

Abstract
Background:
Mutations in SLC6A1, encoding γ-aminobutyric acid (GABA) transporter 1 (GAT-1), have been recently associated with a spectrum of neurodevelopmental disorders ranging from variable epilepsy syndromes, intellectual disability (ID), autism and others. To date, most identified mutations are de novo. We here report a pedigree of two siblings associated with myoclonic astatic epilepsy, attention deficit hyperactivity disorder (ADHD), and ID.
Methods:
Next-generation sequencing identified a missense mutation in the SLC6A1 gene (c.373G>A(p.Val125Met)) in the sisters but not in their shared mother who is also asymptomatic, suggesting gonadal mosaicism. We have thoroughly characterized the clinical phenotypes: EEG recordings identified features for absence seizures and prominent bursts of occipital intermittent rhythmic delta activity (OIRDA). The molecular pathophysiology underlying the clinical phenotypes was assessed using a multidisciplinary approach including machine learning, confocal microscopy, and high-throughput 3H radio-labeled GABA uptake assays in mouse astrocytes and neurons.
Results:
The GAT-1(Val125Met) mutation destabilizes the global protein conformation and reduces transporter protein expression at total and cell surface. The mutant transporter protein was localized intracellularly inside the endoplasmic reticulum (ER) in both HEK293T cells and astrocytes which may directly contribute to seizures in patients. Radioactive 3H-labeled GABA uptake assay indicated the mutation reduced the function of the mutant GAT-1(Val125Met) to ~30% of the wildtype.
Conclusions:
The seizure phenotypes, ADHD, and impaired cognition are likely caused by a partial loss-of-function of GAT-1 due to protein destabilization resulting from the mutation. Reduced GAT-1 function in astrocytes and neurons may consequently alter brain network activities such as increased seizures and reduced attention.
Keywords: Phenotypic heterogeneity, mosaicism, GABA transporter 1, protein stability, ADHD, myoclonic atonic epilepsy (MAE)
Introduction
GABA is the major inhibitory neurotransmitter in the mammalian brain. GABA transporter 1 (GAT-1) encoded by SLC6A1 is one of the principal transporters for GABA in the central nervous system[1]. In contrast with other GABA transporters, GAT-1 is a major transporter subtype of sodium- and chloride-dependent transporters and is localized in GABAergic axons and nerve terminals. GAT-1 retrieves GABA from the synaptic cleft into both presynaptic neurons and astrocytes, by which it can rapidly terminate neurotransmission and modulate the homeostasis of neuronal excitation and inhibition. Given the essential role of GABAergic signaling in neurodevelopment and neurotransmission, it is not surprising that defective GAT-1 resulting from SLC6A1 mutations give rise to a wide spectrum of epilepsy syndromes and neurodevelopmental disorders including myoclonic atonic epilepsy (MAE), childhood absence epilepsy, autism, and intellectual disability or neurodevelopmental delay[2–6].
GAT-1 is a key component of the GABAergic pathway and has complex interactions with GABAA receptors and GABAB receptors. The GABAergic pathway is a converging pathway for epilepsy, autism, and other neurodevelopmental disorders caused by multiple genes and GAT-1-encoding SLC6A1 is one of these genes [6, 7]. Unlike GABAA receptors that directly conduct postsynaptic GABAergic currents, GAT-1 influences GABAergic synaptic transmission by clearance and re-uptake of GABA from the synapse [8]. In epilepsy, an impaired GABAergic pathway has been identified in multiple seemingly unrelated etiologies, including mutations in both ion channel and non-ion channel genes [9]. In experimental animal models and children with absence, the increased ictal inhibition of thalamocortical neurons is enhanced by the loss-of function of GAT-1, suggesting the potential link of impaired GAT-1 function in absence seizures, a commonly identified seizure type in SLC6A1 mutations[2, 3, 10].
Based on previous reports, MAE and absence seizures are common seizure types associated with SLC6A1 mutations, although the less common seizure types are also reported[3, 4, 6, 10]. Besides MAE and absence seizures, developmental and intellectual disability is prevalent among reported SLC6A1 mutations. Approximately ~60% of children with absence seizures have neuropsychiatric comorbidities, with attention deficits as the most common comorbidity (35-40%), followed by mood disorders[11–13]. In absence of epilepsy, neuropsychiatric comorbidities—in particular attention deficits—may precede the first absence seizures and epilepsy diagnosis [14, 15]. This suggests that attention deficit could be a prominent clinical feature for SLC6A1-mediated disorders. Since the first report of SLC6A1 mutations in MAE, several studies have identified a number of mutations in SLC6A1 associated with two prominent features: intellectual disability (ID) and a wide spectrum of epilepsy [3, 7]. In some cases, the patients only display mild developmental delay or learning disorder, nonspecific dysmorphisms, and an electroencephalogram (EEG) pattern closely resembling that of myoclonic-atonic epilepsy with brief absence seizures later on [16]. We previously reported SLC6A1(p.Gly234Ser) associated with Lennox-Gastaut syndrome (LGS) [5] and SLC6A1(p.Pro361Thr) associated with epilepsy and autism[6]. It is likely the neuropsychiatric comorbidity may be independent of seizures and is directly resulted from the defective GAT-1, excitation-inhibition mismatch, and subsequent neural network dysfunction.
In this study, we reported a novel mutation Val125Met in a two sisters but not in their shared mother. We characterized the EEG patterns and neuropsychiatric comorbidities in the affected individuals, including a thorough evaluation of patient disease history, seizure phenotype, EEG, and ADHD phenotype. We evaluated the impact of the disease-causing mutation (Val125Met) by characterizing the mutant protein trafficking and function in different cell types including mouse neurons and astrocytes. We compared the wildtype and mutant transporter with protein structure modeling via machine learning based prediction, 3H radioactive GABA uptake assay, and protein expression and subcellular localizations via confocal microscopy, in both heterologous cells and mouse cortical astrocytes. This study provides molecular mechanisms underlying how a defective GAT-1 can cause ADHD in addition to epilepsy and expands the clinical phenotype of SLC6A1 mutation mediated disorders. We compared the common and differential clinical presentations and drug responses of the siblings, providing important insights into understanding phenotypical heterogeneity of SLC6A1 mutation mediated disorders.
Methods
Patients with epilepsy, ADHD and others
The patients and her unaffected family members were evaluated at the Pediatric Epilepsy Program at the University of Rochester Medical Center. The collected clinical data included age of onset, a detailed developmental history, seizure types and frequency, response to antiepileptic drugs (AEDs), family history, and general and neurological examination results. Video electroencephalography (EEG) was examined repeatedly and the results were reviewed by qualified electroencephalographers. ADHD symptoms were assessed using the Vanderbilt Assessment Tool. Written informed consent for the sharing of clinical and genetic information was obtained from the parents.
Genetic data analysis
Saliva samples of the patients and shared mother were collected and evaluated using the CLIA-approved clinical Invitae Comprehensive Epilepsy Gene Panel (Invitae, USA). Three variants of unknown significance (VUS) were identified in the proband in CACNA1A (c.3547G>A (p.Val1183Ile)), FASN (c.4633C>T (p.Arg1545Cys)), and SLC6A1 (c.373G>A (p.Val125Met)). The VUS in CACNA1A was identified in both the proband’s affected sister and mother, as well as population databases and was reclassified as “Likely Benign.” The VUS in FASN was identified only in the proband and has also been identified in population databases. The SLC6A1 VUS was identified in the proband, her sister, but not their unaffected mother.
The cDNAs for coding GABA transporter 1
The plasmid cDNA encoding enhanced yellow fluorescent protein (EYFP)-tagged rat GAT-1 was sub-cloned into the expression vector pCMV. Replications of patient GAT-1 mutations were cloned via a standard molecular cloning process. QuikChange Site-directed Mutagenesis kit was utilized to introduce the GAT-1(Val125Met) mutation into wildtype GAT-1 coding sequence. The product from polymerase chain reaction was transformed using DHα competent cells and finally plated. A clone was chosen and grown overnight, replicating the cDNA. The GAT-1(Val125Met) mutation was confirmed by DNA sequencing. Both the wildtype and the mutant cDNAs were prepared with Qiagen Maxiprep kit.
