

Abstract
Purpose:
BRCA1 or BRCA2 mutated cancers (BRCAmut) have intrinsic sensitivity to PARP inhibitors due to deficiency in homologous recombination-mediated DNA repair. There are similarities between BRCAmut and BRCAwt ovarian and basal-like breast cancers. This phase I study determined the recommended phase II dose (RP2D) and preliminary efficacy of the PARP inhibitor, veliparib (ABT-888), in these patients.
Patients and Methods:
Patients (n=98) were dosed with veliparib 50–500 mg twice daily (BID). The BRCAmut cohort (n=70) contained predominantly ovarian (53%) and breast (23%) cancers; the BRCAwt cohort (n=28) consisted primarily of breast cancer (86%). The MTD, DLT, adverse events, PK, PD, and clinical response were assessed.
Results:
DLTs were grade 3 nausea/vomiting at 400 mg BID in a BRCAmut carrier, grade 2 seizure at 400 mg BID in a patient with BRCAwt cancer, and grade 2 seizure at 500 mg BID in a BRCAmut carrier. Common toxicities included nausea (65%), fatigue (45%), and lymphopenia (38%). Grade 3/4 toxicities were rare (highest lymphopenia at 15%). Overall response rate (ORR) was 23% (95% CI 13%−35%) in BRCAmut overall, and 37% (95% CI 21%−55%) at 400 mg BID and above. In BRCAwt, ORR was 8% (95% CI 1%−26%), and clinical benefit rate was 16% (95% CI 4%−36%), reflecting prolonged stable disease in some patients. PK was linear with dose and was correlated with response and nausea.
Conclusions:
Continuous veliparib is safe and tolerable. The RP2D was 400 mg BID. There is evidence of clinical activity of veliparib in patients with BRCAmut and BRCAwt cancers.
Keywords: veliparib, phase I, pharmacokinetics, pharmacodynamics, solid tumors, PARP inhibitor, DNA damage, BRCA1, BRCA2, ovarian cancer, triple-negative breast cancer
1. INTRODUCTION
The poly (ADP-ribose) polymerase (PARP) family of enzymes is important for a number of cellular processes, including several DNA repair pathways. PARP1 detects both single- and double-strand DNA breaks, while PARP2 dimerizes with PARP1 to play a role in base excision DNA repair [1]. In addition, PARP1 plays a critical role in stabilizing replication forks [2,3]. Inhibition of PARP is an important treatment strategy for cancers harboring deficiencies in BRCA1 or BRCA2 (BRCA) based on data showing “synthetic lethality” between BRCA deficiency and PARP inhibition [4,5].
In addition to BRCA1 and BRCA2 mutation carriers, there is a larger population of breast (particularly ER/PR and HER2 negative; “triple- negative”) and ovarian cancer patients whose cancers have a BRCA-like phenotype with homologous recombination HR deficiency [6] due to somatic BRCA mutations or deletions, BRCA1 promoter methylation, or deficiencies in other DNA repair genes. Although these mechanisms were not formally tested in this phase 1 study, a cohort of BRCA wild-type patients with triple-negative breast cancer (TNBC) or platinum-refractory ovarian cancer was included as we hypothesized that these patients might be similarly sensitive to single-agent PARP inhibition.
Currently, four PARP inhibitors (olaparib, rucaparib, niraparib, and talazoparib) have received FDA approval for various indications. Compared to olaparib and niraparib, veliparib is a somewhat less potent PARP catalytic inhibitor and a less potent DNA-PARP trapper [7]. While there is clear demonstration of efficacy in BRCA mutant cancers, the optimal use of PARP inhibitors, either as monotherapy or in combination with cytotoxic chemotherapy, remains under investigation. Among the questions yet to be resolved are: (1) whether there are factors in addition to mutations in HR pathway genes such as BRCA1 and BRCA2 that predict response or resistance to PARP inhibitors; and (2) whether PARP inhibitors can be successfully combined with existing chemotherapy agents to enhance efficacy.
Veliparib (ABT-888) is an oral, potent, small molecule inhibitor of PARP1 and PARP2 shown to be a potentiator of DNA damaging agents in various preclinical cancer models [8,9]. A first-in-man phase 0 trial demonstrated >90% PARP 1/2 inhibition in paired tumor biopsies and companion PBMCs 3–6 hours after a 25- or 50-mg dose, and partial recovery of enzymatic activity at 24 hours [10]. This phase I trial, which was previously reported in abstract form [11], aimed to determine the dose-limiting toxicities (DLT), maximum tolerated dose (MTD), and recommended phase II dose (RP2D) of veliparib given as a single agent to patients with advanced cancers with germline BRCA mutation or BRCA wildtype ovarian cancer or TNBC. The secondary endpoints of this study were to evaluate safety and tolerability, pharmacokinetics (PK), pharmacodynamics (PD), and preliminary clinical response. In addition, pre- and post-treatment biopsies from an expansion cohort in patients with germline BRCAmut cancers were evaluated to elucidate biological determinants of response or resistance to veliparib in this patient population.
2. METHODS
2.1. Study Population
This phase I trial (NCI 8282, NCT00892736) enrolled 2 cohorts of patients. The first cohort (referred to as BRCAmut; n=70) included patients with a documented germline mutation in BRCA1 or BRCA2 (Myriad Genetic Laboratories) and a metastatic, BRCA-related malignancy (breast, ovarian, pancreatic, prostate, etc). The second cohort (referred to as BRCAwt; n=28) included patients with TNBC or platinum-refractory ovarian cancer without a known germline BRCA mutation. Patients with unknown BRCA mutation status were screened with BRCAPRO [12] and if the likelihood of mutation was ≥ 20%, were required to undergo BRCA gene testing for cohort allocation.
ECOG PS ≤2 was required as part of the eligibility criteria, as were adequate hepatic, renal and marrow function. Patients with stable and treated CNS metastases were allowed. Patients with history of seizure disorder were excluded, as seizures were seen at higher doses in animal studies. There was no limit to prior therapies, including prior PARP inhibitor or previous platinum-based chemotherapy.
2.2. Study Design
This was an NCI-CTEP-sponsored, multicenter trial performed at six NCI-designated cancer centers, approved by the respective institutional review boards and ethics committees. This study followed a standard 3+3 dose-escalation schema [13]. Veliparib was given orally on a continuous schedule, using a 28-day cycle. There were nine dose levels ranging from an initial dose level (DL) of 50 mg BID (DL1) to a maximum dose of 500 mg BID (DL9) (Table 1).
Table 1.
Dose-escalation and dose limiting toxicities (DLT).
| Dose Level | Veliparib (mg AM/PM) | Enrolled mut/wt | Evaluable mut/wt | DLT mt/wt | Grade – DLT** |
|---|---|---|---|---|---|
| 1 | 50/50 | 7/3 | 7/3 | 1*/0 | Grade 2 thrombocytopenia, later deemed related to disease progression, necessitated hold of veliparib > 2 weeks |
| 2 | 100/50 | 3/3 | 3/3 | ||
| 3 | 100/100 | 3/3 | 3/3 | ||
| 4 | 150/100 | 3/3 | 3/3 | ||
| 5 | 150/150 | 3/3 | 3/3 | ||
| 6 | 200/200 | 4/3 | 3/3 | ||
| 7 | 300/300 | 6/3 | 4/3 | ||
| 8 | 400/400 | 34/7 | 27/6 | 1/1 | G3 nausea / G2 seizure |
| 9 | 500/500 | 7/0 | 6/0 | 1/0 | G2 seizure |
DLT experienced before start of parallel enrollment of BRCAmt and BRCAwt cohorts.
Per protocol, any seizure occurring in a patient on this study was considered a DLT, unless in the setting of CNS metastases.
2.3. Safety Assessments
Dose-limiting toxicity (DLT) was defined as a significant adverse event occurring during cycle 1 considered to be at least possibly drug related, and could be any grade ≥3 non-easily correctable non-hematologic or grade 4 hematologic toxicity (neutropenia being prolonged or febrile), as defined by the Common Terminology Criteria for Adverse Events version 4.0. Toxicity resulting in holding drug for greater than 2 weeks, regardless of attribution or grade, was to be considered a DLT.
Maximum tolerated dose (MTD) was defined as the highest dose level at which ≤ 1/6 patients had a DLT in cycle 1. The recommended phase II dose (RP2D) was defined as the dose at or below the MTD where therapy was determined to be tolerable. After the first DLT, dose escalation was conducted separately in the BRCAmut and the BRCAwt cohorts because, at the time of study inception, it was not known if there would be differential toxicity in those with germline BRCA mutations. A dose-expansion cohort at the RP2D enrolled BRCA mutation carriers and included mandatory research biopsies.
2.4. Tumor Response Assessment
Radiographic assessments were performed every 2 cycles, and the objective response rate (ORR) was determined according to the Response Evaluation Criteria in Solid Tumors (RECIST) 1.0 [14]. Clinical benefit was defined as having complete response, partial response, or stable disease for ≥6 cycles. Patients were considered evaluable for response if they underwent disease re-evaluation or physical examination after receiving at least 1 cycle of therapy or had overt clinical evidence of disease progression.
2.5. Translational Correlative Science
All patients enrolled in the dose escalation had peripheral blood collected to assess PAR as pharmacodynamic (PD) endpoint using a validated assay as described [10], and in-depth PK studies were performed with a validated assay [15]. Archival tumor samples were assessed for BRCA1 protein levels by immunohistochemistry (IHC), basal markers (cytokeratin 5 and EGFR by IHC), BRCA1 promoter methylation, and integrity of the BRCA1/Fanconi Anemia pathway by staining for replication-associated FANCD2 foci in situ. Details are described in the Supplementary File.
After the dose escalation, there was a cohort expansion (n=25) at the RP2D exclusively for patients with a known germline BRCA mutation, with mandatory fresh tumor tissue biopsies prior to study treatment and on Cycle 2, Day 1, 4±1 hours after veliparib administration (Supplementary File). The biopsy specimens were analyzed for PAR and γ-H2AX using validated assays, for DNA repair proteins (RAD51, 53BP1, PARP1) using IHC, and BRCAmut cases were evaluated for BRCA reversion mutations as described [16].
2.6. Statistical Analysis
Patients were considered evaluable for DLT if they received at least 75% of their scheduled doses in cycle 1 or if they experienced a DLT. Response and toxicity rates were compared using Fisher’s exact tests. Statistical methods for PK data are described in the Supplementary File. The association between biomarker and response was assessed by Fisher’s exact tests. Survival data was analyzed using the Kaplan-Meier method. SAS software (version 9.4) was used to analyze demographic, adverse events, efficacy, and exposure-response data.
