

Abstract
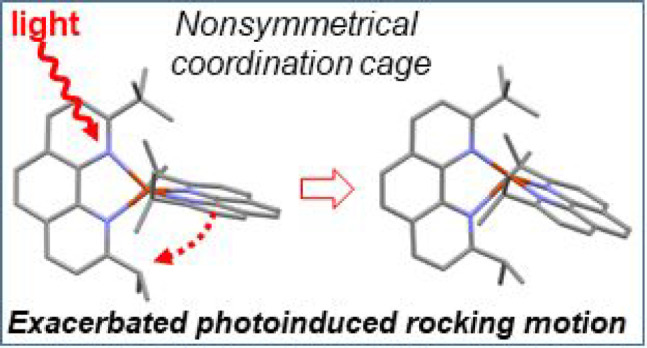
The steric strain around copper(I) in typical [Cu(NNR)2]+ complexes, where NNR is a diimine ligand substituted in α-positions of the nitrogen atoms by R, is known to strongly impact the excited-state properties. Generally speaking, the larger the R, the longer the emission lifetime and the higher the quantum yield. However, the stability of the coordination scaffold can be at stake if the steric strain imposed by R is too large. In this work, we explore a way of fine-tuning the steric strain around Cu(I) to reach a balance between high emission quantum yield and stability in a highly bulky copper(I) complex. Taking stable [Cu(dipp)2]+ and unstable [Cu(dtbp)2]+ (where dipp and dtbp are, respectively, 2,9-diisopropyl-1,10-phenanthroline and 2,9-di-tert-butyl-1,10-phenanthroline) as the boundary of two least and most sterically strained structures, we designed and characterized the nonsymmetrical ligand 2-isopropyl-9-tert-butyl-1,10-phenanthroline (L1) and corresponding complex [Cu(L1)2]+ (Cu1). The key experimental findings are that Cu1 exhibits a rigid tetrahedral geometry in the ground state, close to that of [Cu(dtbp)2]+ and with an intermediate stability between that of [Cu(dipp)2]+ and [Cu(dtbp)2]+. Conversely, the nonsymmetrical nature of ligand L1 leads to a shorter emission lifetime and smaller quantum yield than those of either [Cu(dipp)2]+ or [Cu(dtbp)2]+. This peculiar behavior is rationalized through the in depth analysis of the ultrafast dynamics of the excited state measured with optical transient absorption spectroscopy and theoretical calculations performed on the ground and excited state of Cu1. Our main findings are that the obtained complex is significantly more stable than [Cu(dtbp)2]+ despite the sterically strained coordination sphere. The nonsymmetrical nature of the ligand translates into a strongly distorted structure in the excited state. The distortion can be described as a rocking motion of one ligand, entailing the premature extinction of the excited state via several deactivation channels.
Short abstract
A stable copper(I) complex was prepared, whose steric hindrance is halfway between benchmark [Cu(dipp)2]+ and [Cu(dtbp)2]+. The coordination sphere of the complex is not geometrically symmetric, and this causes a geometric imbalance, which favors rotational and rocking motions of the ligands in the excited state, causing the premature quenching of the excited state.
Introduction
Copper(I) complexes are drawing increased attention as photosensitizers, efficiently replacing heavy metal complexes in lighting devices,1 dye sensitized solar cells,2 solar fuel production3 and organic photochemistry.4 Homoleptic copper(I) complexes of the general formula [CuL2]+ where L is a diimine ligand are particularly interesting. They absorb over a large wavelength range (400–550 nm or farther in the red depending on L)5,6 through a metal-to-ligand charge-transfer transition (MLCT) and emit in the red to near-infrared, with respectable luminescence quantum yields Φ (up to 0.063)7 and lifetimes τ up to a few microseconds.8 Importantly, it is necessary to tether sterically challenging groups in positions 2 and 9 of 1,10-phenanthroline (phen) to promote advantageous excited-state properties.9,10 The rationale is that photoexcitation of the MLCT transition entails electron transfer from copper-centered d orbitals toward π* ligand-centered orbitals. Thus, [CuIL2]+ transiently turns into [CuII(L•–)L]+, leading to a drastic geometry change from pseudotetrahedral D2d toward square planar D2. In other words, the coordination sphere flattens to make the transient CuII center in [CuII(L•–)L]+ more exposed to the solvent molecules, enabling fast quenching of the excited state assuming the solvent–solute interactions lower the energy gap between the MLCT triplet and the ground states.
Grafting bulky groups in α-positions of the nitrogen atoms on the phen ligand hinders the flattening of the copper(I) coordination sphere in the MLCT state and shields the CuII center from solvent quenching, thus enhancing the excited-state lifetime as well as other photochemical properties. In addition, increasing the steric bulkiness around copper(I) elevates the energy of the excited state,11,12 improving its reactivity from a thermodynamic point of view. Thus, the larger steric strain around copper(I) generally leads to larger fluorescence yields Φ and longer excited-state lifetimes τ.7,8,13,14 In dichloromethane (DCM), for example, complexes like [Cu(dipp)2]+13 (where dipp stands for 2,9-diisopropyl-1,10-phenanthroline) or [Cu(dsbtmp)2]+7 (where dsbtmp stands for 2,9-di-sec-butyl-3,4,7,8-tetramethyl-1,10-phenanthroline) exhibit Φ of 0.4 and 6.3%, and τ of 0.37 and 2.8 μs, respectively. [Cu(dmp)2]+ (where dmp stands for 2,9-dimethyl-1,10-phenanthroline) features more modest Φ (0.02%) and τ (ca. 55 ns),10 while [Cu(phen)2]+ is not luminescent at all.
On the other hand, increasing the steric bulk around copper(I) too much can make it difficult to prepare the homoleptic complex.15,16 For example, the synthesis of [Cu(dtbp)2]+ (dtbp stands for 2,9-di-tert-butyl-1,10-phenanthroline) is very challenging,15 and the resulting complex is highly unstable.8 Nevertheless, even if [Cu(dtbp)2]+ is an unstable complex, it exhibits fascinating properties, notably the longest luminescence lifetime recorded for a homoleptic copper(I) complex in solution (ca. 3.3 μs in DCM).8
Structurally, stable [Cu(dipp)2]+ differs from unstable [Cu(dtbp)2]+ by only two methyl groups on the substituents of each phenanthroline ligand. With this in mind, we were curious if removing only one methyl group from dtbp, yielding the nonsymmetrical phenanthroline ligand 2-isopropyl-9-tert-butyl-1,10-phenanthroline (L1), would be sufficient to afford a stable copper(I) complex [Cu(L1)2]+ in the usual mild conditions used for the synthesis of these coordination scaffolds (Figure 1). In addition, we were interested in studying the optical and electrochemical properties of the corresponding complex in both the ground and excited states. In this contribution, we report the synthesis of ligand L1 and associated complex [Cu(L1)2]+ (named Cu1) as well as a detailed optical and electrochemical characterization. The stability and luminescence properties of Cu1 demonstrate that steric strain is not the sole factor governing the excited-state properties of copper(I) complexes.12,17
Figure 1.

Complex Cu1 and models.
Results and Discussion
Synthesis
The synthesis of ligand L1 is performed in two steps (Figure S1). First, nucleophilic attack of anhydrous phen by tert-butyllithium followed by rearomatization with MnO2 afforded monosubstituted 2-tert-butyl-1,10-phenanthroline. The organolithium solution was carefully added (over 20 min) to limit the formation of the bis-substituted ligand. 2-tert-butyl-1,10-phenanthroline was then subjected to the same series of reactions with isopropyllithium as the organolithium reagent, affording L1 in 77% yield over all steps. The homoleptic copper(I) complex [Cu(L1)2]+ was finally assembled by mixing 2 equiv of the ligand with 1 equiv of copper(I) tetrakis acetonitrile hexafluorophosphate in degassed DCM at room temperature.
Structural Properties
After solvent
evaporation under
reduced pressure, the obtained red powder was recrystallized from
hot toluene. We obtained monocrystals which were suitable for a structural
determination by X-ray scattering (Figures 2 and S2). Relevant
angles and distances are reported in Table 1, and nomenclature for the different atoms
is given in Figure S3. The structure reveals
that the copper(I) ion is in a nearly tetrahedral environment. Cu–N
distances are significantly influenced by the nature of the substituent
in the α-position of the corresponding nitrogen atom: tert-butyl groups impose a longer Cu–N distance (ca.
2.16 Å on average), whereas isopropyl allow for a shorter Cu–N
bond length (2.04 Å on average) and likely a tighter coordination
bond. Interestingly, the phenanthroline plane is slightly farther
away from the bulkier tert-butyl group than from
the less challenging isopropyl one (Figure S4), translating to a small waggling distortion (see Figure S5 for the definition of all distortions in homoleptic
copper(I) complexes). Nevertheless, the structures of Cu1 and [Cu(dtbp)2]+ are very close as evidenced by the calculation
of the rocking, waggling, and flattening angles (respectively θx, θy, and
θz) which are close to 90°
in all cases (Figures S6 and S7). For example
θx, θy, and θz values are significantly
different from 90° for [Cu(dipp)2]+ (Figure S7), showing that the addition of one
extra methyl on one isopropyl group of each phen ligand has substantially
brought the structure closer to a tetrahedral geometry, as desired
to promote the excited-state properties. This is confirmed by the
τ4 parameter (τ4 = 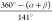 ,
where α and β are the two
largest N–Cu–N angles in the structure of Cu1, namely,
N1A–Cu–N2B and N2A–Cu–N2B, cf. Table 1) which is 0.66, 0.76,
and 0.79 for [Cu(dipp)2]+, Cu1, and [Cu(dtbp)2]+ respectively, with the expected value for a
perfect tetrahedron being 0.93.18
,
where α and β are the two
largest N–Cu–N angles in the structure of Cu1, namely,
N1A–Cu–N2B and N2A–Cu–N2B, cf. Table 1) which is 0.66, 0.76,
and 0.79 for [Cu(dipp)2]+, Cu1, and [Cu(dtbp)2]+ respectively, with the expected value for a
perfect tetrahedron being 0.93.18
Figure 2.
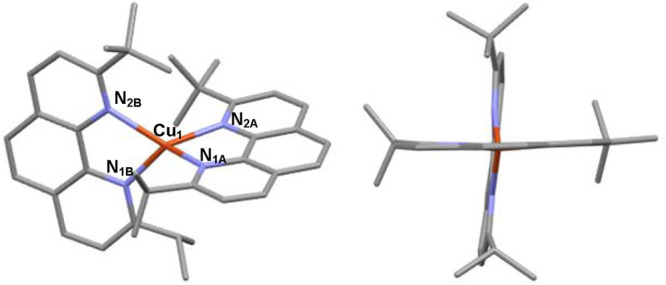
Structure of complex Cu1. Hydrogen atoms and counteranion have been removed for the sake of clarity.