Polyethylenimine (PEI) transfection
Standard transfection protocols were performed using human embryonic kidney 293T (HEK293T) cells [10]. 24 hours before transfection HEK293T cells were split equally into plates. For radiolabeling GABA uptake,1μg of the cDNAs with PEI at a ratio of 1:2.5μl was transfected in 35mm dish. The cDNAs were combined with Dulbecco modified Eagle medium (DMEM) and a PEI/DMEM mixture. For total protein expression, 3μg cDNAs were used while 10 μg cDNAs were used for cell surface protein biotinylation. Transfected HEK293T cells incubated for 48 hours. After incubation, proteins were harvested as described below.
Western blot analysis of total GAT-1 protein
Live transfected HEK293T cells were washed with phosphate buffered saline (1×PBS, pH 7.4) 3 times and then cells were lysed in RIPA buffer (20 mM Tris, 20 mM EGTA, 1 mM DTT, 1 mM benzamidine), supplemented with 0.01 mM PMSF, 0.005 μg/mL leupeptin, and 0.005 μg/mL pepstatin for 30 min at 4º C. The samples were then subject to protein concentration determination and followed by SDS-PAGE. Membranes were incubated with primary rabbit polyclonal antibodies against GAT-1 (Alomone Labs, AGT-001 or Synaptic System, 274102 at 1:200 dilution).
Neuron and astrocyte cultures and transfections
Mouse cortical neuronal cultures and transfection were prepared as previously described [17, 18]. Mouse neurons were cultured from postnatal day 0 mouse pups. The neurons were plated at a density of 2 ×105 for western blot in plating media that contained 420 mL Dulbecco’s Modified Eagle’s Medium (DMEM), 40 mL F12, 40 mL fetal bovine serum, 1 mL penicillin and streptomycin, and 0.2 mL L-Glutamine (200 mM) for 4 hrs. Neurons were then maintained in Neurobasal media that contained B27 supplement (50:1), L-Glutamine (200 mM), and 1 mL penicillin and streptomycin. Neurons were transfected with 2 μg cDNA at day 5-7 in culture with calcium phosphate and were harvested 8-10 days after transfection.
For mouse cortical astrocytes, the cortices of postnatal day 0-3 pups were dissected. The tissues were minced after removing the meninges and then digested with 0.25% trypsin for 10 minutes at 37°C. The debris and large tissue chunks were removed. The cells were then maintained in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. The astrocytes used for experiments were at passage 2 and transfected with PEI for 2 days before experiment. A total of 1μg of cDNAs were transfected in one 35-mm2 dish.
Radioactive 3H-labeled GABA uptake assay
The radioactive 3H-labeled GABA uptake assay in HEK293T, mouse astrocytes and neurons was modified from previous studies [5, 19]. Briefly, cells were cultured in 5 mm2 dishes 3 days before the GABA uptake experiment in DMEM with 10% fetal bovine serum and 1% penicillin/streptomycin. The cells were then transfected with equal amounts of the wildtype or the mutant GAT-1(Val125Met) cDNAs (1 μg) with PEI at a ratio of 1 μg cDNA:2.5 μl of PEI for each condition at 24 hrs after cell seeding. GABA uptake assay was carried out 48 hrs after transfection. The cells were incubated with preincubation solution for 15 min and then incubated with preincubation solution containing 1μci/ml 3H GABA and 10 μM unlabeled GABA for 30 min at room temperature. After washing, the cells were lysed with 0.25 N NaOH for 1 hr. Acetic acid glacial was added and lysates were then determined on a liquid scintillator with QuantaSmart. The flux of GABA (pmol/μg/min) was averaged with at least triplets for each condition at each transfection. The average counting was taken as n = 1. The untransfected condition was taken as a baseline that was subtracted from both the wildtype and the mutant conditions. The pmol/μg/min in the mutant was then normalized to the wildtype from each experiment, which was arbitrarily taken as 100%.
Live cell confocal microscopy and image acquisition
Live cell confocal microscopy was performed using an inverted Zeiss laser scanning microscope (Model 510) with a 63 × 1.4 NA oil immersion lens, 2-2.5 × zoom, and multi-track excitation. HEK 293T cells were plated on poly-D-lysine-coated, glass-bottom imaging dishes at the density of 1-2 × 105 cells and cotransfected with 2 μg of the wildtype or the mutant GAT-1 plasmids and 1 μg pECFP-ER with PEI based on our standard lab protocol. Cells were examined with excitation at 458 nm for ECFP, 514 nm for EYFP. All images were single confocal sections averaged from 8 times to reduce noise, except when otherwise specified. The images were acquired using a LSM 510 invert confocal microscope with 63X objective.
Protein structural modeling and machine learning tools
We simulated the impact of the identified mutation on the transporter protein with multiple machine learning tools. Tertiary structures of both the wildtype and mutated GAT-1(V125M) protein were predicted by I-TASSER [20] and analyzed by MAESTRO web [21]. Details in structural differences between the wildtype and the mutant GAT-1 were illustrated using the modelled structure by DynaMut [22]. Analysis of self-aggregation or co-aggregation was conducted using PASTA 2.0 [23].
Data analysis
Numerical data were expressed as mean ± SEM. Proteins were quantified by Odyssey software and data were normalized to loading controls and then to wildtype transporter proteins, which was arbitrarily taken as 1 in each experiment. The radioactivity of GABA uptake was measured in a liquid scintillator with QuantaSmart. The flux of GABA (pmol/μg/min) in the wildtype GAT-1 samples was arbitrarily taken as 100% each experiment. The fluorescence intensities from confocal microscopy experiments were determined using MetaMorph imaging software and the measurements were carried out in ImageJ as modified from previous description[24–26]. For statistical significance, we used one-way analysis of variance (ANOVA) with Newman-Keuls test or Student’s unpaired t-test. In some cases, one sample t-test was performed (GraphPad Prism, La Jolla, CA), and statistical significance was taken as p < 0.05.
Results
Mutation analysis identified SLC6A1(Val125Met) variation in two sisters and genetic mosaicism in shared mother
Previous studies have identified mutations in SLC6A1 associated with a spectrum of epilepsy syndromes and neurodevelopmental disorders [2–6]. We have reported the mutations in SLC6A1 associated with Lennox-Gastaut syndrome and epilepsy with autism due to partial loss of GAT-1 function. To date, most of the assayed mutations (Figure 1A) had a complete or partial loss of the mutant transporter 1 and are de novo. However, germline transmission has been reported in two other SLC6A1 variants (p.Ala334Pro and p.Ala288Val)[7]. Here we identified 373 G to A mutation in SLC6A1 resulting in a change of valine 125 to methionine in a family (Figure 1B) using a clinical NGS epilepsy genetic panel. This variant of unknown significance was not identified in the shared parent of the half-sisters, suggesting gonadal mosaicism. This variation results in valine, a conserved amino acid across species, being substituted with methionine (Figure 1C), suggesting the variation may have significant impact on the transporter protein function.
Figure 1. A novel SLC6A1 missense mutation GAT-1(Val125Met) in two sisters associated with myoclonic astatic epilepsy.
