn/v2) | The relationships of genes, drugs and compounds |
Note: SVM, support vector machine; DT, Decision tree; k-NN, k-nearest neighbors; RF, random forest; GCN, graph convolutional network; GAT, graph attention network; GAE, graph autoencoder; KG, knowledge graphs; MLE, maximum likelihood estimation; ISLE, identification of clinically relevant synthetic lethality; MiSL, mining synthetic lethals; SiLi, statistical inference-based synthetic lethality identification; IDLE, identifying dosage lethality effects; MNMC, multi-network and multi-classifier; PARIS, PAn-canceR Inferred Synthetic lethalities; GRSMF, graph regularized self-representative matrix factorization; CMF, collective matrix factorization; SGD, saccharomyces genome database; SCNA, somatic copy number alterations.
Table 7.
Performance scores and validation scheme of the methods involved in this review
| Study | Algorithms | Validation scheme | AUROC | AUPRC | ACC | F1 | MCC | Precision | Sensitivity | Specificity |
|---|---|---|---|---|---|---|---|---|---|---|
| Pratapa et al. [22] | SVM | 10-fold cross-validation | 0.796 | |||||||
| Wu et al. [71] | k-NN | 10-fold cross-validation | 0.848 | 0.861 | 0.764 | 0.739 | 0.825 | 0.670 | ||
| Pandey et al. [72] | MNMC | 10-fold cross-validation | 0.897 | |||||||
| Wu et al. [73] | Ensemble learning | 5-fold cross-validation | 0.871 | |||||||
| Li et al. [24] | RF | 10-fold cross-validation | 0.532 | |||||||
| Benstead-Hume et al. [25] | RF | 5-fold cross-validation | 0.889 | |||||||
| Liu et al. [29] | Logistic matrix factorization | 5-fold cross-validation | 0.848 | 0.239 | ||||||
| Huang et al. [28] | Matrix factorization | 5-fold cross-validation | 0.923 | |||||||
| Liany et al. [30] | CMF | 3-fold cross-validation | 0.980 | 0.980 | ||||||
| Wan et al. [41] | Neural network | 5-fold cross-validation | 0.969 | 0.880 | 0.959 | 0.866 | 0.872 | 0.903 | 0.968 | |
| Cai et al. [31] | GCN | 5-fold cross-validation | 0.878 | 0.344 | 0.552 | |||||
| Long et al. [32] | GAT | 5-fold cross-validation | 0.937 | 0.948 | ||||||
| Hao et al. [33] | GAE | 5-fold cross-validation | 0.917 | 0.942 | 0.871 | |||||
| Wang et al. [80] | KG | 5-fold cross-validation | 0.947 | 0.956 | 0.887 |
Notes: AUROC, area under receiver optimizer characteristics curve; AUPRC, area under precision-recall curve; ACC, accuracy; MCC, Matthews correlation coefficient.
Statistical-based methods
This section focuses on the related works of statistical methods on the SL prediction task. Based on the knowledge of systems biology, statistical-based methods learn to fit existing SL data using particular assumptions. The assumptions are based on prior biological knowledge, such as the fact that SL genes are frequently co-expressed, having similar functions, or exhibiting mutual exclusivity with respect to specific genetic events. Models based on these assumptions are usually explainable as they can reveal statistical regularities between gene pairs at the phylogenetic level to some extent, but the accuracy of these models greatly depends on the prior statistical assumptions.
Prediction of SL gene pairs for yeast
Earlier studies mainly focused on identifying SL pairs in yeast, due to the limited access to human SL pairs. For instance, yeast SL pairs can be predicted by maximum likelihood estimation (MLE) method using the domain genetic interaction probabilities [51] or genetic interactions of significant short polypeptide clusters [52]. Furthermore, SL gene pairs of humans or other species can be predicted through yeast orthology mapping [16, 53–55]. However, orthology mapping has two major limitations. First, only a small portion of yeast genes have human orthologs as humans are evolutionarily distant from yeast. Second, SL relationships may develop independently across species [14].
Global prediction of SL gene pairs for human
With the rapidly accumulation of human genome data, global human SL interactions prediction has started to be investigated.
Among them, DAISY is the most representative approach. DAISY is a data-driven computational pipeline based on large amounts of cancer genomic data proposed by Jerby-Arnon et al. [13] in 2014. They identify SL interactions in cancer through three statistical procedures in parallel (Figure 3A): (i) Genomic survival of the fittest. It is based on the observation that the coinactivition of SL pairs leads to cell death. Therefore, SL pairs can be selected by identifying gene coinactivation events that occur substantially less than expected. (ii) shRNA-based functional examination. It is based on the fact that knocking out the SL gene is lethal to cells when its SL partner gene is inactive. This can be implemented by an integrated analysis of shRNA essentiality screening, their somatic copy number alterations (SCNA) and transcriptomic profiles. (iii) Pairwise gene coexpression. SL pairs are likely to be involved in closely associated biological processes and hence tend to be co-expressed [56, 57]. Then a cancer genome-wide SL interactions network is constructed from SL gene pairs identified by all the three procedures. DAISY successfully identifies SL pairs by capturing the results obtained from large-scale genomic data and shRNA screens, but these data are at times noisy and inaccurate.
Figure 3.
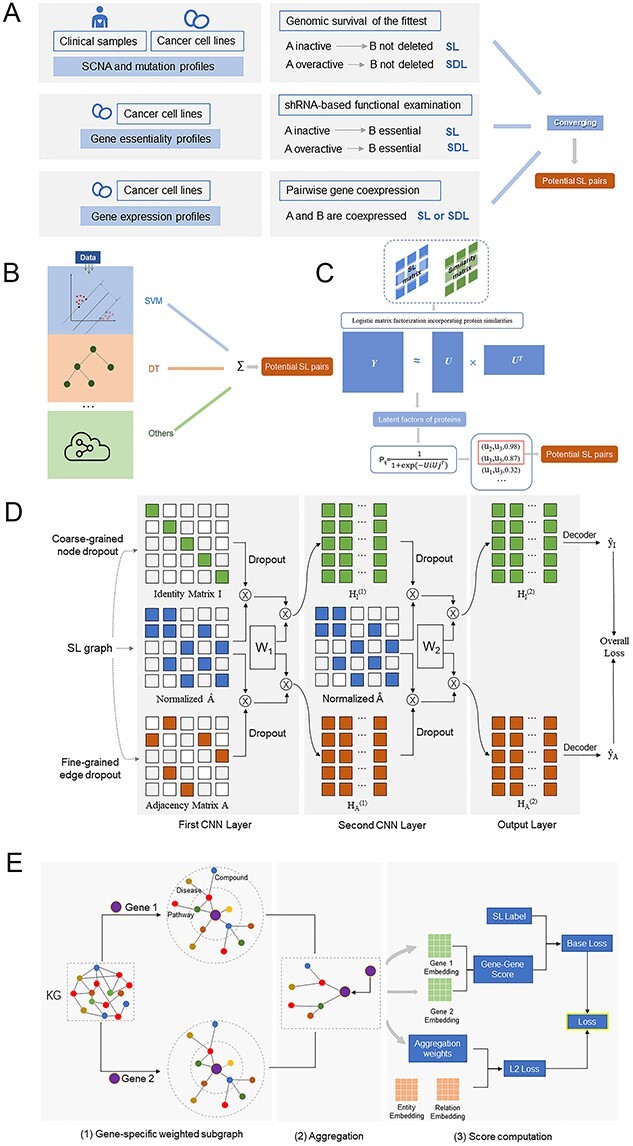
The flowcharts of some selected typical methods reviewed in this manuscript. (A) DAISY [13], a statistical-based method. (B) MNMC [72], an ensemble classifier. (C) SL2MF [29], a logistic matrix factorization method. (D) DDGCN [31], a GCN-based method. (E) KG4SL [80], a KG-based method.
Other researches have also developed some valuable statistical inferring methods and more assumptions have been proposed for SL prediction. For example, gene pairs altered in a mutually exclusive pattern are likely to be SL pairs [58, 59]; SL pairs upon coinactivation may exhibit prolonged patients’ survival [60]; SL pairs tend to have high phylogenetic similarity [60].