Table 1. Bond Lengths and Angles of Interest for Complex Cu1 and Models [Cu(dipp)2]+ and [Cu(dtbp)2]+ Obtained by X-ray Scattering.
| parameter | [Cu(dipp)2]+(BF4–)19 | [Cu(dtbp)2]+(BF4–)15 | Cu1(PF6) |
|---|---|---|---|
| distances (Å) | |||
| Cu–N1A | 2.083 | 2.104(2) | 2.057(4) |
| Cu–N2A | 2.017 | 2.111(2) | 2.122(4) |
| Cu–N1B | 2.067 | 2.108(2) | 2.031(4) |
| Cu–N2B | 2.030 | 2.105(2) | 2.207(4) |
| angles (deg) | |||
| N1A–Cu–N2A | 82.25 | 84.53 | 82.09(14) |
| N1A–Cu–N1B | 107.35 | 123.26 | 120.32(15) |
| N1A–Cu–N2B | 118.71 | 122.92 | 126.36(14) |
| N2A–Cu–N1B | 128.97 | 121.37 | 126.10(15) |
| N2A–Cu–N2B | 138.38 | 124.93 | 126.94(13) |
| N1B–Cu–N2B | 81.52 | 84.82 | 81.06(16) |
| θx | 82.2 | 88.6 | 87.2 |
| θy | 72.5 | 90.0 | 87.4 |
| θz | 85.4 | 91.8 | 87.3 |
| τ4(18) | 0.66 | 0.79 | 0.76 |
One last point concerns the orientation of the tert-butyl groups of each ligand. As shown in Figure 2, tert-butyl groups can be in a staggered or eclipsed conformation (Figure S8). The two phen ligands are thus not equivalent in the crystal phase, possibly a result from crystal packing forces. However, the 1H NMR spectrum of Cu1 clearly shows that both coordinated ligands are equivalent in the solution phase. DFT calculations are a powerful tool to assess the behavior of the complexes in solution, by recomposing the structures with computational chemistry means and defining DCM as the medium.
We computed the structure of the different possible conformers for Cu1 (Figure 3). The first one (Cu1sym1) is symmetric and corresponds to the case where both of the tert-butyl groups on each ligand are in the staggered conformation. The second one (Cu1nosym) is not symmetric, corresponding to the X-ray experimental structure displayed in Figure 2; namely, one tert-butyl is in a staggered conformation and the other one in an eclipsed conformation. Finally, the last calculated conformation (Cu1sym2) corresponds to a symmetric structure where both tert-butyl groups are in an eclipsed conformation. All relevant parameters (bond length, angles for each structure) are given in Table ST1. Cu1sym1 and Cu1sym2 are less stable than Cu1nosym (Figure S9), though the structure of Cu1sym2 is very close in energy. The associated transition states are very low in energy. The barrier from Cu1nosym to Cu1sym1 is 4.1 kcal mol–1 and that from Cu1nosym to Cu1sym2 is only 1.4 kcal mol–1. These results show that there is free rotation of the tert-butyl group at room temperature, as observed in the NMR spectra. The orientation of the tert-butyl group impacts the structural parameters, especially the Cu–N bond length. The difference between Cu–N2A or Cu–N2B (nitrogen atom close to the tert-butyl group) and Cu–N1A or Cu–N1B (nitrogen atom close to isopropyl groups) distances in the same ligand increases from Cu1sym1 (0.07 Å) to Cu1sym2 (0.184 Å), in accordance with the trend observed in the crystal structure.
Figure 3.

Structures of the three most stable rotamers of Cu1. Only the hydrogen of the tBu groups pointing toward the copper are shown. The relative Gibbs Free energies from Gaussian calculations are in kcal/mol.
Absorption Properties of Complex Cu1
Cu1 was dissolved in DCM, yielding orange solutions which remained stable over several days when stored in the dark (Figure S10). The UV–vis absorption spectrum of Cu1 (Figure 4a) is dominated by a strong transition in the UV composed of multiple ligand-centered and high energy MLCT transitions while a broad lower energy MLCT transition spans between 350 and 500 nm. The maximum absorption wavelength of the MLCT transition is 438 nm, between the values measured for [Cu(dipp)2]+ (453 nm) and [Cu(dtbp)2]+ (425 nm).8 This is in line with the steric strain of the coordination sphere in Cu1 which is intermediate between [Cu(dipp)2]+ and [Cu(dtbp)2]+.
Figure 4.

(a) UV–vis (black line) and (b) emission (red line) spectra (λexc = 438 nm) of complex Cu1 in DCM. Inset: zoom-in on the MLCT transition.
The theoretical spectrum (Figure 5) of Cu1nosym reproduces the features of the experimental one. The spectra of the two symmetric rotamers Cu1sym1 and Cu1sym2 are very similar (Figure S11). The nature of the electronic transitions was determined by means of TheoDORE analysis (Figure 6). In this approach, the Cu1nosym complex is divided into three fragments: the metallic cation and the two ligands. The nature of each transition is determined by the contribution of each fragment to either the emptied orbital or the filled one. In the computed spectra, several weak absorption bands are present between 350 and 550 nm corresponding to the broad and weak experimental bands observed in the same domain. The spectrum is dominated by an intense band at 270 nm (exp. 275 nm). The detailed nature of the transitions can be found in Table ST2. The low energy domain (316–526 nm) is composed only of MLCT states (S1–S23, Figure 6). The lowest singlet–singlet transition S0 → S1 is expected at 525 nm but has no oscillator strength, as is commonly observed for Cu(I) complexes with a nearly tetrahedral geometry in the ground state in agreement with the experimental results.
Figure 5.

Theoretical (black line) and experimental (red line) absorption spectra of Cu1nosym in DCM computed with ADF software. Inset: zoom-in on the 350–600 nm domain.
Figure 6.

Nature of the transitions generating the theoretical absorption spectra. Horizontal axis: numbering of the singlet states Sn for the corresponding S0 → Sn transition. Vertical axis: proportions (between 0 and 1) of metal centered states (MC, black), ligand centered π–π* transitions (on the same ligand) (LC, red), metal-to-ligand charge transfer (MLCT, blue), ligand-to-metal charge transfer (LMCT, yellow), and ligand-to-ligand charge transfer (LLCT, green) to the overall calculated S0 → Sn transitions.
The first absorption band is due to the S0 → S3 transition (490 nm, MLCT transition). Several transitions (S0 → S6 at 440 nm, S0 → S7 at 433 nm, and S0 → S8 at 430 nm) generate the weaker band and explain the high-energy tail observed in the experimental spectra between 350 and 450 nm. The small experimental shoulder around 330 nm appearing on the computed spectra at 318 nm can be attributed to the MLCT transition S0 → S23. The most intense band (245–300 nm, exp. 255 to 315 nm) is too often assigned to π–π* LC transitions, whereas it is the combination of numerous electronic transitions (see comments below Table ST2). The nature of the lowest triplet states is identical to that of the corresponding singlet states.
UV–Vis Monitoring of the Stability of Cu1 in the Presence of Acetonitrile
As mentioned above, Cu1 remained stable in aerated solution despite its crowded coordination sphere. In order to appreciate and compare the stabilities of Cu1 and [Cu(dtbp)2]+, we studied the evolution of the UV–vis spectrum of Cu1 in DCM when aliquots of acetonitrile were added in the medium (Figure S12). Indeed, acetonitrile (ACN) is a weak ligand which nevertheless proved to be capable of destroying [Cu(dtbp)2]+ efficiently following the equation:8
The MLCT transition of Cu1 collapses upon addition of ACN, whereas model [Cu(dipp)2]+ is unaffected (Figure S12), showing that the coordination sphere of Cu1 is bulky enough to favor ligand exchange with weakly coordinating acetonitrile for steric relief. On the other hand, Cu1 is much more stable than [Cu(dtbp)2]+ since ca. 5 × 104 equiv of acetonitrile was necessary to destroy half the quantity of Cu1 whereas only one equivalent was sufficient to quantitatively disrupt the coordination scaffold of [Cu(dtbp)2]+.8,15 Conclusively, the removal of one mere methyl group per phenanthroline from the structure of [Cu(dtbp)2]+ is enough to significantly improve the stability of the complex, although the coordination sphere is weakened because of the strong steric strain imposed by the remaining tert-butyl group, as previously observed.12
Study of the Photoluminescence Properties of Complex Cu1
The strong steric bulk surrounding the copper(I) ion is expected to be a positive boon for the promotion of the photoluminescence properties. Indeed, solutions of Cu1 in DCM are brightly luminescent upon excitation of the MLCT transition. The usual broad emission band can be monitored at 678 nm (Figure 4b, red line). The corresponding excitation spectrum (emission at 678 nm) is given in Figure S13 and reproduces the absorption features of the MLCT, confirming that the luminescence originates indeed from the light-induced population of MLCT excited states. We retrieved E00 = 2.14 eV from the emission spectrum (using the tangent method, Figure S14). This value is very close to E00([Cu(dipp)2]+) despite Cu1 being more sterically constrained than [Cu(dipp)2]+. In addition, the emission quantum yield and lifetime are more than twice lower than those of [Cu(dipp)2]+ (Table 2 and Figure S15): these results are in contradiction with the expected trend and show that steric strain is not the sole parameter to influence the excited-state properties of copper(I) complexes. In previous works, it has been noticed that copper(I) complexes built with phenanthroline ligands, which are substituted by two different bulky groups in positions 2 and 9, show decreased luminescence performance due to a high nonradiative rate constant knr.5,12,20 Likewise, a large knr of 7.86 × 106 s–1 was calculated in the case of Cu1 (Table 2). Nevertheless, Cu1 is the most luminescent complex reported in the series of homoleptic copper(I) complexes bearing nonsymmetrical phenanthroline ligands; it is also the most sterically challenged, underlining that steric strain remains an important factor to take into consideration to increase Φ, τ, and E00, all things otherwise being equal.