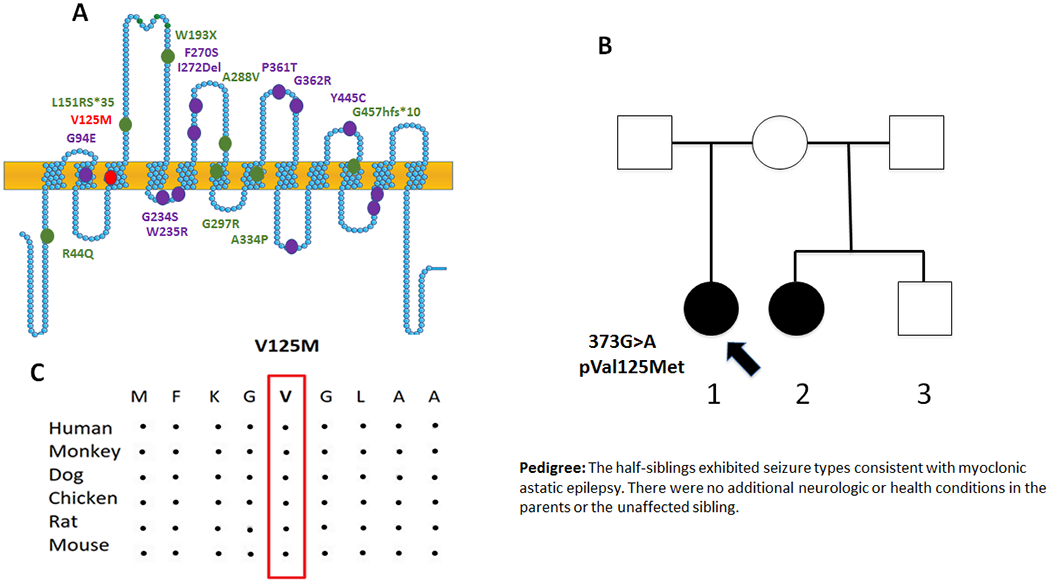
(A). Schematic representation of GAT-1 protein topology and locations of GAT-1 variants previously identified in patients associated with a spectrum of epilepsy syndromes. It is predicted that GAT-1 contains 12 transmembrane domains. Val125Met (V125M) is located at the 3rd transmembrane helix of the GAT-1 protein. The positions of variants are based on the published LeuT crystal structure. (B) Pedigree and the genotype. A missense mutation was found in the proband and the half-sister but not in the rest of the family members. (C). Amino acid sequence homology shows that valine (V) at residue 125 is highly conserved in SCL6A1 in humans (Accession NO.NP_003033.3) and across species as shown in boxed region.
Common and differential clinical features and response to valproic acid
The proband was otherwise normal until onset of epilepsy around 1 year of age. The first seizures were episodes of behavioral arrest with eye fluttering (eyelid myoclonia). The patient experienced the first generalized tonic clonic convulsion at 1 year, with only rare convulsions through the rest of the clinical course. EEG at this time was reported to show 3Hz spike and wave associated with eye flutter and behavioral arrest and the patient was trialed on valproic acid (VPA), which reduced the frequency and intensity of events, but never fully controlled them. With growth, repeat EEG with provoking maneuvers identified slight enhancement of the 3 Hz spike and wave fronto-centrally predominant generalized epileptiform discharges with transition to drowsiness and sleep, but no enhancement of epileptiform discharges with hyperventilation or photic stimulation. EEG showed prominent bursts of occipital intermittent rhythmic delta activity (OIRDA) throughout the otherwise normal waking background. Clinically, seizures were primarily associated with brief behavioral arrest, slight upgaze, eyelid myoclonia and negative myoclonus/loss of tone leading the patient to commonly fall to their knees. The patient was diagnosed with MAE (Doose Syndrome). The prominent presence of eyelid myoclonia shares clinical similarity to Jeavon’s Syndrome phenotype.
Over the years the patient trialed multiple medications with only rare periods of improved seizure control (Table 1). The patient exhibited significant anxiety and periods of selective mutism associated with anxiety in social, particularly medical, settings, prompting speech therapy. In early childhood the patient was able to maintain age-appropriate educational advances but exhibited a progressive educational achievement gap starting after age 8-9, originally attributed to medication side effect. She was diagnosed with ADHD with difficulty with attention and memory more noticeable after age 7-8, for which treatment with stimulants has been trialed with variable benefit. The patient has prominent levo-scoliosis, which was most notable after age 12.
Table 1.
Clinical features of the patients with SLC6A1 (V125M) variant
| Patient ID | Proband (patient 1)/sibling (patient 2) |
| Variant | c.3547G>A (NM_0030423) |
| Protein change | p.V125M |
| Origin | Maternal |
| Sex | Female |
| Current age | 16 years/ 8 years |
| Age at seizure onset | 1 year/ 4 years old |
| Seizure type at onset | Absence, GTCS |
| Seizure frequency at onset | Multiple times daily |
| Further seizure types | Myoclonic-atonic |
| Ictal EEG | 3 Hz SW |
| Interictal EEG | SW, PSW |
| Seizure outcome | Seizure uncontrolled/seizure controlled |
| Duration before seizure free | None for patient 1/4 years for patient 2 |
| Intellectual disability | Moderate ID for patient 1/and for patient 2 |
| Language | Normal |
| Neurological exam | Normal |
| MRI findings | Normal |
| Epilepsy | Myoclonic Astatic seizures |
| SIFT (score) | Damaging (0.02) |
| Polyphen2 (score) | Probably damaging (0.999) |
| MutationTaster(score) | Disease causing (0.999) |
| Frequency in ExAC | - |
EEG = electroencephalography; PSW =polyspike and wave complex; SW= spike and wave complex; GTCS = generalized tonic-clonic seizure; ID= Intellectual Disability; MRI = magnetic resonance imaging.
A half-sibling with shared mother exhibited ADHD symptoms and mild delayed language development. Seizure onset occurred at age 4, presenting with sudden falls while walking or standing. A routine EEG, which did not capture events, identified frequent bursts of high amplitude OIRDA throughout the otherwise normal waking background. A 24-hour ambulatory EEG captured events, which were associated with bursts of fronto-centrally predominant 3Hz spike and wave epileptiform discharges. Discharges and discharge complexes were activated by transition to drowsiness and sleep but were not enhanced with hyperventilation or photic stimulation. Low dose VPA was initiated and seizures have been completely controlled for approximately 4 years. On entry to school, the patient was noted to have significant learning disability and ADHD symptoms and has been placed in special education (Figure 2, Table 1).
Figure 2. Electroencephalogram (EEG) of two sisters carrying the GAT-1(Val125Met) encoding mutation showing high amplitude burst of 3 Hz spike wave discharges and prominent occipital intermittent rhythmic delta activity (OIRDA).
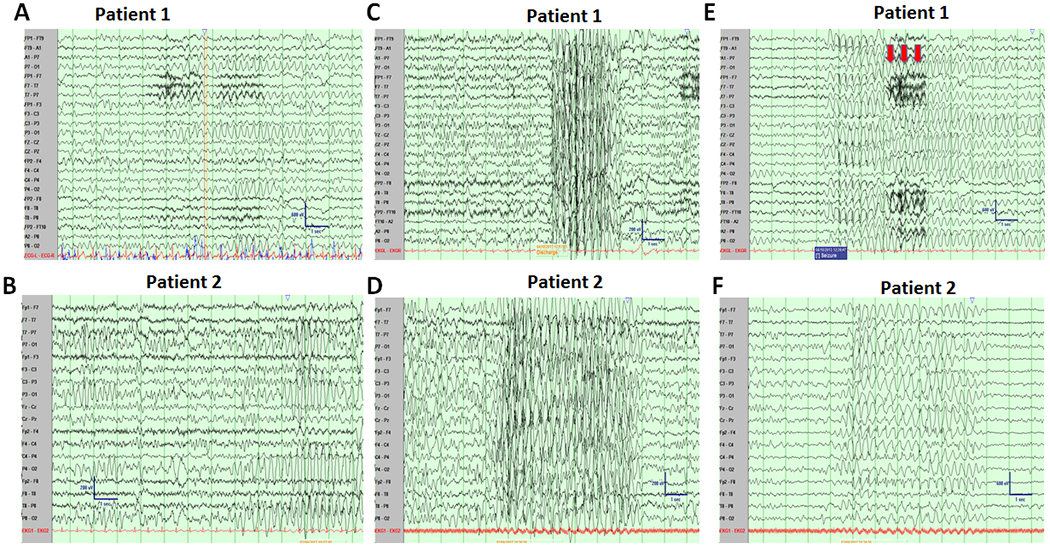
(A, B) Prominent bursts of Occipital Intermittent Rhythmic Delta Activity in the Proband (A, age 8) and Patient 2 (B, age 4) occurred throughout the waking state. (C, D). Video EEG recordings showed sudden burst of generalized high amplitude bursts of 3.0 Hz spike and slow waves in proband (C) and the half-sister (D). (E, F), Typical absence for proband was associated with a myoclonic jerk and loss of tone—noted by muscle artifact (red arrows), sensitivity 15 microvolts (E) and a typical abnormal spike wave discharges for patient 2 (D) arising out of sleep, which was asymptomatic. Sensitivity 15 microvolts.