SL prediction for specific genes or cancers
Wang et al. [61] identified differentially expressed genes between tumors with and without functional p53 mutations by univariate F-test or t-test. The genes which exhibit higher relative expression in p53 mutated tumors were further selected as the candidate SL partner genes for p53. Chang et al. [62] selected lung adenocarcinoma-dependent genes through computing gene expression of lung adenocarcinoma versus nontumorous tissues, and then associated with five clinical factors to obtain predicted SL pairs. Feng et al. [63] developed an integrated computational pipeline based on ISLE (identification of clinically relevant SL) [60], which determine SL partner genes of GNAQ following four aspects: molecular condition (differentially overexpressed genes), clinical condition (genes associated with poor prognosis), phenotypic condition (more essential genes) and druggable condition. Recently, Yang et al. [64] inferred SL gene pairs in liver cancer based on DAISY and ISLE, which contain five inference analyses (functional similarity, differential gene expression, pairwise gene coexpression, pairwise survival and rank aggregation). Sinha et al. [65] proposed a computational pipeline called Mining Synthetic Lethals (MiSL) to identify mutation-specific SL pairs for specific cancers. Their basic assumption is that SL partner genes of a mutated gene tend to be amplified more frequently or deleted at a lower frequency in primary tumor samples containing the mutated gene.
Network-based methods
This section focuses on the network-based methods for SL prediction. Compared with statistical methods, network-based methods provide a more comprehensive understanding of genes in the entire biological network and improve our understanding of the mechanisms of SL. Currently, network-based methods predict SL pairs through constructing biological networks (PPI networks, signaling networks or metabolic networks), then analyzing the topological characteristics of genes in biological networks and assessing the network changes in response to knocking out gene pairs.
PPI network-based methods
Kranthi et al. [15] pointed out that the connectivity of the protein in the PPI network and the structure of the network are related to its functional characteristics. In general, the protein nodes with high degrees are usually functionally basic, and a lack of them would lead to lethality. Based on this, they developed graph information centrality measures in biological systems to identify SL gene pairs. They modified the information centrality method by knocking out two nodes. However, this method does not take the efficiency changes of knocking out a single node in the network into account, as the network changes may be caused by knocking out one gene at times [18]. Jacunski et al. [14] evaluated the connectivity homology by calculating the network parameters in the PPI network and designing an SL prediction model based on connectivity homology. Ku et al. [17] identified functionally distinct KRAS SL subnetworks or modules based on the MCODE clustering algorithm in the PPI network, all of which can be traced back to a specific pathway or protein complex.
Signaling network-based methods
Zhang et al. [19] predicted SL gene pairs by combining a data-driven method with the knowledge of pathway information from signaling networks to mimic the influence of single gene knockdown and double genes knockdown to cell viability. Gene pairs are considered as potential SL pairs when double genes knockdown significantly increase the likelihood of cell death, whereas single gene knockdown does not. Liu et al. [18] constructed human cancer signaling network (HCSN) by calculating the shortest path between no cancer gene and cancer gene pairs. Then they screened SL pairs from HCSN by three procedures: network-based method (according to the distance between cancer genes and noncancer genes), frequency-based method and function-based method. This method screens SL pairs by a multistep strategy, thus it might get better results.
Metabolic network-based methods
Apaolaza et al. [20] developed a genetic minimal cut set (gMCS)-based method to predict SL interactions and revealed a potential mechanism explaining the effect of specific gene knockout to disrupt cell growth. gMCS refers to minimal sets of reactions, the removal of which will invalidate the function of specific metabolic tasks. Megchelenbrink et al. [21] presented a network modeling method called identifying dosage lethality effects (IDLE). IDLE predicts enzymatic SDLs from a genome-scale model of metabolism (GSMM). For each pair of enzymes (A, B) in the human GSMM, they predicted SDL by measuring the growth reduction level caused by changing the enzyme flux of A and B. IDLE identifies SDLs in clinical settings, but it does not integrate more data sources such as patient-specific omics data. In addition, Pratapa et al. [22] developed Fast-SL, an algorithm to rapidly identify SL gene sets in metabolic networks. The algorithm overcomes the issue of computational complexity encountered in previous methods by iteratively narrowing the searching space for SLs, thus substantially reducing the computational time.
Indeed, network-based methods can only integrate one or more interaction networks among genes. Relationships between genes and other entities like patients cannot be directly modeled [30]. In addition, they cannot utilize other data that contain related information about SL, such as sequence and function properties of genes. What is more, they do not use the existing SL samples so the underlying patterns of known SL pairs are not being exploited.
Classic ML methods
This section mainly introduces some classic ML methods for SL prediction tasks. Compared with the network-based methods, ML methods can effectively integrate multidimensional data and achieve feature learning through parameter fitting, providing more comprehensive information for SL prediction. Classical ML methods attempt to reveal the patterns of observed samples that cannot be acquired through principle analysis, in order to achieve reliable prediction of unknown data (Figure 2). There are two main types of classical ML methods: supervised learning and unsupervised learning. Most SL prediction tasks adopt supervised ML models. Support vector machine (SVM), Decision Tree (DT), k-nearest neighbors (k-NN), RF, ensemble classifier and matrix factorization are applied for SL prediction.
Figure 2.

Workflow of ML methods used in SL prediction. SVM refers to support vector machine. DT refers to Decision Tree. RF refers to random forest.
Support vector machine
The principle of SVM is to create an optimal decision boundary that maximizes the distance between two classes [66]. Paladugu et al. [67] proposed an SVM model that uses topological properties of two genes in a PPI network as features for SL prediction in yeast.
Decision Tree
DT model creates tree-like structure for classification purpose, where each internal node corresponds to a test of a feature and each leaf node refers to a classification result [68]. Yin et al. [69] predicted SL interactions in breast cancer based on DT. Two features [mutation coverage and copy number variations (CNV)] are classified and optimized by experimentally validated SL pairs, which are used to predict SL interactions based on DT.
K- nearest neighbors
K-NN algorithm is a nonparametric method [70] that classifies unknown samples by a plurality voting of its neighbors. Wu et al. [71] proposed a k-NN model to achieve the similarity-based classification of gene pairs. The basic hypothesis of this model is that unknown gene pairs which exhibit high levels of similarity to the known SL pairs are more likely to be potential SL pairs.
Ensemble classifiers
Ensemble classifiers achieve predictions by integrating the results of several independently trained weak models on the same samples. The integrated models outperform those of separate models. How to choose the independent weaker models and how to integrate their learning results are the main challenges of this algorithm. Pandey et al. [72] defined a large number of features for characterizing SL interactions from diverse data sources. Then, they design an integrated multi-network and multi-classifier (MNMC) framework composed of six different classifiers to predict yeast SL gene pairs. Wu et al. [73] also developed an ensemble algorithm (MetaSL) that integrates RF, DT, SVM and other ML classifiers based on a variety of biological features. Compared with MNMC [72], MetaSL assigns different weights to different classifiers according to their performances in the training process. Thus the prediction results are based on a weighted consensus from the participating classifiers. However, the limitation of this study is that inter-dependence exists among the input features.
Random forest
RF [74] actually belongs to ensemble classifiers, but all of the integrated classifiers are DTs. RF achieves strong predictive power by combining the simplicity of DTs with the flexibility and powerful functions of ensemble classifiers. Besides, it can cope with high dimensional (containing many features) data without feature selection as it is able to randomly select a subset of features.
Das et al. [23] developed an RF-based R package DiscoverSL to predict SL interactions in cancers using multiomics cancer data. Li et al. [24] encoded genes as enrichment scores based on GO terms and KEGG annotation and a gene pair is represented by numerous features derived from their enrichment scores. Following this, they utilized SL label data to build an RF-based prediction model with optimized functional features. In particular, the maximum relevance and minimum redundancy method [75] is used to generate a ranked feature list and incremental feature selection method is applied to select the most appropriate number of features. Benstead-Hume et al. [25] also extracted features from the graph in the PPI network and use the RF model to predict SL gene pairs. Considering paralog pairs share functionality similarities and are more likely to be SL pairs, De Kegel et al. [26] developed an RF classifier to predict paralog SL pairs. Specifically, they applied TreeExplainer [76] to compute the influence of each feature on a specific prediction, so the classifier is able to make interpretable predictions. Benfatto et al. [27] developed an algorithm called PAn-canceR Inferred Synthetic lethalities (PARIS) that can address the importance of individual gene deficiency in explaining their dependencies in multiple cancer cells. The core of the PARIS algorithm lies in the feature selection step, achieved by RF through assigning importance scores to each mutation and expression feature based on CRISPR screening data across multiple cancer cell lines.