3. RESULTS
3.1. Patients
The study activated in April 2009 and patients enrolled between April 2009 and May 2014. Of the 98 patients who received at least one dose of veliparib, the majority had a germline BRCA mutation (Suppl.Table 1), 86 were evaluable for DLT (59 BRCAmut; 27 BRCAwt), and 87 were evaluable for response (62 BRCAmut; 25 BRCAwt). Eleven patients (8 BRCAmut; 3 BRCAwt) were not evaluable for response because of consent withdrawal, removal from study secondary to toxicity, or inability to swallow the study drug. In the BRCAmut cohort, the range of cycles administered was 1–29, with a mean of 4.8 and median of 2.5. In the BRCAwt cohort, the range of cycles administered was 1–59, with a mean of 5.2 and median of 2.
3.2. DLTs, MTD, and RP2D
The first DLT, grade 2 thrombocytopenia and other complications relating to disease progression, was observed at DL1 for which veliparib was held for >2 weeks. While this was considered a DLT per protocol, the event was confounded by complications of disease progression, and ultimately judged unlikely to be related to study drug. Dose escalation was continued in both cohorts. DLTs were subsequently experienced by two patients at DL8 (one BRCAmut carrier with grade 3 nausea and grade 3 vomiting, one patient with BRCAwt cancer with grade 2 seizure) and one patient at DL9 (BRCAmut carrier with grade 2 seizure). The MTD was 500 mg BID (DL9). Formal criteria for RP2D were not met. There was frequent nausea seen at the higher dose levels often necessitating dose reduction, although it did not formally qualify as a DLT. Seizure activity was seen in preclinical studies, so this was a side effect of particular interest. Of note, one patient had a grade 2 seizure in a dose level higher than the RP2D. Both patients with episodes of seizures had imaging of the brain performed which found no evidence of metastatic disease. After lorazepam and levetiracetam treatment, symptoms improved in both patients. Ultimately, it was determined that the RP2D was 400 mg BID (DL8) based on general toxicity and tolerability.
3.3. Adverse Event Profile
Overall, continuous single-agent veliparib was well-tolerated (Table 2 and Table 3). The incidence of grade 3/4 toxicity was 24% (95% CI 15%−36%) in the BRCAmut group, with the most frequent grade 3/4 toxicity being decreased lymphocyte count (14%). In the BRCAwt group, the incidence of grade 3/4 toxicity was 29% (95% CI 13%−49%) and the most frequent adverse event was decreased lymphocyte count in 18%. There was no significant difference in grade 3/4 toxicity rates between BRCAmut and BRCAwt groups (p=0.8). The most common all-grade adverse events were nausea (65%), fatigue (45%), and decreased lymphocyte count (38%). It is notable that there were 6 patients who came off study after experiencing non-dose limiting nausea (2 in Cycle 1 of DL7, 2 in Cycle 1 of DL8, 1 in Cycle 2 of DL8 and 1 in Cycle 1 of DL9). In general, standard anti-emetics in various dosages and regimens were not consistently effective, and the nausea responded best to dose reduction. There was no signal of secondary malignancies with only one patient with melanoma in situ of the ear, likely unrelated, and no documentation of MDS or leukemia.
Table 2.
Grade 3/4 toxicities by cohort and dose level (number of patients (%)).
| Toxicity (CTCAEv4.0 Term) | Total | DL1 | DL2 | DL3 | DL4 | DL5 | DL6 | DL7 | DL8 | DL9 |
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| BRCAmut cohort | 70 | 7 | 3 | 3 | 3 | 3 | 4 | 6 | 34 | 7 |
|
| ||||||||||
| Any Grade 3/4 toxicity | 17 (24) | 1 (14) | 1 (33) | 0 | 0 | 0 | 0 | 2 (33) | 11 (32) | 2 (29) |
|
| ||||||||||
| Grade 3 toxicity | ||||||||||
| Any Grade 3 toxicity | 15 (21) | 1 (14) | 0 | 0 | 0 | 0 | 0 | 2 (33) | 11 (32) | 1 (14) |
| Lymphocyte count decreased | 9 (13) | 0 | 0 | 0 | 0 | 0 | 0 | 1 (17) | 7 (21) | 1 (14) |
| Thromboembolic event | 2 (3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (6) | 0 |
| Nausea | 2 (3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (6) | 0 |
| Abdominal pain | 2 (3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (6) | 0 |
| Anemia | 1 (1) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (3) | 0 |
| Alkaline phosphatase increased | 1 (1) | 1 (14) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Aspartate aminotransferase increased | 1 (1) | 1 (14) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Appendicitis | 1 (1) | 0 | 0 | 0 | 0 | 0 | 0 | 1 (17) | 0 | 0 |
| Headache | 1 (1) | 0 | 0 | 0 | 0 | 0 | 0 | 1 (17) | 0 | 0 |
| Dehydration | 1 (1) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (3) | 0 |
| Urinary tract infection | 1 (1) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (3) | 0 |
| Fatigue | 1 (1) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (3) | 0 |
| Hypophosphatemia | 1 (1) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (3) | 0 |
| Grade 4 toxicity | ||||||||||
| Any Grade 4 toxicity | 3 (4) | 0 | 1 (33) | 0 | 0 | 0 | 0 | 0 | 1 (3) | 1 (14) |
| Lymphocyte count decreased | 1 (1) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (3) | 0 |
| Thromboembolic event | 1 (1) | 0 | 1 (33) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Anemia | 1 (1) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (14) |
| Dyspnea | 1 (1) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (14) |
| Platelet count decreased | 1 (1) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (14) |
|
| ||||||||||
| BRCAwt cohort | 28 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 7 | 0 |
|
| ||||||||||
| Grade 3 toxicity | ||||||||||
| Any Grade 3 toxicity | 8 (29) | 0 | 1 (33) | 0 | 2 (67) | 0 | 2 (67) | 0 | 3 (43) | - |
| Lymphocyte count decreased | 5 (18) | 0 | 1 (33) | 0 | 2 (67) | 0 | 1 (33) | 0 | 1 (14) | - |
| Basilic vein thrombosis | 1 (4) | 0 | 0 | 0 | 0 | 0 | 1 (33) | 0 | 0 | - |
| Dyspnea | 1 (4) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (14) | - |
| Fatigue | 1 (4) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (14) | - |
| Hypophosphatemia | 1 (4) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (14) | - |
At dose levels 3 and 5, no grade 3/4 toxicities were observed.
Table 3.
Most common all-grade toxicities attributed to veliparib, BRCAmut and BRCAwt combined (number of patients (%)).
| Toxicity (CTCAEv4.0 Term) | Any Grade | Grade 1 | Grade 2 | Grade 3 | Grade 4 |
|---|---|---|---|---|---|
|
| |||||
| Any adverse event | 92 (94) | 86 (88) | 54 (55) | 30 (31) | 3 (3) |
| Nausea | 64 (65) | 42 (43) | 20 (20) | 2 (2) | 0 (0) |
| Fatigue | 44 (45) | 33 (34) | 9 (9) | 2 (2) | 0 (0) |
| Lymphocyte count decreased | 37 (38) | 7 (7) | 15 (15) | 14 (14) | 1 (1) |
| Vomiting | 32 (33) | 25 (26) | 7 (7) | 0 (0) | 0 (0) |
| Anemia | 31 (32) | 19 (19) | 10 (10) | 1 (1) | 1 (1) |
| White blood cell decreased | 27 (28) | 20 (20) | 7 (7) | 0 (0) | 0 (0) |
| Anorexia | 20 (20) | 17 (17) | 3 (3) | 0 (0) | 0 (0) |
| Diarrhea | 19 (19) | 16 (16) | 3 (3) | 0 (0) | 0 (0) |
| Platelet count decreased | 18 (18) | 14 (14) | 3 (3) | 0 (0) | 1 (1) |
| Neutrophil count decreased | 17 (17) | 13 (13) | 4 (4) | 0 (0) | 0 (0) |
| Dysgeusia | 16 (16) | 16 (16) | 0 (0) | 0 (0) | 0 (0) |
| Hyperglycemia | 10 (10) | 9 (9) | 1 (1) | 0 (0) | 0 (0) |
| Aspartate aminotransferase increased | 9 (9) | 8 (8) | 0 (0) | 1 (1) | 0 (0) |
| Constipation | 9 (9) | 8 (8) | 1 (1) | 0 (0) | 0 (0) |
| Dizziness | 9 (9) | 8 (8) | 1 (1) | 0 (0) | 0 (0) |
| Headache | 9 (9) | 7 (7) | 1 (1) | 1 (1) | 0 (0) |
| Dry mouth | 8 (8) | 8 (8) | 0 (0) | 0 (0) | 0 (0) |
| Hypophosphatemia | 8 (8) | 2 (2) | 4 (4) | 2 (2) | 0 (0) |
| Hyponatremia | 7 (7) | 7 (7) | 0 (0) | 0 (0) | 0 (0) |
| Insomnia | 7 (7) | 6 (6) | 1 (1) | 0 (0) | 0 (0) |
| Abdominal pain | 6 (6) | 3 (3) | 1 (1) | 2 (2) | 0 (0) |
| Alkaline phosphatase increased | 6 (6) | 3 (3) | 2 (2) | 1 (1) | 0 (0) |
| Dyspepsia | 6 (6) | 6 (6) | 0 (0) | 0 (0) | 0 (0) |
| Dyspnea | 5 (5) | 2 (2) | 1 (1) | 1 (1) | 1 (1) |
| Myalgia | 5 (5) | 4 (4) | 1 (1) | 0 (0) | 0 (0) |
3.4. Efficacy
There were 62 evaluable BRCAmut carriers (Table 4, Figure 1, and Suppl.Figure 1). Evidence of stable disease was observed at DL2 (100 mg every morning, 50 mg every afternoon) and beyond, with objective responses first observed at DL7 (300 mg BID). The overall ORR was 23% (95% CI 13%−35%), and the ORR at the RP2D and beyond was 37% (95% CI 21%−55%). CBR was 42% (95% CI 29%−55%) including all dose levels, and 51% (95% CI 34%−69%) at the RP2D and beyond. The median progression-free survival (PFS) in the BRCAmut cohort was 3.1 months (95% CI 1.8–5.9 months) (Suppl.Figure 1). All ovarian and peritoneal cancer patients had received prior platinum-based chemotherapy, and 6 of 15 breast cancer patients had prior platinum-based chemotherapy. Three of the 4 breast cancer patients with a response (DL7–8) were platinum-naïve.