Table 2. Ground-State and Excited-State Parameters for Molecules M = Cu1, [Cu(dipp)2]+ and [Cu(dtbp)2]+a.
| M | λabs/nm (ε/M–1·cm–1) | E(M+/M) (V)b | E(M/M–) (V)c | λem/nm (Φ/%)b | E(M*/M–)d | τ/nsb | kr/s–1e | knr/s–1e |
|---|---|---|---|---|---|---|---|---|
| Cu1 | 438 (4460) | 0.60 (120) | –2.13 (100) | 678 (0.17) | 0.01 | 127 | 1.34 × 104 | 7.86 × 106 |
| [Cu(dipp)2]+ | 453 (7260) | 0.65 | –2.10 | 679 (0.4) | 0.03 | 342 | 1.1 × 104 | 2.73 × 106 |
| [Cu(dtbp)2]+, 8 | 425 (3100) | 599 (5.6) | 3260 | 1.72 × 104 | 0.3 × 106 |
Measurements are performed in DCM unless otherwise mentioned.
Degassed medium.
In degassed DMF.
Estimated using the Rehm and Weller equation. Potentials given vs Fc+/Fc couple.
kr = Φ/τ and knr = 1/τ – kr.
The emission properties were computed, and thus, the lowest excited singlet and triplet states were optimized for the three rotamers Cu1sym1, Cu1sym2, and Cu1nosym (Table 3). The excited states (S1 or T1) are MLCT states and therefore can be seen as intramolecular charge-transfer states where a hole is mostly located on copper and one electron is localized either on phenA or phenB. This results in two degenerate electronic states with C1 symmetry for Cu1Sym1 and Cu1sym2 and nearly degenerate electronic states with C1 symmetry for Cu1nosym, corresponding crudely to [CuII(phenA–)(phenB)]+ and [CuII(phenA)(phenB–)]+. Shifting from one potential well to the other is possible through population of a transient state formally described as [CuII(phenA(0.5-))(phenB(0.5-))]+ (Figure S16). The latter has a C2 symmetry in the case of Cu1sym1 and Cu1sym2 (in the case of Cu1nosym, the symmetry of the intermediate state remains in C1 symmetry because of the different orientations of the tert-butyl groups).
Table 3. Emission from the Lowest T1 State (Eem), Distortion Energy Edist (Defined as the Difference between the Energy of the Ground State at Its Optimum and at the Excited State Geometry), and Stabilization Energy Estab (Estab = Edist + Eem)a.
| structure |
||||||
|---|---|---|---|---|---|---|
| Cu1sym1 |
Cu1nosym |
Cu1sym2 |
||||
| C2 | C1 | C1 | C1b | C2 | C1 | |
| Eem | 1.668 | 1.507 | 1.460 | 1.449 | 1.544 | 1.405 |
| λem | 743 | 823 | 849 | 855 | 803 | 882 |
| Edist | 0.281 | 0.371 | 0.365 | 0.387 | 0.333 | 0.404 |
| Estab | 1.949 | 1.878 | 1.824 | 1.837 | 1.877 | 1.810 |
| ΔEST | 0.222 | 0.197 | 0.211 | 0.184 | 0.266 | 0.232 |
| SOC | 0.1 | 19.0 | 33.6 | 15.0 | 0.0 | 25.8 |
| 20.2 | 34.2 | 37.8 | 27.0 | 20.3 | 37.9 | |
| 20.2 | 34.2 | 37.8 | 27.0 | 20.3 | 37.9 | |
Energies in eV, emission wavelength in nm (λem), S1-T1 energy splitting (ΔEST) in eV at T1 geometry and S1-T1 spin orbit coupling in cm–1 of the lowest triplet states T1 at T1 geometry for the three rotamers and group symmetry.
The two phen ligands of Cu1 being inequivalent, the emission properties from both have been computed.
In the C2 point group, the electron density of the excited state is delocalized on both ligands phenA and phenB. Breaking the symmetry leads to its localization on one of the phen, which is associated with a breathing of the Cu–N distance without rotation of the tert-butyl fragment. In the ground state, Cu1nosym was the lowest energy structure. In the excited states, the most stable geometries (in both S1 and T1) were generated by Cu1sym2. All the computed emission wavelengths λem from the T1 states (Table 3) are inconsistent with the experimental maxima at 678 nm. This shows that like other similar copper complexes, the emission likely stems from the singlet S1 state, through thermally activated delayed fluorescence (TADF), where the luminescent S1 state is populated from the weakly luminescent T1 state by thermally activated reverse intersystem crossing. One should notice the strong influence of the tert-butyl group orientations on the T1 state properties (Table 3). This means that a simple rotation of the tert-butyl group (an easy motion at room temperature given the low energy barriers between rotamers) leads to the existence of many different minima on the lowest excited singlet or triplet potential energy surface (PES), each of them with its own set of properties. For example, the spin orbit coupling (SOC) varies impressively from one rotamer to the other. This value is known to strongly impact intersystem crossing and therefore the probability that T1 either repopulates S1 (leading to TADF) or decays to S0 (either radiatively or nonradiatively).
We first computed the lowest excited S1 singlet state retaining the C2 symmetry for Cu1sym1 and Cu1sym2, and then we did the calculation without any symmetry (C1 point group) for the three rotamers. The λem computed for the S1 states in C1 symmetry are too red-shifted (Table 4). However, the λem values of the S1 state of the C2 symmetry of rotamers Cu1sym1 (663 nm) or Cu1sym2 (706 nm) agree well with the experimental values. In what follows, we will not consider rotamer Cu1sym1 because its energy is significantly higher than Cu1sym2, which means that this conformation will be minor in solution compared to the others. As mentioned above, the optimized S1 state of the C1 symmetry of Cu1sym2 rotamer is the most stable structure, though that of Cu1nosym is close in energy. Importantly, the Cu1sym2 rotamer S1 state of C2 symmetry (transition state with the electron delocalized over the two phenanthroline ligands, Figure S16) is also quite low in energy, being only 0.118 eV above the structure without symmetry Cu1nosym.
Table 4. Emission (Eem), Distortion (Edist), and Stabilization (Estab) Energiesa.
| structure |
||||||
|---|---|---|---|---|---|---|
| Cu1sym1 |
Cu1nosym |
Cu1sym2 |
||||
| C2 | C1 | C1 | C1b | C2 | C1 | |
| Eem | 1.870 | 1.650 | 1.589 | 1.597 | 1.756 | 1.530 |
| Edist | 0.280 | 0.396 | 0.404 | 0.399 | 0.344 | 0.452 |
| Estab | 2.149 | 2.046 | 1.992 | 1.996 | 2.100 | 1.982 |
| λem | 663 | 751 | 780 | 776 | 706 | 810 |
| fosc | 3.58 × 10–2 | 3.45 × 10–3 | 3.70 × 10–3 | 2.54 × 10–3 | 3.17 × 10–2 | 1.70 × 10–3 |
Energies in eV, emission wavelength in nm (λem), and oscillator strength (fosc) of the lowest S1 states.
The two phen ligands of Cu1 being inequivalent, the emission properties from each of them have been computed.
From these data, we can deduce the emission properties. After excitation, the complex relaxes in the lowest triplet PES where it hops between the different energetically accessible minima generated by two kinds of motion. The first one is due to the rotation of the tert-butyl groups, and the second is the hopping of the exciton between the two ligands. The S1 PES is thermally repopulated, being accessible due to the small S1–T1 energy difference (Table 3) and significant SOC (as expected for copper(I) complexes with nearly tetrahedral geometries) whatever the geometry considered.21 From the S1 PES, the complex radiatively relaxes to the ground state, generating this broad emission band due to contributions from the various rotamers (Table 4). These multiple transition structures entail that the barriers between the PES minima are low and that the complex evolves freely between them, populating each of these structures with almost equal probability. This tends to “dilute” the excited state over several states, enabling accelerated deactivation channels, as was observed previously.12
In addition, the SOC is significantly smaller for Cu1 than for [Cu(dipp)2]+ (Table ST3). This means that the thermally activated reverse intersystem crossing is less efficient for Cu1 than for [Cu(dipp)2]+. The TADF process is therefore less likely for Cu1, likely participating to the lower luminescence quantum yield and lifetime of the latter compared to model [Cu(dipp)2]+.
One last point concerns the global shape of the T1 and S1 structures. The tetrahedral arrangement around the copper atom is almost lost in the excited state; the coordination sphere approached a trigonal bipyramid with the copper at the center of the equatorial plane and one methyl group of a tert-Butyl group occupying an axial position (see the XYZ file in the Supporting Information (SI), which gathers all calculated structures). Such distortion is particularly pronounced for complexes bearing tert-butyl substituents in the α-position of the nitrogen atoms. It is well-known that extended distortions of the copper(I) complexes coordination sphere lead to high nonradiative rates knr.
Optical Transient Absorption Spectroscopy
Optical transient absorption (TA) spectroscopy experiments were performed on solutions of Cu1 prepared in DCM at a concentration of about 0.5 mM (Figure 7A). Following excitation with 415 nm laser pulses (500 fs, 500 nJ), the difference spectra comprise negative features at wavelengths less than 480 nm and positive features at wavelengths greater than 480 nm, with excited-state absorption (ESA) maxima between 540 and 580 nm. At short time delays, 0–2 ps, the ESA is broad and featureless, centered at 560 nm, and undergoes a subtle red-shift leading to a small decrease in the ESA intensity at 550 nm and a small increase in the ESA intensity at wavelengths greater than 560 nm. The broad ESA feature then becomes more structured over the time delay range of 2–50 ps, with strong maxima at 537 and 574 nm and weak maxima at 678 and 748 nm. The negative and positive features then decay concomitantly to zero over the time delay range of 50 ps to 1 μs. Importantly, the absorbance of the samples did not change during photolysis, proving that Cu1 is photostable during photolysis in degassed samples (Figure S17). The transient absorption data were analyzed using global-kinetic analysis to fit select kinetic traces with a first-order sequential kinetic model, A → B→C → GS (see Experimental Section and Figure S18 for details). The evolution-associated spectra (EAS) were extracted from the raw data using the resultant time-dependent model populations (Figures 7B and S18D).
Figure 7.

(A) Optical transient absorption spectra at select time delays following excitation by 415 nm laser pulses for a solution of Cu1 in DCM and (B) evolution-associated spectra obtained via global-kinetic fitting of the raw data to a first-order sequential kinetic model, A → B → C → GS.