Matrix factorization
The classic ML methods described above are based on a supervised learning frame that requires both positive and negative training samples. However, SL prediction tasks lack real negative samples, as the majority of them are randomly selected from unknown samples, which may pick up false negative data. Matrix factorization methods effectively avoid this defect by capturing the underlying mechanisms of SL samples and integrating relevant information. Matrix factorization aims to decompose an input matrix into the product of two low-rank matrices, and then the data-missing matrix is filled with data obtained through model training.
Huang et al. [28] designed a graph regularized self-representative matrix factorization (GRSMF) model which uses the linear representation of matrix X’s rows and columns to decompose itself. What is more, authors integrate GO similarity matrix data as a graph regularization term to address the sparse input data and improve the prediction accuracy. Compared with the conventional matrix factorization, GRSMF has the ability of data-adaptiveness and avoids determining the dimension of the latent space. To further differentiate the importance weights between SL pairs and unknown pairs, Liu et al. [29] proposed a logistic matrix factorization model, called SL2MF (Figure 3C), to learn latent representations of SL pairs. The combination of the latent vectors determines the probability of SL pairs. Moreover, they apply neighborhood regularization to constrain the latent vector, based on the hypothesis that genes with similar GO or PPI properties should be factorized into similar latent vectors. In addition, conventional matrix factorization methods have limited capability on complicated heterogeneous data. To address this issue, Liany et al. [30] improved the collective matrix factorization (CMF) method through three measures. The first two measures rely on a transformation (principal components analysis and graph features). The third measure is to extend the model by using matrix-specific weights. This modified model figures out the problem that conventional CMF cannot learn the unique representation of each entity when multiple input matrices contain the same entity types.
Deep learning methods
This section discusses the application of deep learning in SL prediction. Deep learning is a subset of ML methods. Compared to classical ML methods that extract features manually based on knowledge, deep network structures can better capture nonlinear and complex relationships between inputs and outputs, allowing them to identify complex patterns behind the data. Interdependent relationships always exist in biological entities and processes, which are often inherently noisy and occur at multiple scales. Therefore, biological data can be well suited for deep learning.
Neural networks are the most commonly employed models in deep learning as they show high significant fit for complex nonlinear problems [111]. Given the fact that most SL pairs are cell-line specific, Wan et al. [41] develop a semisupervised neural network method called EXP2SL to identify SL pairs. For a pair of gene, they use cell line shRNA perturbation (LINCS L1000 project) gene expression profile to construct 978-dimensional features as inputs of the encoding layers and predict the potential SL pairs. EXP2SL is the first model to predict cell-line-specific SL pairs and it makes full use of unlabeled SL data.
Currently, there are plenty of deep learning methods, among which the following two types are the most frequently and effectively used models in SL prediction: GNN and KG embedding.
Graph neural network
GNN can efficiently capture the structures of graph and model complex relations between neighbor nodes in the graph. Three archetypes of GNN are adopted in SL prediction, including graph convolutional network (GCN), graph attention network (GAT) and graph auto-encoder (GAE).
Graph convolutional network
GCN is an extension of convolutional neural network on graph structure. Compared with matrix factorization mentioned in the previous section, GCN can capture the information of neighbor nodes in the graph. Cai et al. [31] apply GCN to SL prediction and propose a model called dual-dropout GCN (DDGCN) (Figure 3D). DDGCN can aggregate information of neighboring genes in a graph by convolution operators. Furthermore, researchers adopt a dual dropout regularization technique [77] during the training process to avoid overfitting due to the sparse SL data. However, there are two limitations in this study. First, DDGCN only utilizes the information of the known SL pairs and lacks information on other features. Second, DDGCN does not assign different weights to different neighbors.
Graph attention network
GAT [78], which is able to assign different weights to each neighbor nodes, adopts attention mechanism to counter the shortcomings of GCN. Long et al. [32] developed a Graph Contextual Attention Network model called GCATSL that effectively integrates multiple biological data for SL prediction. After constructing multiple gene feature graphs with different data source as model’s inputs, a dual attention mechanism (node-level and feature-level) is designed for each feature to capture local and global neighbors’ importance to learn their representations. Multilayer perceptron is further exploited to aggregate the extracted features with original features.
Graph auto-encoder
GAE extends the idea of autoencoder to a graph. The node embeddings in the graph can be obtained through the encoder–decoder structure. In general, GAE uses GCN as the encoder. After inputting the topology and node information of the graph into the encoder, the inner product is adopted as the decoder to reconstruct the original graph. Hao et al. [33] combined GCN with autoencoder to construct a multiview graph autoencoder (SLMGAE) with a variety of data for SL prediction. SLMGAE takes SL graph as main view and graphs of other data (PPI, GO, etc.) as support views. Multiple GAEs are applied to graph reconstruction and GCN is used as the encoder. SLMGAE is able to integrate various data sources of genes in a GNN based framework and differentiate each data source by an attention mechanism.
Knowledge graph
The network embedding-based methods mentioned above integrate the information of multiple or heterogeneous biological networks, but in essence, there is no unified consideration for different relationship types. KG demonstrates excellent performance to this problem, which is a kind of knowledge-rich heterogeneous network composed of interconnected entities and relevant properties. It embeds the rich entities and the relationship information into the continuous vector space with low dimension [79], so as to facilitate computation while retaining the structural information. Due to the complexity and diversity of biological information, KG performs well in biological tasks such as SL prediction.
Wang et al. [80] constructed a KG algorithm (KG4SL) for SL prediction (Figure 3E). The algorithm consists of three modules. First, a gene-specific weighted subgraph is generated for each gene. Second, gene representation is updated by aggregating its neighbors’ representations in its weighted subgraph. Third, SL score can be calculated through the inner product of the two genes’ aggregation result. However, this method may not fully integrate the neighborhood topological structures when generating a gene-specific weighted subgraph due to the large degrees of some nodes. In addition, some neighbors might be uninformative and promiscuous in the process of message passing. Zhang et al. [2] developed the Synthetic Lethality Knowledge Graph (SLKG), which integrates three types of entities (genes, drugs and diseases) and four types of relationships. Drug repositioning is achieved by defining three core scoring functions: SLScore (SDLScore), DrugScore and CancerScore. SLScore is calculated by integrating different SL evidences.
In general, the above four methods have their own characteristics. Statistics-based methods and network-based methods are usually interpretable for novel predictions and do not require known SL samples. Statistics-based methods are based on statistical assumptions on the biological data, thus the accuracy of the assumptions and the quality of the biological data are needed. For example, considering SL gene pairs tend to be coexpression and seldom coinactiviton, DAISY [13] identifies SL gene pairs from large-scale genomic data. Network-based methods are based on a deep knowledge of a single or heterogeneous biological network, often accompanied by some creative concepts to identify the potential nodes that play an important role in the biological network. For instance, Kranthi et al. [15] developed a graph information centrality to identify SL gene pairs from human cancer protein interaction network. ML methods are trained on known SL samples. Classic ML methods tend to get better results than deep learning methods in small and medium-sized data sets (below hundreds or tens of thousands of samples) due to their fewer hyperparameters. Deep learning can achieve a better prediction under a big data set and it can extract high-level features from the data based on its complex network structure and a large number of parameters. However, due to the end-to-end learning process, the intermediate process of deep learning is a black box with good performance but a lack of interpretability [81]. This will lead to great uncertainty and unreliability when applied in biological or medical practice.
Negative sampling methods
SL computational models normally follow a supervised learning framework. Experimental data are composed of positive and negative samples. Positive samples can be extracted from databases in Table 2. How to prepare negative samples is one of the challenges for SL prediction.
Randomly picking up unknown gene pairs is a commonly used method for negative sampling [32, 80]. This approach is relatively simple and can obtain enough negative samples. However, as shown in Figure 4, this method may pick up unidentified positive samples. Mislabeled data would lead to the worse performance of the model.
Figure 4.