Machine learning based protein structural modeling suggests that Val125Met mutation in GAT-1 protein reduces the mutant transporter stability
We then predicted the impact of the mutation on the transporter protein stability via several machine learning tools. Homology modelling of the Val125Met mutation in GAT-1 protein as shown in Figure 3 was conducted using I-TASSER [27] with homology template PDB ID 4m48. Residue valine that is mutated to methionine is colored red, which may trigger several conformational changes on GAT-1. Different from SLC6A1 mutations we reported before[5, 6], this mutation only adds sulfur into the sidechain, not causing drastic changes in polarity and charge, but making the protein less hydrophobic. These hydrophobicity changes may disturb the equilibrium of the transmembrane protein conformation, resulting in protein structure destabilizing. This destabilization hypothesis is also supported by predicting the ΔΔG of the mutation using machine learning-based protein structure stability prediction methods SDM [28], mCSM, DUET [29], INPS [30, 31], DynaMut [22] and MAESTROweb [21]. As indicated in Figure 3C and Supplementary Table 1 [21, 22, 29, 31, 32], nearly all the tools (six out of seven) predicted the V125M mutation destabilized the GAT-1 protein (Supplementary Table 1). Details in structural differences between the wildtype valine and mutated methionine were modelled by DynaMut interatomic interaction predictions. In addition, PASTA 2.0 [23] did not suggest any protein self-aggregation or co-aggregation from the perspective of energy changes.
Figure 3. Modeling of the mutant GAT-1(Val125Met) protein with machine learning tools.
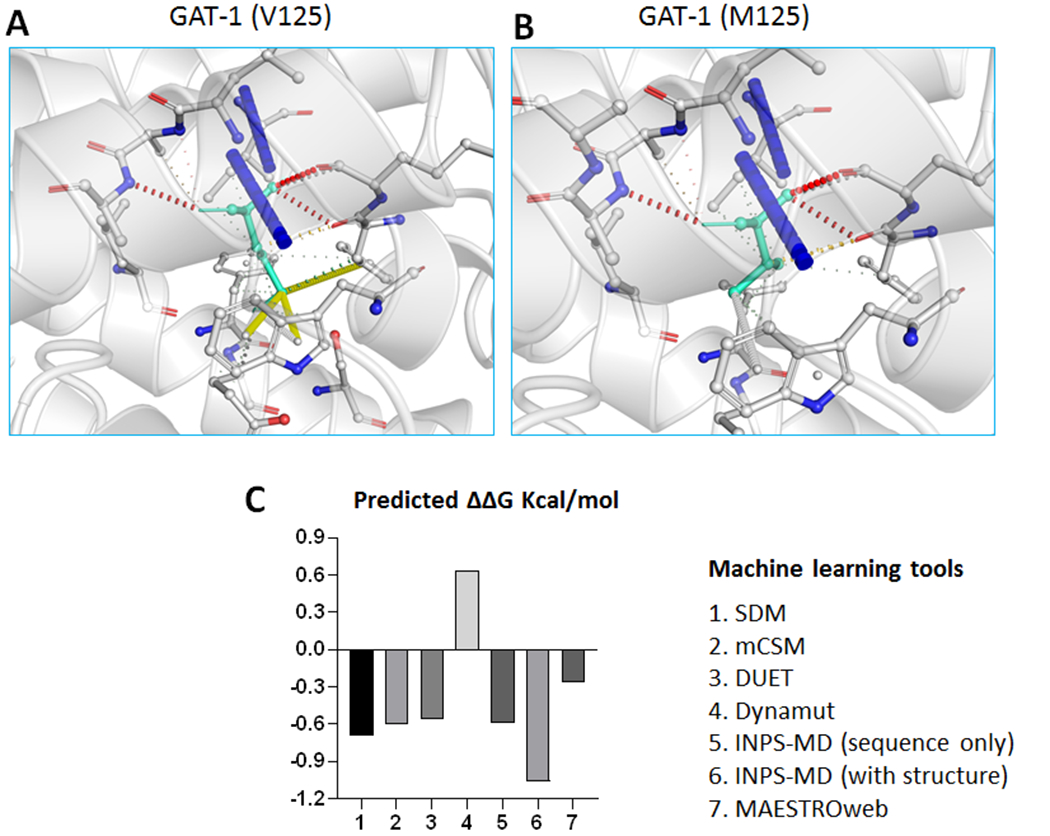
(A-B). Tertiary structures of both the wildtype (A) and Val125Met mutant (B) GAT-1 protein are predicted by I-TASSER and DynaMut. The valine at residue 125 is mutated to methionine, both highlighted in light green, alongside with the surrounding residues. The interatomic interactions were predicted by DynaMut, where halogen bonds are depicted in blue and hydrogen bonds are colored in red. The Val125Met mutation results in the addition of sulfur into the sidechain, not causing drastic changes in polarity and charge, but making the protein less hydrophobic. (C). Machine learning tools predicted ΔΔG (Kcal/mol) of the mutant GAT-1(Val125Met) protein. Bars in the positive direction are predicted as stabilizing while bars in the negative direction are predicted as destabilizing.
GAT-1(Val125Met) had impaired GABA reuptake in HEK293T, mouse astrocytes, and mouse neurons
We then determined the function of the wildtype and the mutant GAT-1 (Val125Met) in HEK cells, cultured mouse astrocytes and cortical neurons by radioactive 3H GABA uptake assay. The measurements in the mutant transporter were then normalized to the cells expressing the wildtype GAT-1YFP which was taken as 100%. Compared with the wildtype, the GAT-1(Val125Met)) had reduced 3H GABA uptake in HEK293T (wt = 100% vs 32.2 ± 3.1%) (Figure 4A), mouse cortical astrocytes (wt = 100% vs 65.40 ± 2%) (Figure 4B) and mouse neurons (wt = 100% vs 53.10 ± 3.4% (Figure 4C). The GAT-1(V125M) transport activity was similar to the activity of wildtype GAT-1 treated with GAT-1 inhibitors Cl-966 (100 μM) for 30 min in HEK293T, but in mouse astrocytes and neurons, the GAT-1(Val125Met)) activity was higher than the Cl-966 conditions. This is likely due to the activity from endogenous GAT-1 expressed in mouse cortical astrocytes and neurons.
Figure 4. The GABA reuptake function of the mutant GAT-1(Val125Met) transporter was reduced in non-neuronal cells and neurons.
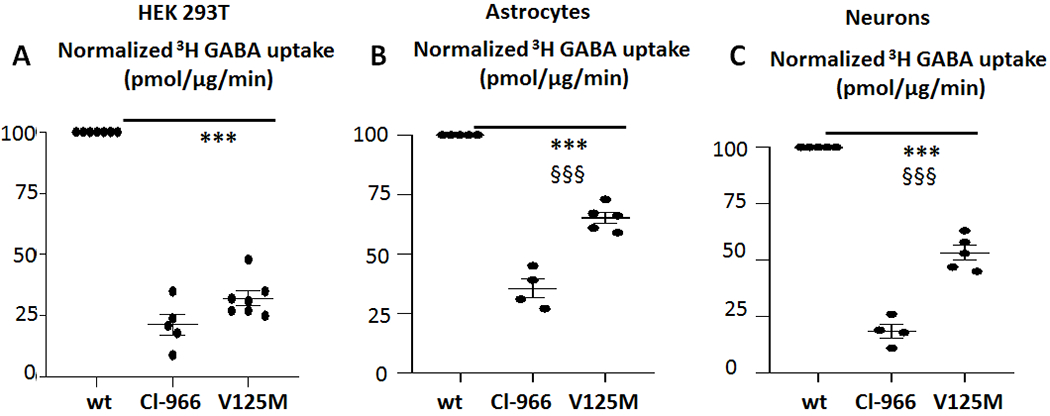
A-B. HEK293T cells or mouse cortical astrocytes were transfected with wildtype GAT-1YFP (wt), or the mutant GAT-1(Val125Met, V125M)YFP cDNAs (1μg per a 35mm2 dish) for 48 hrs before 3H radioactive GABA uptake assay. C. Mouse cortical neurons were transfected with the wildtype or the mutant GAT-1(Val125Met) cDNAs at day 7 in culture. 3H radioactive GABA uptake assay was performed after 8 days of transfection. GABA flux was measured after 30 min transport at room temperature. The influx of GABA, expressed in pmol/μg protein/min, was averaged from duplicates for each condition and for each transfection. The average counting was taken as n = 1. The untransfected condition was taken as baseline flux, which was subtracted from both the wildtype and the mutant conditions in HEK293T cells (A). The pmol/μg protein/min in the mutant was then normalized to the wildtype from each experiment, which was arbitrarily set as 100%. (***p < 0.01 vs. wt, n=4-7 different transfections). Cl-966 (100μm) was applied 30 min before preincubation and removed during preincubation. (One-sample t test. Values were expressed as mean ± S.E.M).