According to the calculations, the 415 nm laser pulses were resonant with the S0 → Sn (n = 3, 6–8, see calculations) MLCT transitions that are optically allowed in the D2d structure adopted by copper(I) diimine complexes in the ground state.22 Prior time-resolved spectroscopy experiments with shorter time resolution and theoretical studies demonstrated that the initial excited-state population rapidly relaxes from Sn to S1 via internal conversion (IC), with a portion of the population undergoing intersystem crossing (ISC), on the 100 fs time scale.23 The subtle red-shift of the broad ESA feature observed here occurs within τ = 1.2 ± 0.6 ps. This process is typically assigned to the pseudo Jahn–Teller flattening distortion commonly observed for photoexcited copper(I) diimine complexes bearing symmetrical phenanthroline ligands: as explained in the introduction, copper(I) is transiently oxidized into copper(II) upon excitation, thus moving from tetrahedral to square planar geometries, and leading to a flattening of the coordination scaffold. However, our calculations of the excited state of Cu1 and of copper(I) and copper(II) variants of Cu1 indicate that this excited-state structural distortion rather corresponds to the rocking motion of a phenanthroline ligand and leads to formation of the trigonal bipyramidal structure (calculated structures for the excited state of Cu1 and the oxidized state of Cu1 are available in the XYZ files in the SI; also see Table ST4 for relevant structural data on the excited states). This rocking motion is well illustrated in Figure 8, where a view of excited Cu1 is given. The flattening distortion was observed on the 200–600 fs time scale in [Cu(tbp)2]+ (tbp stands for 2-tert-butyl-1,10-phenanthroline) and 700–800 fs time scale in [Cu(dipp)2]+ but was not observed in [Cu(dtbp)2]+ due to the increased steric bulk of the tert-butyl substituents in the 2 and 9 positions of the phenanthroline ligands.13,24 The τ ∼ 1 ps time constant for structural distortion in Cu1 is consistent with the intermediate steric bulk of L1 and our calculations, which predicted structural distortion upon oxidation of the copper.
Figure 8.
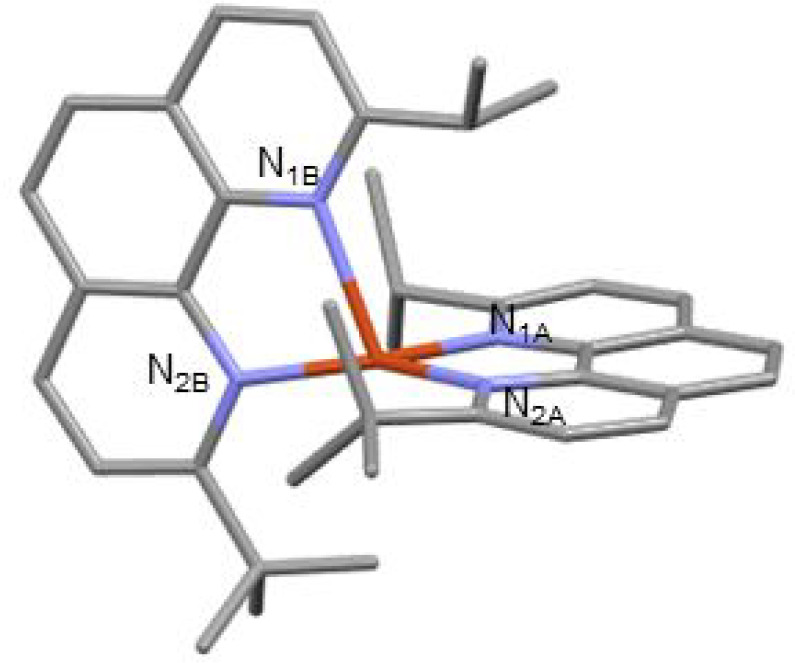
Structure of Cu1 in the excited state (example of Cu1nosym in the S1 state).
Following structural distortion, the ESA blue-shifts and the various maxima become more pronounced with τ = 8.5 ± 0.6 ps, which is assigned to ISC from the 1MLCT (S1) to 3MLCT (T1) state. ISC in Cu1 occurs slightly faster than that in [Cu(tbp)2]+, [Cu(dipp)2]+, and [Cu(dsbp)2]+ (dsbp stands for 2,9-di-sec-butyl-1,10-phenanthroline) but slower than that in the more sterically hindered [Cu(dtbp)2]+, [Cu(diptmp)2]+ (diptmp stands for 2,9-diisopropyl-3,4,7,8-tetramethyl-1,10-phenanthroline), and [Cu(dsbtmp)2]+.13,24,25 Nozaki et al. demonstrated that SOC values for copper(I) bisimine complexes decrease significantly with the degree of the flattening distortion, which explains the trend among the copper(I) complexes with symmetrically substituted phenanthrolines.21 However, our calculations reveal that Cu1 flattens only slightly (89.9° to 86.0°, see Table ST4), but distorts significantly via the rocking motion (160.4° to 148.6°, see Table ST4). Thus, distortions along nuclear coordinates other than flattening similarly decrease the rate of ISC and probably the magnitude of SOC in copper(I) bisphenanthroline complexes. This is corroborated by the lower calculated SOC value for Cu1 compared to [Cu(dipp)2]+ (Table ST3).
While the picosecond dynamics of Cu1 suggest intermediate effects of the structural distortions compared to those of the less sterically hindered [Cu(dipp)2]+ and more sterically hindered [Cu(dtbp)2]+, the 3MLCT lifetime of Cu1, τ = 127 ns, measured with TCSPC, is the shortest of this series of complexes (Table 2). As discussed above, the duration of the 3MLCT state typically decreases with increased flattening in the excited state for two reasons: the flattening distortion (1) enables quenching caused by exciplex-like solvent interactions with the transient copper(II) center and (2) decreases the energy gap between the T1 and S0 states. While Cu1 does not flatten significantly, the rocking distortion has the same two effects on the excited-state lifetime. Given that the picosecond dynamics and absorbance and emission properties correlate with the steric hindrance of the phenanthroline ligands, an additional factor beyond the steric hindrance and structural distortion likely contributes to the abbreviated 3MLCT lifetime in copper(I) complexes bearing different substituents at the 2 and 9 positions. Figure S19 shows optical transient absorption spectra carried out on the nanosecond to microsecond time scales, with a measured 3MLCT lifetime of 126 ns that is in agreement with the TCSPC results (τ = 127 ns). The nonradiative decay rate constant of the 3MLCT state, calculated from the emission decay rate and yield, is larger than that of [Cu(dipp)2]+ and [Cu(dtbp)2]+, which could result from the decreased symmetry of Cu1 and may explain the relatively short excited-state lifetimes of the copper(I) complexes with nonsymmetric substituents. This is in line with the distorted structures calculated for the S1 and T1 states. A secondary nondominant decay component was also observed (τ = 5 ± 1 ns), which may correspond to solvent interactions following the excited-state rocking distortion that increases the exposure of the transient Cu(II) center to solvent molecules in the excited state. This 5 ns component is reproducible across two separate measurements of the Cu1 compound in DCM (Figure S19c). Furthermore, global analysis of the ESA and ground-state bleach (GSB) showed that this secondary decay component (τ = 5 ns) was present with similar relative amplitudes at both features. We assign this secondary decay component to the quenching of the 3MLCT lifetime due to inner shell penetration of DCM and subsequent interaction with the Cu(II) center, drawing from our previous studies of the solute–solvent interactions of [CuI(phen)2]+ in both acetonitrile and DCM.26 In this earlier study, the MLCT triplet state lifetimes of the least sterically strained [CuI(phen)2]+ were significantly shortened in DCM to ∼140 ps and acetonitrile to 16 ps compared to 100 and ∼1 ns for [CuI(dmp)2]+, where methyl groups are attached to the 2,9 positions of the phen ligands. Therefore, when the transient Cu(II) of the MLCT state is exposed to DCM, the lifetime will also be shortened even though the solvent is considered noncoordinating. Thus, we hypothesize that the observation of biexponential 3MLCT quenching results from distinct populations of Cu1 with different orientations of the phenanthroline ligands/substituents that permit different degrees of solute–solvent (i.e., Cu(II)-DCM) interactions. The accelerated decay component (τ = 5 ns) corresponds to the loss of a fraction of the 3MLCT population through enhanced solute–solvent interactions. The relative amplitudes of the 5 and 126 ns decay components indicate about 1/5 to 1/4 of the Cu1 3MLCT population adopted a structure that enabled closer approach of the DCM solvent molecules to the Cu(II) center. Because we observe the ∼5 ns decay component in the GSB, we assume this process corresponds to Cu1 returning back to the ground state without any intervening processes. Thus, we conclude that although excited-state Cu1 was expected to be sterically hindered to solute–solvent interactions, it is still possible to observe large variations in excited-state quenching resulting from a distribution of coordination environments.
Electrochemistry
The electrochemical behavior of Cu1 was investigated by cyclic voltammetry in DCM (anodic part, Figure S20) and dimethylformamide (Figure S21). The voltammograms feature a reversible oxidation wave at 0.60 V and a reversible reduction wave at −2.10 V vs Fc+/Fc, which is consistent with the values usually recorded for this kind of complex. E(Cu1+/Cu1) is slightly below the oxidation potential of [Cu(dipp)2]+ despite the fact that Cu1 is more sterically strained (which should imply the stabilization of the HOMO). Electronic factors could partly explain this result, since tert-butyl is more electron donating than isopropyl: L1 is thus slightly electron richer than dipp and could better stabilize Cu(II). In addition, the coordination scaffold is sterically ill-balanced, and oxidized Cu1 (i.e., [CuII(L1)2]2+) is certainly more warped than [CuII(dipp)2]2+, hence the observed trend of E(CuII/CuI). This is confirmed by DFT calculations (Table ST4): upon oxidation, the phen–Cu–phen angle in [CuII(dipp)2]2+ shifts from 90.0° to 70.3°, evidencing that this complex endures some flattening of about 20°. In addition, all four Cu–N distances shorten equivalently and the X–Cu–X angle stays at 180° (where X is the calculated centroid of the central C6 ring of phenanthroline, Table ST3). On the contrary, the distortions are stronger in oxidized Cu1. Indeed, due to the imbalance, there is a rocking of one of the phen planes with an X–Cu–X angle reduced to 148.6°. The coordination tends toward a trigonal bipyramid (Figure 9) with the copper atom at the center of the structure and one methyl group of the tert-Butyl occupying an axial position.
Figure 9.