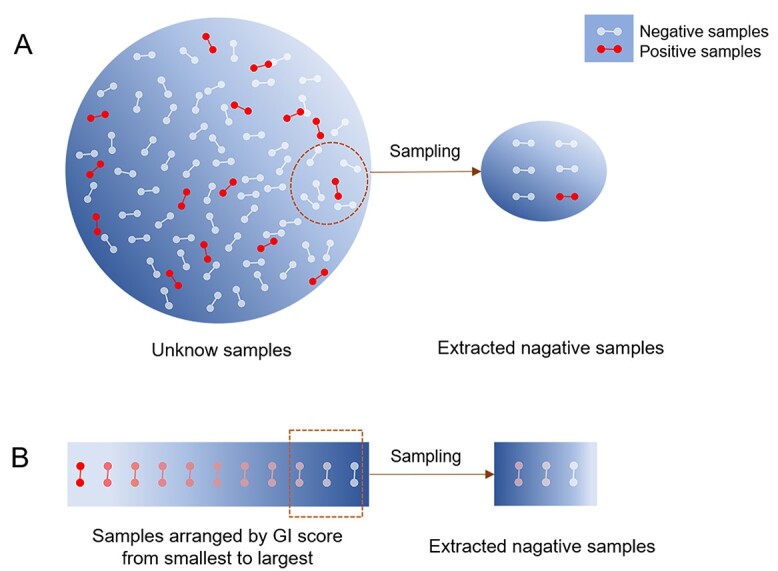
Negative sampling methods. (A) Randomly picking up unknown gene pairs as negative samples. (B) Extracting gene pairs from GI databases with certain GI scores as negative samples.
Another negative sampling method is extracting gene pairs from GI databases with certain GI scores as negative samples [33, 41]. For instance, Hao et al. [33] extracted negative SL samples with GI scores around 0 and positive SL samples with GI scores below −3. This negative sampling method avoids introducing potential positive samples, but the number of negative samples is relatively small.
Tools and applications
To predict or identify SL interactions based on various data, number of easy-to-use tools have been developed. In this section, a brief introduction of these representative tools is presented. Further details of these tools are listed in Table 8.
Table 8.
Tools and applications reviewed in this study
| Tool | Description | Availability | Website |
|---|---|---|---|
| G2G | Predict SL interactions based on mapping genes to GO terms | Online | http://bnet.cs.tau.ac.il/g2g/ |
| SPAGE-Finder | Predict SL interactions from TCGA data | Online | https://amagen.shinyapps.io/spage/ |
| SynLeGG | Predict SL interactions utilizing multiSEp gene expression clusters to Partition CRISPR essentiality scores and mutations from whole-exome sequencing | Online | www.overton-lab.uk/synlegg |
| SL-BioDP | Predict SL interactions from hallmark cancer pathways by mining cancer’s genomic and chemical interactions | Online | https://sl-biodp.nci.nih.gov/sl_index.php |
| DiscoverSL | R package for multiomic data-driven prediction of SL interactions in cancer | Standalone | https://github.com/shaoli86/DiscoverSL/releases/tag/V1.0 |
| ISLE | Identify the most likely clinically relevant SL interactions by mining TCGA cohort | Standalone | https://github.com/jooslee/ISLE/ |
| GEMINI | Identify SL interactions from combinatorial CRISPR experiments | Standalone | https://github.com/sellerslab/gemini |
| Fast-SL | identify synthetic lethal sets in metabolic networks | Standalone | https://github.com/RamanLab/FastSL |
Note: SynLeGG, Synthetic Lethality using Gene expression and Genomics; SL-BioDP, Synthetic Lethality BioDiscovery portal.
G2G
G2G is a web server for the human SL interactions prediction published by Almozlino et al. [82]. The web server provides access to predicting phenotypes of paired gene deletions by an improved algorithm based on RF. Followed by submitting a source gene and a target gene, the phenotype for that gene pair can be computed. Furthermore, users can submit only one gene and then G2G returns all predicted interacting genes according to their neighbor relationships in the PPI network.
Synthetic lethality bio discovery portal and discover SL
Synthetic Lethality Bio Discovery Portal is a comprehensive web tool to predict SL [23] interactions from hallmark cancer pathways through mining genetic and chemical interactions in cancer. The web tool was developed by Deng et al. [83] in 2019 based on the previous statistical approach DiscoverSL (refer to section Statistical-based methods).
Users can search the web tool from three modes: ‘GENES’ (including 623 commonly mutated cancer genes), ‘CANCER’ (including 18 histology types) and ‘DRUG’. In addition, the ‘INFERRED DRUG SYNERGY’ provides potential synergistic drug combinations.
SPAGE finder
Magen et al. [84] define ‘survival-associated pairwise gene expression states’ (SPAGEs) as pairs of genes whose co-expression levels are related to cell survival. They present a data-driven pipeline named SPAGE finder that identifies 71 946 SPAGEs from TCGA data, spanning 12 distinct types and a small portion of which are SL pairs. They provide a webserver visualizing the SPAGEs identified by the original manuscript, and allowing input or upload a gene list file of comma-separated gene names which will be rendered on the left panel.
Synthetic lethality using gene expression and genomics
Synthetic Lethality using Gene expression and Genomics (SynLeGG) is a web server developed by Wappett et al. [85] in 2021. SynLeGG utilizes MultiSEp algorithm to partition gene expression to discover SL-related characteristics. It predicts genetic dependency relationships including SL spanning 30 tissues and 783 cancer cell lines.
GEMINI
GEMINI [47] is an R package based on the variational Bayesian method to identify genetic interactions from combinatorial CRISPR perturbation studies. Scoring systems related to the individual and combined effects are defined to identify SL interactions.
CHALLENGES AND FUTURE WORK
Traditional genetically targeted cancer therapies normally focus on targeting gene products that are mutated or overexpressed in specific cancer types. However, from a drug discovery perspective, the loss-of-function mutations are much harder to target, and the same is true for several undruggable overexpressed genes. Fortunately, SL provides an avenue for the treatments of these targets as they facilitate the indirect targeting of nondruggable genes through the identification of a second druggable target that can interact with the primary genes [10]. Despite a marked increase in the identification of SL gene pairs, relatively few SL drug candidates have entered into clinic, and the field remains largely in its infancy [4]. Computational methods hold great prospects in this field but still remain some challenges. In this section, we will discuss these challenges and possible work in the future, mainly including biological issues and data and algorithm issues.
Biological issues
Expand the concept of SL
The conventional concept of SL is defined as the interaction between two genes. With a deeper understanding of SL, some studies expand the concept of it.
SL interactions among multiple genes
Most present studies focus on identifying SL interactions between two genes. However, the biological genetic interactions are complex and it is imperative to identify multiple genetic interactions. Kuzmin et al. [86] scored trigenic interactions in ~200 000 yeast and identified 3196 trigenic negative interactions. The global trigenic interaction network is estimated nearly 100-fold larger than the digenic network. Pratapa et al. [22] develop Fast-SL to identify high order SL interactions, including triplets and quadruplets. Prediction of SL interactions between multiple genes may be one of the challenges in the future.
Soft SL
Ryan et al. [87] consider that SL can be divided into hard SL and soft SL. Conventional SL is called hard SL. Soft SL exists between gene A and gene B but can be rescued by other genes. These reverse effects are called synthetic rescue (SR) or synthetic viability (SV). Gu et al. [88] identified candidate SR (SV) pairs by applying a statistical-based method and demonstrated that SR (SV) enables the prediction of drug resistance. The integration of SL and SR (SV) may result in higher reproducibility of SL prediction, thus future work for SL prediction should take SR (SV) into consideration.
Phenotype-centric SL
Conventionally, the mutated genes are utilized to distinguish cancer cells from normal cells and pharmacological inhibition of their partner genes is commonly adopted for SL-based cancer therapies. However, this concept can be extended. Akimov et al. [89] point out that the main determinant of any SL interaction is the phenotype alteration caused by a specific mutation or molecular perturbation. Therefore, considering the polygenic nature of the phenotype, they propose that phenotype might be a more robust differentiating context for SL interactions. SL interaction between WRN gene and microsatellite instability phenotype is an example phenotype-centric SL [90, 91]. The identification of more phenotype-centric SLs is a meaningful work in the future.
SL interactions between two signals
The integrated signaling system is critical for cell survival. Within it, various pathways interact with each other for survival and disrupting signals involved in multiple pathways is a practice of SL [92]. From the perspective of signals, we can get a deeper understanding of the biological mechanism of SL. In this regard, SL interactions can expand from genes to any signals, such as epigenetic regulators [93]. Integrating different types of signals to predict SL interactions would be crucial for future researches.