Table 4.
Best responses in BRCAmut and BRCAwt cohorts.
| BRCAmut | BRCAwt | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||
| #Evaluable | CR | PR | SD | PD | ORR (%) | CBR (%)* | #Evaluable | PR | SD | PD | ORR (%) | CBR* (%) | |
|
| |||||||||||||
| All Patients | 62 | 1 | 13 | 20 | 28 | 14/62 (23%) | 26/62 (42%) | 25 | 2 | 7 | 16 | 2/25 (8%) | 4/25 (16%) |
|
| |||||||||||||
| By Primary Tumor | |||||||||||||
| Ovarian | 33 | 0 | 6 | 10 | 17 | 6/33 (18%) | 14/33 (42%) | 3 | 0 | 1 | 2 | 0/3 (0%) | 0/3 (0%) |
| Breast | 15 | 0 | 4 | 4 | 7 | 4/15 (27%) | 5/15 (33%) | 22 | 2 | 6 | 14 | 2/22 (9%) | 4/22 (18%) |
| Fallopian Tube | 4 | 0 | 1 | 2 | 1 | 1/4 (25%) | 2/4 (50%) | ||||||
| Peritoneal | 3 | 1 | 0 | 2 | 0 | 1/3 (33%) | 1/3 (33%) | ||||||
| Prostate | 3 | 0 | 2 | 1 | 0 | 2/3 (67%) | 3/3 (100%) | ||||||
| Pancreatic | 1 | 0 | 0 | 0 | 1 | 0/1 (0%) | 0/1 (0%) | ||||||
| Endometrium | 1 | 0 | 0 | 1 | 0 | 0/1 (0%) | 1/1 (100%) | ||||||
| Kidney | 1 | 0 | 0 | 0 | 1 | 0/1 (0%) | 0/1 (0%) | ||||||
| Unknown | 1 | 0 | 0 | 0 | 1 | 0/1 (0%) | 0/1 (0%) | ||||||
|
| |||||||||||||
| By Dose Level | |||||||||||||
| DL1 | 7 | 0 | 0 | 0 | 7 | 0/7 (0%) | 0/7 (0%) | 3 | 1 | 0 | 2 | 1/3 (33%) | 1/3 (33%) |
| DL2 | 3 | 0 | 0 | 2 | 1 | 0/3 (0%) | 2/3 (67%) | 3 | 0 | 1 | 2 | 0/3 (0%) | 0/3 (0%) |
| DL3 | 3 | 0 | 0 | 1 | 2 | 0/3 (0%) | 1/3 (33%) | 3 | 1 | 0 | 2 | 1/3 (33%) | 1/3 (33%) |
| DL4 | 3 | 0 | 0 | 2 | 1 | 0/3 (0%) | 1/3 (33%) | 3 | 0 | 0 | 3 | 0/3 (0%) | 0/3 (0%) |
| DL5 | 3 | 0 | 0 | 1 | 2 | 0/3 (0%) | 0/3 (0%) | 3 | 0 | 1 | 2 | 0/3 (0%) | 0/3 (0%) |
| DL6 | 4 | 0 | 0 | 3 | 1 | 0/4 (0%) | 2/4 (50%) | 3 | 0 | 2 | 1 | 0/3 (0%) | 1/3 (33%) |
| DL7 | 4 | 0 | 1 | 1 | 2 | 1/4 (25%) | 2/4 (50%) | 2 | 0 | 1 | 1 | 0/2 (0%) | 1/2 (50%) |
| DL8 | 30 | 0 | 12 | 8 | 10 | 12/30 (40%) | 16/30 (53%) | 5 | 0 | 2 | 3 | 0/5 (0%) | 0/5 (0%) |
| DL9 | 5 | 1 | 0 | 2 | 2 | 1/5 (20%) | 2/5 (40%) | ||||||
CBR defined as CR, PR, or SD ≥ 6 cycles
Figure 1.
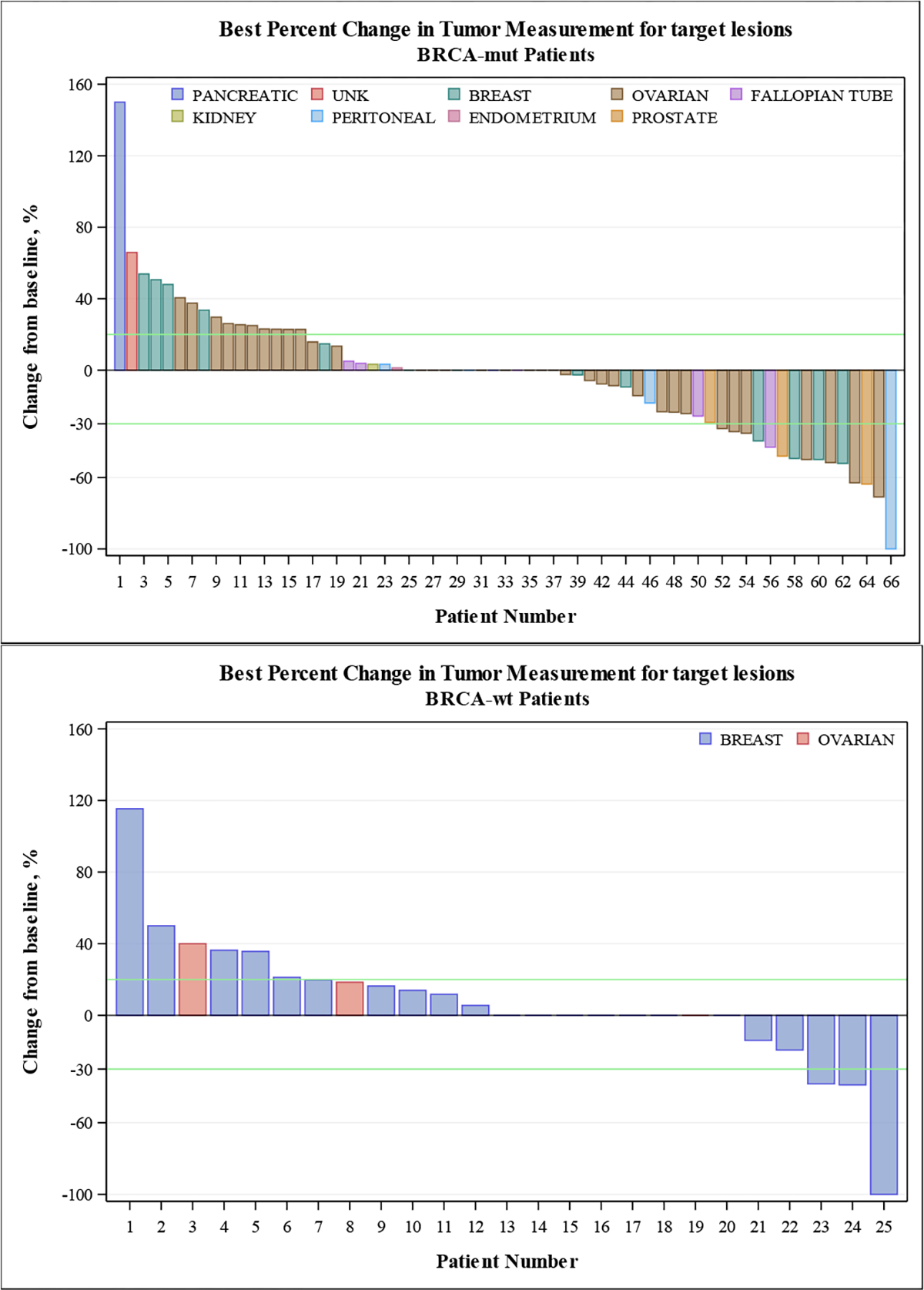
Percent change in tumor measurement from baseline for target lesions for BRCAmut subjects).
Of the 25 evaluable patients with BRCAwt cancer, most had breast cancer (88%) (Table 4, Figure 1, and Suppl.Figure 1). Amongst BRCAwt breast cancers, 14 of 22 (64%) were platinum-naïve. ORR was 8% (95% CI 1%−26%), and CBR was 16% (95% CI 4%−36%). Both rates were lower compared to the BRCAmut cohort (p=0.14 and 0.03 respectively). In contrast to the BRCAmut cohort, objective responses were observed at DL1 and DL3. The median PFS in this cohort was 1.8 months (95% CI 1.5–3.1 months) (Suppl.Figure 1). Of the patients with TNBC, 1 of 11 had stable disease for more than 6 months. This patient (1–7-51) represents an exceptional responder with stable disease on single-agent veliparib for 59 cycles (at the time of data cut-off) after 13 cycles of combined treatment with carboplatin, paclitaxel, and veliparib on NCT00535119[17]. This patient discontinued study therapy after nine years of treatment with stable, minimal disease.
3.5. Translational Correlative Science
3.5.1. Pharmacokinetics
Pharmacokinetic data were available for 67 patients (Suppl.Table 2, Suppl.Table 3, and Suppl.Figure 2). Population pharmacokinetic analysis of these data in aggregate have been previously reported [18], while here we present the first non-compartmental analysis of this dataset. Dose proportionality assessment for veliparib D1 Cmax resulted in a coefficient of 1.066 (95% CI 0.960–1.17; 90% CI 0.978–1.15). Dose linearity assessment for D1 AUC0-inf resulted in a coefficient of 1.059 (95% CI 0.949–1.17; 90% CI 0.967–1.15). The accumulation indices (geometric mean and standard deviation) for Cmax of 1.24 (1.37) and AUC0–8 of 1.30 (1.29) were as expected based on the theoretical accumulation index of 1.34 (1.12) calculated from the D1 half-lives. Similarly, the primary, active metabolite M8 accumulation indices for Cmax of 1.51 (1.34) and AUC0–8 of 1.59 (1.41) were as expected based on the theoretical accumulation index of 1.51 (1.25) calculated from the D1 half-lives. The day 1 M8/ABT-888 metabolic ratio was 0.11 (1.8) and 0.21 (1.9) for Cmax and AUC0-inf, respectively. Exposure-response relationship assessment of day 1 veliparib Cmax and AUC (Figure 2C,D and Suppl.Figure 3) showed increased exposure across response categories from progressive disease (PD) to stable disease (SD), partial response (PR) and complete response (CR), with statistical significance of PD vs SD/PR/CR (Cmax p=0.049, n=58; AUC p=0.019, n=56) by Wilcoxon’s non-parametric test. Within the BRCAmut population, the significance for Cmax was lost while for AUC it was retained (Cmax p=0.114, n=34; AUC p=0.08, n=32). Exposure-toxicity relationship assessment of day 1 veliparib Cmax and AUC (Figure 2A,B and Suppl.Figure 4) showed no relationship with overall toxicity, but statistical significance of grade 0–1 vs grade 2+ nausea (Cmax p=0.003, n=67; AUC p=0.0004, n=65).