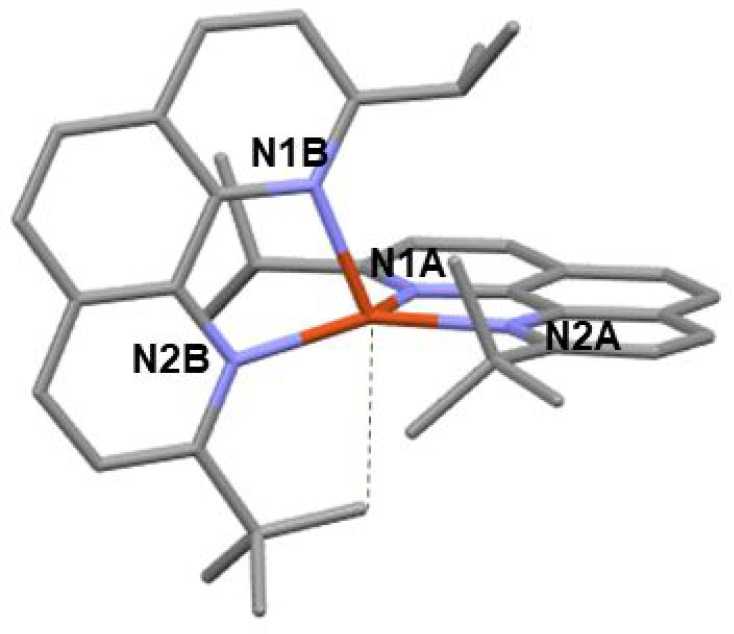
Structure of [CuII(L1)2]2+ (oxidized Cu1) (calculated).
The structure of oxidized Cu1 is similar to the calculated structures of Cu1 in the S1 and T1 states (see the XYZ file in the SI), which is normal since the excited state of copper(I) complexes can be formally described by a copper(II) ion coordinated to a reduced ligand. Such distortions are limited by the four tert-butyl groups in [Cu(dtbp)2]2+. The calculated structures for [CuI(dipp)2]+, [CuII(dipp)2]2+, Cu1+, Cu12+, [CuI(dtbp)2]+, and [CuII(dtbp)2]2+ are given in the SI, and relevant structural parameters are gathered in Table ST4.
Reductive Quenching of the Excited State
Knowing E00 and E(Cu1/Cu1–), one can
estimate the photo-oxidizing power E(Cu1*/Cu1–) at ca. 0.01 V vs Fc+/Fc (Table 1). This value is modest and
compares well with the usual values estimated for homoleptic Cu(I)
complexes. Increasing the steric bulk around the Cu(I) ion did not
entail the expected rise of E00, and therefore,
E(Cu1*/Cu1) and E([Cu(dipp)2]+*/[Cu(dipp)2]+) are similar. Cu1 was put to the test in a reductive
quenching experiment in the presence of decamethylferrocene dmFc as
the electron donor.27 The quenching of
the emission of Cu1 was monitored for different concentrations of
dmFc. Plotting τ0/τ (where τ0 and τ are the emission lifetimes in the absence and presence
of dmFc, respectively) as a function of [dmFc] afforded a straight
line (Figure S22) whose slope gives the
Stern–Volmer constant KSV. The
latter is equal to kQτ0 where kQ is the bimolecular quenching
rate. As stated in previous work,12,28 the quenching
of the excited state of a copper complex by dmFc may occur via two mechanisms: energy transfer (“EnT” from
the MLCT excited state to low lying iron(II) based (d,d) states with
kinetic rate constant kEnT = 2.1 ×
108 s–1)12 and electron transfer (from dmFc to excited copper complexes with
kinetic rate constant kRQ). The quenching
rate constants were anticipated to be rather large,27 and diffusion-limited processes (kinetic rate constant kd = 7 × 109 M–1·s–1 for ferrocene in DCM) cannot be ignored.
Thus, the equation 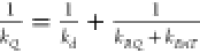 applies.27 From
the Stern–Volmer plots, we retrieved kRQ = 3.9 × 109 M–1.s–1. Interestingly, kRQ(Cu1)
is higher than kRQ([Cu(dpp)2]+) = 8 × 108 M–1.·–1 (where dpp is 2,9-diphenyl-1,10-phenanthroline):28 this is consistent with E(Cu1*/Cu1–) being significantly larger than E([Cu(dpp)2]+*/[Cu(dpp)2]0) (0.01 V vs −0.07 V vs Fc+/Fc respectively).12,28 However, kRQ(Cu1) is smaller than kRQ([Cu(dipp)2]+) (5.9
× 109 M–1·s–1) recorded in the same conditions, despite their similar photo-oxidative
powers. This evidenced that thermodynamic factors are not sufficient
to rationalize the reactivity of Cu1 and kinetic factors must be taken
into account.29 The nonsymmetrical nature
of ligand L1 entails a large reorganization energy, as demonstrated
by the distorted calculated structures of oxidized [CuII(L1)2]2+ (Figure 8) and S1 or T1 excited
states (Tables 3 and 4). Interestingly, Cu1sym2 exhibits the
highest Edist in the series Cu1, [Cu(dipp)2]+, and [Cu(dtbp)2]+ (Tables 4 and ST3). The reorganization energy offsets the thermodynamic
driving force and decreases the electron-transfer kinetics, following
Marcus theory.
applies.27 From
the Stern–Volmer plots, we retrieved kRQ = 3.9 × 109 M–1.s–1. Interestingly, kRQ(Cu1)
is higher than kRQ([Cu(dpp)2]+) = 8 × 108 M–1.·–1 (where dpp is 2,9-diphenyl-1,10-phenanthroline):28 this is consistent with E(Cu1*/Cu1–) being significantly larger than E([Cu(dpp)2]+*/[Cu(dpp)2]0) (0.01 V vs −0.07 V vs Fc+/Fc respectively).12,28 However, kRQ(Cu1) is smaller than kRQ([Cu(dipp)2]+) (5.9
× 109 M–1·s–1) recorded in the same conditions, despite their similar photo-oxidative
powers. This evidenced that thermodynamic factors are not sufficient
to rationalize the reactivity of Cu1 and kinetic factors must be taken
into account.29 The nonsymmetrical nature
of ligand L1 entails a large reorganization energy, as demonstrated
by the distorted calculated structures of oxidized [CuII(L1)2]2+ (Figure 8) and S1 or T1 excited
states (Tables 3 and 4). Interestingly, Cu1sym2 exhibits the
highest Edist in the series Cu1, [Cu(dipp)2]+, and [Cu(dtbp)2]+ (Tables 4 and ST3). The reorganization energy offsets the thermodynamic
driving force and decreases the electron-transfer kinetics, following
Marcus theory.
Conclusion
A sterically strained homoleptic copper(I) complex [Cu(L1)2]+ or Cu1 has been designed, showing an improved stability compared to [Cu(dtbp)2]+ in DCM solutions. The design is based on the synthesis of a nonsymmetrical phenanthroline ligand where positions in α of the nitrogen atoms are occupied by two encumbering branched aliphatic chains, namely tert-butyl and isopropyl groups. The complex has been characterized in the ground and excited states. Early after excitation, structural distortions are observed by ultrafast transient absorption spectroscopy, in contrast to previously studied [Cu(dtbp)2]+. Despite the increased steric strain imposed by the two substituents, the excited-state Cu1 exhibited a shorter lifetime and a weaker emission quantum yield than those of [Cu(dipp)2]+. The rationale behind this observation is twofold. First, many energy minima exist on the potential energy surfaces (triplet and singlet) and the energy barriers between them is small. Statistically, several of these minima can be populated with almost equal probability and decay to the ground state, providing multiple deactivation channels. Second, because the vicinal isopropyl group is smaller than the tert-butyl group, the excited-state distortion corresponds to rocking motion of the ligand, which leads to a trigonal bipyramid structure of the excited state of Cu1 (as verified by calculations of the structures of the excited state and of the oxidized state of Cu1) opening new deactivation channels.
Experimental Section
General
Chemicals were purchased from Sigma-Aldrich or Fisher Scientific and used as received. 2,9-Bis-isopropyl-1,10-phenanthroline (dipp),30 [Cu(dipp)2]+,13 and 2-tert-butyl-1,10-phenanthroline12 were synthesized as previously published. Thin-layer chromatography (TLC) was performed on aluminum sheets precoated with Merck 5735 Kieselgel 60F254. Column chromatography was carried out with Merck 5735 Kieselgel 60F (0.040–0.063 mm mesh). 1H NMR spectra were recorded on an AVANCE 300 UltraShield BRUKER instrument. Chemical shifts for 1H NMR spectra are referenced relative to residual protium in the deuterated solvent (CDCl3 δ = 7.26 ppm). NMR spectra were recorded at room temperature, chemical shifts are written in ppm and coupling constants in Hz. Mass spectrometry was performed with a JEOL JMS-700 B/E spectrometer. Electrochemical measurements were made under an argon atmosphere in CH2Cl2 or in DMF with 0.1 M Bu4NPF6. Cyclic voltammetry experiments were performed by using an Autolab PGSTAT 302N potentiostat/galvanostat. A standard three-electrode electrochemical cell was used. Potentials were referenced vs reversible ferrocenium/ferrocene couple. The working electrode was a glassy carbon disk and the auxiliary electrode was a Pt wire. UV–visible absorption spectra were recorded on an Analytik Jena spectrophotometer, using 1 cm path length cells. Emission spectra were recorded on a Fluoromax-3 Horiba spectrofluorimeter (1 cm quartz cells). E00 values were calculated by the tangent method: a tangent to the emission spectrum was drawn on the blue side of the spectrum. The intersection with the baseline gives the wavelength λ00, used to calculate E00 = 1240/λ00. Luminescence decays were recorded with a DELTAFLEX time correlated single photon counting system (HORIBA) on degassed DCM solutions.
A suitable crystal for X-ray diffraction single crystal experiment was selected and mounted with a cryoloop on the goniometer head of a D8 VENTURE Bruker AXS diffractometer equipped with a (CMOS) PHOTON 100 detector, using Mo K radiation (λ = 0.71073 Å, multilayer monochromator) at T = 150 K. The crystal structure has been described as having monoclinic symmetry and a P21/c space group. Cell parameters were found as follows: a = 16.0924(13), b = 15.3035(11), c = 15.6444(11) Å, β = 110.423(2)°, V = 3610.6(5) Å3. Number of formula units Z is equal to 4, and calculated density d and absorption coefficient μ values are 1.408 g·cm–3 and 0.714 mm–1, respectively. The structure was solved by dual-space algorithm using the SHELXT program,31 and then refined with full-matrix least-squares methods based on F2 (SHELXL program).32 All non-hydrogen atoms were refined with anisotropic atomic displacement parameters. H atoms were finally included in their calculated positions and treated as riding on their parent atom with constrained thermal parameters. A final refinement on F2 with 8267 unique intensities and 470 parameters converged at ωR(F2) = 0.2218 (RF = 0.0869) for 7275 observed reflections with (I > 2σ).