Expand the application of SL
At present, the main application of SL is still focused on the discovery of new anticancer targets. However, some researches indicate that SL would be applied in a wider range. These studies are explained in this section, which may give reference to the researchers in this field.
Nononcological diseases
SL has been successfully applied to identify anti-cancer targets but has found limited use in other diseases. There have been some researches probed into nononcological diseases, such as bacterial infection [94–97], malaria [98] and virus infection [99]. Computational methods may assist further application of SL in more nononcological diseases in the future.
Biological mechanisms revealed by SL
The essence of SL is a kind of genetic interaction and the analysis of SL can provide mechanistic insight into genes. Lippert et al. predict gene function from SL networks [100]. Guell et al. [101] analyze and categorize SL gene pairs in metabolic networks, and unveil plasticity and redundancy are indispensable mechanisms for biological systems. Cheng et al. analyze the role of SL in cancer risk and their findings support a possible role for SL in tumorigenesis [102]. In the future, as more SL pairs would be discovered, more biological mechanisms about genes will be revealed.
Drug repositioning researches
In spite of various promising computational methods that have been developed to identify SL interactions, drug repositioning researches based on SL have seldom been explored. After all, the ultimate goal of identifying novel SL pairs is to develop novel tumor target therapy. Recently, Zhang et al. [2] develop SLKG, a comprehensive KG aimed at providing the computational basis to tumor therapies based on SL. They demonstrate that SLKG is able to identify the optimal repurposing drugs and drug combinations. Future efforts are expected for these pioneer studies to achieve the clinical translation of SL.
Other application
Some researchers explored wider applications of SL. It is reported that [103, 104] SL interaction may be a new approach in chemoprevention of cancer, but this approach is to a great extent in its infancy. Additionally, Lee et al. [105] developed a precision oncology framework to predict patients’ cancer therapy response based on SL and SR interactions.
Data and algorithm issues
Data quality
The quality of training data is crucial to SL prediction. However, high false-positive and false-negative ratios often be observed in SL data generated by high-throughput screenings. In addition, positive SL samples could be negative under certain conditions. All of the abovementioned issues could lead to label inaccuracy and inconsistency. Besides, the performance of current models used to predict SL interactions is difficult to assess due to a lack of a gold standard source of human SL pairs. Therefore, to preprocess the SL data before applying it in computational models, establishing a gold standard source of SL pairs is necessary.
Sparse data and imbalanced samples
Due to the limited technology, known SL pairs are less than 0.1% of all potential pairs [31], which lead to two issues concerning training data.
The first issue is sparse data. When applying these sparse data in ML models, overfitting tends to occur. Cai et al. [31] propose dual forms of dropout in their DDGCN model to avoid overfitting problems. For future work, more SL gene pairs would be identified by the cooperation of biological and computational researchers to address the sparse data. Moreover, computational models fitted better to sparse data should be developed.
The second issue is imbalanced samples. The performance of the model deteriorates as the imbalance between the two classes increases. To address this problem, appropriate evaluation metrics should be adopted. Area under precision-recall curve is a more effective metric than area under receiver optimizer characteristics curve when applied on highly skewed tasks [31, 84]. Matthews correlation coefficient [106] has also been successfully used in SL prediction study [24] of which the samples are highly imbalanced. Besides, Li et al. [24] generated pseudopositive SL samples by synthetic minority oversampling technique method, which is designed to generate a number of predefined new samples from samples of minority class [107]. With these studies, we are one step closer to resolve the issue of imbalanced samples, and researchers would be inspired to explore more innovative solutions in the future researches, such as developing computational models that fit the imbalanced SL data better.
Lack of informative features
The mechanism behind SL is complex and cannot be generalized. Li et al. [9] propose a novel SL classification based on the specificity of its biological mechanism, which contains organelle level, pathway level, gene level and conditional SL. Conducting feature selection according to its biological mechanism before training a model is essential. In this way, informative features could be selected, which would help to explore more efficient computational models.
Lack of interpretability
Most ML approaches have not achieved clinical practice owing to lack of interpretability [108]. These models are regarded as ‘black boxes’, which optimize prediction accuracy without understanding the biological mechanisms behind the predicted results [81]. To resolve these difficulties, model interpretation is now a fast-growing subfield of ML methods [109]. Several efforts have been made on this issue for genetic prediction [110, 111]. For SL prediction, interpretable models have not yet been reported. More attention should be paid in developing interpretable models for SL prediction in the future.
Conclusion
Identification of SL gene pairs is imperative as it can provide novel targets for targeted therapy. However, the search space of gene combinations is too large to be investigated experimentally. Computational methods have been advanced to complement experimental approaches, which can reduce the search space of SL gene pairs. This review provides a comprehensive overview of computational methods, databases and tools for SL prediction. It introduces six types of label databases, three types of feature databases, three types of other related databases and six tools for SL prediction. Moreover, four types of computational methods with a detailed description of strengths and weaknesses have been summarized. In addition, we highlight several challenges in this field, some of which may inspire the future researches.
Key Points
Computational methods for SL can accelerate the discovery of novel SL-based targeted cancer therapies.
This study reviews six types of label databases, three types of feature databases, three types of other related databases and six tools for SL. The related information and links of all databases are provided.
Computational methods including statistical-based methods, network-based methods, classic machine learning methods and deep learning methods are introduced, and their merits and demerits are discussed.
The challenges include biological issues and data and algorithm issues. Expanding the concept of SL and expanding the application of SL are discussed in the section of biological issues. In addition, data quality, sparse data and imbalanced samples, lack of informative features and lack of interpretability require further exploration in future studies.
Supplementary Material
Author Biographies
Jing Wang is a PhD candidate in the School of Medicine, Tsinghua University, Beijing, China, and the Department of Bioinformatics, Institute of Health Service and Transfusion Medicine, Beijing, China.
Qinglong Zhang is a master’s student in the Department of Bioinformatics, Institute of Health Service and Transfusion Medicine, Beijing, China.
Junshan Han is a PhD candidate student in the Department of Bioinformatics, Institute of Health Service and Transfusion Medicine, Beijing, China.
Yanpeng Zhao is a PhD candidate in the Department of Bioinformatics, Institute of Health Service and Transfusion Medicine, Beijing, China.
Caiyun Zhao is an undergraduate student in the Peking University, Beijing, China.
Bowei Yan is a master’s student in the Department of Bioinformatics, Institute of Health Service and Transfusion Medicine, Beijing, China.
Chong Dai is a master’s student in College of Life Science and Technology, Beijing University of Chemical Technology, Beijing, China and the Department of Bioinformatics, Institute of Health Service and Transfusion Medicine, Beijing, China.
Lianlian Wu is a master’s student in the Academy of Medical Engineering and Translational Medicine, Tianjin University, Tianjin, China.
Yuqi Wen is a PhD candidate in the Department of Bioinformatics, Institute of Health Service and Transfusion Medicine, Beijing, China.
Yixin Zhang is a postdoc in the Department of Bioinformatics, Institute of Health Service and Transfusion Medicine, Beijing, China.
Dongjin Leng is a PhD candidate in the Department of Bioinformatics, Institute of Health Service and Transfusion Medicine, Beijing, China.
Zhongming Wang is a master’s student in the Academy of Medical Engineering and Translational Medicine, Tianjin University, Tianjin, China.
Xiaoxi Yang is a postdoc in Beijing Friendship Hospital, Capital Medical University, Beijing, China.
Song He is an associate professor in the Department of Bioinformatics, Institute of Health Service and Transfusion Medicine, Beijing, China.
Xiaochen Bo is a professor in the Department of Bioinformatics, Institute of Health Service and Transfusion Medicine, Beijing, China.
Funding
This work is supported by the National Natural Science Foundation of China (http://www.nsfc.gov.cn; nos. 62103436) to Song He.