Figure 2.
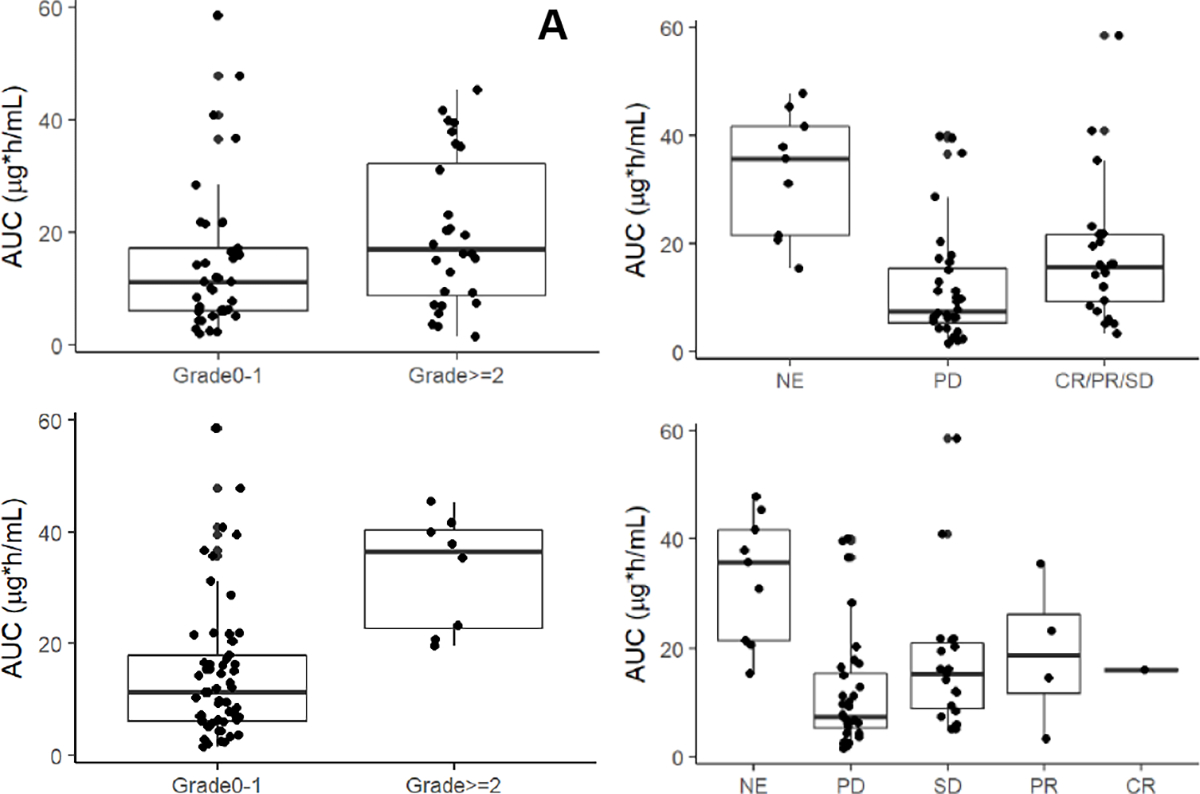
Exposure-response relationship of day 1 veliparib AUC (A) by cycle 1 any toxicity grade 0–1 vs grade 2 and up (no significance); (B) by cycle 1 nausea grade 0–1 vs grade 2 and up (p=0.0004, n=65; per Wilcoxon non-parametric test); (C) by progressive disease (PD) vs stable disease (SD), partial response (PR) and complete response (CR) (p=0.019, n=56; per Wilcoxon non-parametric test); and (D) by individual response categories (patients not evaluable for response are labelled NE).
3.5.2. PAR levels
PBMC PAR levels were evaluated in 45 patients covering all dose levels (Suppl.Figure 5). In general, there was evidence of decreased PAR levels after administration of veliparib, consistent with target inhibition at the doses administered. PAR levels were decreased at 2 hours and remained decreased at 24 hours for most samples tested, particularly at higher dose levels. At DL8, treatment reduced tumor PAR level was ≥99% in the one evaluable patient.
3.5.3. Exploratory analyses of archival tissue for BRCA1 promoter methylation, BRCA1/Fanconi anemia pathway integrity, cytokeratin 5, and EGFR
Archival tissue was available for 48 of the patients (49%; 34 BRCAmut; 14 BRCAwt). Methylation status was investigated by pyrosequencing and methylation specific PCR (MSP) (Suppl.Table 4). Tumors from 12 patients demonstrated BRCA1 promoter methylation by pyrosequencing (12/48, 25%), while six samples demonstrated BRCA1 promoter methylation by MSP (6/43, 14%), three of which were overlapping with the pyrosequencing methylation data (1–7-51, 3–1-09, 4–8-57), with a total concordance of 77%. None of these three patients had a germline BRCA mutation; BRCA1 was lost in one case (1–7-51; IHC data). With regard to response: 1–7-51 had SD, 3–1-09 had PD and 4–8-57 was non-evaluable. The small number of methylated samples precluded further correlative analysis.
Previous studies suggested that FANCD2 foci are formed during normal replication but are not seen in BRCAmut cancer cells because BRCA1 and BRCA2 are upstream of FANCD2 in this pathway [19]. To assess whether the BRCA-Fanconi Anemia (FA) repair pathway was intact and could predict sensitivity to PARP inhibition, the presence of FANCD2 foci was determined in the archival tumor specimens [19]. Among the 48 patients, information on FANCD2 foci was available for 43 (29 BRCAmut;14 BRCAwt). FANCD2 foci were absent in 37.9% of BRCAmut tumors versus 42.9% of BRCAwt cancers. This difference was not significant, and our results do not confirm that absence of FANCD2 foci is a read-out for presumed absence of BRCA1 or BRCA2. Response (CR, PR or SD; evaluable patients only) was demonstrated in 50.0% of FANCD2-negative-BRCAmut cases, 33.3% of FANCD2-negative-BRCAwt cases, 73.3% of FANCD2-positive-BRCAmut cases, and 66.7% of FANCD2-positive-BRCAwt cases. This suggests that FANCD2 status is not a good predictor of response.
Based on data supporting overlapping phenotype between basal-like breast cancer determined by IHC, microarray or genomic sequencing and BRCAmut breast cancer [20], we examined whether the basal breast cancer markers, cytokeratin 5 and EGFR, could be used to identify basal-like TNBC with a BRCA-like phenotype [6] in 20 breast cancer patients (8 BRCAmut; 12 BRCAwt/TNBC). Among the 12 BRCAwt/TNBC breast cancers evaluated, 5 (41.7%) had both, 4 (33.3%) had one, and 3 (25.0%) had no basal marker present. In contrast, among the 8 BRCAmut breast cancers tested, 4 (50.0%) had both and the others (50%) had no basal markers present. In the BRCAwt group with both basal markers present, there were 3 (60.0%) patients with PD and 2 (40%) SD, while among those with none of the basal markers present, 2 had SD as best response while the other was non-evaluable.
3.5.4. BRCAmut biopsy cohort
Of the 25 biopsy cohort patients treated at the RP2D (DL8), 4 had no adequate biopsy specimens, 11 had adequate pre-treatment biopsies only and 10 had both adequate pre- and post-treatment biopsies as determined by touch prep. Of the 11 patients with only pre-treatment biopsies, 3 patients were off study prior to the post-treatment biopsy time point and 4 patients, 3 of which were responders to therapy, did not have adequate specimens for analysis on the post-treatment biopsies. These post-treatment biopsies, although few, provided an opportunity to assess potential mechanisms that result in persistent BRCAmut cancer in the face of veliparib treatment.
Secondary somatic reversion mutations in BRCA have been described as a resistance mechanism to platinum-based chemotherapy and to PARP inhibitors [21]. These are secondary mutations that restore the open reading frame, leading to restoration of BRCA function. These mutations have been reported in 28% of platinum-resistant ovarian cancers in BRCA mutation carriers [16]. We postulated that reversion mutations may lead to PARP inhibitor resistance in the patients treated on this trial as well. In the biopsy expansion cohort, there were 30 neoplastic samples from 20 BRCA mutation carriers (19 pre-treatment and 11 post-treatment) analyzed for the presence of reversion mutations. All tumor samples had the germline mutation that the patient was known to have and 19 of 20 had LOH of the wild-type BRCA allele. There were no clear reversion mutations, prompting us to search for a basis of resistance that is independent of reversion mutations.
Based on preclinical studies suggesting that cells can become PARP inhibitor resistant by downregulation of PARP1 [22,7], loss of 53BP1 in BRCA1-mutated cancer [23], or upregulation of RAD51 [24], these proteins were assessed in biopsies from the expansion cohort using IHC (Supplementary File and Suppl.Figure 6). A wide range of PARP1 expression was observed in pretreatment samples (Suppl.Figure 6A, B). Although the results are limited by the small number of samples, there was no obvious relationship between response and pretreatment PARP1 staining, as summarized by H-score (Suppl.Figure 6A) or percentage of cells with low (0 or 1+) staining (Suppl.Figure 6B). According to the PARP trapping hypothesis [7], cancer cells that persist after PARP inhibitor treatment might be resistant because of low PARP1 expression. In the present study, comparison of paired samples failed to show consistent decreases in PARP1 in neoplastic cells after treatment (Suppl.Figure 6C). Instead, the PARP1 H-score decreased by >50 in two paired cancers and increased by >50 in three pairs and did not correlate with response.
53BP1 staining displayed a similar wide range of staining intensities (Suppl.Figure 6D). In contrast to recent results in a different PARP inhibitor trial [23], there was no obvious relationship between response and pretreatment 53BP1 expression. This lack of correlation persisted even when analysis was limited to the BRCA1-mutant subset of cancers. There was no relationship between response and pretreatment RAD51 expression (Fig. 6E). Although RAD51 overexpression is associated with PARP inhibitor resistance [24], we observed that RAD51 staining was highly similar in tumor cells before and after treatment (r = 0.9, P =0.011); and there was no suggestion that tumor cells overexpressing RAD51 had been selected during treatment (Suppl.Figure 6F).