GAT-1(Val125Met) mutant transporter had reduced total and surface protein expression
Reduced GABA reuptake could be due to the reduced number of functional transporters on the cell surface. Based on our previous study, the mutant GAT-1(Gly234Ser) associated with Lennox-Gastaut syndrome[5] and GAT-1(Pro361Thr) associated with epilepsy and autism had reduced total protein expression [6]. Reduced total expression of the mutant transporters could reduce the cell surface expression and consequently impair the overall function of GAT-1. Altered protein stability and enhanced protein degradation due to protein misfolding are common phenomena caused by mutations in various genes. This has been demonstrated in multiple GABAA receptor mutations across multiple subunits [18] and are now emerging for GAT-1 mutants. We first determined the total expression of the mutant GAT-1(Val125Met) by transfecting HEK293T cells with YFP-tagged wildtype or mutant GAT-1 cDNAs (Figure 5A) for 48 hrs. The wildtype GAT-1YFP or the mutant GAT-1(Val125Met)YFP mainly migrated at 108 KDa, which is predicted for YFP-tagged GAT-1 and is consistent with previous findings [33, 34]. Compared to the wildtype, the GAT-1(Val125Met) had reduced total protein expression (wt=1, Val125Met = 0.63±0.04) for total (Figure 5A, C). Additionally, the GAT-1(V125M) also had reduced cell surface expression (wt=1, Val125Met=0.32 ± 0.013) (Figure 5B, D). This suggests the reduced cell surface protein expression was due to the reduced total protein expression.
Figure 5. Both the total and cell surface expression of the mutant GAT-1(Val125Met) transporter protein was reduced.
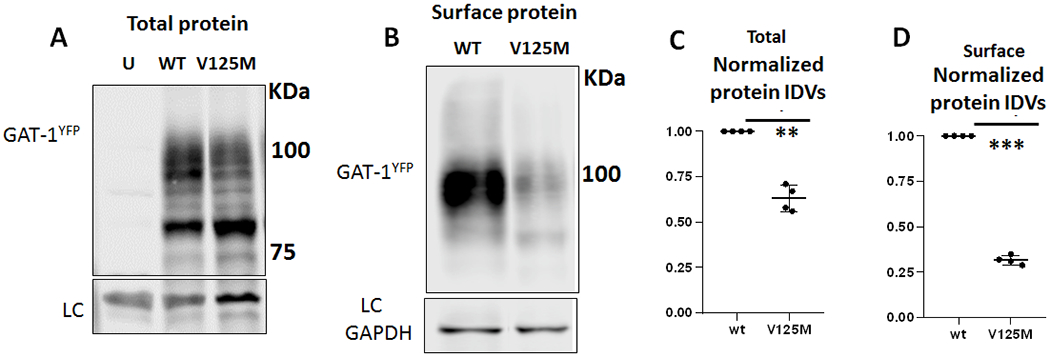
A, B. HEK293T cells were transfected with wildtype GAT-1YFP (wt), or the mutant GAT-1(Val125Met, V125M)YFP cDNAs (3μg/60 mm2) for 48 hrs. The cells (A) were either harvested directly after wash with PBS for total lysates or followed by cell surface biotinylation to isolate the cell surface bound proteins (B). The total lysates (A) or isolated surface protein (B) were then analyzed by SDS-PAGE. Membranes were immunoblotted with rabbit anti-GAT-1 (1:300). (C). The total protein integrated density values (IDVs) from the total lysates (C) or isolated cell surface protein (D) were measured. The abundance of the mutant GAT-1(Val125Met) transporter was normalized to the wildtype cells expressing GAT-1YFP. In C, the total protein abundance was measured by adding up all the bands between 90-110 KDa. The total protein IDVs of either the wildtype or the mutant was normalized to its loading control. The abundance of the mutant transporter was then normalized to the wildtype. (**p < 0.01; ***p < 0.001 vs. wt, n=4 different transfections, One-sample t test, Values were expressed as mean ± S.E.M).
GAT-1(Val125Met) mutant protein had increased endoplasmic reticulum retention in live HEK293T cells
We have previously identified that increased endoplasmic reticulum (ER) retention of mutant protein due to misfolding and glycosylation arrest [18, 35] is a common phenomenon for mutation across genes affecting GABAergic pathway. Those ER retention-prone mutant proteins can have either a higher or lower proportion of the total protein level compared with its wildtype counterpart [18, 35]. To evaluate the subcellular localization of GAT-1(Val125Met), we determined the intracellular localization of the mutant GAT-1(Val125Met) protein by coexpressing GAT-1YFP or GAT-1(Val125Met)YFP with an ER marker, ERCFP[35]. When compared to wildtype, the mutant GAT-1(Val125Met) had a stronger presence intracellularly, colocalizing with the ER marker (Figure 6A). The protein expression pattern was comparable to the expression pattern of the wildtype GAT-1 protein treated with ER stress inducer tunicamycin (10ug/ml for 16 hrs). The percent fluorescence signal of GAT-1 overlapping with ER marker ERCFP was higher in the mutant GAT-1(Val125Met) compared to wildtype (35.6 ± 2.9% for wt vs 75.68 ± 4.7% for Val125Met) (Figure 6B). The fluorescence signal overlapping with ER marker was higher in GAT-1(Val125Met) than cell expressing the wildtype treated with tunicamycin (54.2 ± 3.4%) (Figure 6B).
Figure 6. The mutant GAT-1(V125M) transporters were retained inside the endoplasmic reticulum in HEK 293T cells.
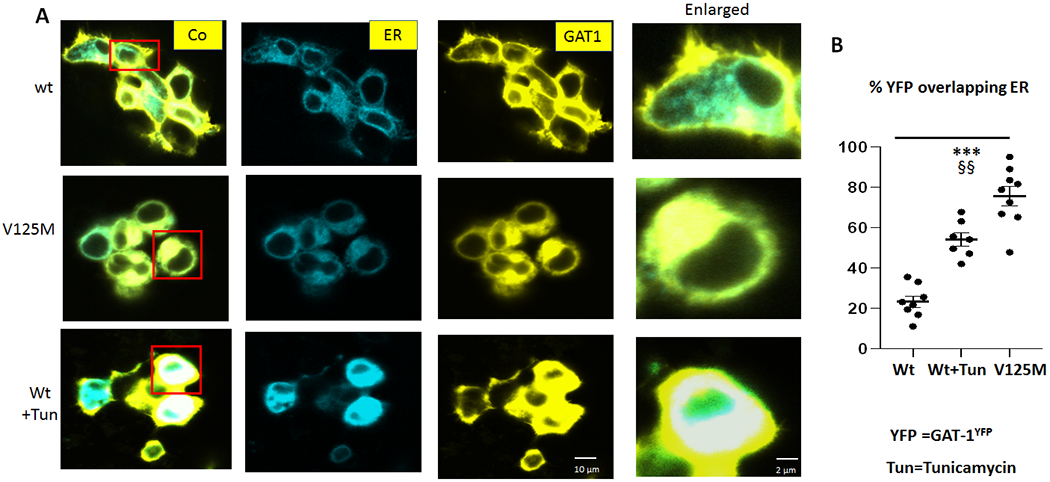
(A) HEK293T cells were transfected with wildtype GAT-1YFP or the mutant GAT-1(Val125Met, V125M)YFP with the pECFP-ER marker (ERCFP) at 2:1 ratio (2 μg:1μg cDNAs) for 48 hrs. Live cells were examined under a confocal microscopy with excitation at 458 nm for CFP, 514 nm for YFP. All images were single confocal sections averaged from 8 times to reduce noise, except when otherwise specified. (B) The GAT-1YFP fluorescence overlapping with ERCFP fluorescence was quantified by Metamorph with colocalization percentage. Tu stands for Tunicamycin (10 μg/ml) treated for 16 hrs. (***p < 0.001 V125M vs. wt, V125M §§ vs wt+Tun. N=7-8 representative fields from 4 different transfections. One-way analysis of variance (ANOVA) and Newman-Keuls test was used to determine significance compared to the wt condition and between mutation and wt+Tu. Values were expressed as mean ± S.E.M).