References
- 1. Huang A, Garraway LA, Ashworth A, et al. . Synthetic lethality as an engine for cancer drug target discovery. Nat Rev Drug Discov 2020;19(1):23–38. [DOI] [PubMed] [Google Scholar]
- 2. Zhang B, Tang C, Yao Y, et al. . The tumor therapy landscape of synthetic lethality. Nat Commun 2021;12(1):1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ashworth A, Lord CJ. Synthetic lethal therapies for cancer: what's next after PARP inhibitors? Nat Rev Clin Oncol 2018;15(9):564–76. [DOI] [PubMed] [Google Scholar]
- 4. Setton J, Zinda M, Riaz N, et al. . Synthetic lethality in cancer therapeutics: the next generation. Cancer Discov 2021;11(7):1626–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bryant HE, Schultz N, Thomas HD, et al. . Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005;434(7035):913–7. [DOI] [PubMed] [Google Scholar]
- 6. Farmer H, Mccabe N, Lord CJ, et al. . Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005;434(7035):917–21. [DOI] [PubMed] [Google Scholar]
- 7. Lord CJ, Ashworth A. PARP inhibitors: synthetic lethality in the clinic. Science 2017;355(6330):1152–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Topatana W, Juengpanich S, Li S, et al. . Advances in synthetic lethality for cancer therapy: cellular mechanism and clinical translation. J Hematol Oncol 2020;13(1):118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li S, Topatana W, Juengpanich S, et al. . Development of synthetic lethality in cancer: molecular and cellular classification. Signal Transduct Target Ther 2020;5(1):241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. O'neil N J, Bailey M L, Hieter P.. Synthetic lethality and cancer. Nat Rev Genet 2017;18(10):613–23. [DOI] [PubMed] [Google Scholar]
- 11. Parameswaran S, Kundapur D, Vizeacoumar FS, et al. . A road map to personalizing targeted cancer therapies using synthetic lethality. Trends Cancer 2019;5(1):11–29. [DOI] [PubMed] [Google Scholar]
- 12. Horlbeck MA, Xu A, Wang M, et al. . Mapping the genetic landscape of human cells. Cell 2018;174(4):953–67 e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jerby-Arnon L, Pfetzer N, Waldman YY, et al. . Predicting cancer-specific vulnerability via data-driven detection of synthetic lethality. Cell 2014;158(5):1199–209. [DOI] [PubMed] [Google Scholar]
- 14. Jacunski A, Dixon SJ, Tatonetti NP. Connectivity homology enables inter-species network models of synthetic lethality. PLoS Comput Biol 2015;11(10):e1004506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kranthi T, Rao SB, Manimaran P. Identification of synthetic lethal pairs in biological systems through network information centrality. Mol Biosyst 2013;9(8):2163–7. [DOI] [PubMed] [Google Scholar]
- 16. Kirzinger MWB, Vizeacoumar FS, Haave B, et al. . Humanized yeast genetic interaction mapping predicts synthetic lethal interactions of FBXW7 in breast cancer. BMC Med Genom 2019;12(1):112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ku AA, Hu HM, Zhao X, et al. . Integration of multiple biological contexts reveals principles of synthetic lethality that affect reproducibility. Nat Commun 2020;11(1):2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu L, Chen X, Hu C, et al. . Synthetic lethality-based identification of targets for anticancer drugs in the human Signaling network. Sci Rep 2018;8(1):8440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang F, Wu M, Li XJ, et al. . Predicting essential genes and synthetic lethality via influence propagation in signaling pathways of cancer cell fates. J Bioinform Comput Biol 2015;13(3):1541002. [DOI] [PubMed] [Google Scholar]
- 20. Apaolaza I, San Jose-Eneriz E, Tobalina L, et al. . An in-silico approach to predict and exploit synthetic lethality in cancer metabolism. Nat Commun 2017;8(1):459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Megchelenbrink W, Katzir R, Lu XW, et al. . Synthetic dosage lethality in the human metabolic network is highly predictive of tumor growth and cancer patient survival. P Natl Acad Sci USA 2015;112(39):12217–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pratapa A, Balachandran S, Raman K. Fast-SL: an efficient algorithm to identify synthetic lethal sets in metabolic networks. Bioinformatics 2015;31(20):3299–305. [DOI] [PubMed] [Google Scholar]
- 23. Das S, Deng X, Camphausen K, et al. . DiscoverSL: an R package for multi-omic data driven prediction of synthetic lethality in cancers. Bioinformatics 2019;35(4):701–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li J, Lu L, Zhang YH, et al. . Identification of synthetic lethality based on a functional network by using machine learning algorithms. J Cell Biochem 2019;120(1):405–16. [DOI] [PubMed] [Google Scholar]
- 25. Benstead-Hume G, Chen X, Hopkins SR, et al. . Predicting synthetic lethal interactions using conserved patterns in protein interaction networks. PLoS Comput Biol 2019;15(4):e1006888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. De Kegel B, Quinn N, Thompson NA, et al. . Comprehensive prediction of robust synthetic lethality between paralog pairs in cancer cell lines. Cell Syst 2021;12(12):1144, S2405-4712(21)00329-X–1159.e6. [DOI] [PubMed] [Google Scholar]
- 27. Benfatto S, Sercin O, Dejure FR, et al. . Uncovering cancer vulnerabilities by machine learning prediction of synthetic lethality. Mol Cancer 2021;20(1):111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huang J, Wu M, Lu F, et al. . Predicting synthetic lethal interactions in human cancers using graph regularized self-representative matrix factorization. BMC Bioinform 2019;20(Suppl 19):657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu Y, Wu M, Liu C, et al. . SL(2)MF: predicting synthetic lethality in human cancers via logistic matrix factorization. IEEE/ACM Trans Comput Biol Bioinform 2020;17(3):748–57. [DOI] [PubMed] [Google Scholar]
- 30. Liany H, Jeyasekharan A, Rajan V. Predicting synthetic lethal interactions using heterogeneous data sources. Bioinformatics 2020;36(7):2209–16. [DOI] [PubMed] [Google Scholar]
- 31. Cai R, Chen X, Fang Y, et al. . Dual-dropout graph convolutional network for predicting synthetic lethality in human cancers. Bioinformatics 2020;36(16):4458–65. [DOI] [PubMed] [Google Scholar]
- 32. Long Y, Wu M, Liu Y, et al. . Graph contextualized attention network for predicting synthetic lethality in human cancers. Bioinformatics 2021;37(16):2432–2440. [DOI] [PubMed] [Google Scholar]
- 33. Hao Z, Wu D, Fang Y, et al. . Prediction of synthetic lethal interactions in human cancers using multi-view graph auto-encoder. IEEE J Biomed Health Inform 2021;25(10):4041–51. [DOI] [PubMed] [Google Scholar]
- 34. Guo J, Liu H, Zheng J. SynLethDB: synthetic lethality database toward discovery of selective and sensitive anticancer drug targets. Nucleic Acid Res 2016;44(D1):D1011–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Oughtred R, Rust J, Chang C, et al. . The BioGRID database: a comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci 2021;30(1):187–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Oughtred R, Stark C, Breitkreutz BJ, et al. . The BioGRID interaction database: 2019 update. Nucleic Acid Res 2019;47(D1):D529–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stark C, Breitkreutz BJ, Reguly T, et al. . BioGRID: a general repository for interaction datasets. Nucleic Acids Res 2006;34(Database issue):D535–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li XJ, Mishra SK, Wu M, et al. . Syn-lethality: an integrative knowledge base of synthetic lethality towards discovery of selective anticancer therapies. Biomed Res Int 2014;2014:196034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schmidt EE, Pelz O, Buhlmann S, et al. . GenomeRNAi: a database for cell-based and in vivo RNAi phenotypes, 2013 update. Nucleic Acid Res 2013;41(Database issue):D1021–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Costanzo M, Vandersluis B, Koch EN, et al. . A global genetic interaction network maps a wiring diagram of cellular function. Science 2016;353(6306):aaf1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wan F, Li S, Tian T, et al. . EXP2SL: a machine learning framework for cell-line-specific synthetic lethality prediction. Front Pharmacol 2020;11:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Laufer C, Fischer B, Billmann M, et al. . Mapping genetic interactions in human cancer cells with RNAi and multiparametric phenotyping. Nat Methods 2013;10(5):427–31. [DOI] [PubMed] [Google Scholar]