Nuclear γ-H2AX staining in tumor from 10 cases, 4 of which were pre-post biopsy pairs, revealed minimal treatment-induced changes without consistent direction.
4. DISCUSSION
Single-agent veliparib was well-tolerated and demonstrated anti-tumor activity in a BRCAmut population at an RP2D of 400 mg BID, while minimal activity was also seen in BRCAwt patients. The most relevant side effect was low-grade nausea, which responded best to dose reductions.
Veliparib monotherapy toxicities are consistent with those of other PARP inhibitors [25–27]. Hematological toxicities are a very common class effect of PARP inhibitors with anemia being the most common. In three phase 3 maintenance trials, grade 3 or 4 anemia was slightly higher for niraparib (25%), followed by rucaparib (19%) and olaparib (19%). In our study, while all-grade anemia was consistent with those of other PARP inhibitors, grade 3 or 4 anemia was only seen in 6% of patients. Unlike other PARP inhibitors, the most frequent hematologic AE in our study was a decrease in lymphocytes (38%).
This single agent trial was started at about the same time as several other veliparib combination trials and explored more and higher doses, and involved numerous correlative components, in part explaining its reporting after several veliparib combination trial have been published. This allows more comprehensive comparison with other trials. Veliparib demonstrated less toxicity in combination with chemotherapy than the other PARP inhibitors, which generally enhance chemotherapy induced myelosuppression limiting the dose or treatment duration of PARP inhibitors and/or chemotherapy [28–31]. One potential advantage with veliparib, therefore, is the ability to use it in combination with a number of other chemotherapeutic regimens [32–34]. The rationale for combining PARP inhibitors with platinum chemotherapy is based on the absence of intact homologous recombination DNA repair due to BRCA dysfunction, which increases sensitivity to both agents. Recent data from a phase II trial (NCT02595905) demonstrated efficacy with the addition of veliparib (300mg BID) to cisplatin which significantly improved PFS and showed a trend towards improved OS for BRCA-like advanced TNBC as well as tolerability with continuous daily dosing of veliparib [35]. According to new results from a phase III trial in patients with ovarian cancer (NCT02470585) in previously untreated stage III or IV high-grade serous ovarian cancer, the frontline regimen of chemotherapy plus veliparib induction therapy followed by veliparib maintenance resulted in significantly longer progression-free survival when compared with chemotherapy plus placebo with placebo maintenance [36]. The observed toxicities were consistent with the known safety profile of veliparib in both combination and maintenance phases. Recent findings from a phase III trial (NCT02163694) showed that the addition of veliparib to carboplatin plus paclitaxel with continuation of veliparib monotherapy at intensified dose and schedule if chemotherapy was withdrawn prior to disease progression led to improved PFS in BRCAmut patients with hormone receptor positive breast cancer and in patients with TNBC [37]. The overall toxicity profile was not substantially different between treatment arms. Veliparib can also be combined with carboplatin using continuous therapeutic daily dosing. Several phase 1 studies showed that daily continuous dosing of olaparib with carboplatin was not tolerable [38,30,31,39], and intensified hematologic toxicity resulting in significant dose reductions [38] and schedule delays [31]. However, veliparib (150 mg BID daily) in combination with weekly carboplatin and paclitaxel was well tolerated with an acceptable safety profile and demonstrated promising anti-tumor activity in a phase 1 study in triple-negative breast cancer [40]. Additionally, in a multicenter phase 2 trial, single-agent veliparib (400 mg BID daily) followed by veliparib (150 mg BID) plus carboplatin at disease progression also showed that safety and efficacy are encouraging in BRCA-associated metastatic breast cancer [41].
The efficacy results seen in this phase I study, with an ORR of 37% and CBR of 51% at dose levels corresponding to the RP2D and beyond, are comparable to those reported in phase I studies with other PARP inhibitors. In particular, response rates of approximately 40% have been reported near the MTDs for cancers associated with BRCA mutations [42–45]. The clinical activity in our overall ovarian and breast cancer patient population is also similar to the 26% response rate in a phase II single-agent veliparib study in ovarian cancer [46].
Our study included a large proportion of patients without BRCA mutations allowing an evaluation of the difference between the BRCAmut and BRCAwt populations. In an early study with olaparib, it was demonstrated that 24% of patients with sporadic ovarian but not patients with sporadic breast cancer showed objective responses [43]. While the findings from our study are relatively similar, we did demonstrate stable disease in a small proportion of breast cancer (n=6) patients who did not have BRCA mutations on germline analysis. In this study, somatic testing was not performed, so it remains possible that some responses were associated with somatic BRCA mutations. One of the six responders with stable disease did not have archival tissue submitted for correlative studies. Of the remaining five responders, FANCD2 foci were absent in tumors of one patient and loss of BRCA1 immunostaining was found in two patients. Patients with sporadic breast and ovarian cancers might derive greater benefit from combining PARP inhibitors with chemotherapy compared to PARP inhibitor alone. This study demonstrated an increase in responses seen at the higher dose levels, consistent with a steep dose response effect as has been described for other PARP inhibitors such as olaparib and niraparib [47]. We started dose escalation at 50 mg based on data from the phase 0 study, and observed response at that first dose level; however, overwhelmingly objective responses and prolonged clinical benefit were seen at doses above 300 mg BID [10].
The values for veliparib pharmacokinetic parameters calculated non-compartmentally in the current study (Cl/F 15.9 (1.40) L/h, Vss/F 123 (1.36) L, t½ 5.9 (1.3) h) are similar to those previously reported (Cl/F 20.9 L/h, V/F 173 L, t½ 6.1 h) in a population model [48], as well as reported (Cl/F 17.3 L/h, V/F 147 L, t½ 4.0 h) in our previous population modeling of the current dataset [18], and veliparib pharmacokinetics reported in combination with cyclophosphamide, vinorelbine, temozolomide, bendamustine, gemcitabine, carboplatin and paclitaxel [49,34,50–52,17,53]. The notable outlier of veliparib PK is in combination with liposomal doxorubicin, where apparent clearance was much lower than in our dataset [54]. Based on both Cmax and AUC0-inf of day 1, we could not reject the null hypothesis of dose proportionality of veliparib over the range of dose levels studied (95% CI of the coefficient included 1). Based on the stringent bioequivalence-derived criteria proposed by Smith et al. [55], we could also not declare dose proportionality (90% CI of the coefficient was not contained within the range of 0.903–1.097). However, these stringent criteria have been deemed impractically strict when applied over a large dose range, and our data indeed meets more lenient criteria proposed subsequently [56] (90% CI of the coefficient was easily contained within the range of 0.699–1.301). Our data, therefore, support veliparib dose proportionality over the dose range of 50–500 mg. We did not observe any unexpected changes in veliparib PK between day 1 and day 15, suggesting there is no time-dependent effect of veliparib dosing on any of its PK parameters. We did find significant relationships between both veliparib Cmax and AUC and response and nausea. The few patients not evaluable for response appeared to have a relatively high exposure, potentially predisposing to toxicity resulting in discontinuation of therapy and inability to derive benefit.
To identify biological features of the cancers that might predict veliparib response, we performed a number of correlative studies in this phase I trial. Decreases in PBMC and tumor PAR levels after veliparib dosing, proved target engagement, while limited tumor γ-H2AX data did not suggest DNA damage after 1 cycle of treatment. It may well be that the clinical responses observed after PARP inhibition in the absence of concomitant cytotoxic therapy is partly associated with many client proteins whose functions is modulated by PAR-ylation [9].
The mandatory biopsy cohort suffered from low biopsy yield (69%), even with onsite pathologic analysis using a touch prep of the specimen. We also learned that while 4 cores were taken, not all contained tumor or could be analyzed. This number is similar to what has been reported in the NCI literature with 60% adequate biopsy yield [57]. In a follow-up study [40] where veliparib was combined with weekly paclitaxel and carboplatin with predominantly breast cancer patients enrolled, the biopsy yield was higher (86%). In our hands, the image-guided biopsies in ovarian cancer patients with carcinomatous implants were low yield, which may be due to their deep location surrounded by large vessels and/or other organs. Iin future, caution should be taken when conducting correlative analyses in this patient population.
Potential resistance mechanisms for PARP inhibitors are under active investigation, and it is postulated that there are similar resistance mechanisms to platinum-based chemotherapy. With both platinum drugs and PARP inhibitors, there is evidence that BRCA reversion mutations can occur, rendering therapy ineffective. In this study, no reversion mutations were identified, likely due to the early timing of the biopsy. More recent data suggest that reversion mutations in circulating tumor DNA (ctDNA) correlate with response to the PARP inhibitor rucaparib [21], and ctDNA may identify subclonal reversion mutations that might not be present in a small needle biopsy.
Low expression of 53BP1 protein has been reported to restore homologous recombination and contribute to PARP inhibitor resistance even in the face of persistent BRCA1 deficiency [58]. Our assays performed in the context of a single-agent trial of the PARP inhibitor ABT-767 demonstrated a negative correlation between 53BP1 expression and the percentage of tumor shrinkage in recurrent BRCA1-mutant ovarian cancer [23]. In contrast, we did not observe a correlation between 53BP1 expression and response in the present trial. It is important to note, however, that the present study included both breast and ovarian cancer. In addition, the endpoint assessed in our study (response vs. no response) was different from the endpoint assayed in the previous trial (% tumor shrinkage). Further studies are required to determine whether expression of 53BP1 and/or other cellular proteins in the same pathway [58] might provide useful information in ovarian cancer patients receiving single-agent PARP inhibitor and/or mechanistic insight into failure of tumors to respond despite the presence of HR defects.
Rigorous analysis of methylation through the use of two orthogonal approaches identified only three samples with methylated BRCA1 by both assays. Samples shown to be methylated by pyrosequencing did not show methylation by MSP and vice versa, potentially also due to false positive results with incomplete conversion of DNA in the bisulfite reaction, as previously reported [59].