Discussion
We report on the first pedigree carrying an SLC6A1 mutation: p.Val125Met, a novel mutation associated with MAE (Doose syndrome) and ADHD.
SLC6A1(p.Val125Met) is a recurring mutation in a conserved amino acid of GAT-1 protein and this is the first pedigree despite numerous previous reports of de novo mutations in SLC6A1. This is also the first report of SLC6A1 mutation associated with ADHD although mutations in SLC6A1 have been associated with other neuropsychiatric conditions. Despite the phenotypic heterogeneity, MAE and ID are consistently to be reported as prominent phenotypes for SLC6A1 mutations [3, 7]. Here we report two sisters who are heterozygous for SLC6A1(p.Val125Met) mutation manifest MAE, further supporting the role of SLC6A1 mutations in MAE as originally reported [2]. This is also consistent with a previous observation that Val125Met is associated with MAE (https://www.ncbi.nlm.nih.gov/clinvar). The shared mother has no seizure history nor any other neuropsychiatric disorders and her saliva-based genetic test is negative for this variant, suggesting gonadic mosaicism. Both sisters exhibit clinical manifestations of ADHD, suggesting that ADHD is likely a strong comorbidity for SLC6A1-mediated disorders.
There exists intrafamilial phenotypical heterogeneity despite common clinical and electrographic features
Both sisters displayed MAE presenting with sudden falls and both had frequent bursts of OIRDA, a strong EEG correlate with absence seizure. However, they have different age onset of seizures and different response to VPA. The proband had first seizures at 1 year old while the sister had first seizures at age 4. Both sisters had 3Hz spike and wave and fronto-centrally dominant generalized epileptiform discharges. The proband had eyelid myoclonia and behavior arrest that is absent in the half-sibling. VPA treatment reduced the frequency and intensity of seizures but never fully controlled the events in the proband but achieved seizure freedom for the sister. However, both siblings have learning difficulty, suggesting impaired cognition may be independent of seizure control. Additionally, both sisters were diagnosed ADHD, expanding the phenotypic presentation of SLC6A1 mutation mediated disorders.
GAT-1 is expressed in both astrocytes and neurons and the mutation reduced GABA reuptake function in both astrocytes and neurons.
It is established that GAT-1 is expressed in both neurons and glial cells and regulates synapse function [36]. Loss-of-function of the astrocytic GAT-1 (but not GAT-3) enhances absence seizures, suggesting additional restraints to the thalamocortical neuron firing from astrocytic GAT-1[37]. There is no prior study on the impact of mutant proteins in astrocytes. Here we identified that the mutant GAT-1 had reduced function in astrocytes, similar to the effect in HEK293T cells and neurons. This is of particular importance given the fact that GAT-1 is exclusively located in astrocytes in thalamus in human and rodents [38] and the loss of GAT-1 function in astrocytes in thalamus may be the direct cause of the pathophysiology that gives rise to absence seizures.
Mutant GAT-1(Val125Met) transporter caused reduced total and surface protein expression due to reduced protein stability
The reduced GABA uptake is likely due to the reduced functional transporter number at the cell surface. The variation from valine 125 to methionine 125 in GAT-1 adds sulfur into the sidechain, which, while not causing drastically changes in polarity and charge of the protein, does make the protein less hydrophobic. These hydrophobicity changes may disturb the equilibrium of the transmembrane protein conformation and destabilize protein structure. The protein simulation data is supported by the biochemical assays as the total and surface GAT-1(Val125Met) transporter protein was reduced.
Mutant GAT-1(Val125Met) transporter was retained inside endoplasmic reticulum and subsequently subject to accelerated disposal
ER retention and enhanced degradation is likely a common molecular mechanism for many disease mutations including the GAT-1(Val125Met). This is based on our extensive evaluation on mutant GABAA receptor subunits associated with variable genetic epilepsy syndromes. Because GAT-1 is a transmembrane protein, it is likely that at least some mutations in GAT-1 cause protein instability and impair trafficking. We evaluated the subcellular colocalization of GAT-1(Val125Met) with coexpression of GAT-1YFP with an ER marker. We compared the expression pattern of the GAT-1(Val125Met) with the wildtype GAT-1 after treatment with an ER stress inducer, tunicamycin (10μg/ml). The GAT-1(Val125Met) expression pattern was highly colocalized with the ER marker. The findings were similar to the expression of the wildtype GAT-1 treated with tunicamycin, suggesting ER retention of the GAT-1(Val125Met) and reduced mutant GAT-1 presence at the cell surface. Our data indicate that the mutant GAT-1(Val125Met) transporter is subject to similar intracellular protein processing as many mutant GABAA receptor subunits and GAT-1(Gly234Ser) and GAT-1(Pro361Thr), due to a conserved protein quality control machinery inside cells [18, 35]. The steady state level of an ER retained mutant protein could be higher or lower than its wildtype counterparts, depending on the intrinsic properties of the mutant protein and the degradation rate of the mutant protein [39, 40] and the cellular capacity of protein degradation machinery. GAT-1(Val125Met) had reduced total and cell surface protein expression, indicating reduced protein stability and enhanced disposal of the mutant protein, and most of the synthesized mutant transporters resided inside the ER. This finding suggests GAT-1 is subject to the same post-translation protein modification as other membrane proteins. Consequently, a misfolded GAT-1 is likely to subject to enhanced degradation without further translocation to other cellular compartments or cell surface [25, 35]. Based on our previous comparison of different GABRG2 loss-of function mutations[24, 25, 41], it is possible that the mutant protein causes mild dominant negative suppression of the wildtype allele during protein folding, oligomerization and trafficking to the cell surface but this merits more extensive studies.
Compromised GAT-1(Val125Met) function in astrocytes could directly contribute to absence seizures
Our data indicate that GAT-1(Val125Met) substantially reduced GABA reuptake in HEK293T cells, neurons and astrocytes. In HEK293T cells the reduction was to the level of cells expressing the wildtype GAT-1 treated with GAT-1 inhibitor Cl-966. In mouse astrocytes and neurons, the mutant GAT-1(Val125Met) had a higher GABA uptake function than the cells treated with Cl-966. This is likely due to the activity of endogenous GAT-1 in astrocytes or neurons. Nevertheless, the cells expressing the mutant GAT-1(Val125Met) had reduced GABA reuptake comparing with the astrocytes or neurons expressing the wildtype GAT-1YFP. It has been established that GAT-1 is exclusively expressed in astrocytes in thalamus and cortico-thalamic-cortical circuitry is involved in absence seizures. The thalamus is a rhythmogenic structure responsible for generating and maintaining oscillatory activity underlying key brain functions such as sleep, sensation, perception and consciousness [42–44]. Thus, malfunctioning GAT-1 in thalamus could also contribute impaired attention besides seizures. It is likely the reduced function of GAT-1 (Val125met) can cause chronic accumulation of GABA, which downregulates the phasic GABA release and increase tonic inhibition based on studies from the GAT-1 knockout mice[45, 46]. Based on this study, regardless of the function of GAT-1(Val125Met) in neurons, malfunctioning astrocytic GAT-1 in thalamus could explain absence epilepsy and related comorbidities.
In summary, this study has reported the first pedigree carrying an SLC6A1 mutation resulting in disruption of a conserved residue valine 125 in GAT-1 and the common and differential clinical features of two affected siblings. We identified the molecular defects with a multidisciplinary approach including 3H GABA uptake assay and confocal microscopy in cells including astrocytes and neurons. The study provides a critical link for understanding SLC6A1 mutation and clinical phenotypes. As loss of GAT-1 function in astrocytes in thalamus could give rise to the key features such as 3-HZ abnormal discharges in absence seizures and the comorbid attention deficit as commonly observed in SLC6A1 mediated disorders [13, 47, 48].
Supplementary Material
Supplementary Video 1. Video of 360° rotation animation of GAT-1 protein model (green) with V125M(red) mutation.
Supplementary Table 1. Protein stability prediction on V125M mutation by machine learning methods
Highlights.
SLC6A1(p.Val125Met) was identified in two sisters but not in their shared mother.
EEGs in both sisters had high amplitude burst of 3 Hz spike wave discharges.