Synthesis
2-isopropyl-9-(tert-Butyl)-1,10-phenanthroline
2-(tert-Butyl)-1,10-phenanthroline (226 mg, 0.96 mmol) was dissolved in dry toluene (15 mL). The solution was cooled to 0 °C before dropwise addition of isopropylithium (0.7 M, 2.1 mL). The resulting solution was allowed to warm to room temperature and then stirred under argon for 18 h. Water (10 mL) was then added followed by a DCM extraction. Organic phases were stirred with activated manganese dioxide (2.15 g, 24 mmol) for 6h. The mixture was filtrated on Celite, dried over magnesium sulfate, and evaporated. The orange oil was purified by chromatography on silica (eluent: 0–5% methanol in DCM). The title compound was obtained in quantitative yield (265 mg). 1H NMR spectrum (300 MHz, CDCl3) δ = 8.14 (m, 2H), 7.73 (m, 3H), 7.54 (d, 3JH,H = 8.4 Hz, 1H), 3.45 (sept, 3JH,H = 6.9 Hz, 1H), 1.60 (s, 9H), 1.55 (d, 3JH,H = 6.9 Hz, 6H). 13C NMR spectrum (CDCl3, 75 MHz) δ= 169.29 (s), 167.36 (s), 136.15 (s), 135.96 (s), 127.20 (s), 126.84 (s), 125.50 (s), 125.31 (s), 120.74 (s), 119.79 (s), 38.73 (s), 37.04 (s), 30.30 (s), 22.48 (s). HRMS (ESI+) for C19H22N2 [M + H]+, m/z 279.1870 found, 279.1861 calcd.
Complex Cu1
Under argon atmosphere, [Cu(ACN)4]PF6 (74 mg, 0.20 mmol) was dissolved in 5 mL of degassed DCM. This solution was transferred into a vial that contains L1 (110 mg, 0.40 mmol). The red resulting solution was stirred overnight at room temperature. The mixture was precipitated in hexane to obtain a red powder. 1H NMR spectrum (CDCl3, 300 MHz) δ= 8.58 (m,4H), 8.13 (d, 3JH,H = 8.7 Hz, 2H), 8.07 (s, 4H), 7.76 (d, 3JH,H = 8.7 Hz, 2H), 3.04 (sept, 3JH,H = 6.9 Hz, 2H), 1.38 (s, 18H), 0.93 (d, 3JH,H = 6.9 Hz, 4H), 0.66 (d, 3JH,H = 6.9 Hz, 4H). 13C NMR spectrum (CDCl3, 300 MHz) δ= 138.55 (s), 138.41 (s), 128.66 (s), 127.03 (s), 126.82 (s), 123.94 (s), 122.02 (s), 38.76 (s), 38.01 (s), 30.66 (s), 22.13 (s), 22.01 (s). HRMS (ESI+) for C38H44CuN4+m/z 619.2861 found, 619.2862 calcd.
Optical Transient Absorption Spectroscopy
Femtosecond transient absorption (fsTA) spectroscopy experiments were performed on a previously described Ti:sapphire laser system (MaiTai oscillator/Spitfire Pro XP amplifier, Spectra-Physics) outputting a 1 kHz train of 830 nm, 100 fs, 2.6 mJ laser pulses.33 Here 0.6 mJ of the fundamental was used to perform the fsTA experiments. A 10% reflective beam splitter directed 60 μJ of the fundamental toward an optical delay line within the transient absorption spectrometer (Helios, Ultrafast Systems). Then 0.54 mJ of the transmitted fundamental was telescoped to about 2 mm diameter and frequency-doubled using a 2 mm thick type I lithium triborate crystal (θ = 90°, φ = 28°). The second harmonic was directed through a λ/2 waveplate and wiregrid polarizer to attenuate the pump pulse energy and set the relative polarization between the pump and probe to magic angle (54.7°). A mechanical chopper operating at 500 Hz was used to block every other pump pulse and the pump pulses were then focused to ∼200–300 μm diameter at the sample with 500 nJ/pulse. The probe was generated by focusing about 3–4 μJ of the fundamental into a 3 mm sapphire crystal after the optical delay line. The probe pulses (450–800 nm) were set to horizontal polarization using a wiregrid polarizer and focused to about 200 μm at the sample. After interacting with the sample, the probe and signal were coupled into a fiber optic cable connected to a CCD detector. The probe spectra and difference signal were recorded and calculated using the Helios software (Ultrafast Systems). The fsTA spectra were measured with 3 s total averaging at each time delay.
Nanosecond transient absorption (nsTA) spectroscopy experiments were performed using a previously described Nd:YAG laser system (PL2210 oscillator and amplifier, Ekspla) coupled to a supercontinuum laser (STM-1-UV, Leukos).33 The amplifier outputs a 0.5 kHz train of 355 nm, 25 ps, 0.3 mJ pulses which pumped an optical parametric generator (PG403, Ekspla) tuned to an output of 415 nm, 25 ps, 25 μJ pulses. The supercontinuum laser was triggered by the 1 kHz master trigger from the amplifier and output a 1 kHz train of broadband (400–1600 nm), 600 ps, 15 μJ pulses. The 415 nm pump was attenuated to 300 nJ/pulse and focused to about 200–300 μm at the sample. The broadband probe was focused to about 150 μm at the sample. After interacting with the sample, the probe and signal were detected using an SP-2150i Acton Series spectrograph (Princeton Instruments) coupled with a Spyder3 SG-14 (Teledyne DALSA) CCD camera. A laboratory-written application (LabVIEW 2014, National Instruments) provided the time delays to the supercontinuum laser via DG535 delay generators (Stanford Research Systems) externally triggered by the 1 kHz master trigger, recorded the probe spectra, and calculated the difference signal.
The fsTA and nsTA data had the background signal (scattered pump photons and long-lived, >1 ms, transient signals) subtracted and were corrected for group delay dispersion in the probe pulses. The nsTA data were corrected for scan-to-scan drift in the pump–probe timing provided by the DG535 delay generators. Samples for the fsTA and nsTA spectroscopy experiments were prepared in DCM with an absorbance of about 0.5 (about 0.5 mM) at the peak of the MLCT transition. Three freeze–pump–thaw cycles were performed on the nsTA samples to degas the solutions and eliminate the effects of oxygen on the measured excited-state dynamics.
Global-kinetic analysis of the transient absorption data was performed using MATLAB programs written by Matthew D. Krzyaniak in Michael R. Wasielewksi’s group. Here, the program simultaneously fit raw kinetic traces at select probe wavelengths (450–700 at 10 nm increments) where the time constants (τi = 1/ki), time zero, and width of the instrument response function were shared and only the amplitudes varied between the kinetic traces. The program numerically solved the first-order kinetic equations for the corresponding rate matrix K (Figure S13) using matrix methods and convolved the solutions with a Gaussian instrument response function. The program employed least-squares fitting with the Levenberg–Marquardt method to find the parameters that best described the data, which were then fed back into the kinetic equations to solve for the time-dependent populations of each state in the kinetic model. The evolution-associated spectra were then extracted from the raw data using the time-dependent populations for each state in the proposed kinetic model.
Computational Details
Two sets of calculation were performed. A first set was performed with ADF 201934 at the DFT level of theory (B3LYP functional). All atoms were described by the TZP basis set. Scalar relativistic effects were introduced through the ZORA Hamiltonian, and the solvent (DCM) was taken into account through a PCM. The UV–visible absorption spectrum was computed by TD-DFT on the basis of ADF optimized structures. The transitions were analyzed by mean of the TheoDORE tool (TheoDORE: Theoretical Density, Orbital Relaxation and Exciton analysis). The emission wavelength of lowest excited singlet and triplet states was computed on the basis of their optimized structures.
A second set of calculations was performed with GAUSSIAN09 at the DFT level of theory (B3LYP functional) including Grimme’s dispersion corrections. All atoms were described by the 6-31+G** basis set, and the solvent (DCM) was introduced through a PCM. All structures were fully optimized, and the nature of the stationary point encountered was determined by frequency calculations within the harmonic approximation. The minima were characterized by a full set of real frequencies.
Acknowledgments
The optical transient spectroscopy part of the work is funded by Chemical, Biological and Geological Sciences Division, Basic Energy Science, Office of Science, the US Department of Energy under Contract No. DE-AC02-06CH11357. The authors wish to thank the ANR (PERCO program n°ANR-16-CE07-0012-01) for financial support, the CEISAM (Nantes University) analysis platforms. The calculations were carried out in part at the IDRIS computer centers through a grant of computer time from GENCI (A0050810629) and in part in the Strasbourg HPC. We would also like to thank Michael Eberhart for his assistance in the freeze–pump–thaw sample preparation procedure.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.1c03901.