5. CONCLUSION
In summary, our study shows that single agent veliparib can be used safely and tolerably at a continuous schedule of 400 mg BID, and that this treatment is associated with clinical activity both in patients with BRCAmut cancers and BRCAwt basal-like breast cancers. Target engagement was observed, and PK exposure correlated with both response and nausea, while none of the other biomarker studies yielded significant relationships. Significant advances have been made in the clinical development of PARP inhibitors for the treatment of patients with BRCA mutations. These agents represent personalized therapy for cancers that have underlying defects in homologous recombination-mediated DNA repair. Since the inception of this study, olaparib, rucaparib, and niraparib have been FDA-approved for ovarian cancers and olaparib and talazoparib have been FDA-approved for germline BRCAmut breast cancer in various settings. There are several ongoing phase 3 trials focused on breast (NCT03150576, NCT02032823) and ovarian (NCT02282020, NCT02446600, NCT02502266, NCT02855944, NCT02655016) cancer. While there is clear demonstration of efficacy in the BRCAmut population, the optimal use of PARP inhibitors, either as monotherapy or in combination with cytotoxic chemotherapy, as well as its potential role as maintenance therapy after chemotherapy is evolving. In addition, the exact role of these agents in the context of BRCAwt cancers remains unclear. With the expansion of PARP inhibitor indications and their incorporation into earlier lines of treatment, further studies are also needed to characterize the mechanisms of resistance that is emerging.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to dedicate this manuscript to the memory of Dr. Merrill Egorin who played a key role in the initial conception and design of this trial. He served as a great mentor in medicine, science, and life, and he is truly missed by those who carried on this work.
We would also like to acknowledge the patients and their families, many of whom traveled a great distance to be a part of this clinical trial, as well as the clinical investigators, research nurses, study coordinators, and clinical research staff who played an important role in the conduct of this trial.
This study was supported in part by NCI grants U01CA099168 (including an ARRA supplement), U24CA247643, and UM1CA186690. This project used the UPMC Hillman Cancer Center (HCC) Cancer Pharmacokinetics and Pharmacodynamics Facility (CPPF) Cancer Biostatistics Facility (CBF) and was supported in part by award P30CA047904. The project described was supported by the National Institutes of Health through Grant Number UL1TR001857. Support was also received from the ASCO Cancer Foundation, the Judith Lese Foundation, the Cancer Fighting Princess, and a BCRF Career Development Award. Correlative studies were supported in part through R01 CA190423 to S.H.K., E.M.S., D.W.V. and A.E.W.H. This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract Number HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
CONFLICT OF INTEREST
Conflict of Interest: Jan Beumer received research support from AbbVie and has consulted as expert witness on behalf of Pfizer and Spectrum Pharmaceuticals. Shannon Puhalla has received research support from AbbVie, Pfizer, Lilly, Novartis, Incyte, Covance-Bayer, AstraZeneca, Genentech, Medivation and has been a consultant for AbbVie, MedImmune, Celldex, Puma, Pfizer, AstraZeneca, Esai, and Nanostring.
Ethical approval: All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. This trial was registered under ClinicalTrials.gov Identifier: NCT01154426.
Informed consent: Informed consent was obtained from all individual participants included in the study.
REFERENCES
- 1.Schreiber V, Ame JC, Dolle P, Schultz I, Rinaldi B, Fraulob V, Menissier-de Murcia J, de Murcia G (2002) Poly(ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision DNA repair in association with PARP-1 and XRCC1. The Journal of biological chemistry 277 (25):23028–23036. doi: 10.1074/jbc.M202390200 [DOI] [PubMed] [Google Scholar]
- 2.Berti M, Ray Chaudhuri A, Thangavel S, Gomathinayagam S, Kenig S, Vujanovic M, Odreman F, Glatter T, Graziano S, Mendoza-Maldonado R, Marino F, Lucic B, Biasin V, Gstaiger M, Aebersold R, Sidorova JM, Monnat RJ Jr., Lopes M, Vindigni A (2013) Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nature structural & molecular biology 20 (3):347–354. doi: 10.1038/nsmb.2501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ, John S, Day A, Crespo AV, Shen B, Starnes LM, de Ruiter JR, Daniel JA, Konstantinopoulos PA, Cortez D, Cantor SB, Fernandez-Capetillo O, Ge K, Jonkers J, Rottenberg S, Sharan SK, Nussenzweig A (2016) Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 535 (7612):382–387. doi: 10.1038/nature18325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T (2005) Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434 (7035):913–917. doi: 10.1038/nature03443 [DOI] [PubMed] [Google Scholar]
- 5.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A (2005) Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434 (7035):917–921. doi: 10.1038/nature03445 [DOI] [PubMed] [Google Scholar]
- 6.Turner N, Tutt A, Ashworth A (2004) Hallmarks of ‘BRCAness’ in sporadic cancers. Nature reviews Cancer 4 (10):814–819. doi: 10.1038/nrc1457 [DOI] [PubMed] [Google Scholar]
- 7.Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, Ji J, Takeda S, Pommier Y (2012) Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer research 72 (21):5588–5599. doi: 10.1158/0008-5472.CAN-12-2753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Donawho CK, Luo Y, Luo Y, Penning TD, Bauch JL, Bouska JJ, Bontcheva-Diaz VD, Cox BF, DeWeese TL, Dillehay LE, Ferguson DC, Ghoreishi-Haack NS, Grimm DR, Guan R, Han EK, Holley-Shanks RR, Hristov B, Idler KB, Jarvis K, Johnson EF, Kleinberg LR, Klinghofer V, Lasko LM, Liu X, Marsh KC, McGonigal TP, Meulbroek JA, Olson AM, Palma JP, Rodriguez LE, Shi Y, Stavropoulos JA, Tsurutani AC, Zhu GD, Rosenberg SH, Giranda VL, Frost DJ (2007) ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin Cancer Res 13 (9):2728–2737. doi: 10.1158/1078-0432.CCR-06-3039 [DOI] [PubMed] [Google Scholar]
- 9.Davar D, Beumer JH, Hamieh L, Tawbi H (2012) Role of PARP inhibitors in cancer biology and therapy. Current medicinal chemistry 19 (23):3907–3921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kummar S, Kinders R, Gutierrez ME, Rubinstein L, Parchment RE, Phillips LR, Ji J, Monks A, Low JA, Chen A, Murgo AJ, Collins J, Steinberg SM, Eliopoulos H, Giranda VL, Gordon G, Helman L, Wiltrout R, Tomaszewski JE, Doroshow JH (2009) Phase 0 clinical trial of the poly (ADP-ribose) polymerase inhibitor ABT-888 in patients with advanced malignancies. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 27 (16):2705–2711. doi: 10.1200/JCO.2008.19.7681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Puhalla S, Beumer JH, Pahuja S, Appleman LJ, Tawbi HA, Stoller RG, Lee JJ, Lin Y, Kiesel B, Yu J, Tan AR, Belani CP, Chew HK, Garcia AA, Morgan R, Giranda VL, Shepherd SP, Chen AP, Chu E Final Results of a Phase 1 Study of Chronically-Dosed, Single-Agent Veliparib (ABT-888) in Patients with Either BRCA 1/2 –Mutated Cancer (BRCA+), Platinum-Refractory Ovarian or Basal-Like Breast Cancer (BRCA-wt). In, 2014. vol 5S. Proceedings of the American Society of Clinical Oncology (Supplement to Journal of Clinical Oncology), [Google Scholar]
- 12.Euhus DM, Smith KC, Robinson L, Stucky A, Olopade OI, Cummings S, Garber JE, Chittenden A, Mills GB, Rieger P, Esserman L, Crawford B, Hughes KS, Roche CA, Ganz PA, Seldon J, Fabian CJ, Klemp J, Tomlinson G (2002) Pretest prediction of BRCA1 or BRCA2 mutation by risk counselors and the computer model BRCAPRO. Journal of the National Cancer Institute 94 (11):844–851 [DOI] [PubMed] [Google Scholar]
- 13.Storer BE (1989) Design and analysis of phase I clinical trials. Biometrics 45 (3):925–937 [PubMed] [Google Scholar]
- 14.Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. Journal of the National Cancer Institute 92 (3):205–216 [DOI] [PubMed] [Google Scholar]