Val125Met mutation reduced protein stability predicted by machine learning tools.
The mutant GAT-1 (Val125Met) had reduced cell surface and total protein expression.
The mutant GAT-1 (Val125Met) had reduced activity with 3H GABA uptake assay.
Acknowledgements
We would like to thank the patients and their family who participated in this study for their cooperation.
Funding
The clinical study was from I.H and was supported by the University of Rochester Medical Center. The study on structural modeling was supported by the United States of America National Institutes of Health (grant No. R35-GM126985 to DX). The study on functional assay was conducted at Vanderbilt University Medical Center and supported by research grants from, SLC6A1 Connect and NINDS R01 082635 (to K.J.Q.). Imaging data were performed in part through the VUMC Cell Imaging Shared Resource.
The funders had no role in study design, data collection and analysis, and interpretation of data, decision to publish or preparation of the manuscript.
List of abbreviations
- GAT-1
GABA transporter 1
- ER
endoplasmic reticulum
- MAE
Myoclonic atonic epilepsy
- ADHD
attention deficit hyperactivity disorder
- OIRDA
occipital intermittent rhythmic delta activity
Footnotes
Competing interests
The authors declare that they are no competing interests.
Availability of supporting data
Any raw data of functional assay can be made available upon request.
Any clinical information can be made available upon request subject to approval by the appropriate ethical board.
Reference List
- (1).Roth FC, Draguhn A. GABA metabolism and transport: effects on synaptic efficacy. Neural Plast 2012;2012:805830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Carvill GL, McMahon JM, Schneider A, Zemel M, Myers CT, Saykally J, Nguyen J, Robbiano A, Zara F, Specchio N, Mecarelli O, Smith RL, Leventer RJ, Moller RS, Nikanorova M, Dimova P, Jordanova A, Petrou S, Helbig I, Striano P, Weckhuysen S, Berkovic SF, Scheffer IE, Mefford HC. Mutations in the GABA Transporter SLC6A1 Cause Epilepsy with Myoclonic-Atonic Seizures. Am J Hum Genet 2015. May 7;96(5):808–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Johannesen KM, Gardella E, Linnankivi T, Courage C, de Saint MA, Lehesjoki AE, Mignot C, Afenjar A, Lesca G, Abi-Warde MT, Chelly J, Piton A, Merritt JL, Rodan LH, Tan WH, Bird LM, Nespeca M, Gleeson JG, Yoo Y, Choi M, Chae JH, Czapansky-Beilman D, Reichert SC, Pendziwiat M, Verhoeven JS, Schelhaas HJ, Devinsky O, Christensen J, Specchio N, Trivisano M, Weber YG, Nava C, Keren B, Doummar D, Schaefer E, Hopkins S, Dubbs H, Shaw JE, Pisani L, Myers CT, Tang S, Tang S, Pal DK, Millichap JJ, Carvill GL, Helbig KL, Mecarelli O, Striano P, Helbig I, Rubboli G, Mefford HC, Moller RS. Defining the phenotypic spectrum of SLC6A1 mutations. Epilepsia 2018. Feb;59(2):389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Mattison KA, Butler KM, Inglis GAS, Dayan O, Boussidan H, Bhambhani V, Philbrook B, da SC, Alexander JJ, Kanner BI, Escayg A. SLC6A1 variants identified in epilepsy patients reduce gamma-aminobutyric acid transport. Epilepsia 2018. Sep;59(9):e135–e141. [DOI] [PubMed] [Google Scholar]
- (5).Cai K, Wang J, Eissman J, Wang J, Nwosu G, Shen W, Liang HC, Li XJ, Zhu HX, Yi YH, Song J, Xu D, Delpire E, Liao WP, Shi YW, Kang JQ. A missense mutation in SLC6A1 associated with Lennox-Gastaut syndrome impairs GABA transporter 1 protein trafficking and function. Exp Neurol 2019. Jun 6;320:112973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Wang J, Poliquin S, Mermer F, Eissman J, Delpire E, Wang J, Shen W, Cai K, Li BM, Li ZY, Xu D, Nwosu G, Flamm C, Liao WP, Shi YW, Kang JQ. Endoplasmic reticulum retention and degradation of a mutation in SLC6A1 associated with epilepsy and autism. Mol Brain 2020. May 12;13(1):76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Carvill GL, McMahon JM, Schneider A, Zemel M, Myers CT, Saykally J, Nguyen J, Robbiano A, Zara F, Specchio N, Mecarelli O, Smith RL, Leventer RJ, Moller RS, Nikanorova M, Dimova P, Jordanova A, Petrou S, Helbig I, Striano P, Weckhuysen S, Berkovic SF, Scheffer IE, Mefford HC. Mutations in the GABA Transporter SLC6A1 Cause Epilepsy with Myoclonic-Atonic Seizures. Am J Hum Genet 2015. May 7;96(5):808–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Durkin MM, Smith KE, Borden LA, Weinshank RL, Branchek TA, Gustafson EL. Localization of messenger RNAs encoding three GABA transporters in rat brain: an in situ hybridization study. Brain Res Mol Brain Res 1995. Oct;33(1):7–21. [DOI] [PubMed] [Google Scholar]
- (9).Kang JQ. Defects at the crossroads of GABAergic signaling in generalized genetic epilepsies. Epilepsy Res 2017. Aug 26;137:9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Cai K, Wang J, Eissman J, Wang J, Nwosu G, Shen W, Liang HC, Li XJ, Zhu HX, Yi YH, Song J, Xu D, Delpire E, Liao WP, Shi YW, Kang JQ. A missense mutation in SLC6A1 associated with Lennox-Gastaut syndrome impairs GABA transporter 1 protein trafficking and function. Exp Neurol 2019. Oct;320:112973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Masur D, Shinnar S, Cnaan A, Shinnar RC, Clark P, Wang J, Weiss EF, Hirtz DG, Glauser TA. Pretreatment cognitive deficits and treatment effects on attention in childhood absence epilepsy. Neurology 2013. Oct 29;81(18):1572–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Glauser TA, Cnaan A, Shinnar S, Hirtz DG, Dlugos D, Masur D, Clark PO, Capparelli EV, Adamson PC. Ethosuximide, valproic acid, and lamotrigine in childhood absence epilepsy. N Engl J Med 2010. Mar 4;362(9):790–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Gencpinar P, Kalay Z, Turgut S, Bozkurt O, Duman O, Ozel D, Haspolat S. Evaluation of Executive Functions in Patients With Childhood Absence Epilepsy. J Child Neurol 2016. Jun;31(7):824–30. [DOI] [PubMed] [Google Scholar]
- (14).Hermann B, Jones J, Dabbs K, Allen CA, Sheth R, Fine J, McMillan A, Seidenberg M. The frequency, complications and aetiology of ADHD in new onset paediatric epilepsy. Brain 2007. Dec;130(Pt 12):3135–48. [DOI] [PubMed] [Google Scholar]
- (15).Jones JE, Watson R, Sheth R, Caplan R, Koehn M, Seidenberg M, Hermann B. Psychiatric comorbidity in children with new onset epilepsy. Dev Med Child Neurol 2007. Jul;49(7):493–7. [DOI] [PubMed] [Google Scholar]
- (16).Posar A, Visconti P. Mild Phenotype Associated with SLC6A1 Gene Mutation: A Case Report with Literature Review. J Pediatr Neurosci 2019. Apr;14(2):100–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Kang JQ, Shen W, Lee M, Gallagher MJ, Macdonald RL. Slow degradation and aggregation in vitro of mutant GABAA receptor gamma2(Q351X) subunits associated with epilepsy. J Neurosci 2010. Oct 13;30(41):13895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Kang JQ, Shen W, Macdonald RL. Two molecular pathways (NMD and ERAD) contribute to a genetic epilepsy associated with the GABA(A) receptor GABRA1 PTC mutation, 975delC, S326fs328X. J Neurosci 2009. Mar 4;29(9):2833–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Keynan S, Suh YJ, Kanner BI, Rudnick G. Expression of a cloned gamma-aminobutyric acid transporter in mammalian cells. Biochemistry 1992. Feb 25;31(7):1974–9. [DOI] [PubMed] [Google Scholar]
- (20).Zhang Y I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 2008. Jan 23;9:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Laimer J, Hiebl-Flach J, Lengauer D, Lackner P. MAESTROweb: a web server for structure-based protein stability prediction. Bioinformatics 2016. May 1;32(9):1414–6. [DOI] [PubMed] [Google Scholar]