Accession Codes
CCDC 2125179 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
Supplementary Material
References
- Roth H. G.; Romero N. A.; Nicewicz D. A. Synlett 2016, 27, 714. 10.1055/s-0035-1561297. [DOI] [Google Scholar]; Keller S.; Prescimone A.; La Placa M.-G.; Junquera-Hernández J. M.; Bolink H. J.; Constable E. C.; Sessolo M.; Ortí E.; Housecroft C. E. The shiny side of copper: bringing copper(i) light-emitting electrochemical cells closer to application. Rsc Advances 2020, 10, 22631–22644. 10.1039/D0RA03824E. [DOI] [PMC free article] [PubMed] [Google Scholar]; Fresta E.; Weber M. D.; Fernandez-Cestau J.; Costa R. D. White Light-Emitting Electrochemical Cells Based on Deep-Red Cu(I) Complexes. Adv. Optical Mater. 2019, 7, 1900830. 10.1002/adom.201900830. [DOI] [Google Scholar]; Marion R.; Sguerra F.; Di Meo F.; Sauvageot E.; Lohier J.-F.; Daniellou R.; Renaud J.-L.; Linares M.; Hamel M.; Gaillard S. NHC Copper(I) Complexes Bearing Dipyridylamine Ligands: Synthesis, Structural, and Photoluminescent Studies. Inorg. Chem. 2014, 53, 9181–9191. 10.1021/ic501230m. [DOI] [PubMed] [Google Scholar]; Zhang Q.; Komino T.; Huang S.; Matsunami S.; Goushi K.; Adachi C. Triplet Exciton Confinement in Green Organic Light-Emitting Diodes Containing Luminescent Charge-Transfer Cu(I) Complexes. Adv. Funct. Mater. 2012, 22, 2327–2336. 10.1002/adfm.201101907. [DOI] [Google Scholar]
- Sandroni M.; Favereau L.; Planchat A.; Akdas-Kilig H.; Szuwarski N.; Pellegrin Y.; Blart E.; Le Bozec H.; Boujtita M.; Odobel F. Heteroleptic copper(i)-polypyridine complexes as efficient sensitizers for dye sensitized solar cells. Journal of Materials Chemistry A 2014, 2, 9944–9947. 10.1039/C4TA01755B. [DOI] [Google Scholar]; Malzner F. J.; Willgert M.; Constable E. C.; Housecroft C. E. The way to panchromatic copper(I)-based dye-sensitized solar cells: co-sensitization with the organic dye SQ2. J. Mater. Chem. A 2017, 5, 13717–13729. 10.1039/C7TA02575K. [DOI] [Google Scholar]; Dragonetti C.; Magni M.; Colombo A.; Fagnani F.; Roberto D.; Melchiorre F.; Biagini P.; Fantacci S. Towards efficient sustainable full-copper dye-sensitized solar cells. Dalton Trans. 2019, 48, 9703–9711. 10.1039/C9DT00790C. [DOI] [PubMed] [Google Scholar]; Karpacheva M.; Malzner F. J.; Wobill C.; Büttner A.; Constable E. C.; Housecroft C. E. Cuprophilia: Dye-sensitized solar cells with copper(I) dyes and copper(I)/(II) redox shuttles. Dyes Pigm. 2018, 156, 410–416. 10.1016/j.dyepig.2018.04.033. [DOI] [Google Scholar]
- Khnayzer R. S.; McCusker C. E.; Olaiya B. S.; Castellano F. N. Robust Cuprous Phenanthroline Sensitizer for Solar Hydrogen Photocatalysis. J. Am. Chem. Soc. 2013, 135, 14068–14070. 10.1021/ja407816f. [DOI] [PubMed] [Google Scholar]; Windisch J.; Orazietti M.; Hamm P.; Alberto R.; Probst B. General Scheme for Oxidative Quenching of a Copper Bis-Phenanthroline Photosensitizer for Light-Driven Hydrogen Production. ChemSusChem 2016, 9, 1719–1726. 10.1002/cssc.201600422. [DOI] [PubMed] [Google Scholar]; Mejía E.; Luo S.-P.; Karnahl M.; Friedrich A.; Tschierlei S.; Surkus A.-E.; Junge H.; Gladiali S.; Lochbrunner S.; Beller M. A Noble-Metal-Free System for Photocatalytic Hydrogen Production from Water. Chem. - Eur. J. 2013, 19, 15972–15978. 10.1002/chem.201302091. [DOI] [PubMed] [Google Scholar]; Heberle M.; Tschierlei S.; Rockstroh N.; Ringenberg M.; Frey W.; Junge H.; Beller M.; Lochbrunner S.; Karnahl M. Heteroleptic copper photosensitizers: Why an extended π-system does not automatically lead to enhanced hydrogen production. Chem. - Eur. J. 2017, 23, 312–319. 10.1002/chem.201604005. [DOI] [PubMed] [Google Scholar]; Fischer S.; Hollmann D.; Tschierlei S.; Karnahl M.; Rockstroh N.; Barsch E.; Schwarzbach P.; Luo S.-P.; Junge H.; Beller M.; Lochbrunner S.; Ludwig R.; Brueckner A. Death and Rebirth: Photocatalytic Hydrogen Production by a Self-Organizing Copper-Iron System. ACS Catal. 2014, 4, 1845–1849. 10.1021/cs500387e. [DOI] [Google Scholar]; Takeda H.; Monma Y.; Sugiyama H.; Uekusa H.; Ishitani O. Development of Visible-Light Driven Cu(I) Complex Photosensitizers for Photocatalytic CO2 Reduction. Front. Chem. 2019, 7, 418. 10.3389/fchem.2019.00418. [DOI] [PMC free article] [PubMed] [Google Scholar]; Gracia L.-L.; Luci L.; Bruschi C.; Sambri L.; Weis P.; Fuhr O.; Bizzarri C. New Photosensitizers Based on Heteroleptic CuI Complexes and CO2 Photocatalytic Reduction with [NiII(cyclam)]Cl2. Chem.—Eur. J. 2020, 26, 9929–9937. 10.1002/chem.202001279. [DOI] [PMC free article] [PubMed] [Google Scholar]; Lennox A. J. J.; Fischer S.; Jurrat M.; Luo S.-P.; Rockstroh N.; Junge H.; Ludwig R.; Beller M. Copper-Based Photosensitisers in Water Reduction: A More Efficient In Situ Formed System and Improved Mechanistic Understanding. Chem.—Eur. J. 2016, 22, 1233–1238. 10.1002/chem.201503812. [DOI] [PubMed] [Google Scholar]; Luo S.-P.; Chen N.-Y.; Sun Y.-Y.; Xia L.-M.; Wu Z.-C.; Junge H.; Beller M.; Wu Q.-A. Heteroleptic copper(I) photosensitizers of dibenzo[b,j]-1,10-phenanthroline derivatives driven hydrogen generation from water reduction. Dyes Pigm. 2016, 134, 580–585. 10.1016/j.dyepig.2016.07.040. [DOI] [Google Scholar]; Takeda H.; Kamiyama H.; Okamoto K.; Irimajiri M.; Mizutani T.; Koike K.; Sekine A.; Ishitani O. Highly Efficient and Robust Photocatalytic Systems for CO2 Reduction Consisting of a Cu(I) Photosensitizer and Mn(I) Catalysts. J. Am. Chem. Soc. 2018, 140, 17241–17254. 10.1021/jacs.8b10619. [DOI] [PubMed] [Google Scholar]
- Baralle A.; Fensterbank L.; Goddard J.-P.; Ollivier C. Aryl Radical Formation by Copper(I) Photocatalyzed Reduction of Diaryliodonium Salts: NMR Evidence for a CuII/CuI Mechanism. Chem.—Eur. J. 2013, 19, 10809–10813. 10.1002/chem.201301449. [DOI] [PubMed] [Google Scholar]; Michelet B.; Deldaele C.; Kajouj S.; Moucheron C.; Evano G. A General Copper Catalyst for Photoredox Transformations of Organic Halides. Org. Lett. 2017, 19, 3576–3579. 10.1021/acs.orglett.7b01518. [DOI] [PubMed] [Google Scholar]; Reiser O. Shining Light on Copper: Unique Opportunities for Visible-Light-Catalyzed Atom Transfer Radical Addition Reactions and Related Processes. Acc. Chem. Res. 2016, 49, 1990–1996. 10.1021/acs.accounts.6b00296. [DOI] [PubMed] [Google Scholar]; Engl S.; Reiser O. Copper Makes the Difference: Visible Light-Mediated Atom Transfer Radical Addition Reactions of Iodoform with Olefins. ACS Catal. 2020, 10, 9899–9906. 10.1021/acscatal.0c02984. [DOI] [Google Scholar]; Hossain A.; Bhattacharyya A.; Reiser O. Copper’s rapid ascent in visible-light photoredox catalysis. Science 2019, 364, eaav9713. 10.1126/science.aav9713. [DOI] [PubMed] [Google Scholar]; Sandoval-Pauker C.; Molina-Aguirre G.; Pinter B. Status report on copper (I) complexes in photoredox catalysis; photophysical and electrochemical properties and future prospects. Polyhedron 2021, 199, 115105. 10.1016/j.poly.2021.115105. [DOI] [Google Scholar]; Hunter C. J.; Boyd M. J.; May G. D.; Fimognari R. Visible-Light-Mediated N-Desulfonylation of N-Heterocycles Using a Heteroleptic Copper(I) Complex as a Photocatalyst. Journal of Organic Chemistry 2020, 85, 8732–8739. 10.1021/acs.joc.0c00983. [DOI] [PubMed] [Google Scholar]; Hernandez-Perez A. C.; Collins S. K. Heteroleptic Cu-Based Sensitizers in Photoredox Catalysis. Acc. Chem. Res. 2016, 49, 1557–1565. 10.1021/acs.accounts.6b00250. [DOI] [PubMed] [Google Scholar]
- Meyer M.; Albrecht-Gary A.-M.; Dietrich-Buchecker C. O.; Sauvage J.-P. π–π Stacking-Induced Cooperativity in Copper(I) Complexes with Phenanthroline Ligands. Inorg. Chem. 1999, 38, 2279–2287. 10.1021/ic981259+. [DOI] [Google Scholar]
- Kern J.-M.; Sauvage J.-P. Photoassisted C-C coupling via electron transfer to benzylic halides by a bis(di-imine) copper(I) complex. J. Chem. Soc., Chem. Commun. 1987, 546–548. 10.1039/C39870000546. [DOI] [Google Scholar]
- McCusker C. E.; Castellano F. N. Design of a Long-Lifetime, Earth-Abundant, Aqueous Compatible Cu(I) Photosensitizer Using Cooperative Steric Effects. Inorg. Chem. 2013, 52, 8114–8120. 10.1021/ic401213p. [DOI] [PubMed] [Google Scholar]
- Green O.; Gandhi B. A.; Burstyn J. N. Photophysical Characteristics and Reactivity of Bis(2,9-di-tert-butyl-1,10-phenanthroline)copper(I). Inorg. Chem. 2009, 48, 5704–5714. 10.1021/ic802361q. [DOI] [PubMed] [Google Scholar]