- 15.Parise RA, Shawaqfeh M, Egorin MJ, Beumer JH (2008) Liquid chromatography-mass spectrometric assay for the quantitation in human plasma of ABT-888, an orally available, small molecule inhibitor of poly(ADP-ribose) polymerase. Journal of chromatography B, Analytical technologies in the biomedical and life sciences 872 (1–2):141–147. doi: 10.1016/j.jchromb.2008.07.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Norquist B, Wurz KA, Pennil CC, Garcia R, Gross J, Sakai W, Karlan BY, Taniguchi T, Swisher EM (2011) Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 29 (22):3008–3015. doi: 10.1200/JCO.2010.34.2980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Appleman LJ, Beumer JH, Jiang Y, Lin Y, Ding F, Puhalla S, Swartz L, Owonikoko TK, Donald Harvey R, Stoller R, Petro DP, Tawbi HA, Argiris A, Strychor S, Pouquet M, Kiesel B, Chen AP, Gandara D, Belani CP, Chu E, Ramalingam SS (2019) Phase 1 study of veliparib (ABT-888), a poly (ADP-ribose) polymerase inhibitor, with carboplatin and paclitaxel in advanced solid malignancies. Cancer chemotherapy and pharmacology 84 (6):1289–1301. doi: 10.1007/s00280-019-03960-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Niu J, Scheuerell C, Mehrotra S, Karan S, Puhalla S, Kiesel BF, Ji J, Chu E, Gopalakrishnan M, Ivaturi V, Gobburu J, Beumer JH (2017) Parent-Metabolite Pharmacokinetic Modeling and Pharmacodynamics of Veliparib (ABT-888), a PARP Inhibitor, in Patients With BRCA 1/2-Mutated Cancer or PARP-Sensitive Tumor Types. Journal of clinical pharmacology 57 (8):977–987. doi: 10.1002/jcph.892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Villalona-Calero MA, Duan W, Zhao W, Shilo K, Schaaf LJ, Thurmond J, Westman JA, Marshall J, Xiaobai L, Ji J, Rose J, Lustberg M, Bekaii-Saab T, Chen A, Timmers C (2016) Veliparib Alone or in Combination with Mitomycin C in Patients with Solid Tumors With Functional Deficiency in Homologous Recombination Repair. Journal of the National Cancer Institute 108 (7). doi: 10.1093/jnci/djv437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cancer Genome Atlas N (2012) Comprehensive molecular portraits of human breast tumours. Nature 490 (7418):61–70. doi: 10.1038/nature11412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin KK, Harrell MI, Oza AM, Oaknin A, Ray-Coquard I, Tinker AV, Helman E, Radke MR, Say C, Vo LT, Mann E, Isaacson JD, Maloney L, O’Malley DM, Chambers SK, Kaufmann SH, Scott CL, Konecny GE, Coleman RL, Sun JX, Giordano H, Brenton JD, Harding TC, McNeish IA, Swisher EM (2019) BRCA Reversion Mutations in Circulating Tumor DNA Predict Primary and Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer discovery 9 (2):210–219. doi: 10.1158/2159-8290.CD-18-0715 [DOI] [PubMed] [Google Scholar]
- 22.Patel AG, Flatten KS, Schneider PA, Dai NT, McDonald JS, Poirier GG, Kaufmann SH (2012) Enhanced killing of cancer cells by poly(ADP-ribose) polymerase inhibitors and topoisomerase I inhibitors reflects poisoning of both enzymes. The Journal of biological chemistry 287 (6):4198–4210. doi: 10.1074/jbc.M111.296475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hurley RM, Wahner Hendrickson AE, Visscher DW, Ansell P, Harrell MI, Wagner JM, Negron V, Goergen KM, Maurer MJ, Oberg AL, Meng XW, Flatten KS, De Jonge MJA, Van Herpen CD, Gietema JA, Koornstra RHT, Jager A, den Hollander MW, Dudley M, Shepherd SP, Swisher EM, Kaufmann SH (2019) 53BP1 as a potential predictor of response in PARP inhibitor-treated homologous recombination-deficient ovarian cancer. Gynecologic oncology 153 (1):127–134. doi: 10.1016/j.ygyno.2019.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chan N, Pires IM, Bencokova Z, Coackley C, Luoto KR, Bhogal N, Lakshman M, Gottipati P, Oliver FJ, Helleday T, Hammond EM, Bristow RG (2010) Contextual synthetic lethality of cancer cell kill based on the tumor microenvironment. Cancer research 70 (20):8045–8054. doi: 10.1158/0008-5472.CAN-10-2352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.LaFargue CJ, Dal Molin GZ, Sood AK, Coleman RL (2019) Exploring and comparing adverse events between PARP inhibitors. The lancet oncology 20 (1):e15–e28. doi: 10.1016/S1470-2045(18)30786-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mirza MR, Monk BJ, Herrstedt J, Oza AM, Mahner S, Redondo A, Fabbro M, Ledermann JA, Lorusso D, Vergote I, Ben-Baruch NE, Marth C, Madry R, Christensen RD, Berek JS, Dorum A, Tinker AV, du Bois A, Gonzalez-Martin A, Follana P, Benigno B, Rosenberg P, Gilbert L, Rimel BJ, Buscema J, Balser JP, Agarwal S, Matulonis UA, Investigators E-ON (2016) Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. The New England journal of medicine 375 (22):2154–2164. doi: 10.1056/NEJMoa1611310 [DOI] [PubMed] [Google Scholar]
- 27.Coleman RL, Oza AM, Lorusso D, Aghajanian C, Oaknin A, Dean A, Colombo N, Weberpals JI, Clamp A, Scambia G, Leary A, Holloway RW, Gancedo MA, Fong PC, Goh JC, O’Malley DM, Armstrong DK, Garcia-Donas J, Swisher EM, Floquet A, Konecny GE, McNeish IA, Scott CL, Cameron T, Maloney L, Isaacson J, Goble S, Grace C, Harding TC, Raponi M, Sun J, Lin KK, Giordano H, Ledermann JA, investigators A (2017) Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 390 (10106):1949–1961. doi: 10.1016/S0140-6736(17)32440-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Balmana J, Tung NM, Isakoff SJ, Grana B, Ryan PD, Saura C, Lowe ES, Frewer P, Winer E, Baselga J, Garber JE (2014) Phase I trial of olaparib in combination with cisplatin for the treatment of patients with advanced breast, ovarian and other solid tumors. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO 25 (8):1656–1663. doi: 10.1093/annonc/mdu187 [DOI] [PubMed] [Google Scholar]
- 29.Dent RA, Lindeman GJ, Clemons M, Wildiers H, Chan A, McCarthy NJ, Singer CF, Lowe ES, Watkins CL, Carmichael J (2013) Phase I trial of the oral PARP inhibitor olaparib in combination with paclitaxel for first- or second-line treatment of patients with metastatic triple-negative breast cancer. Breast Cancer Res 15 (5):R88. doi: 10.1186/bcr3484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oza AM, Cibula D, Benzaquen AO, Poole C, Mathijssen RH, Sonke GS, Colombo N, Spacek J, Vuylsteke P, Hirte H, Mahner S, Plante M, Schmalfeldt B, Mackay H, Rowbottom J, Lowe ES, Dougherty B, Barrett JC, Friedlander M (2015) Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: a randomised phase 2 trial. The lancet oncology 16 (1):87–97. doi: 10.1016/S1470-2045(14)71135-0 [DOI] [PubMed] [Google Scholar]
- 31.van der Noll R, Jager A, Ang JE, Marchetti S, Mergui-Roelvink MWJ, Lolkema MP, de Jonge MJA, van der Biessen DA, Brunetto AT, Arkenau HT, Tchakov I, Beijnen JH, de Bono JS, Schellens JHM (2020) Phase I study of continuous olaparib capsule dosing in combination with carboplatin and/or paclitaxel (Part 1). Investigational new drugs 38 (4):1117–1128. doi: 10.1007/s10637-019-00856-7 [DOI] [PubMed] [Google Scholar]
- 32.Gray HJ, Bell-McGuinn K, Fleming GF, Cristea M, Xiong H, Sullivan D, Luo Y, McKee MD, Munasinghe W, Martin LP (2018) Phase I combination study of the PARP inhibitor veliparib plus carboplatin and gemcitabine in patients with advanced ovarian cancer and other solid malignancies. Gynecologic oncology 148 (3):507–514. doi: 10.1016/j.ygyno.2017.12.029 [DOI] [PubMed] [Google Scholar]
- 33.Atrafi F, Groen HJM, Byers LA, Garralda E, Lolkema MP, Sangha RS, Viteri S, Chae YK, Camidge DR, Gabrail NY, Hu B, Tian T, Nuthalapati S, Hoening E, He L, Komarnitsky P, Calles A (2019) A Phase I Dose-Escalation Study of Veliparib Combined with Carboplatin and Etoposide in Patients with Extensive-Stage Small Cell Lung Cancer and Other Solid Tumors. Clin Cancer Res 25 (2):496–505. doi: 10.1158/1078-0432.CCR-18-2014 [DOI] [PubMed] [Google Scholar]
- 34.Rodler ET, Kurland BF, Griffin M, Gralow JR, Porter P, Yeh RF, Gadi VK, Guenthoer J, Beumer JH, Korde L, Strychor S, Kiesel BF, Linden HM, Thompson JA, Swisher E, Chai X, Shepherd S, Giranda V, Specht JM (2016) Phase I Study of Veliparib (ABT-888) Combined with Cisplatin and Vinorelbine in Advanced Triple-Negative Breast Cancer and/or BRCA Mutation-Associated Breast Cancer. Clin Cancer Res 22 (12):2855–2864. doi: 10.1158/1078-0432.CCR-15-2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharma P, Rodler E, Barlow WE, Gralow J, Huggins-Puhalla SL, Anders CK, Goldstein LJ, Brown-Glaberman UA, Huynh T-T, Szyarto CS, Godwin AK, Pathak HB, Swisher EM, Radke MR, Timms KM, Lew DL, Miao J, Pusztai L, Hayes DF, Hortobagyi GN (2020) Results of a phase II randomized trial of cisplatin +/− veliparib in metastatic triple-negative breast cancer (TNBC) and/or germline BRCA-associated breast cancer (SWOG S1416). JCO 38 (15_suppl):1001–1001. doi: 10.1200/JCO.2020.38.15_suppl.1001 [DOI] [Google Scholar]
- 36.Coleman RL, Fleming GF, Brady MF, Swisher EM, Steffensen KD, Friedlander M, Okamoto A, Moore KN, Efrat Ben-Baruch N, Werner TL, Cloven NG, Oaknin A, DiSilvestro PA, Morgan MA, Nam JH, Leath CA 3rd, Nicum S, Hagemann AR, Littell RD, Cella D, Baron-Hay S, Garcia-Donas J, Mizuno M, Bell-McGuinn K, Sullivan DM, Bach BA, Bhattacharya S, Ratajczak CK, Ansell PJ, Dinh MH, Aghajanian C, Bookman MA (2019) Veliparib with First-Line Chemotherapy and as Maintenance Therapy in Ovarian Cancer. The New England journal of medicine 381 (25):2403–2415. doi: 10.1056/NEJMoa1909707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dieras V, Han HS, Kaufman B, Wildiers H, Friedlander M, Ayoub JP, Puhalla SL, Bondarenko I, Campone M, Jakobsen EH, Jalving M, Oprean C, Palacova M, Park YH, Shparyk Y, Yanez E, Khandelwal N, Kundu MG, Dudley M, Ratajczak CK, Maag D, Arun BK (2020) Veliparib with carboplatin and paclitaxel in BRCA-mutated advanced breast cancer (BROCADE3): a randomised, double-blind, placebo-controlled, phase 3 trial. The lancet oncology 21 (10):1269–1282. doi: 10.1016/S1470-2045(20)30447-2 [DOI] [PubMed] [Google Scholar]
- 38.Lee JM, Hays JL, Annunziata CM, Noonan AM, Minasian L, Zujewski JA, Yu M, Gordon N, Ji J, Sissung TM, Figg WD, Azad N, Wood BJ, Doroshow J, Kohn EC (2014) Phase I/Ib study of olaparib and carboplatin in BRCA1 or BRCA2 mutation-associated breast or ovarian cancer with biomarker analyses. Journal of the National Cancer Institute 106 (6):dju089. doi: 10.1093/jnci/dju089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lampert EJ, Hays JL, Kohn EC, Annunziata CM, Minasian L, Yu M, Gordon N, Sissung TM, Chiou VL, Figg WD, Houston N, Lee JM (2019) Phase I/Ib study of olaparib and carboplatin in heavily pretreated recurrent high-grade serous ovarian cancer at low genetic risk. Oncotarget 10 (30):2855–2868. doi: 10.18632/oncotarget.26869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pahuja S, Beumer JH, Appleman LJ, Tawbi H, Stoller R, Lee JJ, Lin Y, Ding F, Yu J, Belani C, Chen A, Giranda V, Shepherd SP, Chu E, Puhalla SA phase I study of veliparib (ABT-888) in combination with weekly carboplatin and paclitaxel in advanced solid malignancies and enriched for triple negative breast cancer (TNBC). In: Annual Meeting American Society of Clinical Oncology, Chicago, 2015. vol 152623. Proceedings of the American Society of Clinical Oncology (Supplement to Journal of Clinical Oncology), [Google Scholar]