- (22).Rodrigues CH, Pires DE, Ascher DB. DynaMut: predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res 2018. Jul 2;46(W1):W350–W355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Walsh I, Seno F, Tosatto SC, Trovato A. PASTA 2.0: an improved server for protein aggregation prediction. Nucleic Acids Res 2014. Jul;42(Web Server issue):W301–W307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Warner TA, Shen W, Huang X, Liu Z, Macdonald RL, Kang JQ. DIfferential molecular and behavioral alterations in mouse models of GABRG2 haploinsufficiency versus dominant negative mutations associated with human epilepsy. Hum Mol Genet 2016. Jun 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Kang JQ, Shen W, Zhou C, Xu D, Macdonald RL. The human epilepsy mutation GABRG2(Q390X) causes chronic subunit accumulation and neurodegeneration. Nat Neurosci 2015. Jul;18(7):988–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Kang JQ, Macdonald RL. The GABAA receptor gamma2 subunit R43Q mutation linked to childhood absence epilepsy and febrile seizures causes retention of alpha1beta2gamma2S receptors in the endoplasmic reticulum. J Neurosci 2004. Oct 6;24(40):8672–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Zhang Y I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 2008. Jan 23;9:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Pandurangan AP, Ochoa-Montano B, Ascher DB, Blundell TL. SDM: a server for predicting effects of mutations on protein stability. Nucleic Acids Res 2017. Jul 3;45(W1):W229–W235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Pires DE, Ascher DB, Blundell TL. DUET: a server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res 2014. Jul;42(Web Server issue):W314–W319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Allen AS, Berkovic SF, Cossette P, Delanty N, Dlugos D, Eichler EE, Epstein MP, Glauser T, Goldstein DB, Han Y, Heinzen EL, Hitomi Y, Howell KB, Johnson MR, Kuzniecky R, Lowenstein DH, Lu YF, Madou MR, Marson AG, Mefford HC, Esmaeeli NS, O’Brien TJ, Ottman R, Petrovski S, Poduri A, Ruzzo EK, Scheffer IE, Sherr EH, Yuskaitis CJ, Abou-Khalil B, Alldredge BK, Bautista JF, Berkovic SF, Boro A, Cascino GD, Consalvo D, Crumrine P, Devinsky O, Dlugos D, Epstein MP, Fiol M, Fountain NB, French J, Friedman D, Geller EB, Glauser T, Glynn S, Haut SR, Hayward J, Helmers SL, Joshi S, Kanner A, Kirsch HE, Knowlton RC, Kossoff EH, Kuperman R, Kuzniecky R, Lowenstein DH, McGuire SM, Motika PV, Novotny EJ, Ottman R, Paolicchi JM, Parent JM, Park K, Poduri A, Scheffer IE, Shellhaas RA, Sherr EH, Shih JJ, Singh R, Sirven J, Smith MC, Sullivan J, Lin TL, Venkat A, Vining EP, Von Allmen GK, Weisenberg JL, Widdess-Walsh P, Winawer MR. De novo mutations in epileptic encephalopathies. Nature 2013. Sep 12;501(7466):217–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Savojardo C, Fariselli P, Martelli PL, Casadio R. INPS-MD: a web server to predict stability of protein variants from sequence and structure. Bioinformatics 2016. Aug 15;32(16):2542–4. [DOI] [PubMed] [Google Scholar]
- (32).Pires DE, Ascher DB, Blundell TL. mCSM: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 2014. Feb 1;30(3):335–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Bennett ER, Kanner BI. The membrane topology of GAT-1, a (Na+ + Cl−)-coupled gamma-aminobutyric acid transporter from rat brain. J Biol Chem 1997. Jan 10;272(2):1203–10. [DOI] [PubMed] [Google Scholar]
- (34).Cai G, Salonikidis PS, Fei J, Schwarz W, Schulein R, Reutter W, Fan H. The role of N-glycosylation in the stability, trafficking and GABA-uptake of GABA-transporter 1. Terminal N-glycans facilitate efficient GABA-uptake activity of the GABA transporter. FEBS J 2005. Apr;272(7):1625–38. [DOI] [PubMed] [Google Scholar]
- (35).Kang JQ, Shen W, Macdonald RL. The GABRG2 mutation, Q351X, associated with generalized epilepsy with febrile seizures plus, has both loss of function and dominant-negative suppression. J Neurosci 2009. Mar 4;29(9):2845–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Eulenburg V, Gomeza J. Neurotransmitter transporters expressed in glial cells as regulators of synapse function. Brain Res Rev 2010. May;63(1–2):103–12. [DOI] [PubMed] [Google Scholar]
- (37).Cope DW, Di GG, Fyson SJ, Orban G, Errington AC, Lorincz ML, Gould TM, Carter DA, Crunelli V. Enhanced tonic GABAA inhibition in typical absence epilepsy. Nat Med 2009. Dec;15(12):1392–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).De BS, Vitellaro-Zuccarello L, Brecha NC. Immunoreactivity for the GABA transporter-1 and GABA transporter-3 is restricted to astrocytes in the rat thalamus. A light and electron-microscopic immunolocalization. Neuroscience 1998. Apr;83(3):815–28. [DOI] [PubMed] [Google Scholar]
- (39).Kang JQ, Shen W, Lee M, Gallagher MJ, Macdonald RL. Slow degradation and aggregation in vitro of mutant GABAA receptor gamma2(Q351X) subunits associated with epilepsy. J Neurosci 2010. Oct 13;30(41):13895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Kang JQ, Shen W, Macdonald RL. Two molecular pathways (NMD and ERAD) contribute to a genetic epilepsy associated with the GABA(A) receptor GABRA1 PTC mutation, 975delC, S326fs328X. J Neurosci 2009. Mar 4;29(9):2833–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Kang JQ, Shen W, Macdonald RL. Trafficking-deficient mutant GABRG2 subunit amount may modify epilepsy phenotype. Ann Neurol 2013. Oct;74(4):547–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Gent TC, Bassetti C, Adamantidis AR. Sleep-wake control and the thalamus. Curr Opin Neurobiol 2018. Oct;52:188–97. [DOI] [PubMed] [Google Scholar]
- (43).Gent TC, Bandarabadi M, Herrera CG, Adamantidis AR. Thalamic dual control of sleep and wakefulness. Nat Neurosci 2018. Jul;21(7):974–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Avanzini G, Panzica F, de CM. The role of the thalamus in vigilance and epileptogenic mechanisms. Clin Neurophysiol 2000. Sep;111 Suppl 2:S19–S26. [DOI] [PubMed] [Google Scholar]
- (45).Jensen K, Chiu CS, Sokolova I, Lester HA, Mody I. GABA transporter-1 (GAT1)-deficient mice: differential tonic activation of GABAA versus GABAB receptors in the hippocampus. J Neurophysiol 2003. Oct;90(4):2690–701. [DOI] [PubMed] [Google Scholar]
- (46).Chiu CS, Brickley S, Jensen K, Southwell A, Mckinney S, Cull-Candy S, Mody I, Lester HA. GABA transporter deficiency causes tremor, ataxia, nervousness, and increased GABA-induced tonic conductance in cerebellum. J Neurosci 2005. Mar 23;25(12):3234–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Lee S, Kruglikov I, Huang ZJ, Fishell G, Rudy B. A disinhibitory circuit mediates motor integration in the somatosensory cortex. Nat Neurosci 2013. Nov;16(11):1662–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Dlugos D, Shinnar S, Cnaan A, Hu F, Moshe S, Mizrahi E, Masur D, Sogawa Y, Le Pichon JB, Levine C, Hirtz D, Clark P, Adamson PC, Glauser T. Pretreatment EEG in childhood absence epilepsy: associations with attention and treatment outcome. Neurology 2013. Jul 9;81(2):150–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Video 1. Video of 360° rotation animation of GAT-1 protein model (green) with V125M(red) mutation.
Supplementary Table 1. Protein stability prediction on V125M mutation by machine learning methods
Data Availability Statement
Any raw data of functional assay can be made available upon request.
Any clinical information can be made available upon request subject to approval by the appropriate ethical board.