- 43. Vizeacoumar FJ, Arnold R, Vizeacoumar FS, et al. . A negative genetic interaction map in isogenic cancer cell lines reveals cancer cell vulnerabilities. Mol Syst Biol 2013;9:696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shen JP, Zhao D, Sasik R, et al. . Combinatorial CRISPR-Cas9 screens for de novo mapping of genetic interactions. Nat Method 2017;14(6):573–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Najm FJ, Strand C, Donovan KF, et al. . Orthologous CRISPR-Cas9 enzymes for combinatorial genetic screens. Nat Biotechnol 2018;36(2):179–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhao D, Badur MG, Luebeck J, et al. . Combinatorial CRISPR-Cas9 metabolic screens reveal critical redox control points dependent on the KEAP1-NRF2 regulatory Axis. Mol Cell 2018;69(4):699–708 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zamanighomi M, Jain SS, Ito T, et al. . GEMINI: a variational Bayesian approach to identify genetic interactions from combinatorial CRISPR screens. Genome Biol 2019;20(1):137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Han Y, Wang C, Dong Q, et al. . Genetic interaction-based biomarkers identification for drug resistance and sensitivity in cancer cells. Mol Ther Nucleic Acids 2019;17:688–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Srivas R, Shen JP, Yang CC, et al. . A network of conserved synthetic lethal interactions for exploration of precision cancer therapy. Mol Cell 2016;63(3):514–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hu L, Wang X, Huang YA, et al. . A survey on computational models for predicting protein-protein interactions. Brief Bioinform 2021;22(5):bbab036. [DOI] [PubMed] [Google Scholar]
- 51. Li B, Cao W, Zhou J, et al. . Understanding and predicting synthetic lethal genetic interactions in Saccharomyces cerevisiae using domain genetic interactions. BMC Syst Biol 2011;5:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang Y, Li B, Srimani PK, et al. . Predicting synthetic lethal genetic interactions in Saccharomyces cerevisiae using short polypeptide clusters. Proteome Sci 2012;10(Suppl 1):S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Conde-Pueyo N, Munteanu A, Sole RV, et al. . Human synthetic lethal inference as potential anti-cancer target gene detection. BMC Syst Biol 2009;3:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lee SJ, Seo E, Cho Y. Proposal for a new therapy for drug-resistant malaria using plasmodium synthetic lethality inference. Int J Parasitol Drugs Drug Resist 2013;3:119–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Deshpande R, Asiedu MK, Klebig M, et al. . A comparative genomic approach for identifying synthetic lethal interactions in human cancer. Cancer Res 2013;73(20):6128–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Costanzo M, Baryshnikova A, Bellay J, et al. . The genetic landscape of a cell. Science 2010;327(5964):425–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kelley R, Ideker T. Systematic interpretation of genetic interactions using protein networks. Nat Biotechnol 2005;23(5):561–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Srihari S, Singla J, Wong L, et al. . Inferring synthetic lethal interactions from mutual exclusivity of genetic events in cancer. Biol Direct 2015;10:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang R, Han Y, Zhao Z, et al. . Link synthetic lethality to drug sensitivity of cancer cells. Brief Bioinform 2019;20(4):1295–307. [DOI] [PubMed] [Google Scholar]
- 60. Lee JS, Das A, Jerby-Arnon L, et al. . Harnessing synthetic lethality to predict the response to cancer treatment. Nat Commun 2018;9(1):2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wang X, Simon R. Identification of potential synthetic lethal genes to p53 using a computational biology approach. BMC Med Genom 2013;6:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chang JG, Chen CC, Wu YY, et al. . Uncovering synthetic lethal interactions for therapeutic targets and predictive markers in lung adenocarcinoma. Oncotarget 2016;7(45):73664–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Feng X, Arang N, Rigiracciolo DC, et al. . A platform of synthetic lethal gene interaction Networks reveals that the GNAQ uveal melanoma oncogene controls the hippo pathway through FAK. Cancer Cell 2019;35(3):457–72 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yang C, Guo Y, Qian R, et al. . Mapping the landscape of synthetic lethal interactions in liver cancer. Theranostics 2021;11(18):9038–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sinha S, Thomas D, Chan S, et al. . Systematic discovery of mutation-specific synthetic lethals by mining pan-cancer human primary tumor data. Nat Commun 2017;8:15580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Grigoroiu A, Yoon J, Bohndiek SE. Deep learning applied to hyperspectral endoscopy for online spectral classification. Sci Rep 2020;10(1):3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Paladugu SR, Zhao S, Ray A, et al. . Mining protein networks for synthetic genetic interactions. BMC Bioinform 2008;9:426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Che DS, Liu Q, Rasheed K, et al. . Decision tree and ensemble learning algorithms with their applications in bioinformatics. Adv Exp Med Biol 2011;696:191–9. [DOI] [PubMed] [Google Scholar]
- 69. Yin Z B, Qian B W, Yang G W, et al. . Predicting Synthetic Lethal Genetic Interactions in Breast Cancer using Decision Tree. In: Icbbe 2019: 2019 6th International Conference on Biomedical and Bioinformatics Engineering, Shanghai, China 2019. pp. 1–6.
- 70. Altman NS. An introduction to kernel and nearest-neighbor nonparametric regression. Am Stat 1992;46(3):175–85. [Google Scholar]
- 71. Wu LL, Wen YQ, Yang XX, et al. . Synthetic lethal interactions prediction based on multiple similarity measures fusion. J Comput Sci Tech-Ch 2021;36(2):261–75. [Google Scholar]
- 72. Pandey G, Zhang B, Chang AN, et al. . An integrative multi-network and multi-classifier approach to predict genetic interactions. PLoS Comput Biol 2010;6(9):e1000928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wu M, Li X, Zhang F, et al. . In silico prediction of synthetic lethality by meta-analysis of genetic interactions, functions, and pathways in yeast and human cancer. Cancer Inform 2014;13(Suppl 3):71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ho T K. Random decision forests. In: Proceedings of the Third International Conference on, Document Analysis and Recognition, Montreal, QC 1995: 278–282.
- 75. Peng H, Long F, Ding C. Feature selection based on mutual information criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans Pattern Anal Mach Intell 2005;27(8):1226–38. [DOI] [PubMed] [Google Scholar]
- 76. Lundberg SM, Erion G, Chen H, et al. . From local explanations to global understanding with explainable AI for trees. Nat Mach Intell 2020;2(1):56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Srivastava N, Hinton G, Krizhevsky A, et al. . Dropout: a simple way to prevent neural Networks from overfitting. J Mach Learn Res 2014;15(1):1929–58. [Google Scholar]
- 78. Velikovi P, Cucurull G, Casanova A, et al. . Graph attention networks. In: International Conference on Learning Representations, Vancouver, BC, Canada. 2018: 1–12. [Google Scholar]
- 79. Nováček V, Mohamed SK. Predicting polypharmacy side-effects using knowledge graph embeddings. AMIA Jt Summits Transl Sci Proc 2020;2020:449–58. [PMC free article] [PubMed] [Google Scholar]
- 80. Wang S, Xu F, Li Y, et al. . KG4SL: knowledge graph neural network for synthetic lethality prediction in human cancers. Bioinformatics 2021;37(Suppl_1):i418–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ching T, Himmelstein DS, Beaulieu-Jones BK, et al. . Opportunities and obstacles for deep learning in biology and medicine. J R Soc Interface 2018;15(141):20170387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Almozlino YT, Peretz I, Kupiec M, et al. . G2G: a web-server for the prediction of human synthetic lethal interactions. Comput Struct Biotechnol J 2020;18:1028–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Deng X, Das S, Valdez K, et al. . SL-BioDP: multi-cancer interactive tool for prediction of synthetic lethality and response to cancer treatment. Cancers (Basel) 2019;11(11):1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Magen A, Das Sahu A, Lee JS, et al. . Beyond synthetic lethality: charting the landscape of pairwise gene expression states associated with survival in cancer. Cell Rep 2019;28(4):938–48 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Wappett M, Harris A, Lubbock ALR, et al. . SynLeGG: analysis and visualization of multiomics data for discovery of cancer 'Achilles Heels' and gene function relationships. Nucleic Acids Res 2021;49(W1):W613–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kuzmin E, Vandersluis B, Wang W, et al. . Systematic analysis of complex genetic interactions. Science 2018;360(6386):eaao1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ryan CJ, Bajrami I, Lord CJ. Synthetic lethality and cancer - penetrance as the major barrier. Trends Cancer 2018;4(10):671–83. [DOI] [PubMed] [Google Scholar]
- 88. Gu Y, Wang R, Han Y, et al. . A landscape of synthetic viable interactions in cancer. Brief Bioinform 2018;19(4):644–55. [DOI] [PubMed] [Google Scholar]
- 89. Akimov Y, Aittokallio T. Re-defining synthetic lethality by phenotypic profiling for precision oncology. Cell Chem Biol 2021;28(3):246–56. [DOI] [PubMed] [Google Scholar]
- 90. Behan FM, Iorio F, Picco G, et al. . Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature 2019;568(7753):511–6. [DOI] [PubMed] [Google Scholar]