- 41.Somlo G, Frankel PH, Arun BK, Ma CX, Garcia AA, Cigler T, Cream LV, Harvey HA, Sparano JA, Nanda R, Chew HK, Moynihan TJ, Vahdat LT, Goetz MP, Beumer JH, Hurria A, Mortimer J, Piekarz R, Sand S, Herzog J, Van Tongeren LR, Ferry-Galow KV, Chen AP, Ruel C, Newman EM, Gandara DR, Weitzel JN (2017) Efficacy of the PARP Inhibitor Veliparib with Carboplatin or as a Single Agent in Patients with Germline BRCA1- or BRCA2-Associated Metastatic Breast Cancer: California Cancer Consortium Trial NCT01149083. Clin Cancer Res 23 (15):4066–4076. doi: 10.1158/1078-0432.CCR-16-2714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim G, Ison G, McKee AE, Zhang H, Tang S, Gwise T, Sridhara R, Lee E, Tzou A, Philip R, Chiu HJ, Ricks TK, Palmby T, Russell AM, Ladouceur G, Pfuma E, Li H, Zhao L, Liu Q, Venugopal R, Ibrahim A, Pazdur R (2015) FDA Approval Summary: Olaparib Monotherapy in Patients with Deleterious Germline BRCA-Mutated Advanced Ovarian Cancer Treated with Three or More Lines of Chemotherapy. Clin Cancer Res 21 (19):4257–4261. doi: 10.1158/1078-0432.CCR-15-0887 [DOI] [PubMed] [Google Scholar]
- 43.Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K, Hirte H, Huntsman D, Clemons M, Gilks B, Yerushalmi R, Macpherson E, Carmichael J, Oza A (2011) Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. The lancet oncology 12 (9):852–861. doi: 10.1016/S1470-2045(11)70214-5 [DOI] [PubMed] [Google Scholar]
- 44.Sandhu SK, Schelman WR, Wilding G, Moreno V, Baird RD, Miranda S, Hylands L, Riisnaes R, Forster M, Omlin A, Kreischer N, Thway K, Gevensleben H, Sun L, Loughney J, Chatterjee M, Toniatti C, Carpenter CL, Iannone R, Kaye SB, de Bono JS, Wenham RM (2013) The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial. The lancet oncology 14 (9):882–892. doi: 10.1016/S1470-2045(13)70240-7 [DOI] [PubMed] [Google Scholar]
- 45.Kristeleit R, Shapiro GI, Burris HA, Oza AM, LoRusso P, Patel MR, Domchek SM, Balmana J, Drew Y, Chen LM, Safra T, Montes A, Giordano H, Maloney L, Goble S, Isaacson J, Xiao J, Borrow J, Rolfe L, Shapira-Frommer R (2017) A Phase I-II Study of the Oral PARP Inhibitor Rucaparib in Patients with Germline BRCA1/2-Mutated Ovarian Carcinoma or Other Solid Tumors. Clin Cancer Res. doi: 10.1158/1078-0432.CCR-16-2796 [DOI] [PubMed] [Google Scholar]
- 46.Coleman RL, Sill MW, Bell-McGuinn K, Aghajanian C, Gray HJ, Tewari KS, Rubin SC, Rutherford TJ, Chan JK, Chen A, Swisher EM (2015) A phase II evaluation of the potent, highly selective PARP inhibitor veliparib in the treatment of persistent or recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer in patients who carry a germline BRCA1 or BRCA2 mutation - An NRG Oncology/Gynecologic Oncology Group study. Gynecologic oncology 137 (3):386–391. doi: 10.1016/j.ygyno.2015.03.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, Friedlander M, Arun B, Loman N, Schmutzler RK, Wardley A, Mitchell G, Earl H, Wickens M, Carmichael J (2010) Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet 376 (9737):235–244. doi: 10.1016/S0140-6736(10)60892-6 [DOI] [PubMed] [Google Scholar]
- 48.Salem AH, Giranda VL, Mostafa NM (2014) Population pharmacokinetic modeling of veliparib (ABT-888) in patients with non-hematologic malignancies. Clinical pharmacokinetics 53 (5):479–488. doi: 10.1007/s40262-013-0130-1 [DOI] [PubMed] [Google Scholar]
- 49.Kummar S, Ji J, Morgan R, Lenz HJ, Puhalla SL, Belani CP, Gandara DR, Allen D, Kiesel B, Beumer JH, Newman EM, Rubinstein L, Chen A, Zhang Y, Wang L, Kinders RJ, Parchment RE, Tomaszewski JE, Doroshow JH (2012) A phase I study of veliparib in combination with metronomic cyclophosphamide in adults with refractory solid tumors and lymphomas. Clin Cancer Res 18 (6):1726–1734. doi: 10.1158/1078-0432.CCR-11-2821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gojo I, Beumer JH, Pratz KW, McDevitt MA, Baer MR, Blackford AL, Smith BD, Gore SD, Carraway HE, Showel MM, Levis MJ, Dezern AE, Gladstone DE, Ji JJ, Wang L, Kinders RJ, Pouquet M, Ali-Walbi I, Rudek MA, Poh W, Herman JG, Karnitz LM, Kaufmann SH, Chen A, Karp JE (2017) A Phase 1 Study of the PARP Inhibitor Veliparib in Combination with Temozolomide in Acute Myeloid Leukemia. Clin Cancer Res 23 (3):697–706. doi: 10.1158/1078-0432.CCR-16-0984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Soumerai JD, Zelenetz AD, Moskowitz CH, Palomba ML, Hamlin PA Jr., Noy A, Straus DJ, Moskowitz AJ, Younes A, Matasar MJ, Horwitz SM, Portlock CS, Konner JA, Gounder MM, Hyman DM, Voss MH, Fury MG, Gajria D, Carvajal RD, Ho AL, Beumer JH, Kiesel B, Zhang Z, Chen A, Little RF, Jarjies C, Dang TO, France F, Mishra N, Gerecitano JF (2017) The PARP Inhibitor Veliparib Can Be Safely Added to Bendamustine and Rituximab and Has Preliminary Evidence of Activity in B-Cell Lymphoma. Clin Cancer Res 23 (15):4119–4126. doi: 10.1158/1078-0432.CCR-16-3068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stoller R, Schmitz JC, Ding F, Puhalla S, Belani CP, Appleman L, Lin Y, Jiang Y, Almokadem S, Petro D, Holleran J, Kiesel BF, Ken Czambel R, Carneiro BA, Kontopodis E, Hershberger PA, Rachid M, Chen A, Chu E, Beumer JH (2017) Phase I study of veliparib in combination with gemcitabine. Cancer chemotherapy and pharmacology 80 (3):631–643. doi: 10.1007/s00280-017-3409-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tan AR, Chan N, Kiesel BF, Stein MN, Moss RA, Malhotra J, Aisner J, Shah M, Gounder M, Lin H, Kane MP, Lin Y, Ji J, Chen A, Beumer JH, Mehnert JM (2022) A phase I study of veliparib with cyclophosphamide and veliparib combined with doxorubicin and cyclophosphamide in advanced malignancies. Cancer chemotherapy and pharmacology 89 (1):49–58. doi: 10.1007/s00280-021-04350-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pothuri B, Brodsky AL, Sparano JA, Blank SV, Kim M, Hershman DL, Tiersten A, Kiesel BF, Beumer JH, Liebes L, Muggia F (2020) Phase I and pharmacokinetic study of veliparib, a PARP inhibitor, and pegylated liposomal doxorubicin (PLD) in recurrent gynecologic cancer and triple negative breast cancer with long-term follow-up. Cancer chemotherapy and pharmacology 85 (4):741–751. doi: 10.1007/s00280-020-04030-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smith BP, Vandenhende FR, DeSante KA, Farid NA, Welch PA, Callaghan JT, Forgue ST (2000) Confidence interval criteria for assessment of dose proportionality. Pharmaceutical research 17 (10):1278–1283 [DOI] [PubMed] [Google Scholar]
- 56.Hummel J, McKendrick S, Brindley C, French R (2009) Exploratory assessment of dose proportionality: review of current approaches and proposal for a practical criterion. Pharmaceutical statistics 8 (1):38–49. doi: 10.1002/pst.326 [DOI] [PubMed] [Google Scholar]
- 57.Ferry-Galow KV, Datta V, Makhlouf HR, Wright J, Wood BJ, Levy E, Pisano ED, Tam AL, Lee SI, Mahmood U, Rubinstein LV, Doroshow JH, Chen AP (2018) What Can Be Done to Improve Research Biopsy Quality in Oncology Clinical Trials? Journal of oncology practice / American Society of Clinical Oncology:JOP 1800092. doi: 10.1200/JOP.18.00092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Noordermeer SM, Adam S, Setiaputra D, Barazas M, Pettitt SJ, Ling AK, Olivieri M, Alvarez-Quilon A, Moatti N, Zimmermann M, Annunziato S, Krastev DB, Song F, Brandsma I, Frankum J, Brough R, Sherker A, Landry S, Szilard RK, Munro MM, McEwan A, Goullet de Rugy T, Lin ZY, Hart T, Moffat J, Gingras AC, Martin A, van Attikum H, Jonkers J, Lord CJ, Rottenberg S, Durocher D (2018) The shieldin complex mediates 53BP1-dependent DNA repair. Nature 560 (7716):117–121. doi: 10.1038/s41586-018-0340-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Delaney C, Garg SK, Yung R (2015) Analysis of DNA Methylation by Pyrosequencing. Methods in molecular biology 1343:249–264. doi: 10.1007/978-1-4939-2963-4_19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Doyle B, O’Riain C, Appleton K (2011) Pyrosequencing of DNA extracted from formalin-fixed paraffin-embedded tissue. Methods in molecular biology 724:181–190. doi: 10.1007/978-1-61779-055-3_12 [DOI] [PubMed] [Google Scholar]
- 61.Esteller M, Silva JM, Dominguez G, Bonilla F, Matias-Guiu X, Lerma E, Bussaglia E, Prat J, Harkes IC, Repasky EA, Gabrielson E, Schutte M, Baylin SB, Herman JG (2000) Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. Journal of the National Cancer Institute 92 (7):564–569 [DOI] [PubMed] [Google Scholar]
- 62.Duan W, Gao L, Zhao W, Leon M, Sadee W, Webb A, Resnick K, Wu X, Ramaswamy B, Cohn DE, Shapiro C, Andreassen PR, Otterson GA, Villalona-Calero MA (2013) Assessment of FANCD2 nuclear foci formation in paraffin-embedded tumors: a potential patient-enrichment strategy for treatment with DNA interstrand crosslinking agents. Translational research : the journal of laboratory and clinical medicine 161 (3):156–164. doi: 10.1016/j.trsl.2012.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.