- McMillin D. R.; Buckner M. T.; Ahn B. T. A light-induced redox reaction of bis(2,9-dimethyl-1,10-phenanthroline)copper(I). Inorg. Chem. 1977, 16, 943–945. 10.1021/ic50170a046. [DOI] [Google Scholar]; Buckner M. T.; McMillin D. R. Photoluminescence from copper(I) complexes with low-lying metal-to-ligand charge transfer excited states. J. Chem. Soc., Chem. Commun. 1978, 759, 759. 10.1039/c39780000759. [DOI] [Google Scholar]
- Blaskie M. W.; McMillin D. R. Photostudies of copper(I) systems. 6. Room-temperature emission and quenching studies of bis(2,9-dimethyl-1,10-phenanthroline)copper(I). Inorg. Chem. 1980, 19, 3519–3522. 10.1021/ic50213a062. [DOI] [Google Scholar]
- Miller M. T.; Gantzel P. K.; Karpishin T. B. Effects of Sterics and Electronic Delocalization on the Photophysical, Structural, and Electrochemical Properties of 2,9-Disubstituted 1,10-Phenanthroline Copper(I) Complexes. Inorg. chem. 1999, 38, 3414–3422. 10.1021/ic9900399. [DOI] [PubMed] [Google Scholar]
- Gimeno L.; Blart E.; Rebilly J.-N.; Coupeau M.; Allain M.; Roisnel T.; Quarré de Verneuil A.; Gourlaouen C.; Daniel C.; Pellegrin Y. Non-Symmetrical Sterically Challenged Phenanthroline Ligands and Their Homoleptic Copper(I) Complexes with Improved Excited-State Properties. Chem.—Eur. J. 2020, 26, 11887–11899. 10.1002/chem.202001209. [DOI] [PubMed] [Google Scholar]
- Garakyaraghi S.; Crapps P. D.; McCusker C. E.; Castellano F. N. Cuprous Phenanthroline MLCT Chromophore Featuring Synthetically Tailored Photophysics. Inorg. Chem. 2016, 55, 10628–10636. 10.1021/acs.inorgchem.6b01880. [DOI] [PubMed] [Google Scholar]
- Eggleston M. K.; McMillin D. R.; Koenig K. S.; Pallenberg A. J. Steric Effects in the Ground and Excited States of Cu(NN)2+ Systems. Inorg. Chem. 1997, 36, 172–176. 10.1021/ic960698a. [DOI] [Google Scholar]; Kohler L.; Hayes D.; Hong J.; Carter T. J.; Shelby M. L.; Fransted K. A.; Chen L. X.; Mulfort K. L. Synthesis, structure, ultrafast kinetics, and light-induced dynamics of CuHETPHEN chromophores. Dalton Trans. 2016, 45, 9871. 10.1039/C6DT00324A. [DOI] [PubMed] [Google Scholar]; Cunningham C. T.; Cunningham K. L. H.; Michalec J. F.; McMillin D. R. Cooperative Substituent Effects on the Excited States of Copper Phenanthrolines. Inorg. Chem. 1999, 38, 4388–4392. 10.1021/ic9906611. [DOI] [PubMed] [Google Scholar]; Miller M. T.; Gantzel P. K.; Karpishin T. B. A Photoluminescent Copper(I) Complex with an Exceptionally High CuII/CuI Redox Potential: [Cu(bfp)2]+ (bfp = 2,9-bis(trifluoromethyl)-1,10-phenanthroline). Angew. Chem., Int. Ed. 1998, 37, 1556–1558. . [DOI] [PubMed] [Google Scholar]; Miller M. T.; Karpishin T. B. Phenylethynyl Substituent Effects on the Photophysics and Electrochemistry of [Cu(dpp)2]+ (dpp = 2,9-Diphenyl-1,10-phenanthroline). Inorg. Chem. 1999, 38, 5246. 10.1021/ic990588e. [DOI] [Google Scholar]
- Gandhi B. A.; Green O.; Burstyn J. N. Facile Oxidation-Based Synthesis of Sterically Encumbered Four-Coordinate Bis(2,9-di-tert-butyl-1,10-phenanthroline)copper(I) and Related Three-Coordinate Copper(I) Complexes. Inorg. Chem. 2007, 46, 3816–3825. 10.1021/ic0615224. [DOI] [PubMed] [Google Scholar]
- Miller M. T.; Gantzel P. K.; Karpishin T. B. A Highly Emissive Heteroleptic Copper(I) Bis(phenanthroline) Complex: [Cu(dbp)(dmp)]+ (dbp = 2,9-Di-tert-butyl-1,10-phenanthroline; dmp = 2,9-Dimethyl-1,10-phenanthroline). J. Am. Chem. Soc. 1999, 121, 4292–4293. 10.1021/ja9901415. [DOI] [Google Scholar]
- Brown-Xu S.; Fumanal M.; Gourlaouen C.; Gimeno L.; Quatela A.; Thobie-Gautier C.; Blart E.; Planchat A.; Riobé F.; Monnereau C.; Chen L. X.; Daniel C.; Pellegrin Y. Intriguing Effects of Halogen Substitution on the Photophysical Properties of 2,9-(Bis)halo-Substituted Phenanthrolinecopper(I) Complexes. Inorg. Chem. 2019, 58, 7730–7745. 10.1021/acs.inorgchem.9b00042. [DOI] [PubMed] [Google Scholar]
- Yang L.; Powell D. R.; Houser R. P. Structural variation in copper(i) complexes with pyridylmethylamide ligands: structural analysis with a new four-coordinate geometry index, τ4. Dalton Trans. 2007, 955–964. 10.1039/B617136B. [DOI] [PubMed] [Google Scholar]
- Smith C. S.; Mann K. R. Void Space Containing Crystalline Cu(I) Phenanthroline Complexes As Molecular Oxygen Sensors. Chem. Mater. 2009, 21, 5042–5049. 10.1021/cm901109n. [DOI] [Google Scholar]
- Felder D.; Nierengarten J. F.; Barigelletti F.; Ventura B.; Armaroli N. Highly luminescent Cu(I)-phenanthroline complexes in rigid matrix and temperature dependence of the photophysical properties. J. Am. Chem. Soc. 2001, 123, 6291–6299. 10.1021/ja0043439. [DOI] [PubMed] [Google Scholar]
- Siddique Z. A.; Yamamoto Y.; Ohno T.; Nozaki K. Structure-Dependent Photophysical Properties of Singlet and Triplet Metal-to-Ligand Charge Transfer States in Copper(I) Bis(diimine) Compounds. Inorg. Chem. 2003, 42, 6366–6378. 10.1021/ic034412v. [DOI] [PubMed] [Google Scholar]
- Everly R. M.; McMillin D. R. Reinvestigation of the absorbing and emitting charge-transfer excited states of [Cu(NN)2]+ systems. J. Phys. Chem. 1991, 95, 9071–9075. 10.1021/j100176a011. [DOI] [Google Scholar]
- Shaw G. B.; Grant C. D.; Shirota H.; Castner E. W.; Meyer G. J.; Chen L. X. Ultrafast Structural Rearrangements in the MLCT Excited State for Copper(I) bis-Phenanthrolines in Solution. J. Am. Chem. Soc. 2007, 129, 2147–2160. 10.1021/ja067271f. [DOI] [PubMed] [Google Scholar]; Iwamura M.; Takeuchi S.; Tahara T. Real-Time Observation of the Photoinduced Structural Change of Bis(2,9-dimethyl-1,10-phenanthroline)copper(I) by Femtosecond Fluorescence Spectroscopy: A Realistic Potential Curve of the Jahn–Teller Distortion. J. Am. Chem. Soc. 2007, 129, 5248–5256. 10.1021/ja069300s. [DOI] [PubMed] [Google Scholar]; Capano G.; Chergui M.; Rothlisberger U.; Tavernelli I.; Penfold T. J. A Quantum Dynamics Study of the Ultrafast Relaxation in a Prototypical Cu(I)–Phenanthroline. J. Phys. Chem. A 2014, 118, 9861–9869. 10.1021/jp509728m. [DOI] [PubMed] [Google Scholar]
- Gothard N. A.; Mara M. W.; Huang J.; Szarko J. M.; Rolczynski B.; Lockard J. V.; Chen L. X. Strong Steric Hindrance Effect on Excited State Structural Dynamics of Cu(I) Diimine Complexes. J. Phys. Chem. A 2012, 116, 1984–1992. 10.1021/jp211646p. [DOI] [PubMed] [Google Scholar]
- Garakyaraghi S.; Danilov E. O.; McCusker C. E.; Castellano F. N. Transient Absorption Dynamics of Sterically Congested Cu(I) MLCT Excited States. J. Phys. Chem. A 2015, 119, 3181–3193. 10.1021/acs.jpca.5b00901. [DOI] [PubMed] [Google Scholar]
- Lockard J. V.; Kabehie S.; Zink J. I.; Smolentsev G.; Soldatov A.; Chen L. X. Influence of Ligand Substitution on Excited State Structural Dynamics in Cu(I) Bisphenanthroline Complexes. J. Phys. Chem. B 2010, 114, 14521–14527. 10.1021/jp102278u. [DOI] [PubMed] [Google Scholar]
- Cunningham K. L.; McMillin D. R. Reductive Quenching of Photoexcited Cu(dipp)2+ and Cu(tptap)2+ by Ferrocenes (dipp = 2,9-Diisopropyl-1,10-phenanthroline and tptap = 2,3,6,7-Tetraphenyl-1,4,5,8-tetraazaphenanthrene). Inorg. Chem. 1998, 37, 4114–4119. 10.1021/ic980213d. [DOI] [PubMed] [Google Scholar]
- Cunningham K. L.; Hecker C. R.; McMillin D. R. Competitive energy-transfer and reductive quenching of the CT excited states of copper(l) phenanthrolines. Inorg. Chim. Acta 1996, 242, 143–147. 10.1016/0020-1693(95)04859-6. [DOI] [Google Scholar]
- Gushurst A. K. I.; McMillin D. R.; Dietrich-Buchecker C. O.; Sauvage J. P. Comparative studies of the photophysical properties of copper phenanthrolines: from Cu(dmp)2+ to the copper(I) catenates. Inorg. Chem. 1989, 28, 4070–4072. 10.1021/ic00321a008. [DOI] [Google Scholar]
- Pallenberg A. J.; Koenig K. S.; Barnhart D. M. Synthesis and Characterization of Some Copper(I) Phenanthroline Complexes. Inorg. Chem. 1995, 34, 2833–2840. 10.1021/ic00115a009. [DOI] [Google Scholar]
- Sheldrick G. SHELXT - Integrated space-group and crystal-structure determination. Acta Crystallogr., Sect. A 2015, 71, 3–8. 10.1107/S2053273314026370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick G. Crystal structure refinement with SHELXL. Acta Crystallographica Section C 2015, 71, 3–8. 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes D.; Kohler L.; Hadt R. G.; Zhang X.; Liu C.; Mulfort K. L.; Chen L. X. Excited state electron and energy relays in supramolecular dinuclear complexes revealed by ultrafast optical and X-ray transient absorption spectroscopy. Chem. Sci. 2018, 9, 860–875. 10.1039/C7SC04055E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- te Velde G.; Bickelhaupt F. M.; Baerends E. J.; Fonseca Guerra C.; van Gisbergen S. J. A.; Snijders J. G.; Ziegler T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. 10.1002/jcc.1056. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.