- 91. Chan EM, Shibue T, Mcfarland JM, et al. . WRN helicase is a synthetic lethal target in microsatellite unstable cancers. Nature 2019;568(7753):551–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Yu LY, Tseng TJ, Lin HC, et al. . Synthetic dysmobility screen unveils an integrated STK40-YAP-MAPK system driving cell migration. Sci Adv 2021;7(31):eabg2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Yang H, Cui W, Wang L. Epigenetic synthetic lethality approaches in cancer therapy. Clin Epigenetics 2019;11(1):136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Aziz RK, Monk JM, Lewis RM, et al. . Systems biology-guided identification of synthetic lethal gene pairs and its potential use to discover antibiotic combinations. Sci Rep 2015;5:16025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Kalia NP, Hasenoehrl EJ, Ab Rahman NB, et al. . Exploiting the synthetic lethality between terminal respiratory oxidases to kill mycobacterium tuberculosis and clear host infection. Proc Natl Acad Sci U S A 2017;114(28):7426–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Pasquina L, Santa Maria JPJ, Mckay Wood B, et al. . A synthetic lethal approach for compound and target identification in Staphylococcus aureus. Nat Chem Biol 2016;12(1):40–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Xiao S, Guo H, Weiner WS, et al. . Revisiting the beta-lactams for tuberculosis therapy with a compound-compound synthetic lethality approach. Antimicrob Agents Chemother 2019;63(11):e01319–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Subramaniam S, Schmid CD, Guan XL, et al. . Using yeast synthetic lethality to inform drug combination for malaria. Antimicrob Agents Chemother 2018;62(4):e01533–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Mast FD, Navare AT, Van Der Sloot AM, et al. . Crippling life support for SARS-CoV-2 and other viruses through synthetic lethality. J Cell Biol 2020;219(10):e202006159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Lippert C, Ghahramani Z, Borgwardt KM. Gene function prediction from synthetic lethality networks via ranking on demand. Bioinformatics 2010;26(7):912–8. [DOI] [PubMed] [Google Scholar]
- 101. Guell O, Sagues F, Serrano MA. Essential plasticity and redundancy of metabolism unveiled by synthetic lethality analysis. PLoS Comput Biol 2014;10(5):e1003637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Cheng K, Nair NU, Lee JS, et al. . Synthetic lethality across normal tissues is strongly associated with cancer risk, onset, and tumor suppressor specificity. Sci Adv 2021;7(1):e1003637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Huang S, Ren X, Wang L, et al. . Lung-cancer chemoprevention by induction of synthetic lethality in mutant KRAS premalignant cells in vitro and in vivo. Cancer Prev Res (Phila) 2011;4(5):666–73. [DOI] [PubMed] [Google Scholar]
- 104. Walcott FL, Patel J, Lubet R, et al. . Hereditary cancer syndromes as model systems for chemopreventive agent development. Semin Oncol 2016;43(1):134–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Lee JS, Nair NU, Dinstag G, et al. . Synthetic lethality-mediated precision oncology via the tumor transcriptome. Cell 2021;184(9):2487–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Aromolaran O, Aromolaran D, Isewon I, et al. . Machine learning approach to gene essentiality prediction: a review. Brief Bioinform 2021;22(5):bbab128. [DOI] [PubMed] [Google Scholar]
- 107. Chawla NV, Bowyer KW, Hall LO, et al. . SMOTE: synthetic minority over-sampling technique. J Artif Intel Res 2002;16(1):321–57. [Google Scholar]
- 108. Kuenzi BM, Park J, Fong SH, et al. . Predicting drug response and Synergy using a deep learning model of human cancer cells. Cancer Cell 2020;38(5):672–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Murdoch WJ, Singh C, Kumbier K, et al. . Definitions, methods, and applications in interpretable machine learning. Proc Natl Acad Sci U S A 2019;116(44):22071–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Ma J, Yu MK, Fong S, et al. . Using deep learning to model the hierarchical structure and function of a cell. Nat Methods 2018;15(4):290–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Yu MK, Kramer M, Dutkowski J, et al. . Translation of genotype to phenotype by a hierarchy of cell subsystems. Cell Syst 2016;2(2):77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Sayers EW, Cavanaugh M, Clark K, et al. . GenBank. Nucleic Acid Res 2021;49(D1):D92–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Uniprot C. UniProt: the universal protein knowledgebase in 2021. Nucleic Acid Res 2021;49(D1):D480–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. The Gene Ontology C . The gene ontology resource: 20 years and still GOing strong. Nucleic Acid Res 2019;47(D1):D330–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Kanehisa M, Furumichi M, Tanabe M, et al. . KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acid Res 2017;45(D1):D353–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Liberzon A, Birger C, Thorvaldsdottir H, et al. . The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst 2015;1(6):417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Davis AP, Grondin CJ, Johnson RJ, et al. . The comparative Toxicogenomics database: update 2019. Nucleic Acid Res 2019;47(D1):D948–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Subramanian A, Narayan R, Corsello SM, et al. . A next generation connectivity map: L1000 platform and the first 1 000 000 profiles. Cell 2017;171(6):1437–52 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Sadreyev IR, Ji F, Cohen E, et al. . PhyloGene server for identification and visualization of co-evolving proteins using normalized phylogenetic profiles. Nucleic Acid Res 2015;43(W1):W154–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Giurgiu M, Reinhard J, Brauner B, et al. . CORUM: the comprehensive resource of mammalian protein complexes-2019. Nucleic Acid Res 2019;47(D1):D559–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Szklarczyk D, Gable AL, Nastou KC, et al. . The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acid Res 2021;49(D1):D605–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Keshava Prasad TS, Goel R, Kandasamy K, et al. . Human protein reference database--2009 update. Nucleic Acid Res 2009;37(Database issue):D767–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Alanis-Lobato G, Andrade-Navarro MA, Schaefer MH. HIPPIE v2.0: enhancing meaningfulness and reliability of protein-protein interaction networks. Nucleic Acid Res 2017;45(D1):D408–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Tsherniak A, Vazquez F, Montgomery PG, et al. . Defining a cancer dependency map. Cell 2017;170(3):564–76 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Barretina J, Caponigro G, Stransky N, et al. . The cancer cell line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012;483(7391):603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Bamford S, Dawson E, Forbes S, et al. . The COSMIC (catalogue of somatic mutations in cancer) database and website. Br J Cancer 2004;91(2):355–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. O'brien K P, Remm M, Sonnhammer E L.. Inparanoid: a comprehensive database of eukaryotic orthologs. Nucleic Acid Res 2005;33(Database issue):D476–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Chen F, Mackey AJ, Stoeckert CJ, Jr, et al. OrthoMCL-DB: querying a comprehensive multi-species collection of ortholog groups. Nucleic Acid Res 2006; 34(Database issue): D363–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Fischer S, Brunk BP, Chen F, et al. . Using OrthoMCL to assign proteins to OrthoMCL-DB groups or to cluster proteomes into new ortholog groups. Curr Protoc Bioinformatics 2011;Chapter 6:Unit 6.12.1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Cherry JM, Adler C, Ball C, et al. . SGD: saccharomyces genome database. Nucleic Acid Res 1998;26(1):73–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Ye H, Zhang XH, Chen YQ, et al. . Ranking novel cancer driving synthetic lethal gene pairs using TCGA data. Oncotarget 2016;7(34):55352–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Barbie DA, Tamayo P, Boehm JS, et al. . Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009;462(7269):108–U22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Luo J, Emanuele MJ, Li D, et al. . A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 2009;137(5):835–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Reguly T, Breitkreutz A, Boucher L, et al. . Comprehensive curation and analysis of global interaction networks in Saccharomyces cerevisiae. J Biol 2006;5(4):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Pan X, Yuan DS, Xiang D, et al. . A robust toolkit for functional profiling of the yeast genome. Mol Cell 2004;16(3):487–96. [DOI] [PubMed] [Google Scholar]
- 136. Tong AH, Lesage G, Bader GD, et al. . Global mapping of the yeast genetic interaction network. Science 2004;303(5659):808–13. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.